






























































































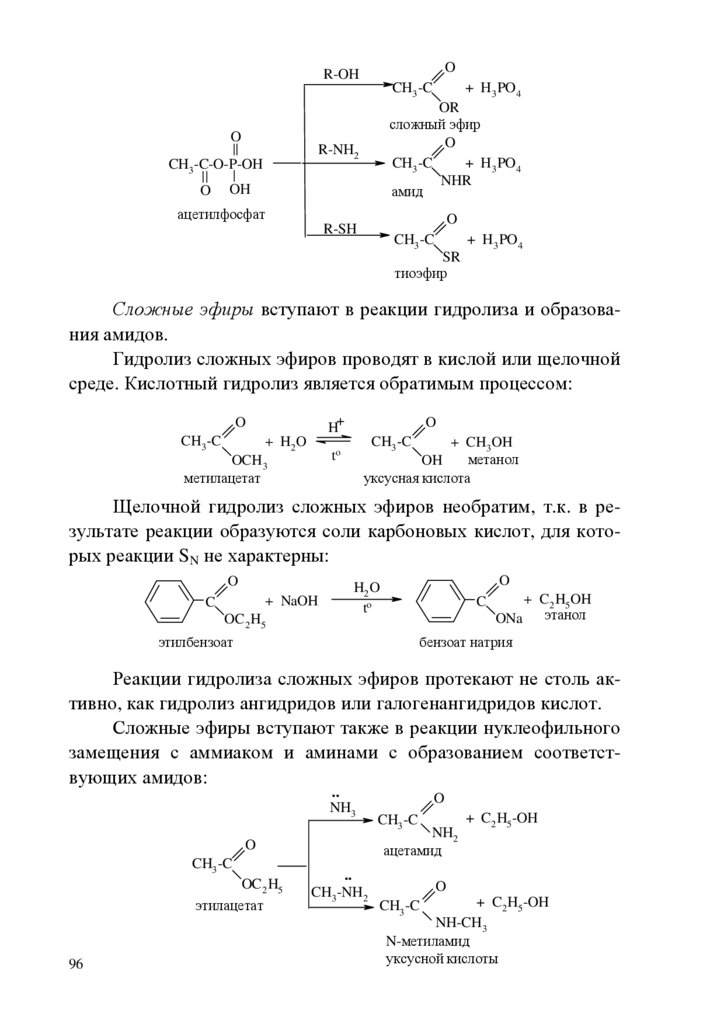





























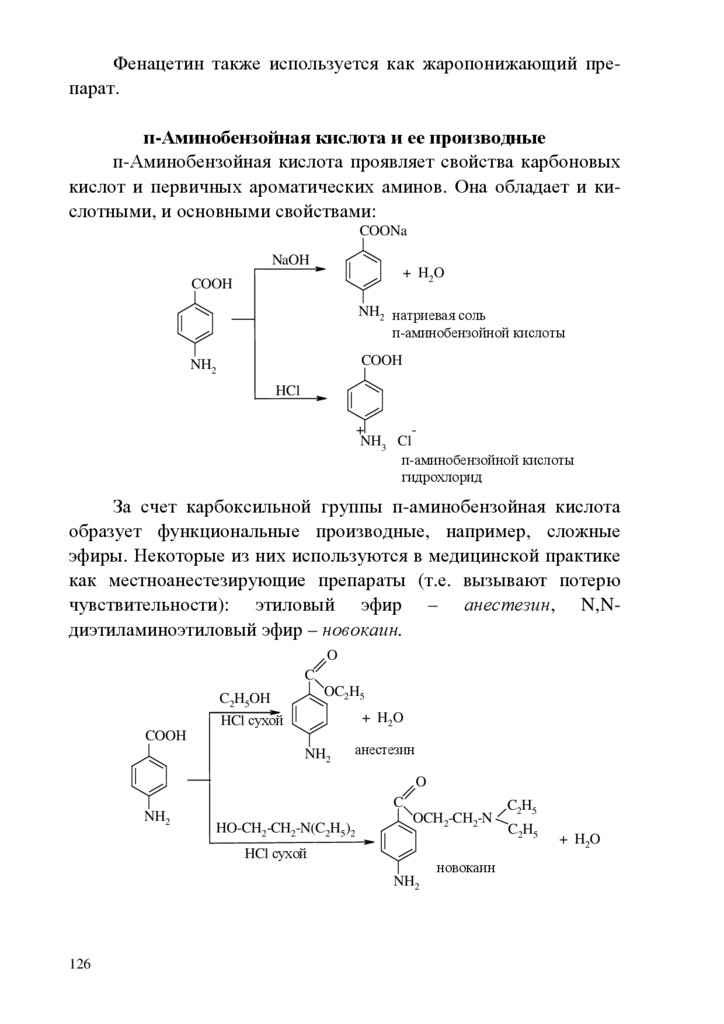











































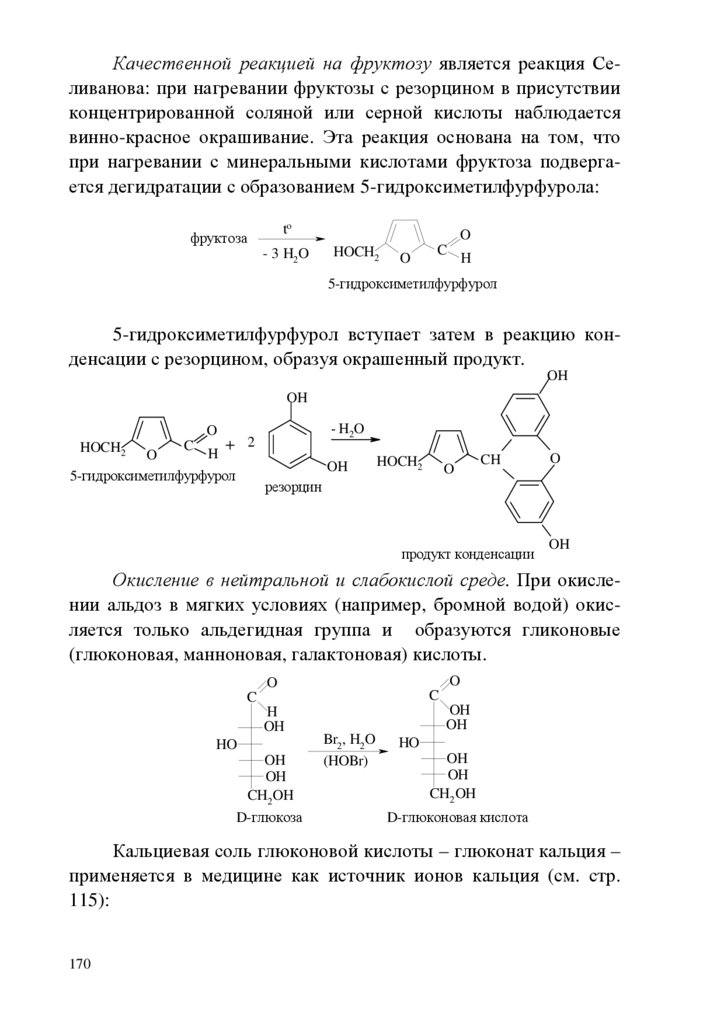










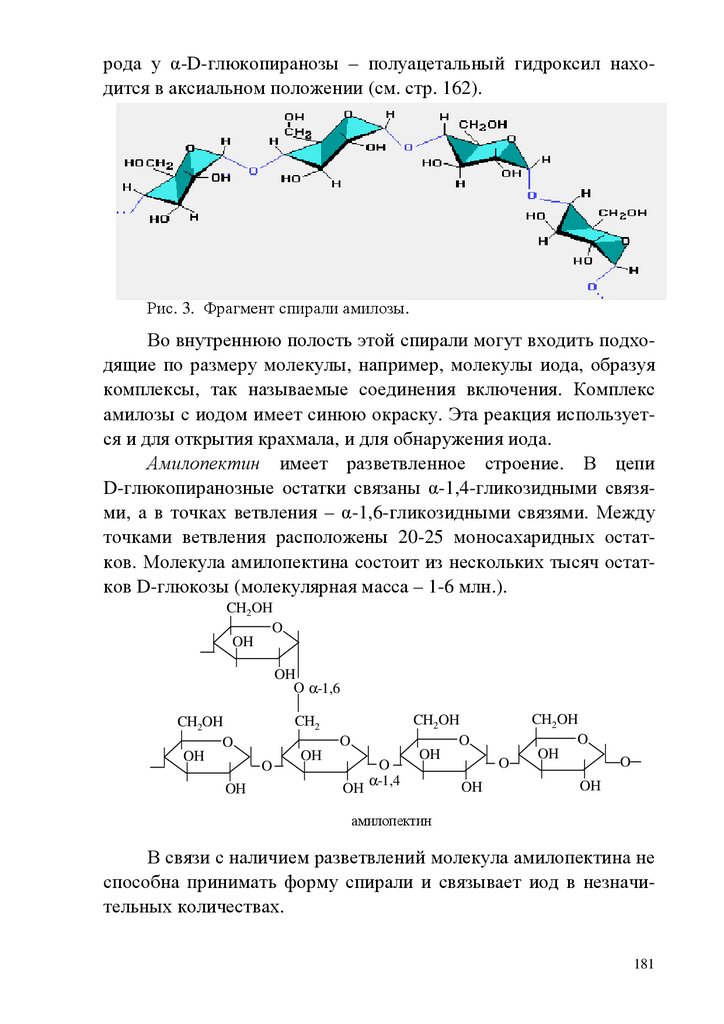































































 chemistry
chemistrySimilar presentations:





")
")



Основы биоорганической химии
1.
Государственное образовательное учреждениевысшего профессионального образования
«Курский государственный медицинский университет
Федерального агентства по здравоохранению
и социальному развитию»
Кафедра биоорганической химии
В.Я. Яцюк, И.В. Зубкова
ОСНОВЫ
БИООРГАНИЧЕСКОЙ ХИМИИ
для студентов лечебного, педиатрического
и медико-профилактического факультетов
Курск – 2010
1
2.
УДК: 577.1(075)ББК: 28.072 я 7
Печатается по решению
редакционно-издательского
совета ГОУ ВПО КГМУ
Росздрава
Яцюк В.Я., Зубкова И.В. Основы биоорганической химии (для студентов лечебного, педиатрического и медико-профилактического факультетов). - Курск: ГОУ ВПО КГМУ Росздрава, 2010. – 248 с.
Общая редакция:
зав. кафедрой биоорганической химии, доктор фармацевтических
наук, профессор Яцюк В.Я.
Рецензенты:
зав. кафедрой фармацевтической химии и фармакогнозии ГОУ ВПО
«Белгородский государственный университет», профессор О.О. Новиков;
профессор кафедры биологической химии ГОУ ВПО «Курский государственный медицинский университет», д.м.н., член-корр. РАЕН
Н.А. Быстрова.
Учебное пособие соответствует программе по биоорганической химии для студентов медицинских вузов (Москва, 2004) и отражает опыт
чтения лекций на лечебном, педиатрическом и медико-профилактическом
факультетах КГМУ.
ISBN
Коллектив авторов, КГМУ, 2010
ГОУ ВПО КГМУ Росздрава, 2010
2
ББК: 28.072 я 7
3.
ВВЕДЕНИЕБиоорганическая химия играет важную роль в развитии современной медицинской науки, поэтому она является важной составляющей общего образования врача.
Целью биоорганической химии как учебной дисциплины
является формирование знаний взаимосвязи строения и химических свойств биологически важных классов органических соединений, биополимеров и их структурных компонентов, т.е. основы
для восприятия биологических, экологических и медицинских
знаний на современном молекулярном уровне. Биоорганическая
химия формирует знания и умения для изучения биохимии, фармакологии, молекулярной биологии, физиологии.
В качестве исходной общетеоретической основы для характеристики свойств и механизма функционирования биологически
активных соединений необходимы знания электронного строения
химических связей, взаимного влияния атомов в молекулах, пространственных эффектов, сведения об электронных механизмах
химических реакций. Важнейшее место отводится овладению
знаниями о реакционной способности функциональных групп.
Для решения вопроса химической совместимости лекарственных средств, выбора пути их введения, предположений о пути метаболизма ксенобиотиков и выведения из организма продуктов их детоксикации необходимо изучение кислотноосновных свойств органических соединений, прогнозирование их
отношения к гидролизу, окислению и т.д., исходя из знания их
химических свойств.
3
4.
ЭЛЕКТРОННОЕ СТРОЕНИЕ ЭЛЕМЕНТОВОРГАНОГЕНОВ. ХИМИЧЕСКАЯ СВЯЗЬВ ОРГАНИЧЕСКИХ МОЛЕКУЛАХ
Биоорганическая химия изучает строение и свойства соединений, участвующих в процессах жизнедеятельности, во взаимосвязи с их биологическими функциями.
Основные объекты изучения биоорганической химии – это
биополимеры (углеводы, пептиды и белки, нуклеиновые кислоты) и биорегуляторы (гормоны, витамины, лекарственные средства).
Биоорганическая химия неразрывно связана с органической
химией, т.к. использует её методы и принципы. В основе теории
органической химии, сформулированной А.М. Бутлеровым, лежит положение о зависимости свойств соединений от их химического строения и взаимного влияния атомов в молекулах. Это
значит, что химические свойства органических соединений обусловлены типом химических связей, природой связываемых атомов и их взаимным влиянием. А это, в свою очередь, определяется электронным строением атомов и взаимодействием их атомных орбиталей.
Рассмотрим электронное строение атомов тех элементов,
которые чаще всего встречаются в структуре органических молекул углерода, водорода, азота и кислорода.
Атомная орбиталь – это область пространства вокруг атомного ядра, в которой возможность нахождения электрона максимальна.
Водород – элемент первого периода, следовательно, его
внешний электронный уровень представлен только s-орбиталью
(она имеет сферическую форму). Углерод, азот и кислород – элементы второго периода, их внешний электронный уровень представлен одной s- и тремя p-орбиталями. Причём эти три pорбитали характеризуются одинаковой формой (объёмной восьмёрки или гантели), энергией, но отличны ориентацией в пространстве (в трёхмерной системе координат – px, py и pz).
4
5.
zy
x
Электронное строение углерода в основном состоянии
1s 2s 2p2. В возбуждённом состоянии (т.е. при образовании связей) – 1s22s2p3.
В органической химии широко используются представления
о гибридных орбиталях. Сущность гибридизации заключается в
том, что из нескольких различных орбиталей, близких по энергии, образуется такое же число атомных орбиталей одинаковых
по форме и энергии. Гибридные орбитали за счёт большего перекрывания затем образуют более прочные связи по сравнению с
негибридными.
Атомы углерода и азота могут находиться в одном из трёх
состояний гибридизации – sp3, sp2, sp. Для атома кислорода характерна sp3- и sp2-гибридизация.
Процесс sp3-гибридизации протекает с участием всех четырёх внешних атомных орбиталей углерода:
2
2
+
s
+
px
+
py
4
pz
гибридные
орбитали
Гибридные орбитали равноценны, имеют форму неправильных объёмных восьмёрок и расположены в
пространстве под углом 109о28’, т.е. направлены к
.
.
.
углам тетраэдра. Поэтому sp3-гибридизованный атом
углерода называют тетраэдрическим. У атома углерода на каждой гибридной орбитали присутствует по одному
электрону.
Электронная конфигурация sp3-гибридизованного атома
азота отличается тем, что на одной из гибридных орбиталей у него присутствуют два электрона (электронная формула
азота
..
2 2
3
1s 2s 2p ):
.
.
.
.
5
6.
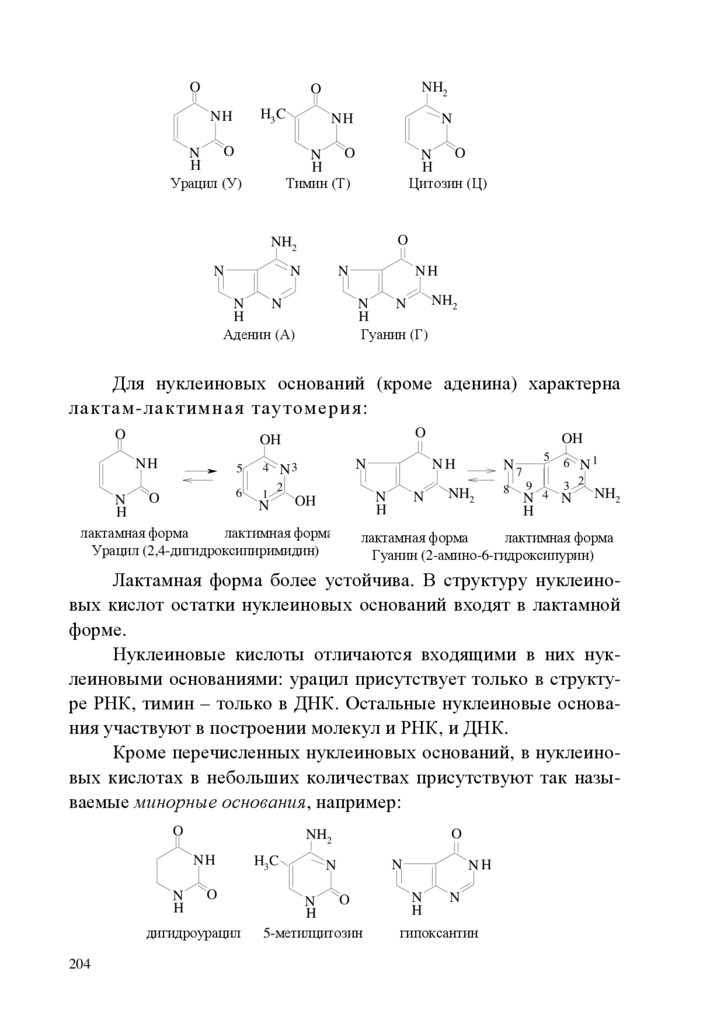
Эту пару электронов называют неподелённой электроннойпарой, т.к. обычно она не участвует в образовании связей. Валентный угол sp3-гибридизованного азота равен 107о.
У sp3-гибридизованного атома кислорода (электронная формула – 1s22s22p4) две гибридные орбитали заняты неподелёнными
..
электронными парами и валентный угол равен 104,5о :
..
.
.
В sp2-гибридизации участвуют s-орбиталь и две p-орбитали:
+
s
+
px
3
py
гибридные
орбитали
Одна pz-атомная орбиталь остаётся негибридизованной.
sp -атомные орбитали расположены в одной плоскости под углом
120о, а pz-орбиталь – перпендикулярно им. p
2
z
У sp2-гибридизованного атома углерода на каждой орбитали
находится по одному электрону:
.
.
.
.
Распределение электронов по орбиталям (электронная конфигурация) у sp2-гибридизованного атома азота может быть различным: неподелённая электронная пара может находиться или
на sp2-атомной орбитали, или на негибридной pz-орбитали.
..
.
.
6
.
.
.
.
:
7.
Известны также два типа sp2-гибридизованных атома кислорода: на pz-орбитали может находиться один электрон или неподелённая электронная пара...
.
:
.
.
..
или
.
:
В sp-гибридизации участвуют s- и p-атомные орбитали:
+
s
2
px
Две гибридные орбитали
расположены под углом
180о.
Оставшиеся негибридными py- и pz-орбитали расположены
перпендикулярно осям гибридных орбиталей и перпендикулярно
pz
друг другу:
гибридные
орбитали
py
У sp-гибридизованного атома углерода каждая орбиталь занята одним электроном:
. .
.
.
Неподелённая электронная пара sp-гибридизованного азота
всегда расположена на одной из гибридных орбиталей:
.
. .
:
Для кислорода sp-гибридизация не характерна.
Очень важно научиться определять тип гибридизации и
электронную конфигурацию атомов. Это необходимо для понимания распределения электронной плотности в молекулах, опре7
8.
деления электронных эффектов, стабильности молекул и промежуточных частиц. А это, в свою очередь, помогает определитьпреимущественное направление реакции, сравнить активность
различных соединений в той или иной химической реакции.
Как же правильно определить тип гибридизации атомов в
молекулах?
Для углерода тип гибридизации определяется по количеству
σ-связей. σ-Связи образуются за счёт гибридных орбиталей, значит, сколько σ-связей у углерода, столько и гибридных орбиталей. Например, определим тип гибридизации атомов углерода в
молекуле пропена CH2=CH-CH3. Первый атом углерода образовал три σ-связи (одну – с соседним углеродом и две – с двумя
атомами водорода), значит, у этого углерода три гибридных орбитали, что соответствует sp2-гибридизации. Второй атом углерода также образовал три σ-связи (две – с двумя соседними атомами углерода и одну – с водородом), т.е. он также находится в
sp2-гибридизации. Третий углерод образовал четыре σ-связи,
имея четыре гибридных орбитали, т.е. он sp3-гибридизован.
Тип гибридизации атомов азота или кислорода определяется
по соседнему углероду, например, в молекуле этиламина
CH3-CH2-NH2 азот имеет sp3-гибридизацию так же, как и соседний углерод. В молекуле виниламина CH2=CH-NH2 азот sp2гибридизован.
Если вы определили, что азот или кислород в какой-либо
молекуле sp2-гибридизован, возникает следующая проблема.
Нужно определить его электронную конфигурацию (распределение электронов по орбиталям). Другими словами, нужно определить число электронов на негибридной орбитали. Запомните, что
если sp2-гибридизованный азот (или кислород) образовал только
одинарные связи, на негибридной орбитали – 2 электрона (неподелённая электронная пара). Если же он образовал двойную
связь, на негибридной орбитали – 1 электрон. Например, электронная конфигурация атома азота в молекуле виниламина
CH2=CH-NH2 :
..
.
.
8
.
9.
а в молекуле имина формальдегида CH2=NH :.
.
.
:
Химическая связь в органических молекулах
Различают два основных типа связи – ковалентную и электровалентную (ионную). Ковалентная связь возникает в результате обобщения неспаренных валентных электронов с противоположными спинами, электровалентная – за счёт передачи неспаренного электрона одного атома другому с образованием противоположно заряженных ионов, которые взаимно притягиваются.
Это можно показать в виде схемы:
A. + . B
A: B
ковалентная связь
A. + . B
A+ + : B
электровалентная связь
В органических молекулах в основном присутствуют ковалентные связи. Для их описания используют два метода: метод
валентных пар и метод молекулярных орбиталей. Коротко рассмотрим их.
Метод валентных пар предполагает, что при образовании
ковалентной связи происходит перекрывание атомных орбиталей
взаимодействующих атомов с образованием общей электронной
пары, посредством которой атомы и удерживаются друг с другом.
При этом электрон продолжает находиться на своей атомной орбитали:
.
+
:
независимые
атомные орбитали
перекрывание
атомных орбиталей
.
В соответствии с методом молекулярных орбиталей при
образовании связи из n атомных орбиталей образуется n молекулярных орбиталей. Молекулярная орбиталь принадлежит не отдельным атомам, а обоим связываемым атомам, т.е. молекулярная орбиталь как минимум двухцентровая (на самом деле, молекулярные орбитали могут быть и многоцентровыми, например, в
так называемых сопряжённых системах). Если связь образуется
между двумя атомами, то при взаимодействии их атомных орбиталей образуются две молекулярные орбитали. На одной из них
(связывающей), с минимальной энергией, находятся два элек9
10.
трона. Другая (разрыхляющая), с максимальной энергией, остаётся вакантной..
+
.
.
независимые
атомные орбитали
.
связывающая
молекулярная орбиталь
Существует два типа ковалентных связей – σ- и π-.
σ-Связью называется ковалентная связь, образованная при
перекрывании атомных орбиталей по оси, соединяющей центры
атомов, с максимумом перекрывания по этой оси. При образовании σ-связей могут перекрываться s-, p- и гибридные орбитали:
s s
s
p
p
p
гибридные орбитали
У элементов-органогенов (C, N, O, S) в образовании σ-связей
участвуют гибридные орбитали, т.к. в этом случае возможно более эффективное перекрывание, и образуется более прочная
связь.
Кроме осевого перекрывания атомных орбиталей возможно
и боковое, приводящее к образованию π-связей.
π-Связь образуется при боковом перекрывании негибридных p-орбиталей с максимумом перекрывания по
обе стороны от оси, соединяющей центры атомов. За
счёт этого π-связь менее прочная, чем σ-связь.
p p
Встречающиеся в органических соединениях кратные связи являются сочетанием σ- и π-связей: двойная – одна σ- и
одна π-связь; тройная – одна σ- и две π-связи.
Свойства ковалентной связи выражаются следующими характеристиками: длина, энергия, полярность, поляризуемость.
Длина связи – это расстояние между центрами связанных
атомов. Длина связи является её важной характеристикой, т.к. от
длины зависит энергия связи. Длина связи приблизительно равна
сумме атомных радиусов связываемых атомов. Чтобы сравнить
длины различных связей, нужно сравнить атомные радиусы атомов, а это можно сделать, используя Периодическую таблицу
Д.И. Менделеева. Например, сравним длины следующих связей:
10
11.
H-H, H-Cl и Cl-Cl. Водород – элемент 1-го периода, хлор – 3-го.Схематично это можно обозначить так (кружки обозначают здесь
не атомные орбитали, а атомы):
l
H H
l
H
l
Cl
Cl
Cl
Двойная связь короче одинарной, тройная – короче двойной.
Это связано с тем, что между ядрами атомов, связанных двойной
связью, находятся две общие электронные пары, значит, ядра
сильнее притянуты друг к другу. В тройной связи силы притяжения ещё больше.
Энергия связи – это энергия (в расчёте на 1 моль), которая
выделилась в результате образования связи. Можно дать и другое
определение: это энергия, необходимая для разрыва связи. Чем
выше энергия связи, тем она прочнее. Энергия связи зависит от
её длины: чем короче связь, тем она прочнее. В нашем примере
(см. выше) самой прочной является H-H-связь.
σ-Связь прочнее π-связи, т.к. при осевом перекрывании
атомных орбиталей площадь перекрывания больше, чем при боковом.
Полярность связи – это неравномерное распределение электронной плотности между двумя атомами из-за различия в их
электроотрицательности. Только связи между двумя одинаковыми атомами являются абсолютно неполярными, т.е. общая электронная пара совершенно в равной степени принадлежит обоим
атомам. Чем больше разница в электроотрицательности атомов,
тем полярнее связь, т.е. тем более общая электронная пара смещена к одному из них. Например, в ряду H-Cl, H-Br, H-I самой
полярной является связь H-Cl, а наименее полярной – связь H-I.
Электроотрицательность характеризует способность атома в
молекуле удерживать свои валентные электроны. Для количественной оценки электроотрицательности применяется шкала Полинга.
Электроотрицательность не является абсолютной константой элемента. В частности, она зависит от типа гибридизации
(sp>sp2>sp3). Поэтому, например, C-C связь в молекуле этана
11
12.
(CH3-CH3) неполярна (оба углерода в sp3-гибридизации), а в молекуле пропена (CH3-CH=CH2) – полярна (sp2-гибридизованныйуглерод более электроотрицателен, чем sp3-гибридизованный).
В формуле для обозначения полярности связи применяют
стрелку, направленную к более электроотрицательному атому,
например:
H3 C
OH
sp3 sp2
H3 C CH=CH2
Полярность связи определяет тип её разрыва, т.е. тип химического взаимодействия.
Поляризуемость – лёгкость, с которой смещаются электроны связи под действием внешних воздействий (электрическое
поле, реагирующая частица, полярные молекулы растворителя).
