










")



 biology
biologySimilar presentations:

")



")
")

")

Пути обмена отдельных аминокислот
1. ПУТИ ОБМЕНА ОТДЕЛЬНЫХ АМИНОКИСЛОТ
2.
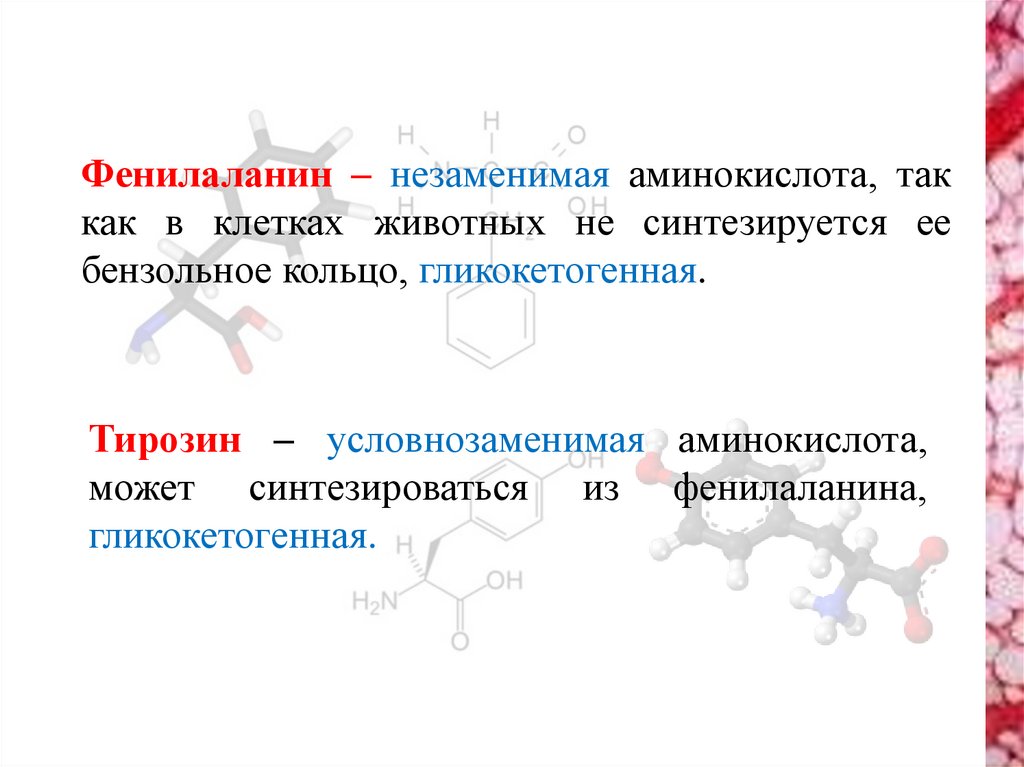
Фенилаланин – незаменимая аминокислота, таккак в клетках животных не синтезируется ее
бензольное кольцо, гликокетогенная.
Тирозин – условнозаменимая аминокислота,
может синтезироваться из фенилаланина,
гликокетогенная.
3. Биологическая роль фенилаланина и тирозина
КатехоламиныФЕНИЛАЛАНИН
ТИРОЗИН
Белки
Дофамин
Норадреналин
Адреналин
Меланины
Йодтиронины
Нервная ткань
Надпочечники
Кожа
Волосы
Радужная
оболочка глаз
Щитовидная
железа
ОПК
Ацетоацетат
Глюкоза
Печень
4.
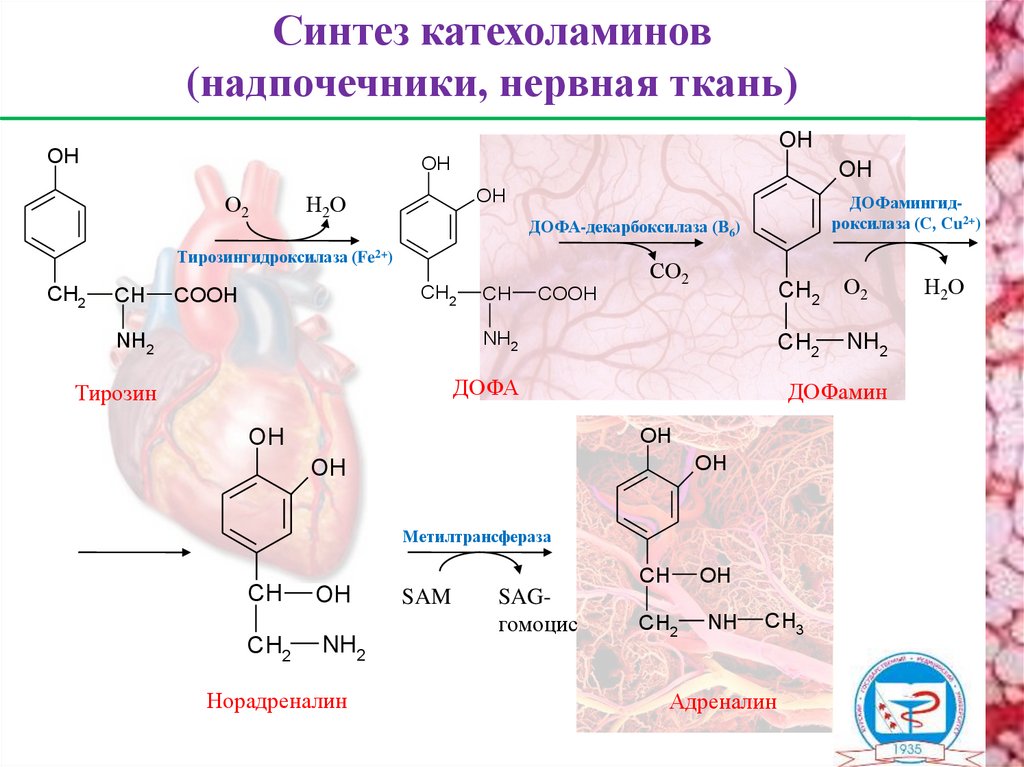
Синтез катехоламинов(надпочечники, нервная ткань)
OH
OH
OH
О2
OH
OH
Н2О
ДОФА-декарбоксилаза (В6)
Тирозингидроксилаза (Fe2+)
CH2
CH
CH2
COOH
ДОФамингидроксилаза (С, Сu2+)
CH
NH2
NH2
Тирозин
ДОФА
COOH
СО2
CH2
О2
CH2
NH2
ДОФамин
OH
OH
OH
OH
Метилтрансфераза
CH
CH2
OH
NH2
Норадреналин
CH
SAM
SAGгомоцис
CH2
OH
NH
CH3
Адреналин
Н2О
5.