Полярность – это статическое явление (то, что уже есть в молекуле), а поляризуемость – динамическое (то, что может быть, если приложить определённые внешние факторы). Рассмотрим это
на следующем примере. Мы знаем, что связь Br-Br неполярна
(связаны два одинаковых атома). Если же молекула брома попадает под действие электрического поля, общая электронная пара
связи сместится в сторону положительного электрода, и связь
станет полярной, т.е. поляризуется.
Br : Br
связь
неполярна
Br: Br
связь
поляризовалась
+
Чем длиннее связь, тем легче она поляризуется, т.к. электроны находятся дальше от ядер атомов и не так прочно ими
удерживаются. Поэтому в ряду H-Cl, H-Br, H-I легче всего поляризуется связь H-I, а труднее всего H-Cl (она самая короткая).
π-Связи поляризуются легче, чем σ-связи, т.к. электронные
облака π-связей находятся дальше от атомных ядер.
Как и полярность, поляризуемость влияет на реакционную
способность молекул, но это влияние ещё более выражено.
12
13.
КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ РЕАКЦИЙ.ХИМИЧЕСКИЕ СВОЙСТВА АЛКАНОВ, АЛКЕНОВ
И АЛКАДИЕНОВ
Для рассмотрения химических свойств конкретных классов
органических соединений необходимо усвоить некоторые общие
понятия, такие, как реакционная способность вещества, реакционные центры молекулы, механизм химической реакции, а также
классификацию химических реакций.
Большинство органических реакций протекает в несколько
элементарных стадий. Механизм реакции – это детальное описание реакции с указанием промежуточных частиц. Иначе говоря,
под механизмом реакции понимают общий путь, по которому
проходит реакция от исходных веществ до её конечных продуктов.
Способность вещества вступать в ту или иную реакцию и
реагировать с большей или меньшей скоростью называется реакционной способностью данного вещества.
В ходе реакции обычно изменяется не вся молекула, а так
называемый реакционный центр. Это атом или группа атомов,
непосредственно участвующих в данной химической реакции.
В реакции участвуют субстрат (само вещество) и реагент
(соединение или частица, действующие на субстрат).
Разнообразные химические реакции классифицируют по
следующим признакам: по конечному результату, по типу разрыва связей и по характеру атакующей частицы.
По конечному результату реакции подразделяют на следующие типы:
1. Реакции замещения (сокращённо их обозначают символом S substitution). Например:
CH3 -Cl + NaOH
CH3 -OH + NaCl
В этой реакции хлор в молекуле хлорметана замещается на
гидроксильную группу.
2. Реакции присоединения (A - addition). Например:
CH2 =CH2 + HCl
CH3 -CH2 -Cl
Молекула хлороводорода присоединяется к молекуле этена.
13
14.
3. Реакции элиминирования (отщепления) (E - elimination).Например:
CH3 -CH 2 -Cl
NaOH
спирт
CH2 =CH2 + NaBr + H2 O
В этой реакции под действием спиртового раствора щёлочи происходит отщепление хлороводорода от молекулы
хлорэтана.
4. Реакции окисления и восстановления, например:
CH3 -OH
[O]
H-C
O
H
В этой реакции произошло окисление метанола до формальдегида (в органической химии под окислением понимают
увеличение количества связей с кислородом).
Классификация по типу разрыва связей
Ковалентные связи могут разрываться по-разному. Рассмотрим типы разрыва связей.
Если при разрыве химической связи у каждого из атомов остаётся по одному электрону, такой тип разрыва называют гомолитическим, или гомолизом (от греческих слов: гомо – одинаковый, лизис – расщепление). Схематично это можно показать следующим образом:
.
A + .B
A:B
Такой тип разрыва характерен для неполярных и малополярных связей. Ему способствуют определённые внешние факторы: облучение, нагревание, проведение реакции в газовой фазе
или в среде неполярного растворителя (чтобы исключить поляризацию связи).
Полученные частицы не заряжены (электронейтральны),
имеют неспаренный электрон, поэтому очень активны. Нейтральные частицы с неспаренным электроном называются свободными радикалами, отсюда другое название гомолитического
типа разрыва связи – радикальный тип.
14
15.
Если при разрыве связи пара электронов остаётся у одногоиз атомов, такой тип разрыва называют гетеролитическим, или
гетеролизом (гетерос – разный, другой).
A+ + B
A :B
Такой тип разрыва характерен для полярных связей. Ему
способствуют наличие полярного растворителя, нагревание, катализаторы (кислоты или щёлочи).
В результате разрыва связи образовались заряженные частицы – ионы, поэтому другое название гетеролитического типа
разрыва связи – ионный тип.
Итак, по типу разрыва связей реакции классифицируют на
гомолитические (радикальные) и гетеролитические (ионные).
Гетеролитические реакции классифицируют дальше в зависимости от электронной природы атакующей частицы. Их
подразделяют на электрофильные и нуклеофильные.
Электрофильные реагенты (электрофилы, Е, «любящие
электроны») – частицы с полным положительным зарядом или
нейтральные молекулы с недостатком электронной плотности на
каком-то из атомов. Например: H+, NO2+, CH3+, Cl+, SO3.
Нейтральная молекула SO3 является электрофилом из-за перераспределения электронной плотности в молекуле.
Три электроотрицательных атома кислорода
δ−
смещают электронную плотность с серы на себя,
δ+ O
O S
и на атоме серы возникает большой по
O
величине
частичный
положительный
заряд
(это обозначается как δ+).
Электрофилы атакуют в молекуле субстрата центры с повышенной электронной плотностью.
Нуклеофильные реагенты (нуклеофилы, Nu, «любящие ядро») – частицы с полным отрицательным зарядом или молекулы с
неподелённой электронной парой. Например:
..
.. ..
HO- , Cl - , H2 O , NH3 , CH3 -OH
15
16.
Нуклеофилы способны образовывать ковалентную связь ссубстратом, атакуя в его молекуле центры с пониженной
электронной плотностью .
При характеристике органических реакций чаще пользуются
критериями различных классификаций, например, радикальное
замещение (SR), электрофильное присоединение (AE), нуклеофильное замещение (SN).
Итак, мы рассмотрели, какие типы реакций вообще возможны в органических соединениях и теперь можем перейти к общим
закономерностям их реакционной способности как химической
основе биологического функционирования органических молекул.
РЕАКЦИОННАЯ СПОСОБНОСТЬ АЛКАНОВ
Алканы – насыщенные углеводороды. Все атомы углерода в
их молекулах sp3-гибридизованы.
Первые представители гомологического ряда алканов (с неразветвлённой цепью):
CH4
метан
CH3-(CH2)4-CH3
гексан
CH3-(CH2)5-CH3
CH3-CH3
этан
гептан
пропан
октан
CH3-CH2-CH3
CH3-(CH2)6-CH3
CH3-(CH2)2-CH3
бутан
CH3-(CH2)7-CH3
нонан
CH3-(CH2)3-CH3
пентан
CH3-(CH2)8-CH3
декан
Для того, чтобы определить, какой тип реакций наиболее
характерен для алканов, нужно рассмотреть электронное строение алкана (на примере этана).
Атомы углерода sp3-гибридиσ
σ
зованы, валентный угол σ
109о28`. C-C-связь образована
σ
o
109 28'
за счёт линейного перекрываσ
ния гибридных орбиталей, т.е.
σ
σ
это σ-связь, C-H-связи являются также σ-связями. C-C-связи неполярны, C-H-связи мало полярны. C-H-связи прочнее, чем C-C-связь, т.к. они короче (0,110 и
0,154 нм соответственно).
16
17.
Так как неполярные и малополярные связи разрываются погомолитическому (радикальному) типу, для алканов характерны
радикальные реакции. Алканы – насыщенные углеводороды, поэтому для них невозможны реакции присоединения, но характерны реакции замещения и отщепления. Наиболее важными реакциями алканов являются реакции радикального замещения (SR).
Типичный пример реакции SR – галогенирование алканов
при облучении. Облучение часто используется для избирательного расщепления относительно слабых связей. При этом образующиеся частицы – радикалы выступают в качестве инициаторов
последующих превращений.
Рассмотрим механизм реакции хлорирования метана.
Это схема реакции. Она поhυ
CH4 + Cl2
CH3 -Cl + HCl казывает, какие соединения
хлорометан
метан
вступили в реакцию и какие
образовались, но не объясняет как это произошло.
Описание механизма реакции (совокупности её элементарных стадий) позволяет понять, какие условия необходимы для
конкретной реакции, а во многих случаях – какой продукт будет
преобладающим.
В реакции хлорирования метана под действием ультрафиолетового облучения (энергии кванта света) происходит гомолиз
молекулы хлора с образованием свободных радикалов хлора:
Cl : Cl
2 Cl
.
Это стадия инициирования.
Следующая стадия реакции – стадия роста цепи. Под действием радикала хлора происходит гомолиз одной из C-H-связей
с образованием нового свободного радикала (метил) и молекулы
хлороводорода:
.
.
H3C : H + Cl
H3C . + Cl : Cl
H3C + HCl
радикал
метил
H3C-Cl + Cl .
хлорметан
Образовавшийся радикал метил атакует следующую молекулу хлора, расщепляя её на радикалы и образуя молекулу хлорметана и новый радикал хлора и т.д. Таким образом, стадии реак17
18.
ции следуют друг за другом как звенья цепи: конец одной реакции (свободный радикал) является началом следующей, один радикал расходуется, но другой образуется. Поэтому реакции галогенированияалканов называют свободнорадикальными цепными реакциями. Отдельные звенья стадии роста цепи могут повторяться сотни раз.
Если встретятся и провзаимодействуют два свободных радикала, произойдёт обрыв цепи: активные частицы расходуются,
а новые не образуются. Например:
.
.
Cl + Cl
Cl2
H3 C . + Cl .
CH3 -Cl
H3 C . + . CH3
CH3 -CH3
В реакции хлорирования метана нам не нужно решать проблему, по какому положению она протекает, т.к. в молекуле
только один атом углерода. При галогенировании пропана в
принципе могут получиться два различных продукта, например,
1-бромпропан и 2-бромпропан:
hυ
CH3 -CH2 -CH3 + Br2
CH3 -CH2 -CH2 -Br + HBr
1-бромпропан
CH3 -CH-CH 3 + HBr
Br
2-бромпропан
Чтобы решить вопрос, какой из продуктов будет преобладать, нужно описать механизм данной реакции. Известно, чем
стабильнее промежуточная частица реакции (интермедиат), тем
больше вероятность её образования. Следовательно, нам необходимо сравнить стабильность двух возможных свободных радикалов, образующихся на стадии роста цепи.
Br2
hυ
2 Br
.
.
CH3 -CH2 -CH3 + Br
.
CH3 -CH2 -CH2 + HBr
радикал пропил
.
CH3 -CH-CH 3 + HBr
радикал изопропил
18
19.
Свободные радикалы в целом очень нестабильны, т.к. имеют неспаренный электрон, они «ищут» пару электрону и легковступают в реакции. Радикал изопропил относительно более устойчив, чем пропил. Это можно объяснить, показав смещение
электронной плотности в данных радикалах.
Изопропил – вторичный радикал. Атом угле.
CH3 CH
CH3 рода с неспаренным электроном находится в
sp2-гибридизации, он более электроотрицателен, чем соседние sp3-гибридизованные атомы. Поэтому электронная плотность смещается к углероду с неспаренным электроном с двух сторон, что способствует стабилизации радикала.
Пропил – первичный радикал. Электронная
.
CH3 CH2
CH2 плотность смещается к углероду с неспаренным электроном с одной стороны. Радикал не
столь стабилен, как изопропил.
Вероятность образования радикала изопропила на стадии
роста цепи выше, а значит, в дальнейшем именно радикал изопропил имеет больше шансов прореагировать с новой молекулой
брома и образовать конечный продукт реакции – 2-бромпропан.
В целом самыми стабильными являются третичные радикалы, менее стабильными – вторичные, наименее – первичные.
Реакции радикального замещения в алканах являются ре гиоселективными . Региоселективность – это преимущественное протекание реакции по одному из нескольких однотипных
реакционных центров. Региоселективность является следствием
различной стабильности образующихся свободных радикалов.
Реакция нитрования алканов также протекает по механизму
радикального замещения, она также региоселективна. Например,
при нитровании 2-метилпропана реакция в первую очередь протекает у третичного атома углерода:
CH3 -CH-CH 3 + HNO3
CH3
2-метилпропан
p, to
NO 2
CH3 -C-CH 3 + H2 O
CH3
2-метил-2-нитропропан
19
20.
Реакцию нитрования проводят разбавленной азотной кислотой при повышенной температуре и давлении. Она известна какреакция Коновалова.
РЕАКЦИОННАЯ СПОСОБНОСТЬ АЛКЕНОВ
Алкены – ненасыщеные углеводороды, содержащие одну
двойную связь. Названия алкенов строятся от названий соответствующих алканов с изменением суффикса –ан на –ен (этен, пропен) и указанием положения двойной связи. Например,
1
2
3
4
CH2 =CH-CH-CH3
3-метилбутен-1
CH3
Рассмотрим электронное строение молекулы этена.
Атомы углерода в этене sp2-гибридизованы. С-С σ-связь образована за счёт осевого перекрывания гибридных орбиталей. CH σ-связь образована за счёт осевого перекрывания гибридной
орбитали углерода и s-орбитали водорода. σ-Связи расположены
в одной плоскости под углом 120о. π-Связь образована за счёт бокового перекрывания негибридных pz-орбиталей в плоскости,
перпендикулярной плоскости σ-связей. Длина C=C-связи 0,134
нм.
Для алкенов характерна структурная изомерия (по положению двойной связи, изомерия цепи), а также пространст венная (геометрическая, или цис,транс-изомерия). Пространственные изомеры (стереоизомеры) имеют одинаковый состав,
одинаковый порядок связывания атомов в молекулах, но различное расположение атомов или атомных групп в пространстве.
20
21.
Например, бутен-2 существует в виде двух стереоизомеров:CH3
CH3
H
H
CH3
C=C
C=C
H
CH3
транс-изомер
и
H
цис-изомер
В цис-изомере метильные группы расположены по одну
сторону от плоскости двойной связи, в транс-изомере – по разные. Цис- и транс-изомеры не могут превратиться друг в друга
самопроизвольно, т.к. вращение вокруг двойной связи невозможно без разрыва π-связи.
Обратите внимание на то, что геометрическая изомерия
возможна лишь в том случае, когда у углерода при двойной связи
есть два различных заместителя. Так, например, бутен-1 не может существовать в виде геометрических изомеров.
Химические свойства алкенов
Алкены проявляют способность к реакциям присоединения
с разрывом π-связей, т.к. π-связь не столь прочная, как σ-связь. За
счёт электронов π-связи в молекулах алкенов имеется область
повышенной электронной плотности, следовательно, они подвергаются атаке электрофильных реагентов. Итак, для алкенов характерны реакции электрофильного присоединения (AE). Примерами реакций являются гидрохлорирование, гидратация, бромирование:
HCl
CH2 =CH2
H2 O, H +
Br2 , H2 O
CH3 -CH2 -Cl
хлорэтан
CH3 -CH2 -OH
этанол
CH2 -CH2
1,2-дибромэтан
Br
Br
Общий механизм реакций электрофильного присоединения
Любой полярный реагент в принципе может быть представлен как результат взаимодействия электрофила E+ и нуклеофила
Nu-:
+
E + :Nu
E : Nu
21
22.
На первой стадии реакции электрофил подходит к электронному облаку π-связи и они притягиваются друг к другу. Образуется так называемый π-комплекс:CH2
CH2 =CH2 + E+
CH2
E+
π -комплекс
Затем электрофил образует новую σ-связь с одним из атомов
углерода. Для этого он использует оба электрона бывшей π-связи.
Образуется так называемый σ-комплекс, или карбкатион:
CH2
CH2
E+
π -комплекс
+
CH2 -CH2
E
σ-комплекс
Так как электрофил использовал на образование новой связи
электроны π-связи, соседний углерод приобретает полный положительный заряд.
σ-Комплекс затем реагирует с оставшимся нуклеофилом,
который предоставляет свои электроны для образования новой
связи:
+
CH2 -CH2 + :Nu
CH2 -CH 2
E
σ-комплекс
E
Nu
Стадия образования карбкатиона является самой медленной
(старая π-связь должна разорваться, это требует затрат энергии).
Скорость этой стадии определяет скорость реакции в целом. Её
называют лимитирующей стадией реакции.
Теперь рассмотрим конкретные примеры реакций электрофильного присоединения.
Реакция гидрогалогенирования (гидрохлорирования, гидробромирования). Эта реакция протекает полностью в соответствии с общим механизмом AE:
CH2 =CH2 + H+Cl этен
22
CH2
CH2
H+
π -комплекс
CH -CH +
3
2
σ-комплекс
Cl -
CH3 -CH2 -Cl
хлорэтан
23.
В результате реакции образуются галогеналканы.Реакция гидратации – присоединение воды. Эта реакция
возможна только в присутствии сильных минеральных кислот
(серной или фосфорной). Чтобы объяснить роль кислоты, необходимо рассмотреть механизм этой реакции.
Молекула воды – плохой источник электрофильных частиц
+
(H ), т.к. степень её диссоциации очень низкая. Фактически роль
электрофилов в реакции гидратации играют протоны, полученные не от молекул воды, а от молекул минеральной кислоты.
Именно они и начинают реакцию.
CH2 =CH2 + H2 SO4
-HSO 4 этен
+ HSO4 -
CH2
CH2
H+
π -комплекс
CH3 -CH2+
σ-комплекс
..
H2 O
+ H
CH3 -CH2 -O
H
протонированная
молекула спирта
CH3 -CH2 -OH + H2 SO4
этанол
На первой стадии реакции протон кислоты образует πкомплекс с электронным облаком π-связи. Затем протон водорода
присоединяется к одному из атомов углерода, при этом на соседнем атоме углерода возникает полный положительный заряд. Образуется σ-комплекс (карбкатион). Вы видите, что эти две стадии
протекают в соответствии с общим механизмом реакций AE. А
вот над следующей стадией этой реакции надо подумать: σкомплекс должен прореагировать с нуклеофилом (см. общий механизм), а в реакционной среде есть два вида нуклеофилов –
анионы кислоты HSO4- и нейтральные молекулы воды с неподелённой электронной парой на кислороде. В общем, анионы – более сильные нуклеофилы, но в этой реакции кислота используется в каталитических количествах (анионов мало), в то время как
воду используют в избытке. Вот почему в этом «соревновании
нуклеофилов» побеждают молекулы воды.
Итак, на следующей стадии реакции молекула воды как
нуклеофил атакует карбкатион, и по донорно-акцепторному механизму образуется новая σ-связь. Так как атом кислорода затра23
24.
тил на образование новой связи два своих электрона (как донор),на нём образовался полный положительный заряд. Получилась
протонированная молекула спирта – нестабильная заряженная
частица. Для стабилизации ей необходимо отщепить протон, что
и делает анион серной кислоты. В результате этой реакции образовались молекулы спирта и серной кислоты. То, что серная кислота в результате реакции не расходуется, подтверждает, что она
является катализатором этой реакции. Итак, роль серной кислоты
в реакции гидратации алкенов – это роль катализатора, источника
электрофильных частиц (H+).
Реакция галогенирования (хлорирования, бромирования).
Алкены в обычных условиях легко присоединяют галогены, например, в реакции бромирования этена образуется 1,2дибромэтан:
H2 O
CH2 =CH2 + Br2
CH2 -CH2
Br
Br
Эта реакция протекает с водным раствором брома, так называемой бромной водой. Под действием полярных молекул воды
неполярная связь в молекуле брома поляризуется:
Br-Br
H2 O
δ+
Br
δ−
Br
Затем эта поляризованная молекула брома взаимодействует
с π-электронным облаком этена, образуя π-комплекс:
δ+
CH2 =CH2 + Br
δBr
CH2
CH2
Brδ+
Brδ−
π -комплекс
Под действием электронов π-облака происходит ещё большая поляризация связи Br-Br, она разрывается гетеролитически,
и Br+ участвует в образовании σ-комплекса:
CH2
CH2
Br δ+
Brδ−
π -комплекс
24
- Br-
+
CH2 -CH2
Br
σ-комплекс
25.
У положительно заряженного атома углерода σ-комплексаесть вакантная p-орбиталь, а у атома брома – неподелённая электронная пара. Они взаимодействуют по донорно-акцепторному
механизму, образуется циклический катион бромония:
δ+ δ+ + Br CH2 -CH 2
+
CH2 -CH 2
Br+
катион
бромония
: Br
.. :
σ-комплекс
Br
CH2 -CH 2
Br
1,2-дибромэтан
Катион бромония подвергается атаке нуклеофила (Br-), и
образуется 1,2-дибромэтан. Поскольку подход Br- со стороны
первого брома затруднён стерически, его атака происходит с противоположной стороны (так называемое транс-присоединение).
В случае образования 1,2-дибромэтана это не столь важно,
т.к. конформеры свободно переходят друг в друга, но при присоединении к циклоалкенам нужно обращать внимание именно
на транс-присоединение, например, при бромировании циклопентена образуется транс-1,2-дибромциклопентан:
+ Br2
циклопентен
H2 O
Br
Br
транс-1,2-дибромциклопентан
Реакцию бромирования используют как качественную реакцию на непредельность (на двойную и тройную связь). Качественными называют реакции, результат которых хорошо виден,
например, выпадение или растворение осадка, изменение окраски, выделение пузырьков газа, появление характерного запаха. В
реакции бромирования алкенов можно наблюдать обесцвечивание бромной воды (исчезновение оранжевой или жёлтооранжевой окраски).
25
26.
Реакции электрофильного присоединенияк несимметричным алкенам
Когда мы рассматривали реакцию гидрогалогенирования
или гидратации на примере этена, у нас не возникало вопроса к
какому из атомов углерода при двойной связи присоединится
протон водорода на стадии образования σ-комплекса: в молекуле
этена (и других симметричных алкенов) оба углерода при двойной связи равноценны. В случае же этих реакций в несимметричных алкенах возможно образование двух продуктов присоединения, а значит, возникает проблема: какой из них будет преобладающим. Рассмотрим это на примере гидрохлорирования пропена:
CH3 -CH-CH 3
CH3 -CH=CH2 + HCl
пропен
Cl
2-хлорпропан
CH3 -CH2 -CH2 -Cl
1-хлорпропан
Теоретически, в реакции гидрохлорирования пропена могут
образоваться 1-хлорпропан и 2-хлорпропан, однако на практике в
основном получается 2-хлорпропан. Эта реакция протекает по
правилу Марковникова: при присоединении реагентов типа
H+X- (например, HCl, HBr, HOH) к несимметричным алкенам
протон водорода присоединяется к более гидрогенизированному
атому углерода при двойной связи.
Чтобы объяснить такой порядок присоединения, необходимо рассмотреть статический и динамический факторы.
Статический фактор – это распределение электронной
плотности в нереагирующей молекуле.
Атом углерода при двойной связи sp2δ+
δCH3
CH=CH2
гибридизован, он более электроотрицателен, чем
3
sp -гибридизованный углерод метильной группы. Поэтому электронная плотность связи CH3-CH= смещена к углероду с двойной
связью. Так как π-связь поляризуется легче, чем σ-связь, электро26
27.
ны π-связи смещаются к соседнему атому углерода, т.е. происходит её поляризация. В конечном итоге на первом атоме углеродавозникает частичный отрицательный заряд (δ-), а на втором –
частичный положительный (δ+). Конечно же, протон водорода
(положительно заряженная частица) будет направляться к тому
углероду, где есть избыток электронной плотности, а он и есть
более гидрогенизированный атом углерода при двойной связи.
Теперь рассмотрим динамический фактор – сравним стабильность промежуточных частиц (карбкатионов). Вы уже знаете: чем стабильнее промежуточная частица (интермедиат), тем
больше вероятность её образования.
CH3 -CH=CH2 + H
+
CH3 -CH
+
CH3 -CH-CH 3
вторичный карбкатион
изопропил
+
CH3 -CH2 -CH2
CH2
H+
π-комплекс
первичный карбкатион
пропил
На стадии образования σ-комплекса теоретически могут образоваться два карбкатиона – пропил (первичный) и изопропил
(вторичный). Атом углерода с полным положительным зарядом
смещает к себе электронную плотность соседней (или соседних)
C-C σ-связи, уменьшая собственный недостаток электронной
плотности. Вторичный карбкатион более стабилен из-за большей
компенсации положительного заряда на углероде:
+
CH3 CH CH3
изопропил
+
CH3 CH2 CH2
пропил
Если рассуждать о стабильности карбкатионов вообще, то
самыми стабильными являются третичные, затем – вторичные,
первичные, а самым нестабильным – катион метил:
R
R
C+ > R
+
CH > R
1o
R
3o
наиболее
стабилен
R
2o
+
+
CH2 > CH3
наименее
стабилен
27
28.
Так как в рассматриваемом примере более стабильным является изопропил-катион, именно он в первую очередь сможетпровзаимодействовать с анионом хлора, а значит преобладающим
продуктом этой реакции является 2-хлорпропан (т.е. протон водорода присоединился к более гидрогенизированному атому углерода):
Cl +
CH3 -CH-CH 3
CH3 -CH-CH 3
изопропил
Cl
2-хлорпропан
Классическое правило Марковникова можно без оговорок
применять только к самим алкенам. В случае их производных
нужно обязательно рассматривать статический и динамический
факторы, влияющие на ход реакции (например, позже мы будем
рассматривать реакции электрофильного присоединения в акриловой кислоте, которые протекают против правила Марковникова).
В современной интерпретации правило Марковникова формулируется так: направление присоединения реагентов типа HX к
производным алкенов определяется относительной устойчивостью промежуточных карбкатионов.
Реакция гидрирования алкенов протекает по механизму радикального присоединения в присутствии катализаторов (никель,
платина), на поверхности которых происходит гомолитический
разрыв молекулы водорода. В результате гидрирования алкенов
образуются алканы:
CH2 =CH2 + H2
этен
Ni, to
CH3 -CH3
этан
Реакции окисления алкенов протекают с образованием различных продуктов в зависимости от условий. Мы рассмотрим
только так называемое мягкое окисление алкенов. Его проводят
разбавленным раствором перманганата калия при комнатной
температуре (реакция Вагнера).
28
29.
В результате реакции образуются гликоль (двухатомныйспирт) и диоксид марганца:
CH2 -CH2 + MnO2 + KOH
CH2 =CH2 + KMnO4 + H2 O
этен
OH OH
этиленгликоль
Эту реакцию используют как качественную на наличие
двойной связи: в результате исчезает малиновое окрашивание
перманганата калия и появляется бурый осадок диоксида марганца.
Реакции присоединения к алкадиенам
Алкадиенами (или просто диенами) называют углеводороды, содержащие две двойные связи. По взаимному расположению двойных связей их классифицируют на:
- кумулированные, в молекулах которых две двойные связи
CH2=C=CH-CH3
расположены рядом, например
бутадиен-1,2
- сопряжённые, в молекулах которых двойные связи
чередуются с одинарными, например CH2=CH-CH=CH2
бутадиен-1,3
- изолированные, в молекулах которых две двойные
связи разделены двумя или более одинарными, например CH2=CH-CH2-CH=CH2
пентадиен-1,4
Реакционная способность кумулированных и изолированных алкадиенов сходна с реакционной способностью алкенов:
каждая двойная связь вступает в реакции присоединения независимо от другой. Единственное отличие этих типов алкадиенов состоит в том, что они могут присоединить два моля реагента. Например, при взаимодействии пентадиена-1,4 с хлороводородом
сначала образуется 4-хлорпентен-1, а затем 2,4-дихлорпентан:
CH2 =CH-CH2 -CH=CH2
пентадиен-1.4
HCl
CH3 -CH-CH 2 -CH=CH2
Cl
4-хлорпентен-1
HCl
CH3 -CH-CH 2 -CH-CH 3
Cl
Cl
2,4-дихлорпентан
29
30.
Сопряжённые алкадиены вступают в реакции электрофильного присоединения особенным образом. Для них характерно такназываемое 1,2- и 1,4-присоединение.
CH3 -CH-CH=CH 2
1
2
3
4
CH2 =CH-CH=CH2
HBr
Br
3-бромбутен-1
(продукт 1,2-присоединения)
CH3 -CH=CH-CH 2 -Br
1-бромбутен-2
(продукт 1,4-присоединения)
При 1,2-присоединении реакция протекает за счёт разрыва
одной π-связи (между С-1 и С-2). При 1,4-присоединении реакция
идёт по концам сопряжённой системы (т.е. к С-1 и С-4) с разрывом обеих π-связей и образованием новой π-связи. Эта особенность в реакциях присоединения связана с тем, что в сопряжённых алкадиенах существуют так называемые сопряжённые системы (подробнее об этом – в следующем разделе).
СОПРЯЖЁННЫЕ СИСТЕМЫ. АРОМАТИЧНОСТЬ.
ЭЛЕКТРОННЫЕ ЭФФЕКТЫ. РЕАКЦИИ
ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ В БЕНЗОЛЕ
И ЕГО ПРОИЗВОДНЫХ
Сопряжение – это выравнивание связей и зарядов в реальной молекуле (или частице) по сравнению с идеальной, но несуществующей структурой. Можно дать и другое определение этому явлению: это процесс дополнительного взаимодействия
p-электронных орбиталей, приводящий к делокализации электронной плотности.
Рассмотрим пример сопряжённой системы – молекулу бутадиена-1,3.
CH2=CH-CH=CH2
Атомы углерода в молекуле бутадиенаH
2
H
H 1,3 sp -гибридизованы. Это значит, что
2
H 1
3
4 H все σ-связи расположены в одной плосH
кости под углом 120о. p-Электронная орбиталь каждого атома углерода расположена перпендикулярно
30
31.
плоскости σ-связей. Вы уже знаете, что за счёт бокового перекрывания p-орбиталей образуются π-связи. В соответствии сформулой бутадиена-1,3 мы можем предположить, что одна πсвязь образуется за счёт перекрывания р-орбиталей С-1 и С-2, а
вторая – С-3 и С-4. Но посмотрите на приведенный выше рисунок: почему р-орбитали С-2 и С-3 не смогут перекрыться? Действительно, происходит их дополнительное перекрывание, и на самом деле в молекуле бутадиена-1,3 не существует двух отдельных областей с повышенной электронной плотностью (две πсвязи), а образуется общее электронное облако, содержащее 4
электрона и принадлежащее четырём атомам углерода. Поэтому
реальную структуру бутадиена-1,3 можно изобразить следующим
образом:
CH2=CH-CH=CH 2
.... ....
или CH....
2 CH CH CH 2
Все углерод-углеродные связи в молекуле выравнены и в
ней практически нет одинарных и двойных связей, а есть общее
делокализованное электронное облако. Это и есть выравнивание
связей в реальной молекуле по сравнению с идеальной, но несуществующей, строение которой мы записали как
CH2=CH-CH=CH2 .
Сопряжение – это энергетически выгодный для молекулы
процесс. Вы знаете, что при образовании связи (т.е. при взаимодействии атомных орбиталей) выделяется энергия, значит, внут ренняя энергия системы понижается , и она становится более стабильной. Так как при сопряжении происходит дополнительное перекрывание орбиталей, то выделяется и дополнительная энергия, и система становится ещё более стабильной. Энергия, выделяющаяся в результате сопряжения, называется энергией сопряжения. Количественно – это разность энергий соединения с сопряжёнными двойными связями и соединения с изолированными двойными связями (где сопряжение невозможно). Для
бутадиена-1,3 она составляет 15 кДж/моль. Чем больше атомов
участвует в сопряжении, тем выше энергия сопряжения, тем стабильнее молекула. Поэтому в природе распространены соединения, молекулы которых содержат длинные сопряжённые цепи
(например, каротиноиды, см. стр. 232).
31
32.
Бутадиен-1,3 является примером π,π-сопряжённой системы, т.к. в сопряжении участвуют электроны π-связей. Признакомπ,π-сопряжённой системы является чередование одинарных и
двойных связей в молекуле.
Другим видом сопряжения является p,π-сопряжение. В
p,π-сопряжении участвуют электронные облака π-связей и
p-орбитали, причём на p-орбитали может находиться 1 электрон,
неподелённая электронная пара или эта орбиталь может быть вакантной. Рассмотрим конкретные примеры.
Это электронное строение молекулы винила.
. H
мина CH2=CH-NH2 . Атомы углерода и азота
H
..
H
C C
sp2-гибридизованы, т.е. все σ-связи лежат в
H
N
H одной плоскости, а негибридные p-орбитали
перпендикулярны этой плоскости. Происходит перекрывание всех трёх р-орбиталей с образованием делокализованного электронного облака; 4 электрона (т.к. у азота на негибридной орбитали неподелённая электронная пара) принадлежат трём атомам сопряжённой системы. На формуле виниламина
это можно обозначить так:
..
CH2=CH-NH2
Количество
электронов,
делокализованных
в
р,π-сопряжённой системе, не всегда совпадает с количеством
атомов, принимающих участие в сопряжении, например:
H
H
.
C
.
H
.
C
H
C
H
H
аллил-радикал
.
H
CH2=CH-CH2
(в сопря жённой системе
делокализовано
3 электрона)
.
.
C
C
H
..
H
H
H
C
H
аллил-анион
CH2=CH-CH2
(в сопря жённой системе
делокализовано
4 электрона)
.
.
C
C
H
H
C
H
аллил-катион
+
CH2=CH-CH2
(в сопря жённой системе
делокализовано
2 электрона)
Признаком
р,π-сопряжённой
системы
является
sp -гибридизация атомов, т.е. если в молекуле есть фрагмент, в
котором три или более атомов подряд sp2-гибридизованы – этот
фрагмент является р,π-сопряжёной системой.
2
32
33.
Во всех сопряжённых системах, которые мы рассмотрели,можно выделить начало и конец, поэтому их называют открытыми сопряжёнными системами. А теперь рассмотрим так называемые замкнутые сопряжённые системы. Примером является молекула бензола.
Формула Кекуле показывает, что бензол – это ненасыщенный углеводород, в молекуле которого есть три πсвязи.
Если же мы рассмотрим электронное строение бензола, то
увидим, что это π.π-сопряжённая система (атомы углерода sp2гибридизованы, все σ-связи лежат в одной плоскости, а pорбитали расположены перпендикулярно этой плоскости и перекрываются не попарно, а образуя общее электронное облако, в
котором делокализовано 6 электронов).
Над и под плоскостью цикла располагаются электронные
облака, так называемые «электронные бублики». Чтобы выразить
это, используют другую формулу бензола:
Длины С-С-связей в бензоле выравнены и равны 0,140 нм
(сравните: длина одинарной С-С-связи – 0,154 нм, двойной С=Ссвязи – 0,134 нм).
Замкнутые сопряжённые системы более стабильны, чем открытые, т.к. энергия сопряжения замкнутых сопряжённых систем
выше (в случае бензола – это 151 кДж/моль).
33
34.
Бензол называют ароматическим соединением. Ароматичность – это совокупность свойств замкнутых сопряжённыхсистем, проявляющаяся в их высокой устойчивости к реакциям присоединения и окисления.
Ароматическими свойствами обладает не только бензол.
Соединения, даже абсолютно не похожие по строению на бензол,
также могут быть ароматическими, если они удовлетворяют
квантово-механическим критериям ароматичности. Эти критерии следующие:
1. Наличие плоского цикла (все атомы цикла должны быть
sp2-гибридизованы).
2. Наличие замкнутой сопряжённой системы (все атомы
должны участвовать в сопряжении).
3. Выполнение правила Хюккеля. Число делокализованных
электронов должно быть равно (4n+2), где n – это любое
целое число.
(Например, в случае бензола 4n+2=6, n=1).
Ароматическими являются, например, нафталин, антрацен,
фенантрен:
нафталин
4n+2=10
n=2
антрацен
4n+2=14
n=3
фенантрен
4n+2=14
n=3
Химические свойства бензола
Бензол является ненасыщенным соединением, но мы выяснили, что в его структуре нет двойных связей, а есть ароматическая связь – делокализованное электронное облако. Типичные реакции непредельных углеводородов – электрофильное присоединение и окисление – для бензола не характерны. Так, он не
обесцвечивает бромную воду, не даёт реакции Вагнера (окисление раствором перманганата калия при комнатной температуре).
Для бензола характерны реакции, не приводящие к нарушению
замкнутой сопряжённой системы, – реакции замещения. Чтобы
34
35.
выяснить, какой тип замещения (радикальное, электрофильное,нуклеофильное) характерен для бензола, вспомните его электронное строение: σ-скелет молекулы плоский, а над и под плоскостью расположено ароматическое облако. Чтобы провзаимодействовать с этим ароматическим облаком, реагент должен быть
электрофильным. Итак, для бензола (и ароматических соединений вообще) характерны реакции электрофильного замещения.
Примерами реакций SE являются:
NO 2
+ H2 O
HNO3 , H2 SO4
Cl2 , AlCl3
Cl
CH3 Cl, AlCl3
CH3
SO3 ( H2 SO4 )
SO3 H
реакция нитрования
реакция галогенирования
+ HCl
реакция алкилирования
+ HCl
реакция сульфирования
Рассмотрим общий механизм реакций электрофильного замещения в бензоле.
H
+ E+ Nu
H
-Nu
π - комплекс
E+
+
H
E Nu
E
+ H+ Nu
σ- комплекс
На первой стадии электрофил подходит к молекуле бензола
и взаимодействует со всем ароматическим облаком (они притягиваются друг к другу). Образуется π-комплекс. Для образования
новой ковалентной связи углерод-электрофил необходима пара
электронов. Электрофил вырывает её из ароматического облака,
образуется σ-комплекс. Он не является замкнутой сопряжённой
системой, т.к. атом углерода, образовавший новую σ-связь, перешёл в sp3-гибридизацию (он вышел из плоскости и больше не
35
36.
имеет негибридной pz-орбитали). Остальные пять атомов углерода продолжают участвовать в сопряжении, образуя общее электронное облако, в котором делокализовано четыре электрона(6-2=4), поэтому положительный заряд в σ-комплексе обозначается не на конкретном атоме углерода, а в центре разомкнутого
кольца. Итак, σ-комплекс не является ароматической структурой.
Для того чтобы вернуть ароматичность, ему необходимо отщепить протон водорода (H+). Его забирает оставшийся в реакционной среде нуклеофил (Nu-). Два электрона связи C-H возвращаются в ароматическое облако, атом углерода вновь становится
sp2-гибридизованным и может участвовать в сопряжении.
Лимитирующей стадией реакции электрофильного замещения является стадия образования σ-комплекса, т.к. при этом происходит потеря ароматичности, что требует затрат энергии.
Различные реакции электрофильного замещения в бензоле
протекают по общему механизму и отличаются только стадией
образования электрофильной частицы.
Реакция нитрования бензола протекает под действием смеси концентрированных азотной и серной кислот (схему реакции
см. выше). Рассмотрим её механизм.
..
H2 SO4 + HO-NO 2
кислота основание
H +
O-NO 2
H
протонированная
азотная кислота
+
H2 O + NO 2
катион
нитрония
На первой стадии реакции азотная кислота взаимодействует
с серной. В данном случае азотная кислота выполняет роль основания, принимая протон от молекулы серной кислоты (по теории
Бренстеда, кислота – это молекула или ион, отдающие протон, а
основание – молекула или ион, принимающие протон водорода).
Образуется протонированная азотная кислота, которая, отщепляя
молекулу воды, превращается в катион нитрония, или нитронийкатион. Это и есть электрофильная частица. Таким образом, серная кислота выполняет роль катализатора, принимая участие в
образовании электрофильного реагента. Вторая роль серной кислоты – это роль водоотнимающего средства. Воду необходимо
36
37.
отводить из сферы реакции, чтобы сместить её равновесие вправо.После образования электрофила – катиона нитрония - реакция протекает по общему механизму, через образование π- и
σ-комплексов:
H +
NO 2
H
+
+ NO 2
+
H
NO 2 + HSO4
σ-комплекс
π -комплекс
NO 2
+ H2 SO4
нитробензол
Обратите внимание: на стадии превращения σ-комплекса в
нитробензол (стадии возвращения ароматичности) протон водорода отщепляется под действием аниона серной кислоты, при
этом вновь образуется серная кислота, что доказывает, что она
являлась катализатором данной реакции.
Катализатором реакции галогенирования являются так называемые кислоты Льюиса (по теории Льюиса, кислоты – это нейтральные молекулы или ионы, способные принимать пару электронов): FeCl3, FeBr3, AlCl3, AlBr3 и т.п. Катализатор необходим,
чтобы поляризовать молекулу галогена. Кислота Льюиса смещает
на себя неподелённую электронную пару хлора, образуя комплекс, в котором на одном из атомов хлора сосредоточивается
частичный положительный заряд:
δ−
δ+ Cl
Cl Cl : + Fe Cl
слабый электрофил
Cl
(неполя рная молекула)
δ+
Cl
Cl :...... FeCl3
более сильный электрофил
(присутствует частичный
положительный заря д)
Далее молекула бензола взаимодействует с образовавшимся
комлексом как с электрофилом:
H
δ+
+ Cl
H δ+
Cl
δ−
Cl : ......FeCl
3
δ−
Cl : ......FeCl3
- FeCl4-
π-комплекс
+
H
Cl
σ-комплекс
+ FeCl4
Cl
+ HCl + FeCl3
хлорбензол
37
38.
На стадии образования π-комплекса происходит дальнейшаяполяризация связи Cl-Cl, и она разрывается гетеролитически,
причём Cl+ сразу участвует в образовании σ-комплекса.
Аналогично протекают реакции алкилирования (реакция
Фриделя-Крафтса).
δ−
δ+ Cl
CH3 -Cl : + Fe
Cl
δ+
CH3
Cl : ......FeCl3
Cl
H
δ+
CH
+
3
H δ+
CH3
δ−
Cl :...... FeCl
3
δ−
Cl :...... FeCl3
- FeCl4-
π-комплекс
+
H
+ FeCl4
CH3
σ-комплекс
CH3
+ HCl + FeCl3
толуол
Связь C-Cl в метилхлориде недостаточно полярна, чтобы
разорваться гетеролитически. Под действием кислоты Льюиса
увеличивается частичный положительный заряд на атоме углерода, и комплекс реагента с катализатором является более сильным
электрофилом, чем исходный метилхлорид.
Реакция сульфирования бензола протекает под действием
олеума (раствор серного ангидрида SO3 в концентрированной
серной кислоте).
Молекула серного ангидрида является электрофилом изδ+ O
S O за большого по величине частичного положительного заO ряда на атоме серы.
H
H
+ SO3
π-комплекс
δ+ O
S O
O
H
+
S
σ-комплекс
O
O
O
SO3H
бензолсульфокислота
При образовании π-комплекса связь S=O (в первую очередь
π-связь) поляризуется и разрывается по гетеролитическому типу,
поэтому при образовании σ-комплекса на атоме кислорода возникает полный отрицательный заряд. Для восстановления арома38
39.
тичности протон водорода отщепляется от атома углерода кольцаи переходит к отрицательно заряженному кислороду. Образуется
бензолсульфокислота.
Когда мы рассматриваем реакции электрофильного замещения в бензоле, перед нами не стоит вопрос, в каком положении
протекает реакция, т.к. все атомы углерода абсолютно равноценны. Другое дело, если в бензольном кольце уже есть заместитель.
В этом случае в результате электрофильного замещения принципиально возможно образование трёх изомеров:
X
E
орто-изомер
X
X
+ E+
мета-изомер
E
X
пара-изомер
E
Чтобы ответить на вопрос, какой из этих возможных продуктов является преобладающим, необходимо рассматривать
электронные эффекты заместителя.
Отвлечёмся от реакций электрофильного замещения в бензоле и его производных и рассмотрим электронные эффекты в
целом.
Взаимное влияние атомов в молекулах органических
соединений. Электронные эффекты
Атомы и атомные группы в молекулах органических соединений влияют друг на друга, причём не только атомы, непосредственно связанные друг с другом. Это влияние каким-то образом
передаётся по молекуле. Передача влияния атомов в молекулах
за счёт поляризации связей называется электронными эффектами. Существует два вида электронных эффектов: индуктивный и мезомерный эффект.
39
40.
Индуктивный эффект - это передача влияния заместителейпо цепи σ-связей за счёт их поляризации. Индуктивный эффект
обозначается символом I. Рассмотрим его на примере
1-хлорбутана:
δ−
δ+'
δ+
δ+'''
Связь C-Cl полярна из-за более
δ+''
CH3 CH 2 CH 2 CH 2 Cl
высокой электроотрицательности хлора. На атоме углерода возникает частичный положительный заряд (δ+). Электронная пара следующей σ-связи смещается
в сторону электронодефицитного атома углерода, т.е. поляризуется. За счёт этого на следующем атоме углерода также возникает
частичный положительный заряд (δ+’) и т.д. Таким образом, хлор
индуцирует поляризацию не только «собственной» σ-связи, но и
последующих в цепи. Обратите внимание, что каждый последующий частичный положительный заряд по величине меньше
предыдущего (δ+>δ+’>δ+’’>δ+’’’), т.е. индуктивный эффект передаётся по цепи с затуханием. Это можно объяснить низкой поляризуемостью σ-связей. Принято считать, что индуктивный эффект распространяется на 3-4 σ-связи. В приведенном примере
атом хлора смещает электронную плотность по цепи связей на
себя. Такой эффект называют отрицательным индуктивным эффектом и обозначают –ICl.
Большинство заместителей проявляют отрицательный индутивный эффект, т.к. в их структуре есть атомы, более электроотрицательные, чем водород (индуктивный эффект водорода принят равным нулю). Например: -F, -Cl, -Br, -I, -OH, -NH2, -NO2,
-COOH, >C=O .
CH3
CH 2
CH3
OH
CH 2
-I OH
CH 2
CH 2
COOH
-I COOH
Если заместитель смещает электронную плотность по цепи
σ-связей от себя, он проявляет положительный индуктивный
эффект (+I). Например:
CH3
40
CH 2
O -Na +
+I O -
41.
Кислород с полным отрицательным зарядом проявляет положительный индуктивный эффект.CH2
CH
CH3
В молекуле пропена углерод метильной группы sp3гибридизован, а атомы углерода при двойной связи sp2гибридизованы, т.е. более электроотрицательны. Поэтому метильная группа смещает электронную плотность от себя, проявляя положительный индуктивный эффект (+ICH3) .
Итак, индуктивный эффект может проявляться в любой молекуле, в которой есть разные по электроотрицательности атомы.
Мезомерный эффект – это передача электронного влияния
заместителей в сопряжённых системах, посредством поляризации
π-связей. Мезомерный эффект передаётся без затухания, т.к. πсвязи поляризуются легко. Обратите внимание: мезомерным эффектом обладают только те заместители, которые сами являются
частью сопряжённой системы. Например:
CH2 =CH-CH=CH-NH 2
p,π-сопряжённая система
CH2=CH-CH=CH-CH2-NH2
π,π-сопря жённая система
Амино-группа является частью
p,π-сопряжённой системы.
Амино-группа не является частью сопряжённой системы.
Мезомерный эффект отсутствует.
Мезомерный эффект может быть как положительный (+М),
так и отрицательный (-М).
В молекуле хлорвинила неподелённая электрон..
ная пара хлора участвует в p,π-сопряжении, т.е.
CH2=CH-Cl
вклад хлора в сопряжённую систему больше, чем
+M Cl
у каждого из атомов углерода. Поэтому хлор проявляет положительный мезомерный эффект.
Молекула акрилового альдегида – это
O
π.π-сопряжённая система. Атом кислорода отдаёт
CH2 =CH-C
-M CHO
H
41
42.
в сопряжение один электрон – столько же, как и каждый атом углерода, но при этом электроотрицательность кислорода выше,чем у углерода, поэтому кислород смещает электронную плотность сопряжённой системы на себя, альдегидная группа в целом
проявляет отрицательный мезомерный эффект.
Итак, заместители, отдающие в сопряжение два электрона,
обладают положительным мезомерным эффектом. К ним относятся:
а) заместители с полным отрицательным зарядом, например, –O-;
б) заместители, в структуре которых есть атомы с неподелёнными электронными парами на pz-орбитали, например: -NH2, -OH,
-F, -Cl, -Br-, -I, -OR (-OCH3, -OC2H5).
Заместители, смещающие электронную плотность по сопряжённой системе на себя, проявляют отрицательный мезомерный эффект. К ним относятся заместители, в структуре которых
есть двойные связи, например:
C
H
O
O
O
,
C
OH
,
S
O
O
OH ,
N
O
.
Заместитель может проявлять и индуктивный, и мезомерный эффекты одновременно. В некоторых случаях направление
этих эффектов совпадает (например, -I и –M), в других – они действуют в противоположных направлениях (например, -I и +M).
Как в этих случаях определить общее влияние заместителя на остальную часть молекулы (другими словами, как определить, является данный заместитель электронодонорным или электроноакцепторным)? Заместители, повышающие электронную плотность в остальной части молекулы, называют электронодонорными, а заместители, понижающие электронную плотность в
остальной части молекулы, – электроноакцепторными.
Чтобы определить общее влияние заместителя, необходимо
сравнить его электронные эффекты по величине. Если преобладает положительный по знаку эффект, заместитель является электронодонорным. Если преобладает отрицательный по знаку эффект, заместитель является электроноакцепторным. Нужно отметить, что, как правило, мезомерный эффект проявляется сильнее,
42
43.
чем индуктивный (из-за большей способности π-связей к поляризации). Однако есть и исключения из этого правила: индуктивный эффект галогенов проявляется сильнее, чем мезомерный.Рассмотрим конкретные примеры:
В этом соединении
..
-I NH , +M NH ; |+M| > |-I | аминогруппа являетCH2=CH NH 2
2
2
ся электронодонорным заместителем, т.к. её положительный мезомерный эффект
проявляется сильнее, чем отрицательный индуктивный.
В этом соединении аминогруппа явля-I NH
CH3-CH2 NH 2
2
ется электроноакцепторным заметителем, т.к. проявляет только отрицательный индуктивный эффект.
В молекуле фенола гидроксильная группа является
..
OH
электронодонорным
за-IOH
, +MOH
; |+M| > |-I |
местителем из-за преобладания положительного мезомерного эффекта над отрицательным индуктивным.
В молекуле бензилового спирта гидроCH2 OH
ксильная группа не участвует в сопряжении
-IOH и проявляет только отрицательный индуктивный эффект. Поэтому она является электроноакцепторным
заместителем.
Эти примеры показывают, что нельзя рассматривать влияние какого-либо заместителя вообще, а нужно рассматривать его
влияние в конкретной молекуле.
Только галогены всегда являются электроноакцепторными
заместителями, т.к. их отрицательный индуктивный эффект проявляется сильнее, чем положительный мезомерный. Например:
..
Cl
CH2 Cl
-ICl
, +MCl
; |+M| < |-I |
-ICl
А теперь вернёмся к реакциям электрофильного замещения
в производных бензола. Итак, мы выяснили, что заместитель, уже
имеющийся в кольце, влияет на ход реакций электрофильного
замещения. В чём же выражается это влияние?
43
44.
Заместитель влияет на скорость реакций SE и на положение второго заместителя, вводимого в кольцо. Рассмотрим обаэтих аспекта влияния.
Влияние на скорость реакции. Чем выше электронная
плотность в кольце, тем легче протекают реакции электрофильного замещения. Понятно, что электронодонорные заместители
облегчают реакции SE (являются активаторами цикла), а электроноакцепторные заместители – затрудняют их (дезактивируют
цикл). Поэтому реакции электрофильного замещения в производных бензола, содержащих электроноакцепторные заместители,
проводят в более жёстких условиях.
Сравним активность фенола, толуола, бензола, хлорбензола
и нитробензола в реакции нитрования.
O
..
OH
|+M| > |-I |
..
Cl
CH3
+I
N
O
|+M| < |-I |
-I , -M
содержат электроноакцепторные
заместители
содержат электронодонорные
заместители
Так как фенол и толуол содержат электронодонорные заместители, они более активны в реакциях SE, чем бензол. Наоборот,
хлорбензол и нитробензол менее активны в этих реакциях, чем
бензол, т.к. содержат электроноакцепторные заместители. Фенол
активнее толуола из-за положительного мезомерного эффекта
OH-группы. Хлор не такой сильный электроноакцепторный заместитель, как нитрогруппа, т.к. нитрогруппа проявляет и отрицательный индуктивный, и отрицательный мезомерный эффекты.
Итак, в данном ряду активность в реакциях электрофильного замещения убывает от фенола к нитробензолу. Экспериментально
установлено, что если скорость реакции нитрования бензола принять за 1, то этот ряд будет выглядеть так:
фенол
толуол бензол хлорбензол нитробензол
скорость
1000
нитрования
44
24,5
1,0
0,033
0,0000001
45.
Вторым аспектом влияния заместителя в ароматическомкольце на ход реакций электрофильного замещения является так
называемое ориентирующее действие заместителей. Все заместители можно подразделить на две группы: орто-, параориентанты (заместители 1 рода) и мета-ориентанты (заместители 2 рода).
К заместителям 1 рода относятся: -OH, -O-, -NH2, алкильные группы
(-CH3, -C2H5 и т.д.) и галогены. Вы можете видеть, что все эти заместители проявляют положительный индуктивный эффект и (или) положительный мезомерный эффект. Все
они, кроме галогенов, повышают электронную плотность в кольце, особенно в орто- и пара-положениях. Поэтому электрофил и
направляется в эти положения. Рассмотрим это на примере фенола:
:OH
За счёт положительного мезомерного эффекта
δ− гидроксильной группы происходит перераспредеδ−
ление электронной плотности по сопряжённой
системе, и в орто- и пара-положениях она особенδ−
но повышена.
При бромировании фенола образуется смесь орто- и парабромфенола:
:OH
OH
Br
δ−
δ−
OH
o-бромфенол
δ−
+ HBr
+
+ Br2
Br
п-бромфенол
Если бромирование проводить в полярном растворителе
(бромной водой) и использовать избыток брома, реакция протекает сразу по трём положениям:
OH
OH
+ 3 Br2
H2O
Br
Br
+ 3 HBr
Br
2,4,6-трибромфенол
45
46.
Заместителями 2 рода являются: -NH3+, -COOH, -CHO(альдегидная группа), -NO2, -SO3H. Все эти заместители понижают электронную плотность в ароматическом кольце, но из-за её
перераспределения в мета-положениях она понижена не так
сильно, как в орто- и пара-. Рассмотрим это на примере бензойной кислоты:
Карбоксильная группа проявляет отрицательный
O
индуктивный и отрицательный мезомерный эффекC
OH ты. За счёт перераспределения по сопряжённой
системе в мета-положениях электронная плотность
δ−
δ− остаётся более высокой, чем в орто- и пара-, поэтому электрофил будет атаковать мета-положения:
O
O
C
C
OH
δ−
OH
+ Cl2
δ−
FeCl3
to
+ HCl
Cl
м-хлорбензойная кислота
Реакции электрофильного замещения в нафталине
Мы уже выяснили, что нафталин является ароматической
системой. Однако электронная плотность в системе нафталина
распределена не столь равномерно, как в бензоα
α
β ле: она выше в α-положениях, поэтому реакции
β
электрофильного замещения протекают именно
β
β
по α-положениям. Например, при бромировании
α
α
нафталина образуется α-бромнафталин:
Br
+ Br2
CH3 COOH
+ HBr
α-бромнафталин
46
47.
Реакция сульфирования нафталина может давать разныепродукты в зависимости от условий: при более низкой температуре (80оС) образуется α-нафталинсульфокислота, а при температуре около 160оС – β-нафталинсульфокислота.
SO3 H
80o C
+ H2 O
α-нафталинсульфокислота
+ H2 SO4
SO3 H
160o C
+ H2 O
β-нафталинсульфокислота
Реакции окисления гомологов бензола
Сам бензол очень устойчив к реакциям окисления. Однако
его гомологи окисляются относительно легко по боковой цепи. В
качестве окислителя обычно используют перманганат калия в кислой среде.
Например, при окислении толуола образуется бензойная кислота:
CH3
COOH
KMnO4
to
толуол
бензойная кислота
Обратите внимание, что и другие гомологи бензола (этилбензол, пропилбензол и т.д.) окисляются тоже в бензойную кислоту, т.е. окисление протекает по α-положению боковой цепи:
CH2 -CH2 -CH3
COOH
KMnO4
to
пропилбензол
+ CH3 COOH
бензойная кислота
47
48.