Значение катехоламиновДОФамин – нейромедиатор среднего отдела мозга.
Норадреналин – тормозный медиатор симпатической нервной системы и разных отделов головного мозга, может выполнять функцию возбуждающего медиатора в гипоталямусе, участвует в регуляции гемодинамики сердечно-сосудистой системы.
Адреналин – гормон интенсивной физической
работы, синтезируется при стрессе, регулирует
основной обмен, усиливает сокращение сердечной
мышцы.
6.
Нарушение обмена ДОФаминаБолезнь Паркинсона – развивается при гипосекреции дофамина в черной субстанции мозга (в среднем
отделе мозга). Частота 1:200 среди людей старше 60 лет.
Дефект ферментов тирозингидроксилазы или ДОФАдекарбоксилазы. Основные симптомы заболевания :
акенизия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание).
Гиперсекреция дофамина в височной доле мозга
обнаруживается при шизофрении.
ДОФА-декарбоксилаза
тирозингидроксилаза
Тир
ДОФА
О2
Н2 О
ДОФамин
СО2
7.
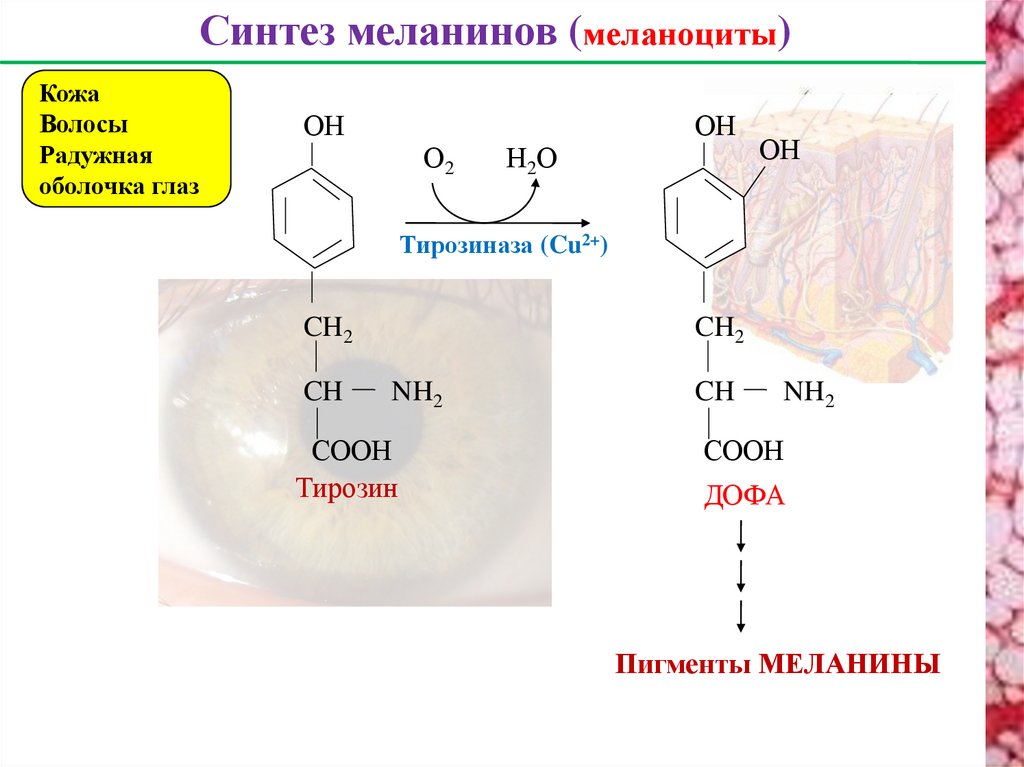
Синтез меланинов (меланоциты)Кожа
Волосы
Радужная
оболочка глаз
ОH
ОH
О2
Н2О
ОH
Тирозиназа (Cu2+)
CH2
CH
CH2
NH2
CООH
Тирозин
CH
NH2
CООH
ДОФА
Пигменты МЕЛАНИНЫ
8.
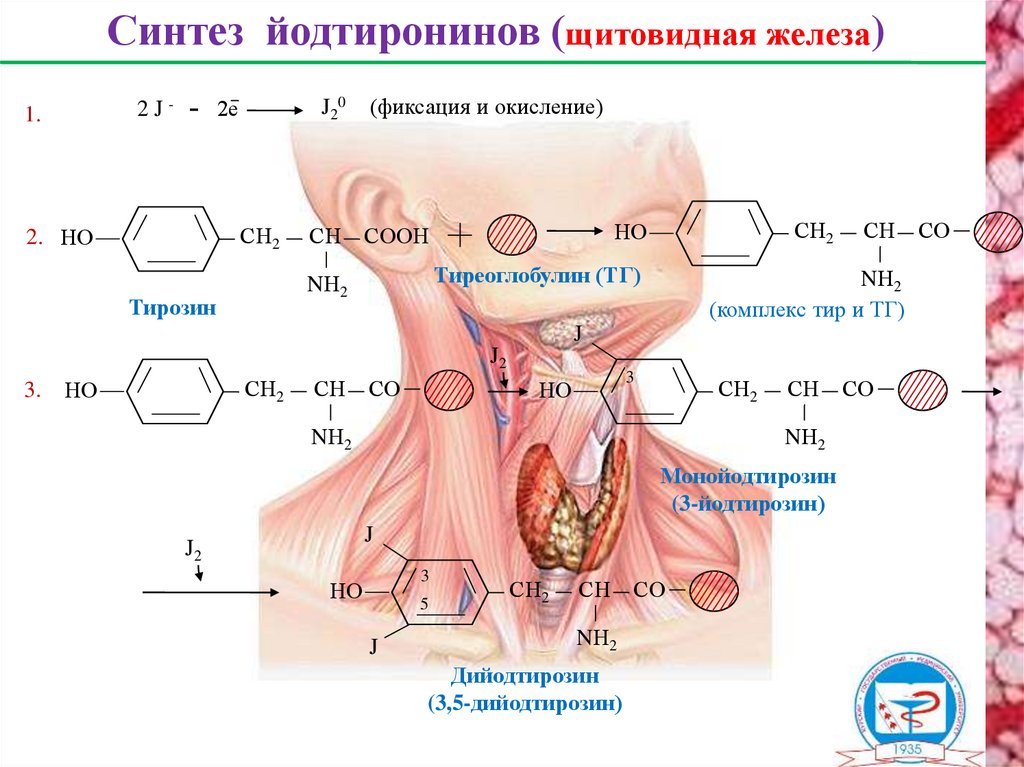
Синтез йодтиронинов (щитовидная железа)2е
2J-
1.