ХИМИЧЕСКИЕ СВОЙСТВА ГАЛОГЕНАЛКАНОВ,СПИРТОВ И ФЕНОЛОВ
Функциональной группой галогеналканов является атом
галогена (фтор, хлор, бром или иод). Отсюда первый признак их
классификации – по природе галогена:
CH3-I
иодметан
CH3-CH2-Br
бромэтан
Другой вид классификации – по положению атома галогена
(по характеру атома углерода, связанного с галогеном). Галогеналканы классифицируют на первичные, вторичные и третичные:
CH3
CH3-CH2-Cl
CH3-CH-CH3
CH3-C-CH2-CH3
хлорэтан
(первичный
галогеналкан)
Br
2-бромпропан
(вторичный
галогеналкан)
I
2-иод-2-метилбутан
(третичный
галогеналкан)
Для названия галогеналканов используют радикалофункциональную (для несложных молекул) и заместительную
номенклатуру IUPAC.
По радикало-функциональной номенклатуре название галогеналкана состоит из названия соответствуюшего углеводородного радикала и слова хлорид, бромид и т.п. Например:
CH3-CH2-Cl
CH3-CH-CH3
этилхлорид
Br
изопропилбромид
По заместительной номенклатуре галогеналкан рассматривают как производное соответствующего углеводорода. Наличие
галогена обозначают префиксом (если их несколько, используют умножающие частицы ди-, три-, тетра-) и обозначают положение цифрой.
48
49.
Например:CH3
CH3-C-CH2-CH3
CH2-CH2
Br
2-бром-2-метилбутан
Cl Cl
1,2-дихлорэтан
Для галогеналканов характерны реакции нуклеофильного
замещения (SN). Это связано с распределением электронной
плотности в молекуле.
δ+
δ− Хлор смещает электронную плотность на себя
CH3 -CH 2 Cl
вследствие более высокой электроотрицательности.
На атоме углерода возникает частичный положительный заряд.
Этот электронодефицитный углерод является электрофильным
центром и может быть атакован каким-либо нуклеофилом.
С помощью реакций нуклеофильного замещения можно
превратить галогеналканы в спирты, тиолы, амины, простые
эфиры:
R-NH2 + HCl
первичный
амин
R-NH-R' + HCl
вторичный
амин
R-SH + NaCl
тиол
(тиоспирт)
Na+OH
NH3
R'-NH2
R-Cl
H2O
- +
R'-O Na
Na HS
R-OH + NaCl
спирт
R-OH + HCl
спирт
R-O-R' + NaCl
простой
эфир
Общая схема реакции нуклеофильного замещения может быть представлена следующим образом:
R :X
субстрат
+
:Nu
нуклеофил
R : Nu
продукт
реакции
+: Х
уходя щая
группа
Связь углерод-галоген разрывается гетеролитически. Новая связь
образуется за счет электронной пары нуклеофила (нуклеофил замещает галоген в молекуле галогеналкана). Частица Х- уходит, унося электроны
бывшей σ-связи. Эту частицу называют «уходящей группой». Реакции нулеофильного замещения часто бывают обратимыми, т.к. любая уходящая
группа также является нуклеофилом. Чтобы сместить равновесие реакции
вправо, необходимы «хороший» нуклеофил и «хорошая» уходящая группа.
49
50.
«хороший» нуклеофил – это активный нуклеофил. Как же сравнить силуразличных нуклеофилов?
1) Анионы более активные нуклеофилы, чем соответствующие нейтральные молекулы, т.к. анионы легче отдают электроны. Например, анион
OH- является более сильным нуклеофилом, чем нейтральная молекула воды H2O. Алкоголят-анион R-O- активнее, чем молекула спирта R-OH, тиолят-анион R-S- активнее, чем молекула тиола R-SH.
2) Чем ниже электроотрицательность атома нуклеофильного центра,
тем активнее нуклеофил, т.к. такой атом легче отдает электронную пару на
образование новой σ-связи. Поэтому молекула аммиака NH3 является более сильным нуклеофилом, чем молекула воды H2O (электроотрицательность азота ниже, чем кислорода).
«Хорошая» уходящая группа, наоборот, должна быть слабым нуклеофилом. Например, нейтральная молекула воды лучшая уходящая группа, чем гидроксид-анион. Анионы галогенов являются «хорошими» уходящими группами вследствие высокой электроотрицательности атомов галогена.
Реакции нуклеофильного замещения в галогеналканах могут
конкурировать с реакциями отщепления (элиминирования).
Например, при нагревании галогеналкана с водным раствором щелочи протекает реакция нуклеофильного замещения:
CH3-CH2-Cl + NaOH
хлорэтан
H2 O
to
CH3-CH2-OH + NaCl (SN)
этанол
Если же в реакции использовать не водный, а спиртовой
раствор щелочи, протекает реакция элиминирования с образованием соответствующего алкена:
CH3-CH2-Cl + NaOH
хлорэтан
спирт
to
CH2=CH2 + NaCl + H2O (E)
этен
Обратите внимание, что реакции элиминирования в галогеналканах и спиртах протекают по правилу Зайцева (см. стр. 56).
Медико-биологическое значение галогеналканов
Галогеналканы широко используются как алкилирующие
реагенты в органическом синтезе, в том числе и при получении
лекарственных веществ. Некоторые галогеналканы применяются
в медицине.
50
51.
Этилхлорид (C2H5Cl) используют как местноанестезирующее средство. Его действие основано на быстром испарении, чтовызывает сильное охлаждение.
Хлороформ (CHCl3) долгое время использовался в качестве
средства для наркоза. В настоящее время в связи с появлением
более эффективных и менее токсичных препаратов он используется в лабораторной практике для наркоза животным.
Иодоформ (CHI3) используют как наружное антисептическое средство.
Фторотан (CF3-CHClBr) является эффективным средством
для общего наркоза.
Спирты и фенолы
И спирты, и фенолы являются гидроксилсодержащими производными углеводородов, т.е. их функциональной группой является гидроксильная группа –OH.
Спирты – это производные углеводородов, в молекулах которых один или несколько атомов водорода замещены на гидроксильную группу.
Спирты классифицируют по количеству гидроксильных
групп на одноатомные и многоатомные (двухатомные, трёхатомные и т.д.). Например:
CH3-CH2-OH
одноатомный спирт
(этиловый спирт, этанол)
CH2-CH2
CH2-CH-CH2
OH OH
двухатомный спирт
(этиленгликоль, этандиол)
OH OH OH
трёхатомный спирт
(пропантриол, глицерин)
Другой вид классификации – по положению гидроксильной
группы (по характеру углерода, связанного с гидроксильной
группой). Спирты классифицируют на первичные, вторичные и
третичные. Например:
CH3
CH3 -CH 2 -OH
этанол
(первичный)
CH3 -CH-CH 3
OH
изопропиловый спирт,
пропанол-2 (вторичный)
CH3 -C-CH 3
OH
трет-бутиловый спирт,
2-метилпропанол-2 (третичный)
51
52.
Cпирты называют по двум видам номенклатуры: радикалофункциональной и заместительной.По радикало-функциональной номенклатуре название спирта составляется из названия соответствующего радикала с добавлением «овый спирт», например, метиловый спирт, этиловый
спирт, изопропиловый спирт.
По заместительной номенклатуре спирт рассматривают как
производное соответствующего углеводорода. Наличие гидроксильной группы обозначают суффиксом –ол (если их несколько,
используют умножающие частицы ди-, три-, тетра-) и обозначают её положение цифрой. Например, метанол, этанол, пропанол2, этандиол.
Некоторые спирты называют также тривиальными названиями, например, пропантриол – это глицерин.
Чтобы выяснить, какие типы реакций характерны для спиртов, необходимо рассмотреть, какие реакционные центры присутствуют в их молекулах.
Связь O-H полярна изэлектрофильный центр
основный и нуклеофильный
за большей электроотцентр
δрицательности кисло.. δ+
δ+
R- CH2 O H
рода и может разорваться гетеролитичеOH-кислотный центр
ски с отщеплением
протона водорода. Способность отщеплять протон водорода известна как кислотность, значит, в молекулах спиртов присутствует кислотный центр.
У атома кислорода есть неподелённая электронная пара, к
которой может присоединиться протон водорода. Это свойство
известно как основность. Значит, атом кислорода является основным центром.
Вы уже знаете, что нейтральные молекулы, имеющие неподелённые электронные пары, могут являться нуклеофильными
реагентами. Атом кислорода за счёт своей неподелённой электронной пары является нуклеофильным центром.
Связь C-O полярна, электронная пара связи смещена к кислороду, а на атоме углерода возникает частичный положитель52
53.
ный заряд (δ+). Такой электронодефицитный углерод («любящийэлектроны») является электрофильным центром.
А теперь рассмотрим реакции спиртов по каждому из реакционных центров.
Кислотные свойства спиртов. Одноатомные спирты являются слабыми кислотами. Они образуют соли только с щелочными металлами, но не с щелочами.
CH3 -CH 2 -OH
этанол
+ Na
CH 3 -CH 2 -O Na+ + H 2
этоксид натрия
(этанолят натрия)
Даже вода, являясь более сильной кислотой, вытесняет
спирт из его соли:
CH3 -CH2 -O Na+ + H2 O
этанолят натрия
CH3 -CH2 -OH + NaOH
этанол
Многоатомные спирты являются более сильными кислотами, чем одноатомные. Это связано с электроноакцепторным
влиянием второй гидроксильной группы на кислотный центр.
Многоатомные спирты образуют соли и с концентрированными
щелочами. Они могут реагировать также с гидроксидом меди (II).
Эта реакция используется как качественная реакция на многоатомные спирты:
HO
CH2 -OH
+
CH2 -OH
Cu
HO-CH 2
CH2 -O
HO-CH 2
CH2 -O
H
+
OH
Cu
H
O-CH 2
O-CH 2
+ 2 H2 O
Качественным признаком этой реакции является растворение голубого осадка гидроксида меди и образование ярко-синего
раствора хелатного медного комплекса.
Основные свойства спиртов слабые (из-за высокой электроотрицательности атома кислорода) и проявляются только при
взаимодействии с сильными кислотами. Например, этанол образует этилоксония гидросульфат при взаимодействии с концентрированной серной кислотой при комнатной температуре:
53
54.
..CH3 -CH 2 -OH + H 2 SO 4
+ H
CH3 -CH 2 -O
H
HSO 4
этилоксония гидросульфат
Реакции по электрофильному центру. За счёт наличия электрофильного центра (электронодефицитного атома углерода) для
спиртов характерны реакции нуклеофильного замещения (SN).
Примером такой реакции является замещение гидроксильной
группы на галоген:
CH3 -CH 2 -OH + HCl
этанол
CH3 -CH 2 -Cl + H2 O
этилхлорид
Нуклеофильной частицей в этой реакции является хлориданион, однако для её проведения используют не соли хлороводородной кислоты (NaCl, KCl), а именно хлористый водород (он
образуется при взаимодействии NaCl с серной кислотой непосредственно в момент реакции). Это значит, что протоны водорода также играют определённую роль в этой реакции. Рассмотрим
её механизм.
HCl
CH3 -CH2 -OH
-Cl
этанол
+ H
CH3 -CH2 -O
H -H2 O
протонированный
этанол
+
CH3 -CH2
карбкатион
Cl
CH3 -CH2 -Cl
этилхлорид
На первой стадии реакции этанол взаимодействует с хлороводородом как основание, присоединяя протон водорода. Образуется протонированная молекула этанола, которая далее отщепляет молекулу воды. На атоме углерода (электрофильном центре)
появляется полный положительный заряд (а в исходной молекуле
он был частичным). Таким образом, активность электрофильного
центра возрастает. Дальше полученный карбкатион активно
взаимодействует с анионом хлора, образуя молекулу конечного
продукта – этилхлорида.
Реакцию образования простых эфиров (реакцию межмолекулярной дегидратации спиртов) также относят к реакциям нуклеофильного замещения, хотя в ней принимают участие различ54
55.
ные реакционные центры. Рассмотрим эти реакции на примереобразования диэтилового эфира.
Эта реакция протекает при нагревании этанола с концентрированной серной кислотой при температуре около 140оС:
2 CH3 -CH 2 -OH
этанол
c.H2 SO 4
t=140o C
CH 3 -CH 2 -O-CH 2 -CH 3 + H2 O
диэтиловый эфир
Чтобы понять роль серной кислоты в этой реакции, необходимо описать её механизм.
.
+
+ H
HO-CH2 -CH 3
CH3 -CH 2 -OH
CH3 -CH 2 -O
CH 3 -CH 2
-.HSO
-H
H
этанол
4
2
карбкатион
протонированный
этанол
+
.HSO4
CH3 -CH 2 -O-CH 2 -CH 3
CH3 -CH 2 -O-CH 2 -CH 3 + H2 SO4
диэтиловый эфир
H
протонированный эфир
Начинается эта реакция точно так же, как и предыдущая: с
образования оксониевого катиона (протонированной молекулы
спирта) за счёт основного центра спирта. Далее идёт отщепление
молекулы воды и образуется этилкарбкатион. Вы уже знаете, что
при этом увеличивается активность электрофильного центра. Далее вторая молекула спирта выступает в роли нуклеофила по отношению к первой (нуклеофильным центром является атом кислорода с неподелённой электронной парой). Новая ковалентная
связь C-O образуется по донорно-акцепторному механизму за
счёт этой пары электронов. Образуется протонированный простой эфир (положительный заряд на кислороде появляется потому, что кислород затратил свою электронную пару на образование общей связи). Для стабилизации катиону необходимо отщепить протон. Он отщепляется под действием аниона серной кислоты. Таким образом, в результате реакции вновь образовалась
серная кислота, значит, она играла роль катализатора (увеличивая
активность электрофильного центра). Концентрированная серная
кислота является также водоотнимающим средством, которое необходимо для смещения равновесия реакции вправо.
55
56.
Реакция внутримолекулярной дегидратации (реакция элиминирования) протекает при нагревании спирта с концентрированной серной кислотой при температуре 200оС и выше (кислотуберут в избытке). При этом образуются соответствующий алкен и
вода, например:
CH3 -CH 2 -OH
этанол
c.H2 SO 4
t=200o C
CH2 =CH2 + H2 O
этен
Первые две стадии этой реакции те же, что и при образовании простых эфиров:
.H2 SO4
CH3 -CH2 -OH
-.HSO4этанол
+ H
CH3 -CH2 -O
H
этилоксония
катион
+
CH3 -CH2
-H2 O этил карбкатион
Затем карбкатион как CH-кислота реагирует с анионом серной кислоты (основанием) и отдаёт ему протон водорода. Образуется этен и молекула серной кислоты:
+
CH3 -CH2 + HSO4
карбкатион основание
CH2 =CH2 + H2 SO4
этен
Таким образом, реакции меж- и внутримолекулярной дегидратации (другими словами, реакции нуклеофильного замещения
и элиминирования) конкурируют друг с другом, и конечный результат реакции зависит от условий её проведения.
Реакции элиминирования в спиртах протекают по правилу
Зайцева: протон водорода отщепляется от менее гидрированного атома углерода, соседнего с тем, от которого отщепляется гидроксильная группа.
Например, при внутримолекулярной дегидратации бутанола-2 более вероятным является образование бутена-2, а не бутена-1: более
менее
гидрированный
CH3 -CH-CH 2 -CH3
OH
бутанол-2
56
CH3 -CH=CH-CH 3 + H2 O
бутен-2
гидрированный
c.H2 SO4
t=200o C
CH2 =CH-CH2 -CH3 + H2 O
бутен-1
57.
Примером реакции, в которой спирты проявляют тольконуклеофильные свойства, является этерификация (образование
сложных эфиров). Эту реакцию можно также назвать ацилированием спиртов.
δ+ O
CH3 -C
+
OH
уксусная кислота
..
HO-CH 3
метанол
O
к.H2 SO4
CH3 -C
+ H2 O
OCH3
метиловый эфир
уксусной кислоты
to
Реакция этерификации протекает при нагревании карбоновых кислот со спиртами в присутствии концентрированной серной кислоты. Это реакция нуклеофильного замещения.
Спирты могут образовывать сложные эфиры и с минеральными кислотами, например, при взаимодействии глицерина с
концентрированной азотной кислотой в присутствии серной кислоты образуется тринитрат глицерина:
CH2 -OH
CH -OH
CH2 -O-NO 2
+ 3 HNO3
H2 SO4
CH2 -OH
CH -O-NO 2
+ 3 H2 O
CH2 -O-NO 2
тринитрат глицерина
глицерин
1% спиртовой раствор тринитрата глицерина под названием
«Нитроглицерин» применяется в медицине при приступах стенокардии (он расширяет сосуды сердца).
Реакции окисления спиртов. Первичные и вторичные спирты способны к окислению под действием таких сильных окислителей, как перманганат калия или бихромат калия, в кислой среде, при нагревании. Третичные спирты устойчивы к окислению в
этих условиях.
При окислении первичных спиртов образуются соответствующие альдегиды, которые в свою очередь окисляются в карбоновые кислоты, например, этанол окисляется сначала в уксусный
альдегид, а затем в уксусную кислоту:
CH3 -CH2 -OH
этанол
[O]
O
CH3 -C
[O]
H
уксусный альдегид
O
CH3 -C
OH
уксусная кислота
57
58.
Вторичные спирты окисляются в соответствующие кетоны,например, пропанол-2 – в ацетон:
CH3 -CH-CH 3
[O]
CH3 -C-CH 3
O
пропанон
(ацетон)
OH
пропанол-2
Реакция окисления этанола подкисленным раствором бихромата калия используется, как его качественная реакция:
CH2 -CH2 -OH + K 2 Cr2 O7 + H2 SO4
O
to
CH3 -C
оранжевый
+ Cr 2 (SO4 )3 + K2 SO4 + H2 O
H синевато-зелёный
При нагревании реакционной смеси наблюдается изменение
окраски с оранжевой в синевато-зелёную, а выделяющийся уксусный альдегид имеет запах перезревших яблок.
Другой качественной реакцией на этанол является так называемая иодоформная реакция. При лёгком нагревании этанола с
раствором иода в присутствии щёлочи образуется иодоформ
CHI3, обладающий характерным «аптечным» запахом. Если концентрация его достаточна высока, можно наблюдать также выпадение желтоватого осадка.
CH2 -CH2 -OH + I2 + NaOH
to
O
CHI3
+
иодоформ
H-C
ONa
формиат натрия
Тиолы (тиоспирты, меркаптаны) являются тиоаналогами спиртов.
Номенклатура тиолов. Наличие тиольной группы (-SH) в структуре
обозначается суффиксом -тиол.
CH3-CH2-SH
этантиол
CH3-CH-CH3
SH
пропантиол-2
Тиолы являются более сильными кислотами, чем соответствующие
спирты. Они могут реагировать не только со щелочными металлами, но и
со щелочами:
CH3-CH2-SH + NaOH
этантиол
58
CH3-CH2-SNa + H2O
этантиоля т
натрия
59.
Тиолы проявляют нуклеофильные свойства, они могут алкилироваться и ацилироваться:CH3-SH
метантиол
NaOH
CH3-CH2-Cl
CH3-S Na+
метантиоля т
натрия
CH3-S-CH2-CH3 + NaCl
метилэтилсульфид
Тиолы окисляются легче, чем соответствующие спирты. В зависимости от условий реакции при окислении тиолов можно получить разные
продукты. При «мягком» окислении (O2, H2O2) образуются диалкилдисульфиды:
H2O2
2 CH3-CH2-SH
этантиол
CH3-CH2-S-S-CH2-CH3 + 2 H2O
диэтилдисульфид
Подобным образом происходит окисление остатков аминокислоты
цистеин в организме с образованием дисульфидных мостиков, которые
фиксируют третичную структуру белка.
При использовании более сильных окислителей («жесткое окисление») образуются соответствующие сульфоновые кислоты (сульфокислоты):
O
CH3-CH2-SH
этантиол
[O]
KMnO4
CH3-CH2-S-OH
O
этансульфокислота
Фенолы – это гидроксилпроизводные ароматических углеводородов, в молекулах которых гидроксильная группа непосредственно связана с ароматическим кольцом. Фенолы бывают
одно- и многоатомными, например:
OH
OH
OH
OH
CH3
фенол
одноатомные
фенолы
CH3
орто-
мета-
CH3
пара-
крезолы (метилфенолы)
59
60.
OHOH
OH
OH
OH
резорцин
пирокатехин
Многоатомные
(двухатомные)
фенолы
OH
гидрохинон
Сравним свойства спиртов и фенолов.
Фенолы проявляют более сильные, чем спирты, кислотные
свойства. Это можно объяснить большей стабильностью фенолят-аниона из-за наличия p,π-сопряжения (подробнее об этом в
O
следующей теме):
Фенолы являются более сильными кислотами, чем вода, они
могут взаимодействовать не только с щелочными металлами (как
спирты), но и с щелочами:
OH
ONa
+ NaOH
фенол
+ H2 O
фенолят натрия
Однако фенолы по кислотным свойствам слабее угольной
кислоты, поэтому не могут вытеснить её из солей и не взаимодействуют с гидрокарбонатом натрия.
Основные свойства у фенолов отсутствуют, т.к. неподелённая электронная пара кислорода, ответственная за основные
свойства, участвует в p,π-сопряжении с ароматическим облаком
: OH
бензольного кольца:
По этой же причине снижены и нуклеофильные свойства
фенолов. Например, фенолы не могут быть ацилированы карбоновыми кислотами (в реакциях получения сложных эфиров). С
этой целью используют более активные производные карбоновых
60
61.
кислот, например, ангидриды. Так, фенилацетат (сложный эфирфенола и уксусной кислоты) получают при взаимодействии фенола с уксусным ангидридом:
OH
O
O-C-CH 3
CH3 -C
O
+
фенол
+ CH3 -COOH
O
CH3 -C
O
уксусный ангидрид
фенилацетат
(сложный эфир)
Для получения простых эфиров фенолы предварительно переводят в феноляты, а уже потом подвергают реакции алкилирования алкилгалогенидами:
OH + NaOH
ONa + H2 O
фенолят натрия
ONa + C 2 H5 -I
этил иодид
фенолят натрия
OC 2 H5 + NaI
метилфениловый эфир
Реакции окисления фенолов протекают легче, чем спиртов.
Особенно легко окисляются многоатомные фенолы (даже кислородом воздуха).
При окислении фенола образуется смесь орто- и парахинонов:
O
OH
O
K2 Cr2 O7 , H2 SO4
+
фенол
O
п-хинон
OH
пирокатехин
о-хинон
O
[O]
о-хинон
O
OH
O
OH
O
[O]
OH
гидрохинон
O
п-хинон
61
62.
Хиноны широко распространены в природе, они участвуют в окислительно-восстановительных процессах, сопровождающих дыхание. Принимая два электрона и два протона, хиноны восстанавливаются в соответствующие гидрохиноны:O
OH
+
+2e, +2H
-2e, -2H +
OH
гидрохинон
O
п-хинон
Окислительно-восстановительные свойства системы хинон – гидрохинон играют важную роль в организме. По отношению к большинству
органических субстратов эта система выполняет роль окислителя. Принимая электроны от субстрата, производное хинона in vivo превращается в
соответствующий гидрохинон, который в свою очередь передаёт электроны кислороду (через цитохромную систему) и опять окисляется в хинон.
Таким образом, система хинон – гидрохинон участвует в переносе электронов от субстрата к кислороду.
Биологически важными производными хинонов являются витамины
группы К (см. стр. 233 ) .
Реакции фенола по ароматическому кольцу. Фенол вступает
в реакции электрофильного замещения легче, чем бензол, т.к. фенольный гидроксил, являясь электронодонорным заместителем
(+M>-I), повышает электронную плотность в кольце, особенно в
орто- и пара-положениях.
Нитрование фенола протекает при действии разбавленной
азотной кислоты, без катализатора (бензол нитруют смесью концентрированных азотной и серной кислот):
OH
OH
+
+ HNO3
фенол
OH
NO 2
O2 N
п-нитрофенол
о-нитрофенол
Бромирование фенола в неполярном или малополярном растворителе приводит к образованию смеси о- и п-бромфенолов:
OH
+ Br2
фенол
62
CHCl3
OH
OH
+
Br
о-бромфенол
Br
п-бромфенол
63.
Если же бромирование проводят в избытке бромной воды,образуется 2,4,6-трибромфенол:
Br
OH
+ 3 Br2
фенол
OH
H2O
+ 3 HBr
Br
Br
2,4,6-трибромфенол
Эта реакция используется как качественная реакция на фенол: 2,4,6-триброфенол образует белый осадок.
Другой качественной реакцией на фенол (реакция на фенольный, а точнее, енольный, гидроксил =C-OH) является реакция с раствором FeCl3. Различные фенолы при этом образуют
продукты разной окраски, например, фенол – фиолетовое, крезолы – голубое.
Медико-биологическое значение спиртов и фенолов
Метанол является высокотоксичным соединением, вызывает ацидоз и слепоту, связанные с накоплением в организме формальдегида и муравьиной кислоты – продуктов окисления метанола.
Этанол используется как обеззараживающее средство, для
приготовления настоек, как растворитель и реагент в органическом синтезе.
Глицерин применяется как компонент кремов и мазей для
смягчения кожи, является структурной основой молекул жиров.
Фенолы являются антисептическими средствами. Фенол –
первый антисептик, использованный в хирургии.
Крезолы используются как дезинфицирующие средства.
Резорцин применяют при лечении кожных заболеваний.
Тимол (2-изопропил-5-метилфенол) содержится в эфирных
маслах многих растений. В медицине он применяется как антисептическое и антигельминтное средство.
63
64.
КИСЛОТНЫЕ И ОСНОВНЫЕ СВОЙСТВАОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. РЕАКЦИОННАЯ
СПОСОБНОСТЬ АМИНОВ
Кислотные и основные свойства органических соединений
являются важными аспектами их реакционной способности. Перенос протона водорода наблюдается в ходе многих биохимических реакций.
Современные представления о природе кислот и оснований
основаны на теории Бренстеда.
По Бренстеду, кислоты – это нейтральные молекулы и ионы,
способные отдавать протоны водорода (доноры протонов). Основаниями являются нейтральные молекулы и ионы, способные
присоединять протоны водорода (акцепторы протонов).
Кислотно-основное взаимодействие может быть представлено схемой:
A-H + B
кислота основание
A+
B+-H
сопряжённые
основание кислота
Кислота A-H, теряя протон, превращается в основание A-,
которое называют сопряжённым основанием данной кислоты.
Наоборот, основание B, принимая протон, превращается в сопряжённую кислоту B+-H.
В кислотно-основном равновесии существует важная закономерность: чем сильнее кислота, тем слабее сопряжённое с ней
основание.
Понятие кислотности и основности взаимосвязаны: кислотные свойства проявляются только в присутствии оснований и наоборот.
Например, газообразный хлороводород не проявляет кислотных свойств, для этого необходимо присутствие основания:
HCl + H2O
кислота основание
Cl - +
H3O+
сопряжённые
основание
кислота
Некоторые соединения в зависимости от условий могут
проявлять и кислотные, и основные свойства, т.е. понятия «кислота» и «основание» относительны. Например, в присутствии
64
65.
воды как основания уксусная кислота проявляет кислотные свойства, а в присутствии концентрированной серной кислоты – основные:O
O
+ H2 O
CH3 -C
OH основание
кислота
CH3 -C
OH
основание
-
+
OH
O
CH3 -C
O
+
+ H3 O
CH3 -C
+ H2 SO 4
OH
+ HSO4
кислота
Так как в биохимических процессах растворителем обычно
является вода, то дальше мы будем рассуждать о кислотноосновных свойствах соединений по отношению к воде.
Кислоты Бренстеда
Сила кислот количественно выражается константой равновесия реакции (К), заключающейся в переносе протона водорода
от кислоты к основанию – молекуле воды.
A-H + H2 O
A + H3 O+
Используя значение константы равновесия этой реакции и
учитывая, что концентрация воды практически постоянна, можно
определить произведение K.[H2O], называемое константой кислотности (Ka) (от слова acid – кислота).