J2 0
СН2
2. НО
СН
(фиксация и окисление)
Тиреоглобулин (ТГ)
NН2
Тирозин
СН2
НО
СООН
СН
NН2
(комплекс тир и ТГ)
J
J2
3.
СН2
НО
СН
СО
3
НО
СН2
NН2
СН
NН2
Монойодтирозин
(3-йодтирозин)
J
J2
3
НО
5
J
СН2
СН
NН2
Дийодтирозин
(3,5-дийодтирозин)
СО
СО
СО
9.
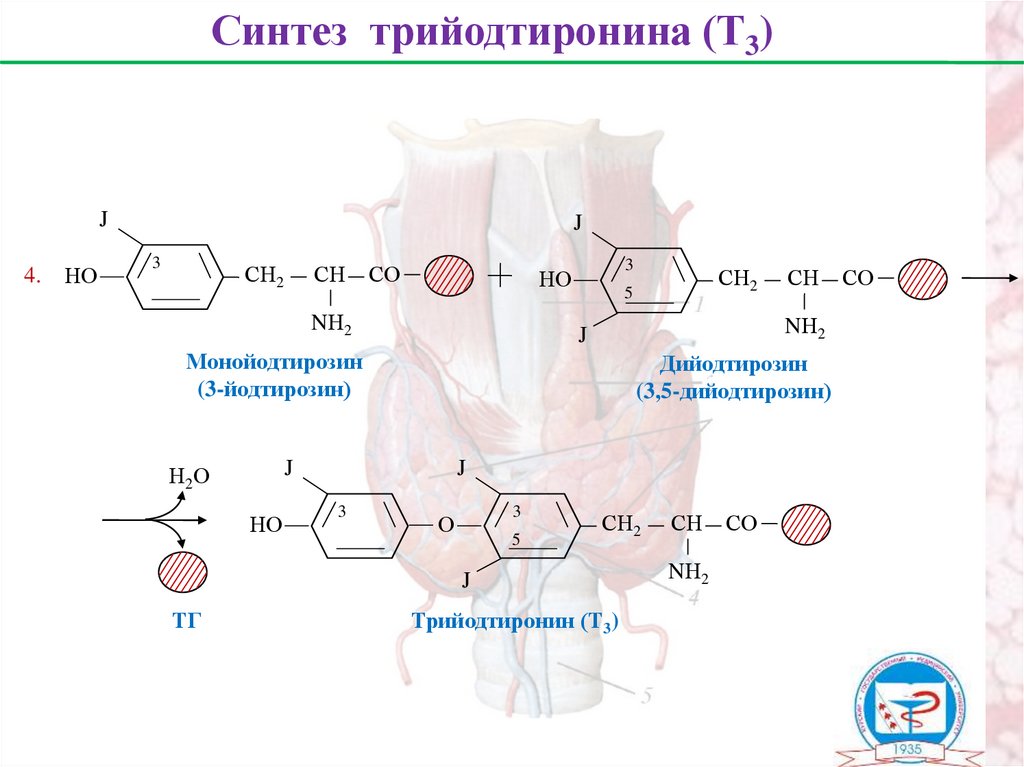
Синтез трийодтиронина (Т3)J
4.
НО
J
3
СН2
СН
СО
3
НО
NН2
Дийодтирозин
(3,5-дийодтирозин)
J
J
НО
3
3
О
5
СН2
J
ТГ
СН
NН2
J
Монойодтирозин
(3-йодтирозин)
Н2О
СН2
5
Трийодтиронин (Т3)
СН
NН2
СО
СО
10.
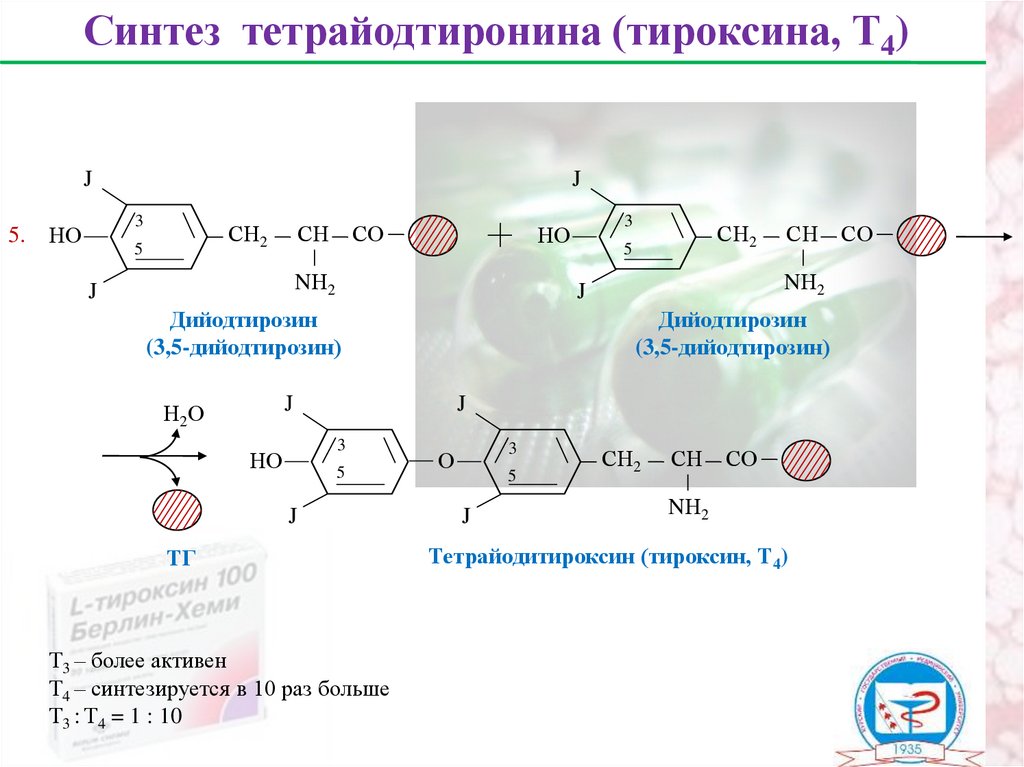
Синтез тетрайодтиронина (тироксина, Т4)J
5.