K=
[A- ][H3 O+]
[A-H] [ H2 O]
;
K a= K . [H2 O] =
[A- ][H3 O +]
[A-H]
Чем больше Ka, тем сильнее кислота. Но величины Ka очень
малы (например, Ka уксусной кислоты 1,75. 10-5 ), пользоваться
ими неудобно, поэтому введено понятие «показатель константы
кислотности» (её отрицательный логарифм) - pKa. pKa=-lg Ka.
Например, для уксусной кислоты pKa=4,75. Чем меньше величина pKa, тем сильнее кислота.
65
66.
В зависимости природы кислотного центра (атома, отщепляющего протон водорода) различают следующие типы кислот:- OH-кислоты (вода, спирты, фенолы, карбоновые кислоты);
- SH-кислоты (тиоспирты, тиофенолы, тиокислоты);
- NH-кислоты (аммиак, амины, амиды кислот, пиррол);
- CH-кислоты (углеводороды и их производные).
Сила кислот определяется стабильностью сопряжённых оснований – чем стабильнее сопряжённое основание (анион кислоты), тем сильнее кислота.
Стабильность сопряжённого основания определяется степенью делокализации заряда, которая зависит от следующих факторов: природа атома в кислотном центре, влияние радикала, степень сольватации.
Рассмотрим каждый из этих факторов.
Природа атома в кислотном центре. Для делокализации
отрицательного заряда наибольшее значение имеют электроотрицательность атома и его поляризуемость.
Чем выше электроотрицательность атома, получившего отрицательный заряд после отщепления протона, тем сильнее он
этот отрицательный заряд удерживает и труднее предоставляет
протону, а значит равновесие
A-H
А- + H+ смещается
вправо.
В периодах слева направо электроотрицательность возрастает. Поэтому OH-кислоты сильнее NH-кислот, а NH-кислоты
сильнее CH-кислот (при прочих равных условиях).
Так как электроотрицательность атомов зависит от типа
гибридизации, то среди CH-кислот при переходе от алканов к алкенам и далее к алкинам кислотность возрастает:
CH3-CH3 < CH2=CH2 < CH≡CH.
Так, этан и этилен не реагируют с амидом натрия, а ацетилен – реагирует:
CH CH + NaNH
CH CNa + NH
2
3
В группах сверху вниз увеличивается поляризуемость. Это
связано с тем, что увеличивается число энергетических уровней,
т.е. объем электронных оболочек. Чем выше поляризуемость, тем
более делокализован заряд, тем стабильнее анион, а значит, выше
66
67.
сила кислоты. Поэтому SH-кислоты сильнее, чем OH-кислоты.Например, спирты не реагируют со щелочами, а тиолы – реагируют:
C2 H5 -OH + NaOH
этанол
C2 H5 -SH + NaOH
этантиол
C 2 H5 -SNa + H 2 O
этантиолят
натрия
Влияние радикала (заместителя). Кислотность органических соединений в значительной степени зависит от природы заместителя, связанного с реакционным центром. Особенно сильно
повышает кислотные свойства наличие сопряженной системы в
анионе.
Сравним кислотные свойства спиртов и фенолов.
-
OH
O
CH3 -OH
фенолят анион
CH O - + H +
+ H+
3
алкоголят
анион
В случае фенолят-аниона (p,π-сопряженная система) степень
делокализации заряда гораздо выше, поэтому фенолы более
сильные OH-кислоты, чем спирты. Например, фенолы в отличие
от спиртов реагируют с щелочами:
OH + NaOH
ONa + H2 O
фенолят натрия
Разницу в кислотных свойствах можно подтвердить и величинами pKa: для фенола pKa=10,00; для метанола pKa=16,00.
Фенолы – более сильные кислоты, чем спирты, но и они являются слабыми: фенол не вытесняет угольную кислоту из её солей, т.е. является более слабой кислотой, чем угольная.
Сравнивая кислотные свойства фенолов и карбоновых кислот, нужно отметить, что и фенолят-, и ацилат-анион являются
p,π-сопряженными системами, но в ацилат-анионе наблюдается
более полная делокализация отрицательного заряда за счёт двух
электроотрицательных атомов кислорода:
67
68.
OCH3 -C
O
O
CH3 -C
OH
- + H
+
O
ацетат анион
или CH3 -C
O
-1/2
-1/2
O- + H +
OH
фенолят анион
В отличие от фенолов карбоновые кислоты взаимодействуют с гидрокарбонатом натрия:
CH3 -COOH + NaHCO 3
CH3 -COONa + CO 2 + H2 O , т.е. они являуксусная
кислота
ацетат натрия
ются более сильными кислотами, чем угольная кислота.
Заместители в радикале также влияют на кислотные свойства. Если заместитель способствует делокализации отрицательного заряда в анионе (электроноакцепторный заместитель), то кислотность будет выше. И наоборот, электронодонорные заместители затрудняют делокализацию заряда и поэтому понижают кислотные свойства.
Сравним кислотные свойства следующих соединений: масляная кислота, α-хлормасляная и β-хлормасляные кислоты.
CH3 -CH2 -CH2 -C
масляная кислота
pKa=4,80
CH3 -CH 2 -CH-C
O
OH
O
O
CH3 CH2 CH2 C
O
+
- +H
O
CH3 -CH2 -CH
OH
Cl
α-хлормасляная кислота
pKa=2,84
O
CH3 -CH
CH3 -CH-CH 2 -C
OH
Cl
Cl
β-хлормасляная кислота
pKa=4,06
C
O
Cl
O
CH2
C
O
- +H
+
+
- +H
Анион масляной кислоты стабилизируется только за счёт
слабого индуктивного эффекта углеводородного радикала. Атом
хлора проявляет сильный отрицательный индуктивный эффект,
68
69.
поэтому гораздо более сильно стабилизирует анион кислоты. Таккак индуктивный эффект передается с затуханием, влияние хлора
в α-положении проявляется сильнее. Итак, самой сильной является α-хлормасляная кислота, затем – β-хлормасляная и, наконец,
самая слабая кислота – масляная. Это подтверждается и значениями pKa данных кислот.
В ароматическом ряду электроноакцепторные заместители
также способствуют делокализации заряда, таким образом увеличивая кислотные свойства. Электронодонорные заместители оказывают обратное влияние.
O
-
O
-
N
-
: NH2
+M NH > -I NH
2
2
O
O
-I NO , -M
NO 2
2
pKa=7,16
O
pKa=10,0
pKa=10,68
Суммируя все наши рассуждения, можно выстроить ряд
убывания кислотных свойств OH-кислот:
карбоновые кислоты > угольная кислота > фенолы > спирты.
(Причем в каждой группе электронодонорные заместители
понижают кислотность, а электроноакцепторные – повышают).
Основания Бренстеда
Основания, по Бренстеду, это нейтральные молекулы и ионы, способные присоединять протон водорода. Для образования
ковалентной связи с протоном основания Бренстеда должны предоставлять или неподелённую электронную пару, или электроны
π-связи. В соответствии с этим основания Бренстеда делятся на
n-основания и π-основания.
n-Основания – это анионы или нейтральные молекулы,
имеющие атом с неподелённой электронной парой. Их классифицируют по центрам основности следующим образом:
69
70.
- оксониевые основания: спирты R-ÖH, простые эфирыR-Ö-R, сложные эфиры R-C O : , альдегиды и кетоны R-C O :
O-R'
- аммониевые основания: амины
соединения, например, пиридин
R'(H)
..
R-NH2 , гетероциклические
N
..
..
- сульфониевые основания: тиоспирты R-SH, тиоэфиры R-S-R'.
π-Основания – соединения, имеющие π-связи, т.е. алкены,
алкадиены, алкины, арены. Это очень слабые основания, т.к. протонируемые электронные пары несвободны. Например, этен
проявляет π-основные свойства при образовании π-комплекса с
протоном водорода:
CH =CH + H+
CH
CH
2
2
2
2
H+
π -комплекс
Для количественной характеристики основности используют величину pKa сопряжённой с данным основанием кислоты
(BH+). Эту величину обозначают pKBH+. Чем pKBH+ больше, тем
сильнее основание.
Влияние природы атома в основном центре и связанных с
ним заместителей на основность противоположно рассмотренному ранее их влиянию на кислотность:
- с увеличением электроотрицательности атома основного
центра основность уменьшается (атом труднее отдаёт свою неподелённую электронную пару для присоединения протона), т.е.
аммониевые основания сильнее оксониевых. Так, этанол способен взаимодействовать только с концентрированными минеральными кислотами:
..
CH3 -CH 2 -OH + H 2 SO 4
+ H
CH3 -CH 2 -O
H
HSO 4
,
этилоксония гидросульфат
а этиламин проявляет основные свойства даже при взаимодействии с водой:
..
+
C2 H5 -NH2 + H2 O
этиламин
70
C2 H5 -NH3 OH
этиламония гидроксид
.
71.
- с увеличением поляризуемости атома основного центраосновность уменьшается, т.е. оксониевые основания сильнее
сульфониевых;
- электронодонорные заместители повышают основность, а
электроноакцепторные – понижают (чем выше электронная плотность на основном центре, тем легче он предоставит свою электронную пару протону). Так, в ряду п-нитроанилин, анилин,
п-толуидин основность повышается: нитро-группа является
электроноакцепторным заместителем, а метильная группа – электронодонорным. Это подтверждают и значения pKBH+.
..
NH2
..
NH2
..
NH2
<
<
N
CH3
O
O
-I NO , -M
NO 2
2
+I
CH3
pKBH+=1,00
pKBH+=4,60
pKBH+=5,10
Итак, самыми сильными основаниями являются аммониевые. Основность различных типов аминов мы обсудим, рассматривая химические свойства аминов в целом.
АМИНЫ
Амины – это производные аммиака, в молекуле которого
один, два или три атома водорода замещены углеводородными
радикалами. Отсюда первый тип классификации аминов: по количеству радикалов амины подразделяют на первичные, вторичные и третичные.
CH3 -NH2
первичный
метиламин
CH3 -NH-CH2 -CH3
вторичный
метилэтиламин
CH3 -N-CH 3
CH3
третичный
триметиламин
71
72.
Другой вид классификации аминов – по природе радикалов.Амины подразделяют на алифатические и ароматические. Например, приведенные выше амины являются алифатическими, а
анилин (аминобензол) – ароматическим:
NH2
анилин
Для
названия
аминов
применяют
радикалофункциональную и заместительную номенклатуру IUPAC. По
радикало-функциональной номенклатуре называют радикал или
радикалы, если их несколько (в алфавитном порядке), и добавляют слово «амин», например, метиламин, метилэтиламин, диметиламин и т.д. Те амины, которые нельзя назвать по радикалофункциональной номенклатуре (сложные радикалы), называют
по заместительной номенклатуре, например: 4 3 2 1
CH3 -CH-CH-CH 3
CH3 NH2
3-метилбутанамин-2
Ароматические амины обычно рассматривают как производные анилина, например:
NH2
CH3
2-метиланилин
(о-метиланилин,
о-толуидин)
NH-CH3
N-метиланилин
За счёт неподелённой электронной пары азота амины проявляют основные и нуклеофильные свойства.
Сравним основные свойства различных групп аминов.
Алифатические амины являются более сильными основаниями, чем ароматические. Это связано с тем, что неподелённая
электронная пара азота в ароматических аминах участвует в p,πсопряжении и менее доступна для атаки протона водорода. В
алифатических же аминах электронная плотность на атоме азота
аминогруппы повышена за счёт электронодонорного влияния алкильных групп:
72
73.
..NH2
CH3
..
NH2
Алифатические амины взаимодействуют с минеральными
кислотами, карбоновыми кислотами и даже с водой (очень слабой
кислотой):
+
CH3 -NH 3 Cl
метиламмония
хлорид
CH3 -NH 2 + HCl
метиламин
CH3 -NH 2 + CH3 COOH
метиламин
CH3-NH2 + H2O
метиламин
O
- +
O NH 3 -CH 3
метиламмония ацетат
CH3 -C
+
CH3-NH3 OH
метиламмония
гидроксид
Ароматические амины как слабые основания взаимодействуют с минеральными кислотами:
+
NH3 Cl
NH2 + HCl
анилин
анилиния хлорид
(фениламмония хлорид)
Заместители в ароматическом кольце влияют на основные
свойства аминов: электронодонорные заместители повышают основные свойства, а электроноакцепторные – понижают (см.
стр.71).
Сравним основные свойства различных типов алифатических аминов – первичных, вторичных и третичных, например,
метиламина, диметиламина и триметиламина.
..
CH3
NH2
первичный
амин
..
CH3 NH CH3
вторичный амин
pK BH+=10,62
pK BH+=10,77
H3 C
..
N
CH3
CH3
третичный амин
pK BH+=9,80
73
74.
Чем выше электронная плотность на атоме азота, тем вышеосновные свойства амина. Каждая метильная группа смещает
электронную плотность к атому азота, поэтому можно было бы
предположить, что самым сильным основанием является третичный амин. Однако, сравнивая значения pKBH+, можно увидеть,
что это не так: самым сильным основанием является вторичный
амин. Этот факт можно объяснить с позиций пространственной
доступности неподелённой электронной пары азота: в триметиламине три крупных заместителя «прикрывают» её. Таким образом, ряд убывания основности алифатических аминов:
вторичные > первичные > третичные.
За счет неподелённой электронной пары азота амины проявляют также нуклеофильные свойства. Амины являются нуклеофилами, например, в реакциях алкилирования и ацилирования.
Алкилирование – это введение в молекулу алкила (метил,
этил, пропил и т.п.). В качестве алкилирующих реагентов обычно
используют алкилгалогениды (этилхлорид, метилбромид, например). Так, в реакции этиламина с метилхлоридом образуется метилэтиламин:
..
C2 H5 -NH 2
этиламин
δ+
+ CH3 Cl
метилхлорид
C2 H5 -NH-CH 3 + HCl
метилэтиламин
Это реакция нуклеофильного замещения. Она позволяет получить вторичный амин из первичного и третичный – из вторичного.
..
δ+
C2 H5 -NH-CH3 + CH3 -Cl
метилэтиламин
C2 H5 -N-CH 3 + HCl
CH3
диметилэтиламин
Ароматические амины также вступают в реакции алкилирования, но менее активно, т.к. их нуклеофильные свойства понижены (неподелённая электронная пара, отвечающая за них, участвует в p,π-сопряжении).
..
NH2 + CH3 -Cl
анилин
74
NH-CH3 + HCl
N-метиланилин
(метилфениламин)
75.
Ацилированием называют введение в молекулу ацила – остатка карбоновой кислоты (например, ацетил – это остаток уксусной кислоты, пропионил – пропионовой). Для ацилированияаминов обычно используют ангидриды соответствующих кислот.
δ+
CH3 -C
CH3 -C
O
O
O
..
+ H2 N-CH 3
метиламин
CH3 -C
+ CH3 -COOH
NH-CH3
уксусная кислота
N-метиламид
уксусной кислоты
O
уксусный ангидрид
Подробнее мы будем рассматривать эти реакции, изучая тему «Карбоновые кислоты и их функциональные производные».
Для ароматических аминов характерны также реакции, протекающие за счёт ароматического кольца – реакции электрофильного замещения. Ранее мы уже рассматривали
влияние
аминогруппы на ход реакций SE (см. стр.44,45): аминогруппа, являясь электронодонорным заместителем (+MNH2 >> -INH2), облегчает эти реакции в сравнении с бензолом и является орто-, параориентантом. Например, бромирование анилина бромной водой
приводит к образованию белого осадка 2,4,6-триброманилина:
NH2
NH2
+ 3 Br2
анилин
H2 O
Br
Br
+ 3 HBr
Br
2,4,6-триброманилин
Эту реакцию используют для качественного обнаружения
анилина.
РЕАКЦИОННАЯ СПОСОБНОСТЬ ОКСОСОЕДИНЕНИЙ
Функциональной группой оксосоединений является карбонильная или оксогруппа C=O. В молекулах альдегидов оксогруппа связана с радикалом и атомом водорода, поэтому общая формула альдегидов
. R-C O
H
75
76.
В кетонах оксогруппа связана с двумя радикалами, общаяформула кетонов R-C-R' .
O
Классификация и номенклатура оксосоединений
Оксосоединения классифицируют по природе радикала на
алифатические (предельные и непредельные) и ароматические.
Рассмотрим номенклатуру первых представителей предельных алифатических альдегидов. По заместительной номенклатуре IUPAC их названия строятся с использованием названий алканов с соответствующим количеством атомов углерода в цепи с
добавлением суффикса –аль. Часто используют и тривиальные
названия альдегидов.
Таблица 1
Названия алифатических альдегидов
Строение
Тривиальное название
O
H-C
муравьиный
альдегид метаналь
(формальдегид)
H
O
CH3 -C
H
O
CH3 -CH2 -C
уксусный альдегид
(ацетальдегид)
этаналь
пропионовый альдегид
пропаналь
масляный альдегид
бутаналь
валериановый альдегид
пентаналь
H
O
CH3 -CH2 -CH 2 -C
Название по IUPAC
H
CH3 -CH2 -CH2 -CH2 -C
O
H
Альдегиды с разветвлёнными радикалами также можно называть, используя два вида номенклатуры. Рассмотрим это на
конкретном примере:
β
3
α
2
1
CH3 -CH-CH 2 -C
CH3
76
O
H
3-метилбутаналь (IUPAC)
β-метилмасляный альдегид
77.
По номенклатуре IUPAC это соединение рассматриваетсякак производное бутаналя, положение заместителя обозначается
цифрой (первым является атом углерода альдегидной группы):
3-метилбутаналь. Если за основу принимается тривиальное название альдегида (масляный), то положение заместителя обозначается греческой буквой (α-, β-, γ- и т.д.), при этом α-положением
является атом углерода, ближайший к альдегидной группе: βметилмасляный альдегид.
Простейшим непредельным альдегидом является акриловый
альдегид, или акролеин (тривиальное название). По номенклатуре
IUPAC это пропеналь:
O
CH2 =CH-C
H
Примером ароматических альдегидов является бензальдегид:
O
C
H
Кетоны называют по двум видам международной номенклатуры – заместительной и радикально-функциональной.
По заместительной номенклатуре название кетона рассматривают как производное соответствующего углеводорода, обозначая оксогруппу суффиксом –он и указывая её положение. По
радикало-функциональной номенклатуре название кетона складывается из названий соответствующих радикалов (в алфавитном
порядке) с добавлением слова кетон. Например:
1
CH3 -C-CH 3
O
пропанон
диметилкетон
ацетон
2 3
4
5
CH3 -C-CH 2 -CH2 -CH 3
O
пентанон-2
метилпропилкетон
Диметилкетон часто называют тривиальным названием –
ацетон.
77
78.
Реакционные центры в молекулах оксосоединенийδ−
основный центр
H
:
O
α δ+
R C C
H
H
CH-кислотный
центр
электрофильный
центр
Более электроотрицательный атом кислорода смещает к себе электронную плотность, создавая на атоме углерода карбонильной группы частичный положительный заряд. Углерод карбонильной группы становится электрофильным центром. За счёт
неподеленной электронной пары кислород является основным
центром. Альдегидная группа смещает электронную плотность с
соседнего атома углерода (α-атом), создавая на нем частичный
положительный заряд. За счёт этого C-H-связи поляризуются,
возникает CH-кислотный центр.
Электрофильный центр в молекулах оксосоединений может
быть атакован нуклеофильным реагентом. Реакции протекают с
разрывом π-связи (присоединение). Значит, для альдегидов и кетонов характерны реакции нуклеофильного присоединения (AN).
Легкость протекания реакций нуклеофильного присоединения зависит от величины δ+ на электрофильном центре и от его
пространственной доступности.
Сравним активность муравьиного, уксусного, трихлоруксусного альдегидов и ацетона.
δ+
H-C
O
H
муравьиный
альдегид
CH3
δ+
C
Cl
O
H
уксусный
альдегид
Cl
δ+
C C
O
H
Cl
трихлоруксусный
альдегид
CH3
δ+
C CH3
O
ацетон
С позиций величины заряда на электрофильном центре наибольшей реакционной способностью обладает трихлоруксусный
альдегид, т.к. три электроноакцепторных заместителя способствуют его увеличению. В молекуле муравьиного альдегида замес78
79.
титель отсутствует. В молекуле уксусного альдегида метильнаягруппа понижает частичный положительный заряд на углероде
карбонильной группы, а значит, понижает и активность. В молекуле ацетона таких электронодонорных заместителей два, а значит, активность ещё ниже. Таким образом, электроноакцепторные заместители в радикале повышают активность оксосоединений в реакциях AN, а электронодонорные заметители – понижают.
С позиций доступности реакционного центра ацетон также
является наименее реакционноспособным, т.к. возникают пространственные затруднения для атаки нуклеофилом (реакционный центр «экранирован» двумя крупными углеводородными радикалами).
В целом, альдегиды более реакционноспособны в реакциях
нуклеофильного присоединения, чем кетоны.
Механизм реакции нуклеофильного присоединения может
быть описан следующим образом:
δ+ O
+ E+Nu
R-C
H (R')
-E +
O
R-C Nu
H (R')
1
2
E+
R-C
OE
Nu
H (R')
3
-
Нуклеофильная частица (Nu ) атакует электрофильный
центр молекулы оксосоединения (1). π-Связь разрывается гетеролитически, и оба её электрона уходят к атому кислорода. Новая
связь C-Nu образуется за счёт электронов нуклеофила. Образовавшаяся отрицательная частица (2) дальше взаимодействует с
электрофилом, давая продукт нуклеофильного присоединения
(3).
По приведенному механизму протекает ряд важных реакций
альдегидов и кетонов, в которых нуклеофилами являются, например, H2O, R-OH, HCN, H-, NH3, R-NH2.
79
80.
Примеры реакций нуклеофильного присоединенияВзаимодействие со спиртами. Альдегиды при взаимодействии со спиртами образуют полуацетали, а при избытке спирта –
ацетали. Реакция протекает в присутствии катализатора – газообразного хлороводорода.
CH3
δ+
C
O:
H
HCl газ
-Cl
1
CH3
+
OH
CH3
C
H
2
OH
+
C O CH3
H H
Cl -
4
+
C
CH3
CH3
OH
..
HO-CH3
H
3
OH
C O CH3
H
полуацеталь
Молекула этанола не является достаточно активным нуклеофилом для этой реакции. Чтобы увеличить активность электрофильного центра (карбонильный углерод с δ+) и используют
кислотный катализ. На первой стадии реакции альдегид (1) взаимодействует с хлороводородом по основному центру (атом кислорода с неподеленной электронной парой), образуя катион, который существует в виде двух резонансных структур (2 и 3). В
карбкатионе (3) на электрофильном центре уже полный положительный заряд (а не частичный, как в исходном альдегиде). Таким
образом, активность электрофильного центра увеличилась, и он
может взаимодействовать со слабым нуклеофилом – молекулой
спирта. Новая связь C-O образуется за счет неподеленной электронной пары кислорода, поэтому на нем появляется положительный заряд (4). Для стабилизации этому катиону необходимо
отщепить протон водорода. Его принимает анион Cl-. Образуется
молекула конечного продукта реакции – полуацеталя.
По такому же механизму происходит дальнейшее превращение полуацеталя в ацеталь:
CH3
OH
C O CH3 + CH3 -OH
H
полуацеталь
80
HCl газ
-
CH3
OCH3
C O CH3 + H2 O
H
диметилацеталь
уксусного альдегида
81.
Эти реакции обратимы, в кислой среде полуацетали и ацетали гидролизуются до соответствующих альдегидов и спиртов.Вот почему катализатором в реакциях их образования является
газообразный хлороводород, а не его водный раствор.
Реакцию образования ацеталей часто используют в органическом синтезе для защиты альдегидной группы от окисления.
После проведения реакции окисления защиту «снимают», т.е.
проводят кислотный гидролиз.
Кетоны в реакцию со спиртами практически не вступают.
Присоединение синильной кислоты HCN. Эта реакция характерна и для альдегидов, и для кетонов. Рассмотрим её на примере ацетона.
OH CH3 -C-CH 3 + HCN
CH 3 -C-CH 3
HO
CN
оксинитрил (циангидрин)
ацетона
O
ацетон
Реакция протекает в присутствии щелочи в качестве катализатора.
Синильная кислота – слабая кислота, с малой степенью диссоциации, т.е. плохой источник нуклеофилных частиц (цианиданионов CN-). Для активизации нуклеофила и используют катализатор:
HCN + OH
-
CN - + H2 O
Далее реакция протекает по общему механизму нуклеофильного присоединения:
CH3 -C-CH 3
O
CN -
CH3 -C-CH 3
O - CN
1
H2 O
CH3 -C-CH 3 + OH HO
CN
2
Нуклеофил атакует электрофильный центр молекулы ацетона, π-связь C=O разрывается гетеролитически, оба ее электрона
уходят к атому кислорода, поэтому там появляется отрицательный заряд (1), новая связь C-C образуется за счет электронов
нуклеофила. Анион (1) присоединяет протон водорода от молекулы воды, образуя оксинитрил (2) и гидроксид-анион, что доказывает, что щелочь являлась катализатором этой реакции.
Циангидрины способны к гидролизу с образованием соответствующих оксосоединений и синильной кислоты. Некоторые
81
82.
циангидриды встречаются в природе (синтезируются растениями). Употребление их в пищу (косточки сливы, вишни, горькогоминдаля) может привести к отравлению.
Присоединение воды. Эта реакция обратима. Её равновесие
контролируется стерическими и электронными факторами.
δ+
R C
O
H
OH
R C OH
H
гидрат
..
+ H2 O
Гидраты большинства альдегидов и особенно кетонов неустойчивы, существуют только в растворах. Гидрат трихлоруксусного альдегида (хлораля) очень устойчив, что связано с электроноакцепторным действием атомов хлора:
Cl
Cl
δ+
C C
O
H
OH
CCl3 C OH
H
хлоралгидрат
..
+ H2 O
Cl
хлораль
Хлоралгидрат используется в медицине как снотворное и
успокаивающее средство.
Присоединение гидридов металлов. В результате этой реакции происходит восстановление альдегидов и кетонов. В качестве
реагентов используют гидриды лития и натрия (LiH, NaH) или
алюмогидрид лития (LiAlH4). В качестве нуклеофильной частицы
выступает гидрид-ион (H-).
На первой стадии реакции образуется алкоголят, при последующем гидролизе которого получается соответствующий спирт.
При восстановлении альдегидов образуются первичные спирты:
δ+
R C
O
H
+ + Na H
ONa
R C H
H
алкоголят
натрия
H2 O
R-CH2 -OH + NaOH
первичный
спирт
Восстановление кетонов приводит к получению вторичных
спиртов, например:
CH3 -C-CH 3
O
ацетон
82
[H]
CH3 -CH-CH3
OH
пропанол-2
83.
Взаимодействие с аммиаком и аминами. Амины и их производные типа X-NH2 реагируют с альдегидами и кетонами в двестадии. Сначала протекает реакция нуклеофильного присоединения, продукты которой неустойчивы и отщепляют молекулу воды. Поэтому такие реакции характеризуют как присоединениеотщепление.
Рассмотрим механизм реакции на примере взаимодействия
ацетальдегида с аммиаком.
..
δ+ O
CH3 -C
+ NH 3
H
ацетальдегид
O-
OH
+
CH3 -C-NH 3
CH3 -C-NH 2
H
H
продукт реакции
присоединения
CH3 -CH=NH + H2 O
имин
Имины называют также основаниями Шиффа.
Имины являются промежуточными продуктами во многих
ферментативных
процессах,
например,
в
биосинтезе
α-аминокислот в организме.
Основания Шиффа легко гидролизуются водными растворами минеральных кислот с образованием исходных продуктов.
В организме гидролиз иминов происходит в процессе окислительного дезаминирования α-аминокислот.
Помимо аммиака, в реакцию просоединения-отщепления с
альдегидами и кетонами вступают первичные амины (R-NH2),
гидроксиламин (NH2-OH), гидразин (NH2-NH2), фенилгидразин
(C6H5-NH-NH2), семикарбазид (NH2-NH-CO-NH2):
R"-NH2
NH2 -OH
O
R
C
NH2 -NH 2
R' (H)
C6 H5 -NH-NH 2
O
H2 N-NH-C-NH 2
R-C=N-R"
R' (H)
замещенный имин
R-C=N-OH
R' (H)
оксим
R-C=N-NH 2
R' (H)
гидразон
R-C=N-NH-C 6 H5
R' (H)
O
фенилгидразон
R-C=N-NH-C-NH 2
R' (H)
семикарбазон
83
84.