J
3
НО
СН2
5
СН
СО
NН2
J
СН
NН2
Дийодтирозин
(3,5-дийодтирозин)
J
J
3
НО
СН2
5
J
Дийодтирозин
(3,5-дийодтирозин)
Н2О
3
НО
5
J
ТГ
Т3 – более активен
Т4 – синтезируется в 10 раз больше
Т3 : Т4 = 1 : 10
3
О
5
J
СН2
СН
СО
NН2
Тетрайодитироксин (тироксин, Т4)
СО
11.
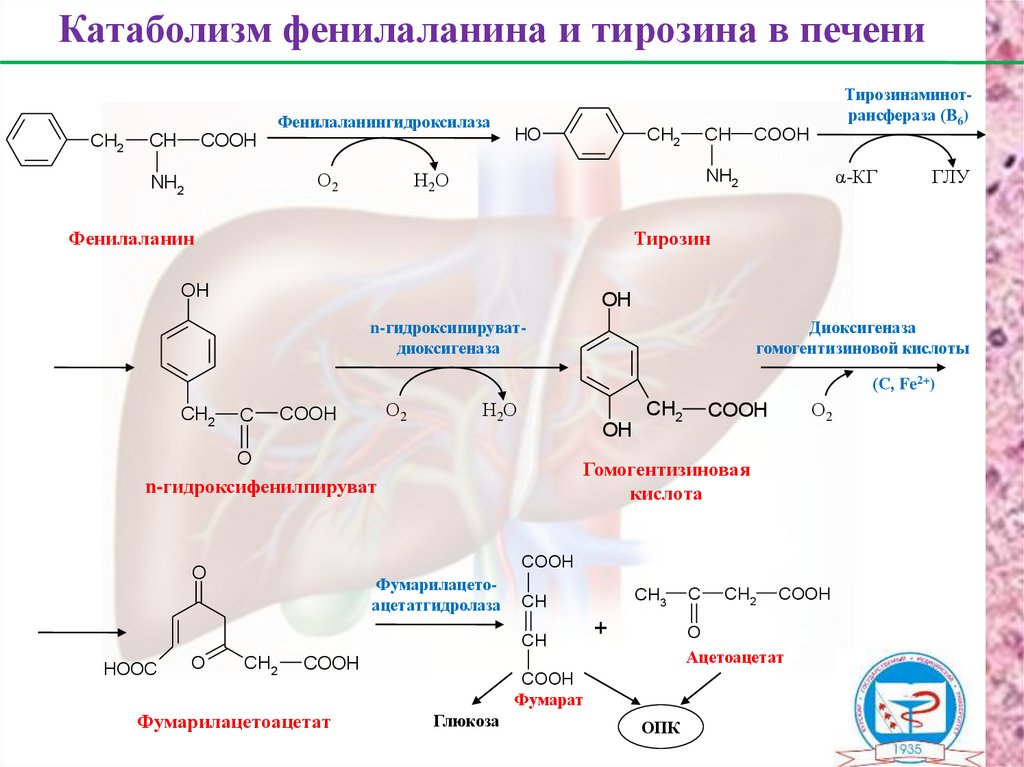
Катаболизм фенилаланина и тирозина в печениФенилаланингидроксилаза
CH2
CH
COOH
О2
NH2
Тирозинаминотрансфераза (В6)
HO
CH2
CH
Н2О
COOH
α-КГ
NH2
Фенилаланин
ГЛУ
Тирозин
OH
OH
Диоксигеназа
гомогентизиновой кислоты
n-гидроксипируватдиоксигеназа
(С, Fe2+)
CH2
C
О2
COOH
Н2О
OH
O
COOH
O
Фумарилацетоацетатгидролаза
CH2
COOH
Фумарилацетоацетат
CH3
CH
CH
O
О2
COOH
Гомогентизиновая
кислота
n-гидроксифенилпируват
HOOC
CH2
+
CH2
COOH
O
Ацетоацетат
COOH
Фумарат
Глюкоза
C
ОПК
12. Альтернативные пути катаболизма фенилаланина
α - КГФен
Глу
ПФ
NADH+H+
NAD+
СН2 – С – СООН
Фенилпируват
NAD+
H2O
NADH+H+
СО2
СН2 – СН – СООН
ОH
О
Фениллактат
Глн
СН2 – СООН
H2O
Фенилацетилглутамин
Кровь
Фенилацетат
Почки
13. Врожденные нарушения обмена ФЕН и ТИР
Белки (пищи и тканей)Фенилаланингидроксилаза
Фен
Фенилкетонурия
Фенилпируват
Фенилактат
Фенилацетат
Тирозиназа
(меланоциты)
Тир
Альбинизм
ДОФА
Тирозинемия II
Тирозинаминотрансфераза
Парагидроксифенилпируват
Семейный гипотиреоз (кретинизм)
n-гидрокТирозинемия III сифенилпируватдиоксигеназа
Гомогентизиновая к-та
Алкаптонурия
Диоксигеназа гомогенизированной кислоты
Фумарилацетоацетат
Тирозинемия I (тирозиноз)
Фумаровая
Ацетоацетат
Меланины
Фумарилацетоацетатгидролаза
Гормоны
щитовидной
железы
14.
Фенилкетонурия(пировиноградная олигофрения)
Фенилкетонурия – наследственное заболевание, наследуется по
аутосомно-рецессивному типу, частота 1:10 тыс. новорожденных.