Все эти производные – кристаллические соединения с чёткими температурами плавления, поэтому их получение можетиспользоваться для идентификации альдегидов и кетонов. А так
как эти производные так же, как и имины способны к гидролизу с
образованием исходных соединений, то данные реакции могут
использоваться и для выделения оксосоединений из смесей с другими соединениями.
Через стадию образования альдимина при взаимодействии
пиридоксальфосфата (см. стр. 195) и α-аминокислоты протекает
реакция переаминирования белковых α-аминокислот (основной
путь биосинтеза α-аминокислот).
Реакция альдольной конденсации характерна только для
альдегидов, в структуре которых есть α-CH-кислотный центр. Реакция катализируется щелочами.
O
2 CH3 -C
HO
O
CH3 -CH-CH 2 -C
H
OH
альдоль
H
Конденсацией называется реакция, приводящая к усложнению углеродного скелета и возникновению новой углеродуглеродной связи, при этом из двух или более относительно простых молекул образуется новая более сложная молекула.
Эта реакция начинается по CH-кислотному центру альдегида:
CH2
H
δ+ O
C
H
HO
-
O
+ H2 O
CH2 C
H
Образующийся анион CH-кислоты стабилизирован за счёт
p,π-сопряжения. Он является нуклеофилом по отношению к другой молекуле альдегида, и далее реакция протекает по общему
механизму нуклеофильного присоединения:
δ+
CH3-C
84
O
H
+ CH2 C
O
H
OCH3-C-CH2-C
H
O
H
OH
H2 O
CH3-CH-CH2-C
O
H
+ HO
-
85.
В результате реакции образуется соединение, являющеесяальдегидом и спиртом (алкоголь) – альдоль.
Альдольная конденсация имеет место и в биологических
системах. Например, биосинтез лимонной кислоты, нейраминовой кислоты протекает по механизму альдольной конденсации.
Те альдегиды, которые не имеют атомов водорода при αуглеродном атоме, т.е. не проявляют CH-кислотных свойств, в
присутствии щелочей реагируют по-иному. Для них характерна
реакция Канниццаро. Другое название этой реакции – диспропорционирование, или реакция оксидоредукции: одна молекула альдегида при этом окисляется, а другая – восстанавливается.
Реакция Канниццаро характерна, например, для бензальдегида:
O
2
50% NaOH
O
C
CH2 -OH +
H
бензальдегид
бензиловый спирт
C
ONa
бензоат натрия
В случае формальдегида реакция Канниццаро протекает в
водном растворе без катализатора:
O
2 H-C
O
H2 O
CH3 -OH + H-C
OH
метанол
H
формальдегид
муравьиная
кислота
В водных растворах формальдегида накапливается муравьиная кислота, поэтому эти растворы имеют кислую реакцию.
За счёт CH-кислотного центра протекают также реакции галогенирования, например, так называемая иодоформная реакция:
CH3-C-CH3 + I2
O
ацетон
(пропанон)
NaOH
CI3-C-CH3
NaOH
O
1,1,1-трииодопропанон
O
CHI3 + CH3-C
иодоформ
ONa
ацетат натрия
Эту реакцию дают все оксосоединения, в структуре которых
есть группа
CH3-C- , т.е. уксусный альдегид и все метилкетоны (ацетон, меO тилэтилкетон, метилпропилкетон и т.п.). Образующийся
85
86.
иодоформ обладает характерным «аптечным» запахом, а при достаточной концентрации выпадает в виде желтоватого осадка.Иодоформную реакцию используют как качественную, чтобы отличить уксусный альдегид от всех других альдегидов. В медицинской практике иодоформную реакцию используют для обнаружения ацетона в моче больных сахарным диабетом.
Реакции окисления альдегидов. Альдегиды окисляются
очень легко. Даже такие слабые окислители, как аммиачный раствор гидроксида серебра (реактив Толленса) и гидроксид меди
(II), окисляют альдегиды в соответствующие карбоновые кислоты. Обе эти реакции используют как качественные для обнаружения альдегидной группы.
AgNO3
NH4 OH
AgOH
O
CH3 -C
H
CH3 -C
CuOH
to
+ Ag(NH3 )2 OH
O
+ Cu(OH)2
H
голубой
осадок
2 NH4 OH
to
+
[Ag(NH3 )2 ] OH
реактив Толленса
O
CH3 -C
ONH 4
+ Ag + H2 O
серебряное
зеркало
O
CH3 -C
+ CuOH
OH
желтый
осадок
Cu2 O + H2 O
кирпичнокрасный осадок
Медико-биологическое значение альдегидов и кетонов
Муравьиный альдегид (формальдегид) обладает способностью свертывать белки. Его 40% водный раствор (формалин)
применяется в медицине как дезинфицирующее средство и консервант анатомических препаратов.
86
87.
При взаимодействии формальдегида с аммиаком образуетсягексаметилентетрамин, или уротропин:
O
6 H-C
+ 4 NH3
H
to
N
CH2
H2C
N
CH2
N
C H2
CH2
N
N
N
N
N
CH2
уротропин
Уротропин используют в качестве дезинфицирующего средства при заболеваниях мочевыводящих путей. Его действие основано на способности расщепляться в кислой среде с выделением
формальдегида.
Хлоральгидрат CCl3CH(OH)2 применяется в медицине как
успокаивающее и снотворное средство.
ХИМИЧЕСКИЕ СВОЙСТВА КАРБОНОВЫХ КИСЛОТ
И ИХ ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
Функциональной группой карбоновых кислот является карбоксильная группа : -C O .
OH
Таблица 2
Названия предельных алифатических монокарбоновых кислот
Строение
O
H-C
OH
O
CH3 -C
OH
O
CH3 -CH2 -C
OH
O
CH3 -CH2 -CH2 -C
OH
O
Название
по IUPAC
Тривиальное
название
метановая
кислота
муравьиная
кислота
формиаты
этановая
кислота
уксусная
кислота
ацетаты
Название солей
пропановая пропионовая
кислота
кислота
пропионаты
бутановая
кислота
масляная
кислота
бутираты
валериановая
кислота
валераты
пентановая
CH3 -CH2 -CH2 -CH2 -C
OH кислота
87
88.
Представителями непредельных алифатических кислот являютсяO
O
CH3 -CH=CH-C
CH2 =CH-C
OH
бутен-2-овая кислота
кротоновая кислота
OH
пропеновая кислота
акриловая кислота
Простейшей ароматической кислотой является бензойная:
C
O
OH
Реакционные центры в молекулах карбоновых кислот
электрофильный
центр
основный
δ
−
центр
H
:
O
δ'+ δ+
R C C ..
O H
H
CH-кислотный
OH-кислотный
центр
центр
Карбоксильная группа является
p,π-сопряженной системой. Неподеленная электронная пара кислорода OH-группы участвует в
сопряжении с π-электронным
облаком
C=O-связи.
Общая
электронная плотность смещена
к более электроотрицательному атому кислорода, поэтому на углероде карбоксильной группы возникает частичный положительный заряд. Этот углерод является электрофильным центром. За
счет p,π-сопряжения C-H-связь еще более поляризуется, водород
может отщепиться в виде протона – это OH-кислотный центр.
Неподеленная электронная пара кислорода C=O-группы не участвует в сопряжении, к ней может присоединяться протон водорода – это оксониевый основный центр. За счет электроноакцепторного действия карбоксильной группы на α-углеродном атоме
возникает частичный положительный заряд, CH-связи поляризуются. α-Углеродный атом является CH-кислотным центром.
88
89.
Кислотные свойства карбоновых кислотРанее (стр.67,68) мы сделали вывод о том, что карбоновые
кислоты проявляют более сильные кислотные свойства, чем
спирты и фенолы. Это связано с высокой стабильностью карбоксилат-аниона, являющегося p,π-сопряженной системой:
R-C
O
O
R-C
OH
O
- + H
+
O
карбоксилат анион
или R-C
O
-1/2
-1/2
Отрицательный заряд в карбоксилат-анионе делокализован
и распределен между двумя атомами кислорода.
Карбоновые кислоты способны взаимодействовать с щелочами, карбонатами и гидрокарбонатами, образуя соли:
CH3-CH2-CH2-COOH + NaOH
C6H5-COOH + NaHCO3
CH3-CH2-CH2-COONa + H2O
натрия бутират
C6H5-COONa + CO2 + H2O
натрия бензоат
Электроноакцепторные заместители в радикале карбоновых
кислот повышают их кислотные свойства, электронодонорные –
понижают. В гомологическом ряду предельных алифатических
карбоновых кислот кислотность понижается.
Реакции нуклеофильного замещения
За счет электрофильного центра в молекулах карбоновых
кислот протекают реакции нуклеофильного замещения. Замещение, а не присоединение с разрывом π-связи характерно потому,
что при этом не нарушается p,π-сопряженная система.
Рассмотрим механизм реакции SN в общем виде:
OE
O
R-C
O+
+ E Nu
OH
-
R-C-Nu
-E+
OH
присоединение
R-C-Nu
OH
E+
замещение
O
R-C
+ E +OH
-
Nu
Новая связь C-Nu образуется за счет пары электронов нуклеофила. π-Связь C=O разрывается гетеролитически, пара элек89
90.
тронов уходит к кислороду, на нем возникает отрицательный заряд. На следующей стадии реакции электрофил E+ может присоединиться к O-, но при этом сопряженная система нарушится. Этотпуть реакции для карбоновых кислот не характерен. Электрофил
E+ может также отщепить группу HO-, при этом вновь образуется
p,π-сопряженная система. Поэтому данный путь реакции (замещение) и характерен для карбоновых кислот.
По механизму нуклеофильного замещения протекает, например, реакция образования сложных эфиров (реакция этерификации).
В этой реакции участвуют карбоновая кислота и спирт в
присутствии концентрированной серной кислоты. Например:
O
O
H+
CH3 -C
+ H2 O
+ CH3 OH
CH3 -C
OCH3
OH метанол
метилацетат
уксусная
кислота
Серная кислота необходима как водоотнимающее средство
и как катализатор.
Рассмотрим механизм реакции этерификации:
δ+
R-C
O:
H2 SO4
-HSO 4
OH
+
O-H
+
R-C
R-C
OH
OH
..
HO-CH3
OH
+
R-C O CH3
OH H
OH
1
2
3
OH
R-C
O CH3
+
- H2 O
O-H
H
OH
+
R-C
HSO4
O
R-C
OCH3
OCH3
5
+ H2 SO4
6
4
Молекула спирта – это слабый нуклеофил. Поэтому для успешного протекания реакции необходимо активизировать электрофильный центр в молекуле карбоновой кислоты. С этой целью
и применяют серную кислоту как катализатор. Реакция начинается по основному центру карбоновой кислоты, к которому присоединяется протон водорода. Протонированная карбоновая кислота
90
91.
может существовать в виде двух резонансных структур (1 и 2). Вкатионе (2) на электрофильном центре существует полный положительный заряд, а не частичный, как в исходной молекуле карбоновой кислоты, т.е. электрофильный центр активизировался.
Далее он атакуется нуклеофильным реагентом – молекулой спирта. Новая связь C-O образуется за счёт неподеленной электронной пары кислорода, поэтому на нем появляется положительный
заряд (3). Кислород отщепляет протон водорода, чтобы компенсировать положительный заряд, а протон присоединяется к неподеленной электронной паре другого атома кислорода. Он также
получает положительный заряд (4). Далее происходит отщепление молекулы воды и образование катиона (5), для стабилизации
которого необходимо отщепление протона водорода (под действием гидросульфат-аниона, оставшегося в реакционной среде).
Образуется молекула сложного эфира (6) и серная кислота – катализатор реакции.
Сложные эфиры являются функциональными производными
карбоновых кислот, т.е. производными, полученными путем замещения гидроксила в карбоксильной (функциональной) группе
на нуклеофил.
Другими функциональными производными карбоновых кислот являются:
O
O
R-C
O
R-C
Cl
хлорангидриды
R-C
O
ангидриды
O
R-C
NH2
амиды
O
R-C
NH-NH2
гидразиды
Все эти производные могут быть получены из карбоновых
кислот.
91
92.
Так, хлорангидриды кислот получают при взаимодействиикарбоновых кислот с тионилхлоридом (SOCl2) или хлоридом
фосфора (PCl5), например:
O
O
CH3 -C
OH
уксусная
кислота
CH3 -C
+ SOCl2
+ SO 2 + HCl
Cl
ацетилхлорид
O
O
+ PCl5
C
C
Cl
OH
бензойная кислота
+ POCl3 + HCl
бензоилхлорид
Ангидриды образуются при нагревании карбоновых кислот
с водоотнимающими средствами, например с P2O5:
O
CH3 -C
+
CH3 -C
O
CH3 -C
P2 O5 , to
OH
CH3 -C
OH
O + H2 O
O
уксусный ангидрид
O
Амиды могут быть получены при взаимодействии карбоновых кислот с аммиаком с последующим термическим разложением образующихся аммониевых солей:
CH3 -CH2 -C
пропионовая
кислота
O NH
3
O
CH3 -CH2 -C
OH
ONH4
пропионат
аммония
to
O
CH3 -CH2 -C
NH2
+ H2 O
амид пропионовой
кислоты
Функциональные производные карбоновых кислот можно
также получить одни из других (менее активные из более активных).
Сравним активность функциональных производных карбоновых кислот в реакциях нуклеофильного замещения. Для этого
необходимо сравнить величины частичных положительных заря92
93.
дов на электрофильном центре, т.е. рассмотреть влияние заместителей на δ+.O
O
O
δ+
R-C ..
Cl
хлорангидрид
-I Cl > +M Cl
δ+
R-C
R-C ..
O-R'
сложный эфир
-I OR'< +M OR'
O
:
δ+ O
R-C
O
ангидрид
δ+
R-C ..
OH
карбоновая кислота
-I
< +M OH
OH
O
..
NH2
δ+
R-C
амид
-I NH << +M NH
2
2
-I OCOR < +M OCOR
Атом хлора в хлорангидриде оказывает электроноакцепторное действие, т.е увеличивает величину δ+ на электрофильном
центре (активность также увеличивается). Гидроксильная группа
в молекуле кислоты и алкокси-группа (OR’) в сложном эфире
оказывают примерно одинаковое электронодонорное действие,
понижая величину частичного положительного заряда на электрофильном центре, значит, в сравнении с хлорангидридом активность также будет снижаться. Заместитель в молекуле ангидрида кислоты также является электронодонорным, но его влияние
распределяется пополам между двумя электрофильными центрами. Величина δ+ уменьшается, но не так сильно, как в случае
карбоновой кислоты или сложного эфира. И, наконец, в молекуле
амида кислоты электронодонорное действие NH2-группы проявляется наиболее сильно, и активность амидов в реакциях нуклеофильного замещения самая низкая.
Таким образом, ряд убывания активности функциональных
производных карбоновых кислот в реакциях нуклеофильного замещения следующий:
хлорангидрид > ангидрид > сложный эфир ≈ карбоновая кислота > амид.
Сравнивая активность функциональных производных карбоновых
кислот с позиций «уходящей группы» (см. стр. 50), можно сделать тот же
93
94.
вывод (ведь чем лучше «уходящая группа», тем успешнее протекает реакция).O
Cl
-
R-C
R'
-
O
лучшая
уходя щая группа
(из-за высокой
электроотрицательности
хлора)
хорошая
уходя щая группа
(из-за высокой
степени делокализации заря да
в сопря женной
системе)
-
H 2N
H-O
O-
плохая
уходя щая группа
(делокализация
заря да только
за счет индуктивного эффекта)
-
очень плохая
уходя щая группа
(делокализации
заря да нет, азот
не столь электроотрицателен как
кислород)
плохая
уходя щая группа
(делокализации
заря да нет)
Исходя из этого, из хлорангидридов кислот можно получить
все остальные функциональные производные, из ангидридов кислот – сложные эфиры, карбоновые кислоты и амиды.
Рассмотрим реакции хлорангидридов кислот:
O
R'-COO Na+
R-C
O
+ HCl
реакция ацилолиза
(или ацидолиза)
+ HCl
реакция гидролиза
+ HCl
реакция алкоголиза
R'-C
O
..
H2 O
O
R-C
OH
O
R-C
..
R'-OH
R-C
OR'
Cl
..
NH3
..
R'-NH2
94
O
O
R-C
NH2
+ HCl
реакция аммонолиза
+ HCl
реакция аминолиза
O
R-C
NH-R'
95.
Все эти реакции протекают по механизму нуклеофильногозамещения, нуклеофилами являются, соответственно, карбоксилат-анион, вода, спирт, аммиак и амин. Реакции протекают активно, не требуют нагревания.
По отношению к хлорангидриду кислоты эти реакции называют гидролиз, аминолиз и т.д. («лизис» – расщепление). С другой стороны, по отношению к спирту, амину и т.д. – это реакции
ацилирования (введение в молекулу остатка карбоновой кислоты – ацила). Поэтому ряд убывания активности функциональных
производных карбоновых кислот в реакциях нуклеофильного замещения можно также назвать рядом убывания ацилирующей
способности.
Из ангидридов карбоновых кислот можно получить все другие функциональные производные, кроме хлорангидридов:
..
H2 O
O
2 R-C
OH
O
..
R'-OH
O
+ R-COOH
R-C
OR'
R-C
O
R'-C
O
..
NH3
..
R'-NH2
O
R-C
NH2
+ R-COOH
O
R-C
+ R-COOH
NH-R'
Реакции протекают менее активно, чем с хлорангидридами
кислот.
В биохимических процессах важную роль играют смешанные ангидриды карбоновых кислот и фосфорной кислоты –
ацилфосфаты.
С их помощью в организме осуществляется перенос ацильных групп к гидроксильным, тиольным и аминогруппам различных соединений:
95
96.
OR-OH
O
OH
ацетилфосфат
+ H3 PO4
OR
сложный эфир
O
CH3 -C
+ H3 PO4
NHR
амид
R-NH2
CH3 -C-O-P-OH
O
CH3 -C
O
R-SH
CH3 -C
SR
тиоэфир
+ H 3 PO4
Сложные эфиры вступают в реакции гидролиза и образования амидов.
Гидролиз сложных эфиров проводят в кислой или щелочной
среде. Кислотный гидролиз является обратимым процессом:
H+
O
CH3 -C
+ H2 O
O
CH3 -C
+ CH3 OH
метанол
OH
уксусная кислота
to
OCH3
метилацетат
Щелочной гидролиз сложных эфиров необратим, т.к. в результате реакции образуются соли карбоновых кислот, для которых реакции SN не характерны:
O
+ NaOH
C
OC 2 H5
O
H2 O
to
C
этилбензоат
+ C 2 H5 OH
этанол
ONa
бензоат натрия
Реакции гидролиза сложных эфиров протекают не столь активно, как гидролиз ангидридов или галогенангидридов кислот.
Сложные эфиры вступают также в реакции нуклеофильного
замещения с аммиаком и аминами с образованием соответствующих амидов:
..
NH3
OC 2 H5
этилацетат
96
CH3 -C
NH2
ацетамид
O
CH3 -C
O
..
CH3 -NH 2
+ C2 H5 -OH
O
+ C2 H5 -OH
NH-CH3
N-метиламид
уксусной кислоты
CH3 -C
97.
Амиды карбоновых кислот являются наименее активнымифункциональными производными. Они подвергаются гидролизу,
но только в достаточно жестких условиях (кипячение с водными
растворами кислот или щелочей в течение длительного времени).
O
CH3-C
+ H2O + HCl
CH3-C
to
O
NH2
ацетамид
O
CH3-C
O
to
+ NaOH
+ NH4Cl
OH
+ CH3NH2
ONa
CH3-C
NHCH3
N-метиламид
уксусной кислоты
За счёт p,π-сопряжения основные свойства амидов понижены в сравнении с аминами. Амиды кислот образуют соли с сильными минеральными кислотами. Предполагают, что при этом
амид образует соль за счёт оксониевого основного центра:
O:
δ+
CH3 -C .. + H +
NH2
+
O-H
CH3 -C
NH2
Амиды могут реагировать также с сильными основаниями,
например, амидом натрия, проявляя кислотные свойства:
δ+
CH3 -C
O
.. + NaNH 2
N H
O
CH3 -C
NHNa
+ NH3
H
Реакции карбоновых кислот по радикалу
Для насыщенных алифатических карбоновых кислот характерны реакции замещения (галогенирования) по α-положению
радикала. Эта реакция известна как реакция Гелль-ФольгардаЗелинского и проводится в присутствии фосфора:
CH3 -CH2 -COOH + Cl2
пропионовая кислота
P
CH3 -CH-COOH + HCl
Cl
α-хлорпропионовая
кислота
97
98.
Ненасыщенные карбоновые кислоты вступают в реакцииэлектрофильного присоединения по π-связи, например, для них
характерны реакции галогенирования, гидрогалогенирования.
CH2 =CH-COOH + Br 2
пропеновая кислота
H2 O
CH2 -CH-COOH
Br Br
2,3-дибромпропановая
кислота
Реакции гидрогалогенирования акриловой кислоты протекают против правила Марковникова. Это связано с перераспределением электронной плотности в π,π-сопряженной системе:
δ+
δ−
CH2 =CH-C
O
+ HBr
OH
пропеновая кислота
CH2 -CH2 -COOH
Br
3-бромпропановая
кислота
Ароматические кислоты вступают в реакции электрофильного замещения (галогенирования, нитрования) по бензольному
кольцу. Реакции протекают в жестких условиях, т.к. карбоксильная группа является электроноакцепторным заместителем и дезактивирует кольцо.
COOH
COOH
+ Br2
FeBr3
to
бензойная
кислота
бензойная
кислота
98
+ HBr
м-бромбензойная
кислота
COOH
COOH
+ HNO3
Br
H2SO4
to
+ H2O
NO2
м-нитробензойная
кислота
99.
ДИКАРБОНОВЫЕ КИСЛОТЫВ структуре дикарбоновых кислот присутствуют две карбоксильные группы. Алифатические дикарбоновые кислоты
классифицируют на насыщенные и ненасыщенные.
Таблица 3
Названия насыщенных алифатических
дикарбоновых кислот
Название по
IUPAC
Тривиальное
название
Название
солей
этандиовая
кислота
щавелевая
кислота
оксалаты
пропандиовая малоновая
кислота
кислота
малонаты
бутандиовая
кислота
сукцинаты
Строение
O
O
C-C
HO
OH
O
O
HO
C-CH 2 -C
OH
O
O
C-CH2 -CH2 -C
HO
OH
C-CH 2 -CH2 -CH2 -C
HO
пентандиовая глютаровая
кислота
кислота
O
O
янтарная
кислота
OH
глютараты
Примерами ненасыщенных дикарбоновых кислот являются
стереоизомеры
бутендиовых
кислот: малеиновая (цисбутендиовая кислота) и фумаровая (транс-бутендиовая кислота).
H
H
COOH
HOOC
COOH
H
C
C
малеиновая кислота
H
C
C
COOH
фумаровая кислота
Для дикарбоновых кислот характерны все свойства монокарбоновых кислот: они проявляют кислотные свойства, образуют функциональные производные. Кроме того, для дикарбоновых
кислот характерны и специфические свойства.
99
100.
Кислотные свойства дикарбоновых кислот проявляютсясильнее, чем у монокарбоновых. Это можно объяснить электроноакцепторным влиянием одной карбоксильной группы на другую. Среди дикарбоновых кислот самой сильной кислотой является щавелевая. В гомологическом ряду кислотные свойства понижаются, т.к. электронное влияние двух карбоксильных групп
затухает, передаваясь по цепи.
Карбоновые кислоты образуют два ряда солей: кислые (за
счёт одной карбоксильной группы) и средние (за счёт обеих).
O
O
C-C
OH
HO
O
NaOH
O
ONa
-H2 O
NaO
O
NaOH
C-C
-H2 O
HO
щавелевая
кислота
кислый оксалат натрия
(мононатриевая соль)
O
C-C
ONa
оксалат натрия
(динатриевая соль)
Дикарбоновые кислоты способны образовывать и два ряда
функциональных производных: неполные и полные галогенангидриды, моно- и диэфиры, моно- и диамиды).
O
O
C-CH 2 -CH 2 -C
OH
HO
янтарная кислота
CH3 OH
H+
O
O
C-CH 2 -CH 2 -C
HO
CH3 OH
H+
OCH3
монометилсукцинат
O
O
C-CH 2 -CH 2 -C
H3 CO
O
O
C-C
OH
HO
щавелевая
кислота
100
OCH3
диметилсукцинат
SOCl2
O
O
C-C
HO
Cl
неполный
хлорангидрид
SOCl2
O
O
C-C
Cl
Cl
полный
хлорангидрид
101.
Специфическим свойством щавелевой и малоновой кислотявляется их способность к декарбоксилированию при нагревании:
O
O
O
to
C-C
H-C
OH
HO
OH
+ CO 2
щавелевая
муравьиная
кислота
кислота
O o
O
O
t
CH3 -C
+ CO 2
C-CH 2 -C
OH
OH
HO
малоновая кислота
уксусная
кислота
Реакция декарбоксилирования характерна для дикарбоновых кислот, у которых две карбоксильные группы оказывают
друг на друга наиболее сильное влияние.
Янтарная, глутаровая и малеиновая кислоты при нагревании
образуют циклические ангидриды. Это связано, во-первых, с тем,
что при нагревании их молекулы принимают клешневидную
конформацию, и две карбоксильные группы оказываются пространственно сближенными, а во-вторых, с тем, что образуются
пяти- или шестичленные циклы, которые являются наиболее стабильными.
O
H2 C
O
C
OH
H2 C
H2C
to
C
O + H2 O
OH
H2C
O
O
янтарный ангидрид
C
C
янтарная кислота
O
CH2 -C
H2 C
OH
OH
CH2 -C
O
глютаровая кислота
O
to
H2 C C
O + H2 O
H2C
H2 C C
O
глютаровый ангидрид
101
102.
Медико-биологическое значение карбоновых кислоти их производных
Соли уксусной кислоты применяются в медицине. Ацетат
калия (CH3COOK) – диуретическое средство; ацетат свинца
((CH3COO)2Pb . 3 H2O) – вяжущее средство, применяется при
воспалительных воспалениях кожи и слизистых оболочек.
Изовалериановая кислота (CH3)2CHCH2COOH входит в состав валидола, присутствует в настойке валерианы. Оказывает
успокаивающее действие.
Бензоат натрия C6H5COONa применяется как отхаркивающее средство.
Сложные эфиры карбоновых кислот широко распространены в природе. Жиры являются сложными эфирами высших карбоновых кислот и спирта глицерина. Запах плодов и цветов в
значительной степени обусловлен присутствием сложных эфиров.
Некоторые лекарственные средства химически модифицируют, получая их сложные эфиры (так называемые «пролекарства»). Этерификация приводит к снижению или исчезновению некоторых отрицательных свойств исходных лекарств (горький
вкус, раздражающее действие в месте инъекции). В организме
под действием ферментов эстераз происходит гидролиз сложных
эфиров с выделением исходного лекарственного соединения. Например, в отличие от левомицетина его стеарат не обладает горьким вкусом, что дало возможность применять его в педиатрической практике. В организме левомицетина стеарат гидролизуется
до свободного левомицетина:
NH-CO-CHCl 2
O 2 N-
-CH-CH-CH 2 -O-C-(CH 2 )16 -CH 3
гидролиз
O
OH
левомицетина стеарат
NH-CO-CHCl 2
O 2 N-
-CH-CH-CH 2 -OH
OH
левомицетин
102
+ C 17 H35 COOH
стеариновая кислота
103.
ГЕТЕРОФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯАЛИФАТИЧЕСКОГО РЯДА – МЕТАБОЛИТЫ
И БИОРЕГУЛЯТОРЫ
Большинство органических веществ, участвующих в процессах метаболизма, относятся к гетерофункциональным соединениям, т.е. имеющим в структуре несколько различных функциональных групп. Наиболее распространенными гетерофункциональными соединениями являются аминоспирты, аминокислоты, оксикислоты и оксокислоты.
Химические свойства гетерофункциональных соединений
нельзя рассматривать как сумму свойств, обусловленных наличием каждой функциональной группы. Так как функциональные
группы влияют друг на друга, то у гетерофункциональных соединений появляются и специфические химические свойства.
Аминоспирты
Аминоспиртами называют соединения, содержащие одновременно амино- и окси-группы.
Простейшим представителем аминоспиртов является 2аминоэтанол, или коламин:
CH -CH .
2
2
OH NH2
Как амин коламин образует соли с сильными кислотами:
CH2 -CH2 Cl -
CH2 -CH2 + HCl
OH NH2
+
OH NH3
коламина гидрохлорид
Основные свойства его понижены по сравнению с алифатическими аминами, т.к. OH-группа является электроноакцепторным заместителем.
Коламин проявляет нуклеофильные свойства за счет неподеленной электронной пары азота и подвергается реакции алкилирования:
CH
CH2 -CH2
OH NH2
3 CH3 I
OH -
+
3
CH2 -CH2 -N-CH 3
OH
OH -
CH3
холин
103
104.
В результате реакции образуется другой аминоспирт – холин.Коламин и холин участвуют в построении молекул фосфолипидов – основных компонентов клеточных мембран
(см.
стр. 225).
За счет наличия гидроксильной группы холин способен к
реакциям ацилирования:
O
CH3
CH2 -CH 2 -N-CH 3 +
CH3 -C
OH
CH3 -C
+
CH3
холин
CH3
CH2 -CH2 -N-CH 3
+
O
O-C-CH 3
O
CH3
O
ацетилхолин
(в организме этот процесс осуществляется с участием ацетилкоэнзима А). Ацетилхолин является нейромедиатором.
Важную роль в организме играют аминоспирты, содержащие в качестве структурного фрагмента остаток пирокатехина.
Их общее название катехоламины. Катехоламины синтезируются
в организме из фенилаланина через образование дофамина:
HO
HO-
окисление
-CH2 -CH2 -NH2
HO
HO-
OH
норадреналин
дофамин
метилирование
HO
HO-
-CH-CH2 -NH2
-CH-CH2 -NH-CH 3
OH
адреналин
Адреналин является гормоном надпочечников, участвует в
регуляции сердечной деятельности, обмена углеводов. При стрессах он выделяется в кровь, его называют «гормоном страха».
Структурно близки катехоламинам некоторые природные и
синтетические биологически активные вещества, применяемые в
качестве лекарственных средств. Так, алкалоид эфедрин обладает
сосудосуживающим действием, расширяет бронхи, повышает артериальное давление. Его используют в виде гидрохлорида при
бронхиальной астме, для уменьшения воспалительных процессов
при ринитах.
-CH-CH-NH-CH 3
OH CH3
эфедрин
104
105.
АминокислотыАминокислоты – гетерофункциональные соединения, содержащие амино- и карбоксильную группу. По их взаимному
расположению аминокислоты классифицируют на α-, β-, γ-, δ-, εаминокислоты. Например:
CH3 -CH-CH2 -COOH
CH3 -CH-COOH
NH2
NH2
β-аминомасляная кислота
3-аминобутановая кислота
α-аминопропионовая кислота
2-аминопропановая кислота
Аминокислоты обладают всеми свойствами карбоновых кислот - они способны образовывать соли, сложные эфиры и другие
функциональные производные:
NaOH
CH3 -CH-COONa + H 2 O
NH2
CH3 -CH-COOH
NH2
натриевая соль
CH3 OH
H+
CH3 -CH-COOCH 3 + H2 O
NH2
метиловый эфир
Аминокислоты проявляют также свойства аминов (основные свойства, способность к реакциям ацилирования и алкилирования):
HCl
CH3 -CH-COOH
CH3 -CH-COOH
+
NH3 Cl
гидрохлорид
(CH3 CO)2 O
NH2
CH3 -CH-COOH + CH 3 -COOH
NH-C-CH 3
O
N-ацетильное производное
CH3 I
CH3 -CH-COOH + HI
NH-CH3
N-метильное производное
105
106.
Амфотерность аминокислот приводит к тому, что они могутсуществовать в виде внутренних солей:
CH3 -CH-COO
+
NH3
внутренняя соль
CH3 -CH-COOH
NH2
Специфические свойства аминокислот зависят от взаимного
расположения амино- и карбоксильной групп.
α-Аминокислоты вступают в реакции декарбоксилирования
при нагревании с гидроксидом бария с образованием соответствующих аминов, например:
CH3 -CH-COOH
Ba(OH)2
to
CH3 -CH2 -NH2 + BaCO3 + H2 O
этиламин
NH2
α-аминопропионовая
кислота
Сходным образом декарбоксилирование природных αаминокислот протекает и in vivo с участием ферментов, например, при декарбоксилировании серина образуется коламин:
CH2 -CH-COOH
фермент
CH2 -CH 2 + CO 2
OH NH2
OH NH2
серин
коламин
Другим специфическим свойством α-аминокислот является
их способность к образованию межмолекулярных циклических
амидов (дикетопиперазинов):
O
CH3 -CH-C
OH
HN
H + H
NH
HO
C-CH-CH 3
O
O
to
CH3 -HC
HN
C
NH
C
+ 2 H2 O
CH-CH3
O
дикетопиперазин
Так как дикетопиперазины фактически являются амидами,
они способны к гидролизу и в кислой, и в щелочной среде.
Характерной особенностью β-аминокислот являются реакции внутримолекулярного элиминирования: при нагревании они
отщепляют молекулу аммиака. Например, при нагревании
106
107.
β-аминомасляной кислоты образуется кротоновая (бутен-2-овая)кислота:
H
α
CH3 -CH-C-COOH
to
CH3 -CH=CH-COOH + NH 3
NH2 H
Способность к этой реакции связана с подвижностью протона водорода при α-углеродном атоме из-за электроноакцепторного влияния двух функциональных групп.
За счет существования молекул γ-аминокислот в клешневидной конформации функциональные группы оказываются пространственно сближенными, и между ними может происходить
взаимодействие с образованием циклического внутримолекулярного амида (лактама):
O
O
α C
to
OH
H2 C
NH
H2 C γ H
CH
CH3
γ -аминовалериановая кислота
C
H2C
H2C
NH + H2 O
CH
CH3
γ -валеролактам
Пятичленные циклы устойчивы, поэтому реакция протекает
достаточно легко.
Как амиды лактамы подвергаются гидролизу и в кислой, и в
щелочной среде:
H2 O, HCl
O
C
H2C
NH
C H2
H2C
γ-бутиролактам
NaOH, H2 O
+
H3 N-CH 2 -CH 2 -CH 2 -COOH
Cl
γ-аминомасляной кислоты
гидрохлорид
H2 N-CH 2 -CH 2 -CH 2 -COONa
натриевая соль γ-аминомасляной
кислоты
O
R-C-NH
S
N
O
CH3
CH3
COOH
пенициллины
Некоторые природные соединения и синтетические лекарственные средства содержат в
своей структуре лактамный цикл. Например, четырехчленный β-лактамный цикл
присутствует в молекулах пенициллинов.
107
108.
Из-за его способности к гидролизу, что приводит к потере биологической активности, пенициллины не стерилизуют в водныхрастворах.
Гидроксикислоты (оксикислоты)
Примерами гидроксикислот являются:
CH3 -CH-CH 2 -COOH
CH3 -CH-COOH
OH
2-гидроксипропановая кислота
α-оксипропионовая кислота
молочная кислота
OH
3-гидроксибутановая кислота
β-оксимасляная кислота
Для гидроксикислот характерны свойства карбоновых кислот и спиртов. Как карбоновые кислоты они образуют соли и
сложные эфиры:
NaOH
CH3 -CH-COONa + H 2 O
OH
лактат натрия
CH3 -CH-COOH
OH
молочная кислота
CH3 OH
H+
CH3 -CH-COOCH 3 + H2 O
OH
метиловый эфир
молочной кислоты
Как спирты оксикислоты способны к реакциям окисления и
ацилирования:
[O]
O
2-оксопропановая
(пировиноградная) кислота
CH3 -CH-COOH
OH
молочная кислота
CH3 -C-COOH
(CH3 CO) 2 O
CH3 -CH-COOH + CH 3 -COOH
O-C-CH 3
O
O-ацетильное производное
молочной кислоты
108
109.
Специфические свойства α-оксикислот. α-Оксикислоты принагревании в присутствии концентрированной серной кислоты
разлагаются с образованием карбонильных соединений и муравьиной кислоты. Например:
O
c.H2 SO 4
CH3 -CH-COOH
CH3 -C
+ H-COOH
H
муравьиная
уксусный
кислота
альдегид
to
OH
молочная кислота
CH3 -CH 2 -CH-COOH
c.H2 SO4
to
OH
α-оксимасляная
кислота
O
CH3 -CH 2 -C
H
пропионовый
альдегид
+ H-COOH
муравьиная
кислота
Другим специфическим свойством α-оксикислот является
их способность к реакциям образования межмолекулярных циклических сложных эфиров – лактидов:
O
O
CH3 -CH-C
OH
HO
+
OH
HO
C-CH-CH 3
O
to
CH3 -HC
O
C
O
C
+ 2 H2 O
CH-CH3
O
лактид молочной кислоты
молочная кислота
Лактиды способны к гидролизу и в кислой, и в щелочной
среде.
Для β-оксикислот характерны реакции внутримолекулярного элиминирования, при нагревании они отщепляют воду:
H
α
CH3 -CH-C-COOH
to
OH H
CH3 -CH=CH-COOH + H 2 O
кротоновая (бутен-2овая) кислота
β-оксимасляная кислота
109
110.
γ-Оксикислоты способны к реакциям внутримолекулярнойэтерификации, приводящим к образованию циклических сложных эфиров, например,
O
O
α C
to
OH
H2 C
β
OH
H2 C γ
CH2
γ -оксимасляная кислота
C
H2C
H2C
O + H2 O
C H2
γ -бутиролактон
Как сложные эфиры лактоны гидролизуются и в кислой, и в
щелочной среде:
H2 O, H+
O
C
H2C
OH
γ-оксивалериановая кислота
O
CH
CH3
γ-валеролактон
H2C
CH3 -CH-CH 2 -CH2 -COOH
NaOH, H2 O
CH3 -CH-CH 2 -CH2 -COONa
OH
натриевая соль
γ-оксивалериановой кислоты
Примерами многоосновных оксикислот являются яблочная
(соли – малаты), винная (соли – тартраты) и лимонная (соли –
цитраты) кислоты:
COOH
HOOC-CH 2 -C-CH 2 -COOH
HOOC-CH-CH 2 -COOH
HOOC-CH-CH-COOH
OH
яблочная кислота
2-гидроксибутандиовая
кислота
OH OH
винная кислота
2,3-дигидроксибутандиовая
кислота
OH
лимонная кислота
2-гидроксипропантрикарбоновая кислота
Как дикарбоновая кислота, винная кислота дает два ряда солей – кислые и средние, причем кислая калиевая соль нерастворима в воде, а средняя – растворима.
110
111.
На этом основано качественное определение винной кислоты.HOOC-CH-CH-COOH
KOH
HOOC-CH-CH-COOK
KOH
OH OH
кислая калиевая соль
(белый осадок)
OH OH
винная кислота
KOOC-CH-CH-COOK
OH OH
средняя я калиевая соль
(растворима в воде)
Второй качественной реакцией винной кислоты является ее
взаимодействие с гидроксидом меди (II). Это реакция на диольный фрагмент молекулы (происходит растворение голубого осадка Cu(OH)2 с образованием ярко-синего раствора:
COOK
COOK
CH-OH HO
HO-CH
+
+ Cu
CH-OH
OH HO-CH
COOK
COOK
COOK
COOK
H
O-CH
CH-O
CH-O
H
COOK
Cu
O-CH
+ 2 H2 O
COOK
хелатный медный комплекс
тартрат калия
Как α-оксикислота лимонная кислота разлагается при нагревании в присутствии концентрированной серной кислоты, давая
ацетондикарбоновую (3-оксопентандиовую) кислоту и муравьиную кислоту. Для β-кетонокислот характерны реакции декарбоксилирования, поэтому ацетондикарбоновая кислота превращается
в ацетон:
COOH
HOOC-CH 2 -C-CH 2 -COOH
OH
лимонная кислота
H2 SO 4 , to
HOOC-CH 2 -C-CH 2 -COOH + H-COOH
муравьиная
O
кислота
ацетондикарбоновая кислота
to
CH3 -C-CH 3 + 2 CO 2
O
ацетон
111
112.
Ацетон можно обнаружить с помощью иодоформной пробы(см. стр. 85).
Оксокислоты
К оксокислотам относятся альдегидо- и кетонокислоты.
Примером альдегидокислоты является глиоксиловая или
глиоксалевая кислота (2-оксоэтановая кислота):
O
H
C-COOH
Важную роль в биохимических процессах играют кетонокислоты, например:
CH3-C-COOH
CH3-C-CH2-COOH
HOOC-C-CH2-COOH
O
пировиноградная
α-кетопропионовая
2-оксопропановая
O
ацетоуксусная
β-кетомасля ная
3-оксобутановая
O
щавелевоуксусная
кетоя нтарная
2-оксобутандиовая
Они участвуют в организме в цикле трикарбоновых кислот
(цикле Кребса) и образуются при окислении соответствующих
оксикислот:
[O]
CH3-CH-COOH
CH3-C-COOH
OH
молочная кислота
O
пировиноградная
кислота
CH3-CH-CH2-COOH
[O]
OH
β-оксимасля ная кислота
CH3-C-CH2-COOH
O
ацетоуксусная кислота
Подробнее остановимся на свойствах кетонокислот.
Для оксокислот характерны свойства карбоновых кислот,
кетонов, а также специфические свойства.
NaOH
CH3 -C-COONa + H 2 O
O
пируват натрия
CH3 -C-COOH
O
пировиноградная
кислота
CH3 OH, H +
CH3 -C-COOCH 3 + H2 O
O
метилпируват
NH2 OH
CH3 -C-COOH + H 2 O
N-OH
оксим
112
113.
Как карбоновые кислоты оксокислоты образуют соли, проявляя кислотные свойства, и функциональные производные, например, сложные эфиры (реакция SN). Как кетоны они вступаютв реакции нуклеофильного присоединения и присоединенияотщепления (например, образуя оксимы, гидразоны и т.п.).
Специфическим свойством β-кетокислот является их способность к декарбоксилированию, например, при ферментативном декарбоксилировании ацетоуксусной кислоты образуется
ацетон:
ферменты
CH3 -C-CH 2 -COOH
O
ацетоуксусная кислота
CH3 -C-CH 3 + CO 2
O
ацетон
Ацетоуксусная кислота и ацетон накапливаются в крови
больных сахарным диабетом (т.н. «кетоновые тела»). К кетоновым телам относят также и предшественницу ацетоуксусной кислоты – β-оксимасляную кислоту.
Другим специфическим свойством β-кетонокислот (а точнее, их сложных эфиров) является их способность к существованию в двух таутомерных формах.
Таутомерия – равновесная динамическая изомерия. Это значит, таутомеры самопроизвольно превращаются друг в друга и
существуют в подвижном равновесии. Таутомеры превращаются
друг в друга за счет переноса какой-либо подвижной группы. Если такой группой является протон водорода, то таутомерию называют прототропной.
Рассмотрим это на примере этилового эфира ацетоуксусной
кислоты (часто его называют просто ацетоуксусным эфиром).
CH-кислотный
центр
H
CH3 -C
C
:O
H
основный
центр
O
O
C
OC 2 H5
кето-форма (92.5%)
CH3 -C=CH-C
OH
OC 2 H5
енольная форма (7.5%)
За счет электронодонорного влияния сложноэфирной и кето-групп α-атом углерода становится CH-кислотным центром. Он
отщепляет протон водорода, который присоединяется к неподеленной электронной паре кислорода оксо-групы (оксониевому
113
114.
основному центру). Кето-таутомер превращается в енольный(ен – двойная связь, -ол – гидроксильная группа). Енольная гидроксильная группа проявляет кислотные свойства, она отщепляет
протон, который присоединяется к электронному облаку π-связи
– енольный таутомер превращается в кето-таутомер. Эти превращения протекают самопроизвольно, и таутомеры существуют в
динамическом равновесии.
При действии на ацетоуксусный эфир определенного реагента в реакцию вступает один из таутомеров. Так, кето-форма
вступает в реакции, характерные для кетонов (нуклеофильного
присоединения с синильной кислотой, присоединенияотщепления с гидразином или гидроксиламином):
OH
HCN
O
CH3 -C-CH 2 -C
OC 2 H5
CN
гидроксинитрил
O
CH3 -C-CH 2 -C
O
OC 2 H5
NH2 OH
O
CH3 -C-CH 2 -C
N-OH
оксим
+ H2O
OC 2 H5
За счет енольной таутомерной формы ацетоуксусный эфир
взаимодействует с хлоридом железа (III) (реакция, характерная
для всех енолов), с бромной водой (реакция на двойную связь):
FeCl3
вишневое окрашивание
O
CH3-C=CH-C
OH
OC2H5
Br2, H2O
Br
CH3-C-CH-C
O
OC2H5
OHBr
этиловый эфир
2,3-дибром-3-гидроксибутановой кислоты
114
115.
Обратите внимание, поскольку второй таутомер за счет равновесия восполняет убыль реагирующего таутомера, таутомернаясмесь реагирует в данном направлении как единое целое.
Обычно енольная форма менее устойчива, поэтому доля её в
таутомерном равновесии низкая. Иногда более энергетически выгодным является существование молекулы именно в енольной
форме, как, например, для диэтилового эфира щавелевоуксусной
кислоты:
H
O
C2 H5 O
C-C
C
:O
H
O
O
O
C-C=CH-C
C
OC 2 H5
кето-форма (15%)
C2 H5 O
OH
OC 2 H5
енольная форма (85%)
Это связано с тем, что енольная таутомерная форма представляет собой единую сопряженную систему.
Медико-биологическое значение гетерофункциональных
производных карбоновых кислот
Молочная кислота широко распространена в природе как
продукт молочнокислого брожения углеводов (см. стр. 173). В
организме она является одним из продуктов превращения глюкозы (гликолиза). Молочная кислота накапливается в мышцах при
интенсивной работе, вследствие чего в них возникает характерная боль.
Некоторые соли молочной кислоты (лактаты) применяются
в медицине. Лактат железа (II) назначают при гипохромных
анемиях. Он легче, чем неорганические соединения железа всасывается из ЖКТ, не вызывает раздражения слизистых оболочек.
Лактат кальция является источником ионов кальция, которые
необходимы для осуществления процесса передачи нервных импульсов, формирования костной ткани, процессов свертывания
крови. Применяется также как антиаллергическое средство. Лактат кальция переносится лучше, чем хлорид кальция, т.к. не раздражает слизистую оболочку.
115
116.
β-Гидроксимасляная кислота накапливается в организмебольных сахарным диабетом, являясь предшественницей ацетоуксусной кислоты, относится к «кетоновым телам».
Натриевая соль γ-гидроксимасляной кислоты (натрия оксибутират) применяется как снотворное средство, а также в анестезиологии в качестве средства для неингаляционного наркоза.
γ-Аминомасляная кислота (ГАМК) принимает участие в обменных процессах головного мозга, является нейромедиатором.
В медицинской практике применяется под названиями гаммалон,
аминалон при лечении нервно-психических заболеваний. В медицине применяются и производные лактама γ-аминомасляной
кислоты (пирролидона). Поливинилпирролидон используют как
заменитель плазмы крови, амид (1-пирролидон-2-ил)-уксусной
кислоты (пирацетам, ноотропил) применяют при нарушениях обменных процессов и кровообращения мозга.
N
O
(-CH-CH 2 -)n
поливинилпирролидон
N
O
CH2 CONH 2
пирацетам
ε-Аминокапроновая кислота оказывает кровоостанавливающее действие.
Яблочная и лимонная кислоты принимают участие в цикле
Кребса – универсальном этапе окислительного катаболизма углеводов, липидов и других соединений в присутствии кислорода.
Соли лимонной кислоты (цитраты) применяются в медицине. Например, цитрат натрия препятствует свертыванию крови.
Соли винной кислоты (тартраты) используются как мягкие
слабительные средства.
Ацетоуксусный эфир широко применяется в органическом
синтезе как исходное вещество для получения кетонов, карбоновых кислот, производных гетероциклов, представляющих интерес в качестве лекарственных средств.
116
117.
ОПТИЧЕСКАЯ ИЗОМЕРИЯОптическая изомерия – один из видов пространственной
или стереоизомерии.
Стереоизомеры – соединения, молекулы которых имеют
одинаковое химическое, но различное пространственное строение. Пространственное строение взаимосвязано не только с физическими и химическими свойствами веществ, но и с их биологической активностью. Название «оптическая» данный вид стереоизомерии получил оттого, что стереоизомеры могут обладать
оптической активностью. Другое название оптической изомерии – «зеркальная», т.к. изомеры похожи как предмет и его зеркальное отображение.
Оптическая активность – способность вещества отклонять
плоскость поляризации поляризованного луча света.
Колебания вектора электрического поля обычного луча света происходят в разных плоскостях (рис. 1а). После прохождения
через специальную призму-поляризатор луч становится плоскополяризованным, т.е. вектор электрического
поля колеблется только в одной плоскости,
перпендикулярной направлению распростраа
б
нения луча (рис. 1б). Эта плоскость и называется плоскостью поляризации света. При прорис.1
хождении поляризованного луча света через
раствор оптически активного соединения происходит отклонение
плоскости поляризации света на определенный угол по часовой
стрелке (тогда вещество называется правовращающим, обозначается «+») или против часовой стрелки (левовращающим, обозначается «-»). Оптическую активность можно определить с помощью прибора – поляриметра. Схематично принцип работы поляриметра можно описать следующим образом (рис. 2):
Рис. 2. Схема строения поляриметра.
117
118.
Поляриметр состоит из двух призм, между которыми помещается трубка с раствором исследуемого вещества. Первая призма – поляризатор поляризует луч, исходящий из источника света.Вторая призма – анализатор пропускает плоскополяризованный
луч, выходящий из поляриметрической трубки. Если вещество
оптически неактивно, то при одинаковой ориентации двух призм
свет через анализатор проходит полностью. Если же при выходе
из поляриметрической трубки плоскость поляризации света изменилась, то для полного прохождения такого луча света нужно
повернуть анализатор на определенный угол (α), который и соответствует углу вращения плоскости поляризации света оптически
активным веществом.
Оптической активностью обладают только хиральные
молекулы. Хиральность – это свойство объекта (не обязательно
молекулы!) не совмещаться со своим зеркальным отображением. Любой симметричный объект совместится со своим отражением в зеркале. Значит, хиральность – это несимметричность.
Сам термин «хиральность» произошел от греческого слова «рука», так как наши руки являются примером хиральных объектов.
Они похожи как в зеркале, но их невозможно совместить, не вынося из плоскости. Наглядным примером их несовместимости
служит невозможность надеть на правую руку левую перчатку,
соответствующую пространственному строению левой руки.
Чтобы установить, является ли молекула хиральной, необходимо построить её модель и модель её зеркального отображения и попытаться их совместить. Если это удается – молекула
ахиральна, не удается – хиральна. Есть и другой прием: признаком хиральности является наличие в молекуле асимметрического
атома углерода. Асимметрическим называют атом углерода,
связанный с четырьмя различными заместителями. Обратите
внимание: этот признак является необходимым, но не всегда достаточным для проявления оптической активности! Молекула в
целом должна быть асимметричной.
118
119.
Рассмотрим стереоизомерию на примере молочной кислоты.В молекуле молочной кислоты один асимметриH
ческий атом углерода (обозначается звездочкой).
*
CH3 -C-COOH
По формуле N=2 n, где n – число асимметричеOH
ских атомов углерода, рассчитываем количество
стереоизомеров: 21=2. Эти стереоизомеры похожи как предмет и
его зеркальное отражение:
зеркало
COOH
COOH
C
OH
CH3
H
HO
H3 C
C
1
H
2
Стереоизомеры, похожие как предмет и несовместимое с
ним зеркальное отражение, называются энантиомерами (1 и 2).
Для изображения структур стереоизомеров на плоскости
пользуются проекционными формулами Фишера. При их построении пользуются определенными правилами. Углеродную
цепь располагают вертикально таким образом, чтобы вверху оказалась старшая группа (в нашем примере – карбоксильная группа). Хиральный атом углерода находится на перекрестье вертикальной и горизонтальной линий и символом не обозначается.
Внизу на вертикальной линии обозначается углеродсодержащий
заместитель (CH3-группа). На горизонтальной линии располагают
оставшиеся группы (в нашем примере – водород и гидроксильная
группа). Расположенные на горизонтали заместители должны
быть направлены к наблюдателю, а на вертикали – от наблюдателя, т.е. находятся за плоскостью бумаги:
COOH
H
C
OH
CH3
проекция модели
на плоскость
COOH
COOH
OH
H
CH3
проекционная формула
Фишера
HO
C
COOH
H
CH3
проекция модели
на плоскость
H
HO
CH3
проекционная
формула Фишера
119
120.
Энантиомеры обладают одинаковыми химическими и физическими свойствами (температура кипения, температура плавления, плотность, показатель преломления и т.д.). Они проявляютодинаковую реакционную способность по отношению к ахиральным реагентам.
Энантиомеры отличаются следующими свойствами:
1. Они отклоняют плоскость поляризации поляризованного
луча на одинаковый угол, но в противоположных направлениях,
т.е. один из них является правовращающим (+), а другой - левовращающим (-).
2. Они с разной скоростью реагируют с другими хиральными молекулами. А так как ферменты являются хиральными молекулами, то они будут взаимодействовать только с одним из стереоизомеров. Отсюда различная биологическая активность энантиомеров.
Предсказать направление и величину угла вращения
поляризованного луча света для данного стереоизомера нельзя! Оптическая активность определяется только опытным
путем с помощью поляриметра. Но если определена оптическая
активность одного из энантиомеров (например, -20о ), то оптическую активность второго энантиомера можно не определять, она
будет равна +20о.
Определение абсолютной конфигурации, т.е. действительного расположения заместителей вокруг хирального центра, стало возможным только с появлением метода рентгеноструктурного анализа. До этого конфигурации стереоизомеров сравнивали
с конфигурацией эталонных (ключевых) соединений, т.е. определяли относительную конфигурацию. В 1906 г. по предложению
М.А. Розанова за конфигурационный стандарт был принят глицериновый альдегид.
CHO
CHO
H
OH
HO
H
CH2 OH
CH2 OH
I
D-глицериновый
альдегид
II
L-глицериновый
альдегид
120
Стереоизомерам глицеринового
альдегида были предписаны определенные конфигурации, которые обозначили как D- и L-.
121.
Поэтому стереоизомеры, имеющие конфигурацию как Dглицериновый альдегид, относят к D-стереохимическому ряду, аимеющие конфигурацию как L-глицериновый альдегид – к Lстереохимическому ряду, например:
H
COOH
COOH
COOH
H
OH
H2 N
NH2
CH3
CH3
CH3
D-молочная
кислота
H
D-α-аминопропионовая
кислота
L-α-аминопропионовая
кислота
Правильность произвольно приписанной D-глицериновому
альдегиду конфигурации в дальнейшем была подтверждена экспериментально и приобрела силу абсолютной конфигурации.
Это значит, что можно продолжать сравнивать конфигурации
данного изомера с конфигурациями D- и L-глицеринового альдегида.
Сравнение конфигурации исследуемого соединения с глицериновым альдегидом проводят путем серии химических превращений, не затрагивающих центр хиральности. Например, (-)молочную кислоту можно получить из D-(+)-глицеринового альдегида. Значит, конфигурация хирального атома углерода у неё
будет одинаковой с исходным альдегидом, т.е. полученная (-)молочная кислота будет принадлежать к D-стереохимическому
ряду.