дефект фермента фенилаланингидроксилазы.
В печени здоровых людей около 10% фенилаланина превращается в
фениллактат и фенилацетилглутамат. При ФКУ в крови и моче
повышается содержание метаболитов альтернативного пути:
фенилпирувата, фенилацетата, фениллактата, которые токсичны для
мозга.
Концентрация фенилаланина повышается в крови в 20-30 раз (в
норме 1,0-2,0 мг/дл), в моче –в 100-300 раз по сравнению с нормой
(30 мг/дл). Концентрация фенилпирувата и фениллактата в моче
достигает 300-600 мг/дл при полном отсутствии в норме.
Фенилацетат
Фенилаланингидроксилаза
Фенилпируват
Фениллактат
Фен
Тир
15.
Фенилкетонурия•Проявления ФКУ – нарушения умственного и физического развития,
судорожный синдром, нарушение пигментации. Больные не доживают до
30 лет. Большие концентрации фенилаланина ограничивают транспорт
тирозина и триптофана через гематоэнцефалический барьер и тормозят
синтез нейромедиаторов (дофамина, норадреналина, серотонина).
•Для выявления ФКУ разработана скрининг-программа (наличие простого
метода обнаружения, опасные последствия, частота не менее 1:20 тыс.,
есть способы предупреждения или лечения).
•Используют качественные и количественные методы обнаружения
патологических метаболитов в моче (фенилпируват, фениллактат),
определение фенилаланина в крови и моче.
•Лечение: содержание ребенка 10-12 месяцев на диете с малым
содержанием фен (не более 10-12 мг в сутки) с повышенным содержанием
тир. Прием глу, который быстро поступая в мозг в реакции
переаминирования переводит фенилпировиноградную кислоту в
фенилаланин.
16.
АльбинизмНаследуется по аутосомно-рецессивному типу,
частота 1:20 тыс. новорожденных.
Причина метаболического нарушения - врожденный дефект
тирозиназы, катализирующей превращение тирозина в
диоксифенилаланин в меланоцитах, что приводит к нарушению
синтеза пигментов меланинов.
Клинические проявления альбинизма – снижение до
отсутствия пигментации кожи, волос, снижение остроты
зрения, светобоязнь. Длительное пребывание таких больных на
солнце приводит к раку кожи.
Помощь – генетическая консультация.
Тирозиназа
( в меланоцитах)
Тир
ДОФА
Меланины
(кожа, волосы, радужная оболочка)
17.
Алкаптонурия («черная моча»)Наследуется по аутосомно-рецессивному типу, частота
встречаемости – 2-5 : 1 млн. новорожденных.
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. С мочой выделяется большое
количество гомогентизиновой кислоты (до 0,5 г/сут), которая
кислородом окисляется с образованием темных пигментов
алкаптонов. Кроме потемнения мочи, характерна пигментация
соединительной ткани (охроноз) и артрит.
Диоксигеназа гомогенизированной кислоты
Гомогентизиновая к-та
Фумарилацетоацетат
Фумарат
Ацетоацетат
18.
ТирозинемииНарушения катаболизма тирозина в печени приводит к
тирозинемии и тирозинурии. Различают 3 типа тирозинемии:
1) Тирозинемия типа 1 (тирозиноз). Причина – дефект
фермента фумарилацетоацетатгидролазы.
Клинические проявления у новорожденных – диарея,
рвота, задержка в развитии. Без лечения дети погибают в
возрасте 5-8 месяцев из-за развивающейся недостаточности
печени.
Для лечения используют диету с пониженным
содержанием тирозина и фенилаланина.
Фумарилацетоацетатгидролаза
Фумарилацетоацетат
Фумарат
Ацетоацетат
19.
Тирозинемии2) Тирозинемия типа 2 (Синдром Рихнера –Ханхорта).
Причиной является дефект фермента тирозинаминотрансферазы. Для заболевания характерны поражения глаз и кожи,
умеренная умственная отсталость, нарушения координация
движений.
Тирозинаминотрансфераза
ТИР
n-гидроксифенилпируват
3) Тирозинемия новорожденных (кратковременная). Причина
– дефект фермента п–гидроксифенилпируватдиоксигеназы. В
крови повышается концентрация п-гидроксифенилацетата,
тирозина и фенилаланина.
n-гидроксифенилпируватдиоксигеназа
n-гидроксифенилпируват
Гомогентизиновая к-та
При лечении назначают малобелковую диету и витамин С.
20. Биологическая роль триптофана (незаменимая, гликокетогенная)
ЭпифизБелок
(активные центры)
Мелатонин (гормон)
Триптофан
Серотонин
(медиатор)
Нервная ткань,
гладкая мускулатура, кишечник
Никотинамид
(витамин РР)
Ацетоацетат
Глюкоза
Печень
21.
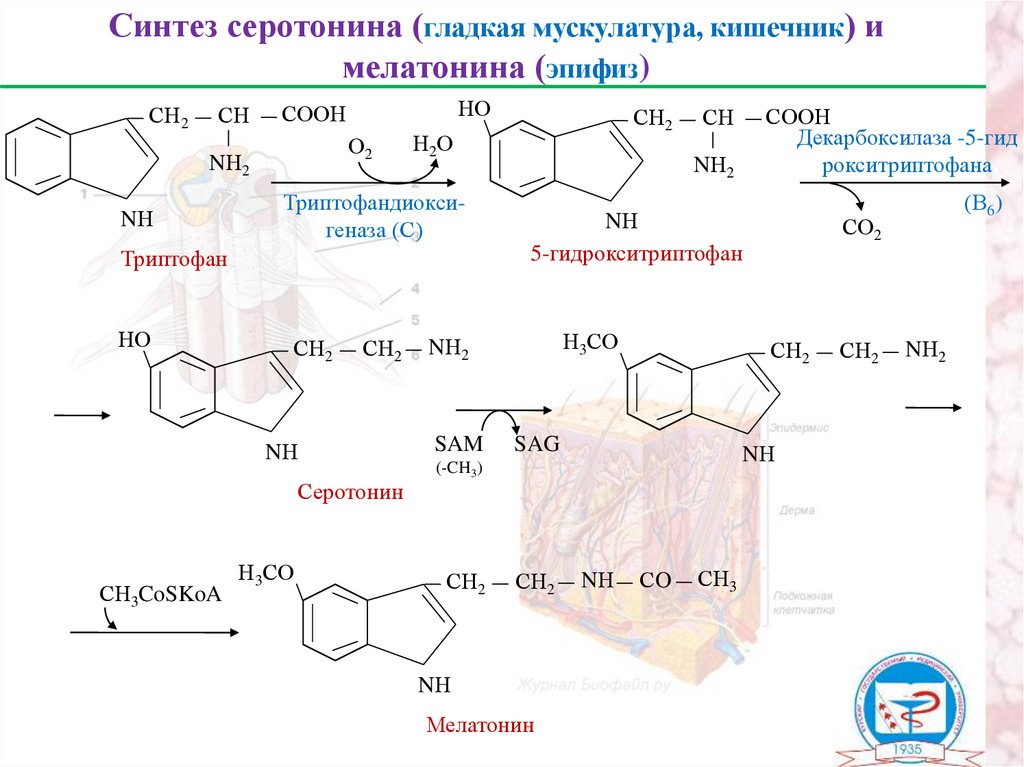
Синтез серотонина (гладкая мускулатура, кишечник) имелатонина (эпифиз)
СН2
СН
О2
NН2
NН
НО
СООН
Н2О
Триптофандиоксигеназа (С)
Триптофан
НО
СН2
СН2
NН
Серотонин
СН3CoSKoA
Н3СО
СН2
NН2
NН
5-гидрокситриптофан
Н3СО
NН2
SAM
SAG
СН2
NН
NН
Мелатонин
NН
СO
СН3
(В6)
СО2
СН2
(-CH3)
СН2
СООН
Декарбоксилаза -5-гид
рокситриптофана
СН
СН2
NН2
22.
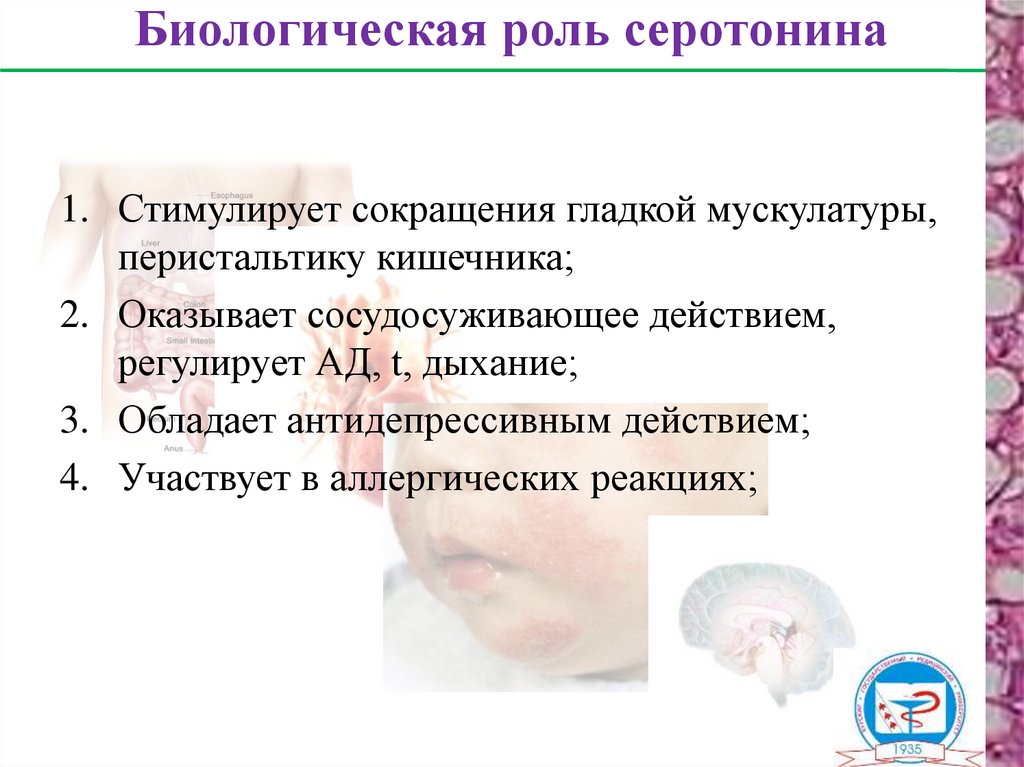
Биологическая роль серотонина1. Стимулирует сокращения гладкой мускулатуры,
перистальтику кишечника;
2. Оказывает сосудосуживающее действием,
регулирует АД, t, дыхание;
3. Обладает антидепрессивным действием;
4. Участвует в аллергических реакциях;
23. Биологическая роль серотонина
Синтез витамина РРСН2
СН
NН2
NН
Триптофан
СООН
Н2О
О2
НО
Триптофандиоксигеназа (С)
СН2
СН
СООН
СН
NН2
О
С
СН2
СООН
NН
5-гидрокситриптофан
АЛА
О
NН2
С
Кинуренин
NН2
N
Никотинамид
24. Синтез витамина РР
Врожденное нарушение обмена триптофана болезнь ХартнупаВозникает метаболический дефект связан с генетическим
дефектом
фермента
триптофандиоксигеназы
или
врожденным нарушением всасывания триптофана в
кишечнике и реабсорбции в почках.
Основными
клиническими
и
лабораторными
проявлениями
являются
пеллагроподобные
кожные
проявления (дерматит), диарея, задержка умственного
развития (дименция) (гиповитаминоз 3Д), психические
расстройства, аттаксия, гипераминоацидурия.
триптофандиоксигеназа
Три
5-гидрокситриптофан
25.
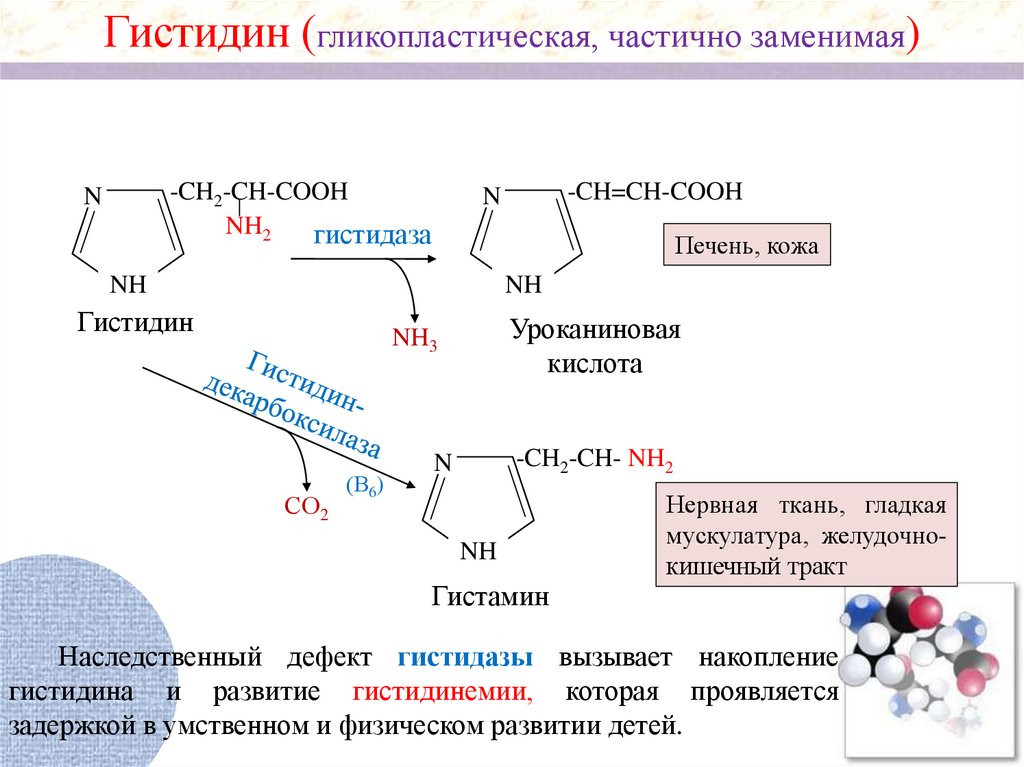
Гистидин (гликопластическая, частично заменимая)-CH2-CH-COOH
NH2
гистидаза
N
-CH=CH-COOH
N
Печень, кожа
NH
NH
Гистидин
Уроканиновая
кислота
NH3
СО2
(В6)
-CH2-CH- NH2
N
NH
Гистамин
Нервная ткань, гладкая
мускулатура, желудочнокишечный тракт
Наследственный дефект гистидазы вызывает накопление
гистидина и развитие гистидинемии, которая проявляется
задержкой в умственном и физическом развитии детей.
26.
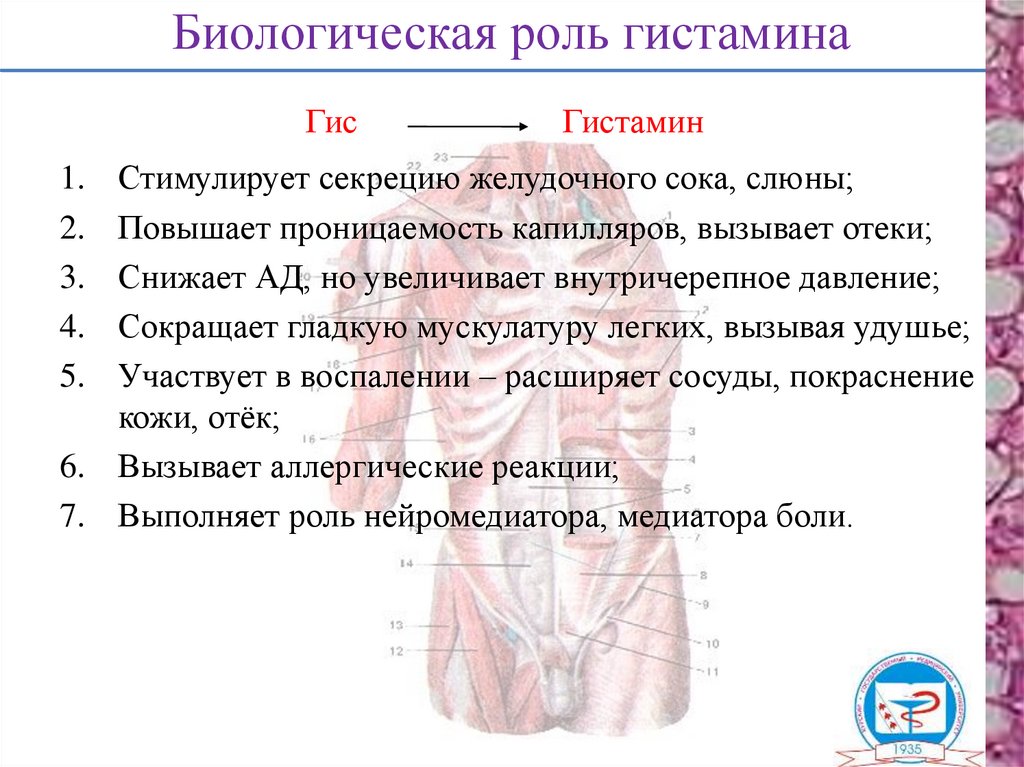
Биологическая роль гистаминаГис
Гистамин
Стимулирует секрецию желудочного сока, слюны;
Повышает проницаемость капилляров, вызывает отеки;
Снижает АД, но увеличивает внутричерепное давление;
Сокращает гладкую мускулатуру легких, вызывая удушье;
Участвует в воспалении – расширяет сосуды, покраснение
кожи, отёк;
6. Вызывает аллергические реакции;
7. Выполняет роль нейромедиатора, медиатора боли.
1.
2.
3.
4.
5.
27. Биологическая роль гистамина
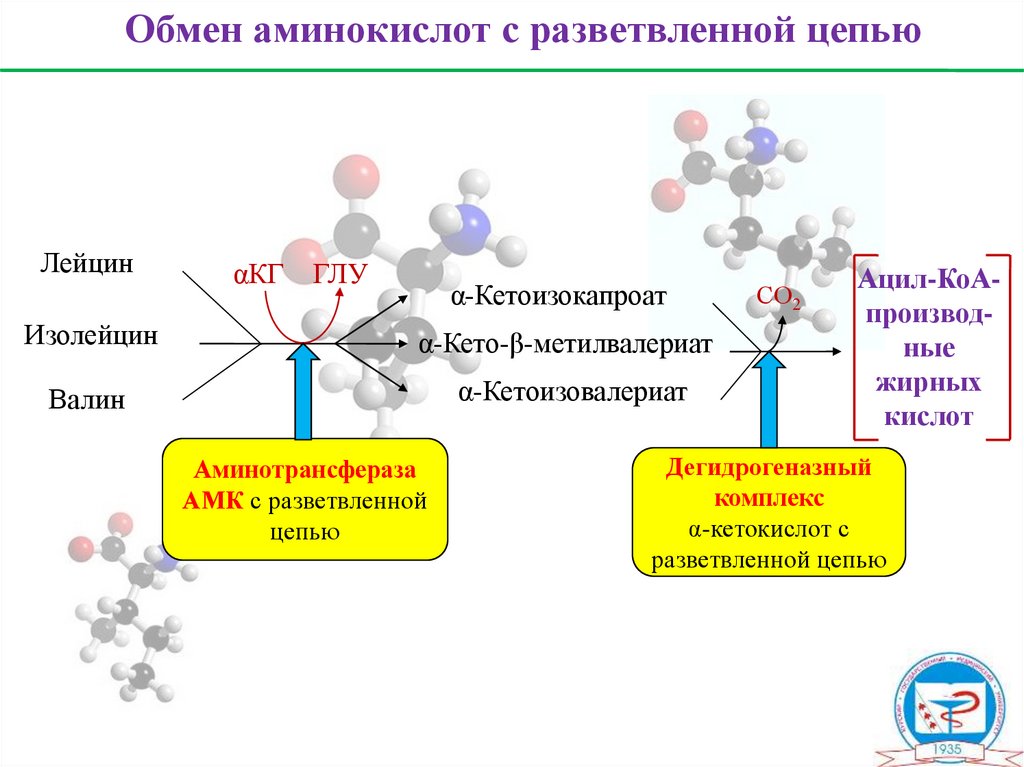
Валин, лейцин, изолейцинНезаменимые аминокислоты
Вал
Лей
Илей
гликогенная (пропионил-КоА
кетогенная (ацетил-КоА
сукцинил-КоА
кетоновые тела)
гликокетогенная (ацетил-КоА + пропионил-КоА)
глю)
28.
Обмен аминокислот с разветвленной цепьюЛейцин
αКГ
ГЛУ
α-Кетоизокапроат
Изолейцин
α-Кето-β-метилвалериат
Валин
α-Кетоизовалериат
Аминотрансфераза
АМК с разветвленной
цепью
CО2
Ацил-КоАпроизводные
жирных
кислот
Дегидрогеназный
комплекс
α-кетокислот с
разветвленной цепью
29.
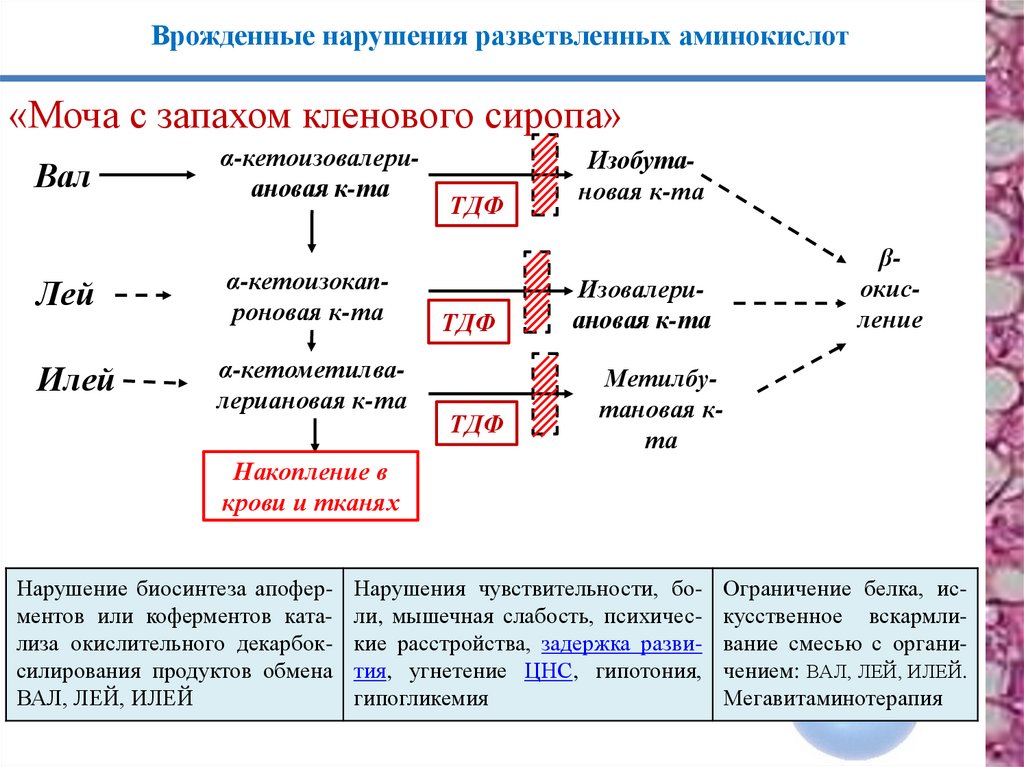
Врожденные нарушения разветвленных аминокислот«Моча с запахом кленового сиропа»
Вал
Лей
Илей
α-кетоизовалериановая к-та
α-кетоизокапроновая к-та
α-кетометилвалериановая к-та
ТДФ
ТДФ
ТДФ
Изобутановая к-та
βокисление
Изовалериановая к-та
Метилбутановая кта
Накопление в
крови и тканях
Нарушение биосинтеза апоферментов или коферментов катализа окислительного декарбоксилирования продуктов обмена
ВАЛ, ЛЕЙ, ИЛЕЙ
Нарушения чувствительности, боли, мышечная слабость, психические расстройства, задержка развития, угнетение ЦНС, гипотония,
гипогликемия
Ограничение белка, искусственное вскармливание смесью с органичением: ВАЛ, ЛЕЙ, ИЛЕЙ.
Мегавитаминотерапия