CHO
H
OH
COOH
Br2
H2 O
H
OH
COOH
COOH
PBr3
H
OH
Zn,H+
H
OH
CH2 OH
CH2 OH
CH2 Br
CH3
D-(+)-глицериновый
альдегид
D-(-)-глицериновая
кислота
D-(-)-3- бром-2-гидроксипропановая
кислота
D-(-)-молочная
кислота
Знак вращения (+ или -) не имеет связи с конфигурацией
(D- или L-). Например, D-глицериновый альдегид является правовращающим стереоизомером, а D-молочная кислота – левовращающим.
121
122.
Смесь равных количеств энантиомеров называется рацемической смесью. Рацемическая смесь не обладает оптической активностью. Существуют методы, позволяющие разделить рацемическую смесь на оптически активные энантиомеры.Стереоизомерия молекул с несколькими центрами хиральности
В молекуле 2,3-дибромбутановой кислоты два асимметрических
атома углерода. Количество стереоизомеров
* *
CH3 -CH-CH-COOH N=22=4. Приведём их проекционные формулы
Br Br
Фишера:
COOH
COOH
Br
Br
H
H
Br
H
Br
H
COOH
H
Br
CH3
CH3
CH3
I
II
III
COOH
Br
Br
H
H
H
Br
CH3
IV
Относительную конфигурацию определяют по верхнему
асимметрическому атому углерода, сравнивая её с конфигурацией стандарта – стереоизомеров глицеринового альдегида. Пары
стереоизомеров I, II и III, IV являются зеркальным отображением
друг друга, т.е. энантиомерами. Стереоизомеры I и III, II и IV, I и
IV не являются зеркальным отражением друг друга, их называют
диастереомерами. Диастереомеры отличаются по физическим и
химическим свойствам.
Рассмотрим стереоизомерию винной кислоты.
* *
HOOC-CH-CH-COOH
N=22=4
OH OH
COOH
COOH
H
H
H
OH
HO
H
H
OH
H
OH
HO
H
COOH
COOH
COOH
COOH
I
II
III
IV
энантиомеры
122
COOH
OH HO
H
HO
COOH
мезо-форма
123.
Теоретически возможно существование четырёх стереоизомеров. На самом деле, III и IV идентичны (при повороте III на180о получается IV). Стереоизомеры I и II являются энантиомерами, I и III, II и III – диастереомерами.
Мезо-форма оптически не активна, т.к. её молекула симметрична (ахиральна), а оптическая активность – это свойство хиральных молекул.
Рацемическая смесь энантиомеров винной кислоты называется виноградной кислотой.
π-Диастереомерия
Цис-, трансизомеры не являются зеркальным отображением
друг друга, поэтому их можно рассматривать как диастереомеры.
H
H
Cl
H
Cl
C
C
C
C
Cl
цис-1,2-дихлорэтен
Cl
H
транс-1,2-дихлорэтен
Действительно, они обладают различными физическими и
химическими свойствами. Так как хиральным центром у них является π-связь, то их называют π-диастереомерами.
Стереоизомерия и биологическая активность
В организме реакции протекают с участием биокатализаторов – ферментов. Молекулы ферментов построены из хиральных
молекул α-аминокислот (аминокислоты, участвующие в построении белков относятся к L-ряду). Проявление биологической активности лекарственных препаратов связывают со строгим соответствием строения препарата и активных центров ферментов
(они должны подходить как ключ к замку). Биохимические процессы являются стереоспецифичными. Часто один стереоизомер
лекарственного препарата активен, а другой – нет. Например, D(-)-стереоизомер левомицетина широко применяется в медицинской практике как антимикробное средство. Рацемат левомице123
124.
тина – синтомицин менее активен, применяется только для наружных целей.L-(-)-изомер адреналина в 12 раз более активен, чем его
правовращающий изомер.
D-(-)-эфедрин применяется в медицинской практике как сосудосуживающее средство, а его правовращающий изомер – не
применяется.
Правовращающая камфора (природная) применяется как
сердечный стимулятор, а левовращающий стереоизомер камфоры
или рацемическая смесь (синтетическая) – для наружных целей
для растираний при ревматизме.
Противоопухолевое средство сарколизин является левовращающим изомером, правовращающий изомер не активен.
Молекулы многих применяемых в настоящее время лекарственных средств имеют асимметрический центр: более 90% адреномиметиков, адреноблокаторов, антикоагулянтов, противоэпилептических средств, более 50% антигистаминных препаратов
и местных анестетиков.
Несмотря на то, что клинический эффект этих лекарственных веществ обусловлен лишь одним из энантиомеров, а другой
можно рассматривать как загрязнение («изомерный балласт»),
вследствие технологических проблем, связанных с разделением
энантиомеров, они выпускаются в виде рацематов. Лишь немногие препараты выпускаются в виде индивидуальных стереоизомеров.
ГЕТЕРОФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
БЕНЗОЛЬНОГО РЯДА
Многие гетерофункциональные производные бензола применяются в медицинской практике в качестве лекарственных
препаратов. Это производные п-аминофенола – парацетамол и
фенацетин; п-аминобензойной кислоты – анестезин и новокаин;
сульфаниловой кислоты – сульфаниламиды и т.п.
124
125.
п-Аминофенол и его производныеКак фенол п-аминофенол проявляет кислотные свойства,
как ароматический амин – основные:
ONa
NaOH
+ H2 O
OH
NH2 натриевая соль
п-аминофенола
OH
HCl
NH2
+
NH3 Cl
п-аминофенола
гидрохлорид
п-Аминофенол проявляет нуклеофильные свойства и способен к реакциям ацилирования. При взаимодействии его с уксусным ангидридом образуется N-ацетил-п-аминофенол (пацетамидофенол), который используется в медицине под названием парацетамол (жаропонижающий препарат).
O
OH
+
CH3 -C
OH
+ CH3 -COOH
O
CH3 -C
NH2
O
NH-C-CH 3
O
парацетамол
Фенацетин – этиловый эфир п-ацетамидофенола – получают в результате следующих превращений:
ONa
OH
OC2H5
NaOH
C2H5Br
(CH3CO)2O
- H2O
- HBr
- CH3COOH
NH2
п-аминофенол
NH2
п-аминофеноля т
натрия
NH2
этиловый эфир
п-аминофенола
(фенитидин)
OC2H5
NH-C-CH3
O
этиловый эфир
п-ацетамидофенола
(фенацетин)
125
126.
Фенацетин также используется как жаропонижающий препарат.п-Аминобензойная кислота и ее производные
п-Аминобензойная кислота проявляет свойства карбоновых
кислот и первичных ароматических аминов. Она обладает и кислотными, и основными свойствами:
COONa
NaOH
+ H2 O
COOH
NH2 натриевая соль
п-аминобензойной кислоты
COOH
NH2
HCl
+
NH3 Cl
п-аминобензойной кислоты
гидрохлорид
За счет карбоксильной группы п-аминобензойная кислота
образует функциональные производные, например, сложные
эфиры. Некоторые из них используются в медицинской практике
как местноанестезирующие препараты (т.е. вызывают потерю
чувствительности): этиловый эфир – анестезин, N,Nдиэтиламиноэтиловый эфир – новокаин.
O
C
COOH
OC2H5
C2H5OH
HCl сухой
+ H2O
NH2
анестезин
O
NH2
C
HO-CH2-CH2-N(C2H5)2
OCH2-CH2-N
HCl сухой
NH2
126
новокаин
C2H5
C2H5
+ H2O
127.
В основе своей структуры новокаин имеет те же фрагменты,что и широко применяющийся ранее в медицине кокаин, и хотя
по силе анестезирующего действия новокаин несколько уступает
кокаину, но не вызывает привыкания (при длительном применении кокаина развивается лекарственная зависимость – кокаинизм).
Новокаин применяется в виде соли – гидрохлорида, что повышает его растворимость в воде:
COOCH 2 -CH2 -N
C2 H5
C2 H5
HCl
NH2
+ CH
2 5
COOCH 2 -CH2 -N
C2 H5
Cl
H
NH2
новокаина гидрохлорид
Производные сульфаниловой кислоты
Сульфаниловая
кислота
(п-аминобензолNH2
сульфокислота) проявляет сильные кислотные
свойства за счет сульфогруппы и слабые основные свойства за счет ароматической амиSO 3 H
ногруппы. Сульфаниловая кислота образует
сульфаниловая
соли с щелочами и сильными минеральными
кислота
кислотами:
NH2
NaOH
+ H2 O
NH2
натриевая соль
SO3 Na
SO3 H
сульфаниловая
кислота
HCl
+
NH3
Cl
гидрохлорид
SO3 H
127
128.
За счет присутствия и кислотного, и основного центров вмолекуле сульфаниловая кислота существует в виде биполярного
иона:
NH2
SO3 H
+
NH3
SO3
биполярный ион
Амид сульфаниловой кислоты (сульфаниламид, стрептоцид)
является родоначальником группы лекарственных средств антибактериального действия – сульфаниламидов:
NH2
SO2 NH2
амид сульфаниловой
кислоты
NH2
SO2 NH-R
общая формула
сульфаниламидных
препаратов
Наиболее активные сульфаниламиды содержат в структуре
остатки гетероциклических соединений (табл. 4).
128
129.
Таблица 4Сульфаниламидные препараты
R
Препарат
R
Препарат
N
H
Стрептоцид
Сульфазин
N
Норсульфазол
S
N N
Этазол
C2 H5
S
Сульфапиридазин
CH3
N
N
Сульфадимезин
N
Сульфадиметоксин
N
Сульфален
N N
CH3
OCH3
N
OCH3
H3 CO
N
OCH 3
N
Антибактериальное действие сульфаниламидов основано на
том, что являются антиметаболитами по отношению к паминобензойной кислоте, которую микроорганизмы используют
в синтезе фолиевой кислоты, необходимой для их жизнедеятельности. Амид сульфаниловой кислоты имеет структурное сходство
с п-аминобензойной кислотой и конкурирует с ней за активные
центры ферментов в синтезе фолиевой кислоты:
H2 N
N
HN
N
N
O
CH2 -NH-
-C-NH-CH-COOH
O
CH2 -CH 2 -COOH
остаток п-аминобензойной кислоты
129
130.
Как ароматические амины сульфаниламиды способны к реакциям ацилирования:NH-CO-CH 3
NH2
(CH3 CO)2 O
+ CH3 -COOH
SO2 NH-R
SO2 NH-R
ацетильное производное
сульфаниламида
В организме подобная реакция протекает с участием ацетилкоэнзима А. Ацетильные производные сульфаниламидов хуже
растворимы в воде, чем исходные препараты, и могут кристаллизоваться в почечных канальцах, вызывая кристаллурию. Вот почему при длительной терапии сульфаниламидами назначают щелочное питьё.
Салициловая кислота и ее производные
Салициловая (о-оксибензойная кислота) проявляCOOH
ет свойства карбоновых кислот и фенолов. Она
OH
является более сильной кислотой, чем бензойная
кислота. Салициловая кислота проявляет жаропонижающий и антиревматический эффект, но как сильная кислота обладает раздражающим действием на ЖКТ и не используется для внутреннего употребления.
Повышенные кислотные свойства салициловой кислоты
связаны с устойчивостью ее аниона, стабилизированного за счет
образования внутримолекулярной водородной связи:
O
O
C
COOH
OH
-
водородная связь
H
O
H2 O
+ H3 O+
Салициловая кислота образует соли при взаимодействии с
щелочами и с гидрокарбонатом натрия:
COOH
COONa
OH
натрия салицилат
130
NaOH
OH
салициловая
кислота
COONa
NaHCO 3
OH
натрия салицилат
131.
Салицилат натрия используется при лечении ревматизма.В медицине используются сложные эфиры салициловой
кислоты по карбоксильной группе – метилсалицилат и
фенилсалицилат. Метилсалицилат получают при взаимодействии
салициловой кислоты с метиловым спиртом в присутствии
концентрированной серной кислоты (обычные условия
этерификации):
COOH
COOCH 3
OH
+ CH3 OH
салициловая
кислота
OH
H+
+ H2 O
метилсалицилат
Для получения фенилсалицилата карбоксильную группу салициловой кислоты предварительно активируют (фенолы не ацилируются карбоновыми кислотами из-за своей пониженной нуклеофильности):
COOH
COCl
OH
COOC 6 H5
OH
PCl5
OH
C6 H5 OH
фенилсалицилат
хлорангидрид
салициловой
кислоты
Метилсалицилат оказывает противовоспалительное, жаропонижающее и обезболивающее действие, но применяется только
наружно в составе мазей. Фенилсалицилат (салол) применяется
при кишечных инфекциях. Его действие связано со способностью
гидролизоваться в щелочной среде кишечника с высвобождением
фенола.
Ацетилсалициловая кислота (аспирин) – это сложный эфир
салициловой кислоты по фенольному гидроксилу. Ее получают
ацилированием салициловой кислоты уксусным ангидридом:
COOH
COOH
OH
+ (CH3 CO)2 O
салициловая
кислота
O-C-CH 3
O
+ CH3 COOH
ацетилсалициловая
кислота
131
132.
Ацетилсалициловая кислота обладает жаропонижающим ипротивовоспалительным действием.
При неправильном хранении (повышенная влажность и
температура) ацетилсалициловая кислота способна гидролизоваться, т.е. в препарате появляется примесь свободной салициловой кислоты.
COOH
COOH
O-C-CH 3
O
ацетилсалициловая
кислота
+ H2 O
OH
фенольный
гидроксил
+ CH3 COOH
салициловая
кислота
Такой препарат применять нельзя, т.к. салициловая кислота
раздражает слизистую ЖКТ и обладает ульцерогенным действием. Определить доброкачественность ацетилсалициловой кислоты (т.е. отсутствие в ней примеси салициловой кислоты) можно,
используя качественную реакцию на фенольный гидроксил. Если
при взаимодействии с хлоридом железа (III) появилось фиолетовое окрашивание, препарат недоброкачественный.
COOH
Из производных салициловой кислоты в
медицине применяется также п-аминосаOH
лициловая кислота (ПАСК). Она используется при лечении туберкулеза и дейстNH2
вует как антагонист п-аминобензойной
п-аминосалициловая
кислоты (см. стр.129).
кислота
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ.
ПРОИЗВОДНЫЕ ПЯТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
Производные гетероциклических соединений, особенно
ароматических, широко распространены в природе. Они составляют основу таких природных веществ, как витамины, хлорофилл, гемоглобин, входят в структуру различных пигментов, алкалоидов. Более 50% лекарственных препаратов как растительного, так и синтетического происхождения содержат в своей структуре гетероциклические фрагменты.
132
133.
Гетероциклическими называют соединения, в цикле которых помимо атомов углерода содержатся и другие атомы (такназываемые гетероатомы), чаще всего азот, кислород, сера.
Гетероциклы классифицируют по размеру цикла на трёх-,
четырёх-, пяти-, шестичленные; по характеру гетероатома (азот-,
кислородсодержащие и т.д.); по количеству гетероатомов в цикле
(гетероциклы с одним гетероатомом, с двумя гетероатомами и
т.д.).
Пятичленные гетероциклы с одним гетероатомом
Представителями пятичленных гетероциклов с одним гетероатомом являются пиррол, фуран и тиофен:
β
α
β
α
N
H
пиррол
O
S
фуран
тиофен
Все они являются ароматическими соединеH
.
. ниями, т.к. отвечают критериям ароматичности. Все атомы цикла sp2-гибридизованы,
H
H
.
..
цикл плоский. Имеется замкнутая p,πN H
H
сопряженная система. Гетероатом отдает в
сопряжение неподеленную электронную пару, т.е. 4n+2=6; n=1.
Пиррол, фуран и тиофен относят к π-избыточным гетероциклам, т.к. электронная плотность ароматического кольца повышена в сравнении с бензолом (шесть электронов ароматического облака делокализована между пятью атомами).
Как для всех ароматических соединений для пиррола, фурана и тиофена характерны реакции электрофильного замещения.
Из-за π-избыточности циклов эти реакции протекают легче, чем в
бензоле. Кроме того, в отличие от бензола электронная плотность
в их молекулах распределена неравномерно, она наиболее высокая в α-положениях. Поэтому реакции SE протекают по
α-положениям.
.
133
134.
Например, реакцию бромирования пиррола проводят прикомнатной температуре, без катализатора:
..
N
H
α + Br2
..
Br
+ HBr
N
H
α-бромпиррол
Реакции нитрования и сульфирования пиррола и фурана
проводят в специальных условиях, что связано с ацидофобностью
этих циклов. Ацидофобность в переводе с греческого – «боязнь
кислот». При действии на ацидофобные гетероциклы концентрированных кислот происходит реакция полимеризации, и образуется смолоподобная масса (происходит осмоление). Рассмотрим
причины ацидофобности на примере пиррола.
Неподеленная электронная пара азота, отвечающая за основные свойства, является частью ароматического облака, поэтому пиррол не проявляет основных свойств при взаимодействии с
разбавленными кислотами. Протоны концентрированных минеральных кислот «вырывают» пару электронов атома азота из сопряжения, тем самым нарушая ароматичность. Полученный продукт подвергается полимеризации:
+ HNO3
..
N
H
+
N
NO 3 -
полимеризация
H H
H
+
N
H
n
Для нитрования и сульфирования пиррола и фурана необходимо использовать реагенты, не проявляющие сильных кислотных свойств: ацетилнитрат и пиридинсульфотриоксид.
O
+ CH3 -C
O
..
N
H
O-NO 2
ацетилнитрат
+
N . SO3
пиридинсульфотриоксид
134
NO 2 + CH3 -COOH
O
α-нитрофуран
уксусная кислота
.. SO H +
3
N
N
H
α-пирролсульфопиридин
кислота
135.
Свойства пиррола и его производныхПиррол проявляет крайне слабые основные свойства при
взаимодействии с концентрированными минеральными кислотами (см. ацидофобность пиррола). За счёт полярности NH-связи
проявляются слабые кислотные свойства. Пиррол образует соли
при взаимодействии с щелочными металлами или их амидами:
K
..
- H2
N
H
H2 O
..
N
K+
пирролкалий
N
H
+ KOH
Даже вода, являясь более сильной кислотой, чем пиррол,
вытесняет его из солей.
Реакции восстановления (гидрирования) пиррола протекают
в довольно жёстких условиях, ступенчато:
[H]
[H]
..
N
H
N
H
пирролин
пиррол
N
H
пирролидин
Пирролидин – сильное основание (как вторичный алифатический амин). Ядро пирролидина входит в структуру алкалоида
никотина, аминокислоты пролина:
N
N
CH3
COOH
N
H
пролин
никотин
..
N
H
N
N
H
N
..
Важную группу азотсодержащих природных веществ составляют так называемые
тетрапиррольные соединения, т.е. соединения, в структуру которых входят четыре
пиррольных кольца. К тетрапиррольным соединениям относится порфин, производными которого являются такие биологически
порфин
135
136.
важные соединения как хлорофилл, гем, цитохром. Порфин является устойчивой ароматической системой (энергия сопряжения –840 кДж/моль). В замкнутой p,π-сопряженной системе делокализовано 26 электронов.
Порфины, имеющие заместители в пиррольных циклах, называются порфиринами. Заместители могут содержать кратные
связи, способные вступать в сопряжение с ароматическим облаком, что еще более стабилизирует молекулу. Пример порфирина – протопорфирин, входящий в структуру гемоглобина крови.
CH=CH2
H3 C
..
N
H
H3 C
N
HOOC-CH 2 -CH2
CH3
N
CH=CH2
H
N
..
HOOC-CH 2 -CH2
..
N
CH3
протопорфирин
N
Fe
N
HOOC
N
..
HOOC
гем
Порфирины находятся в природе в виде комплексов с металлами. Комплекс порфирина с железом – гем – небелковая
часть молекулы гемоглобина и цитохромов. Комплекс с магнием
является основой молекулы хлорофилла.
При биологическом окислении гемоглобина образуются окрашенные вещества с линейной тетрапиррольной структурой –
билирубиноиды, например, билирубин, имеющий оранжевую окраску.
COOH COOH
[O]
Гемоглобин
O
N
H
CH
N
H
CH2
N
H
билирубин
136
CH
N
H
O
137.
Индол (бензопиррол) – конденсированноегетероциклическое соединение. В его молекуле
3β
5
сконденсированы бензольное и пиррольное
.. 2 α
6
кольца. Индол ароматичен. В его p,πN1
7
H
сопряженной системе делокализовано 10 элекиндол
тронов.
По свойствам индол подобен пирролу. Его основные свойства практически не проявляются, он ацидофобен, обладает слабыми NH-кислотными свойствами. Индол вступает в реакции
электрофильного замещения легче, чем бензол. Реакции SE протекают по β-положению пиррольного кольца:
4
Br
β
N
H
α
+ Br2
+ HBr
N
H
β-броминдол
индол
Наиболее важными биологически активными производными
индола являются α-аминокислота триптофан и продукты его метаболических превращений.
Триптофан входит в структуру белков. В организме он подвергается метаболическим превращениям по двум направлениям:
неокислительное и окислительное декарбоксилирование:
CH2 -CH2
ферменты
NH2 + CO 2
N
H
триптамин
CH2 -CH-COOH
NH2
N
H
триптофан
[O]
HO
CH2 -CH-COOH
NH2
ферменты
N
H
5-гидрокситриптофан
ферменты
HO
CH2 -CH2
NH2 + CO 2
N
H
5-гидрокситриптамин
(серотонин)
137
138.
В результате неокислительного декарбоксилирования образуется токсичный биогенный амин триптамин, который затемподвергается дальнейшему превращению в β-индолилуксусную
кислоту.
При окислительном декарбоксилировании вначале образуется 5-гидрокситриптофан, который затем декарбоксилируется в
5-гидрокситриптамин (серотонин). Серотонин является одним из
нейромедиаторов головного мозга. Нарушение его нормального
обмена в организме может привести к шизофрении. В норме серотонин окисляется в (5-гидрокси-β-индолил)уксусную кислоту и
выводится с мочой.
Другим важным производCH2 -COOH
CH3 O
ным индола является нестероидный противовосCH3
N
палительный препарат инCl
O=C
дометацин.
индометацин
Свойства фурана и его производных
Фуран является ацидофобным соединением, нитруется и
сульфируется в специальных условиях (стр. 134) по
α-положениям. Фуран способен к реакции гидрирования с образованием тетрагидрофурана:
2 H2 , Ni
O
фуран
O
тетрагидрофуран
Тетрагидрофуран используется как растворитель в органических синтезах, в т.ч. при получении лекарственных препаратов.
4
3
Фурфурол (фуранкарбальдегид) является
O
2
5
одним из наиболее важных производных
1
C
O
H
фурана.
фурфурол
Фурфурол проявляет свойства альдегида: он легко окисляется, участвует в реакции Канниццаро (т.к. в его структуре отсутствует α-CH-кислотный центр).
138
139.
При окислении фурфурола образуется фуранкарбоновая кислота:C
O
O
+ Ag(NH3 )2 OH
H
to
O
+ Ag + NH3 + H2 O
O
OH
фуранкарбоновая кислота
C
В реакции Канниццаро одна молекула фурфурола окисляется в фуранкарбоновую кислоту, а вторая – восстанавливается в
фурфуриловый спирт:
2
C
O
NaOH
O
C
O
H
фурфурол
O
+
O
ONa
натриевая соль
фуранкарбоновой
кислоты
CH2 OH
фурфуриловый
спирт
Из фурфурола получают 5-нитропроизводные фуранового
ряда, проявляющие антибактериальное действие. Для этого фурфурол нитруют азотной кислотой в уксусном ангидриде (он необходим для защиты альдегидной группы от окисления), а затем
проводят реакцию присоединения-отщепления с различными
аминами или производными гидразина. Например, в синтезе фурацилина (семикарбазона 5-нитрофурфурола) используют семикарбазид:
O
O
C
O
H
O2 N
HNO3
O
C
O
H
5-нитрофурфурол
(CH3 CO)2 O O2 N
O
5
H2 N-NH-C-NH 2
семикарбазид
CH=N-NH-C-NH 2
O
семикарбазон 5-нитрофурфурола
К производным 5-нитрофуранового ряда также относятся
фуразолидон, фурадонин, фуразолин и др. препараты.
Производным тиофена, а точнее тетрагидротиофена является биотин (витамин H), недостаток которого ведет к кожным
заболеваниям:
139
140.
ONH
HN
S
биотин
(CH2 )4 -COOH
Пятичленные гетероциклы с двумя гетероатомами
Примерами пятичленных гетероциклических соединений с
двумя гетероатомами являются:
3
3
3
4
5
4
N
1
2
N
H
имидазол
4
3
1 N2
N
H
пиразол
5
5
4
N
1
S
5
2
тиазол
N
1
O
2
оксазол
Все эти соединения ароматические. Они вступают в реакции
электрофильного замещения с меньшей скоростью, чем пятичленные гетероциклы с одним гетероатомом (электронная плотность ароматического кольца ниже из-за электроноакцепторного
влияния пиридинового атома азота). Для имидазола и пиразола
характерны также таутомерные превращения и образование
межмолекулярных водородных связей.
3
В молекуле имидазола два неравноN:
пиридиновый
ценных атома азота: пиррольный в по..
азот
N1
ложении 1 и пиридиновый в положеH
нии 3. Пиррольный азот отдает в сопиррольный
пряжение неподеленную электронную
азот
пару, пиридиновый азот – один электрон. Его неподеленная электронная пара в сопряжении не участвует. Пирольный азот отвечает за слабые кислотные свойства,
пиридиновый – за основные:
+
N:
..
N
H
140
кислотный
центр
NH
HCl
основный
центр
N
N
H
Cl -
N
H
K
имидазолия хлорид
N
имидазол калий
N
+
K
141.
Для имидазола характерная прототропная таутомерия засчет перехода протона водорода от кислотного центра к основному и наоборот, поэтому положения 4 и 5 в молекуле равноценны:
1
4 3
5
N:
5
1
4
2
NH
3
2
N
N
H
таутомерия имидазола
За счет наличия в молекуле и кислотного, и основного центров служит причиной образования межмолекулярных ассоциатов:
:N
N
δ+
H ........ :N
N
δ+
H ........ :N
N
δ+
H ........ :N
N
δ+
H ........
Из производных имидазола наиболее важными являются
аминокислота гистидин и продукт ее декарбоксилирования гистамин:
N
N
H
CH2 -CH-COOH
гистидин
NH2
- CO 2
N
N
H
CH2 -CH 2 -NH 2
гистамин
Биологические функции гистидина и гистамина в организме
связаны с их амфотерностью, за счет чего они могут выполнять
роль донора и акцептора протонов.
Гистидин входит в состав белков, в том числе глобина. В
гемоглобине за счет пиридинового атома азота имидазольного
фрагмента гистидина белок глобин связывается с атомом железа
гема.
Гистамин участвует в регуляции жизненно важных функций
организма (снижает артериальное давление, усиливает секрецию
желудочного сока). Повышенный уровень гистамина в организме
ведет к аллергическим заболеваниям.
Кольцо имидазола входит в структуру алкалоида пилокарпина.
141
142.
CH2H5 C2
O
CH3
N
Пилокарпин
используется
при лечении глазных заболеваний.
N
O
пилокарпин
Ядро бензимидазола входит в структуру витамина B12 и лекарственного препарата дибазола (2-бензилбензимидазола), снижающего артериальное давление:
N
N
N
H
CH2
N
H
бензимидазол
дибазол
Пиразол является изомером имидазола. В их свойствах много общего. Пиразол также амфотерен, для него характерна прототропная таутомерия:
HCl
.. N :
N
H
основный
центр
N
H
кислотный
центр
+
NH Cl -
N
H
N
K
пиразолия хлорид
- N
пиразол калий
N
+
K
За счет прототропной таутомерии равноценны положения 3
и 5 пиразольного кольца: 4 3
5
4
1
2
1 N:
N
H
5
3
2N
..
NH
таутомерия пиразола
Наиболее известным производным пиразола является пиразолон, который существует в различных таутомерных формах:
4
5
O
3
4
2
1 N
3
O
N
H
5
1
2 NH
N
H
пиразолон
Так как положения 3 и 5 в молекуле пиразола равноценны,
его называют и пиразолон-3, и пиразолон-5, и пиразолон-3(5).
142
143.
На основе пиразолона-3(5) созданы лекарственные препараты – амидопирин, анальгин, бутадион:H3 C
H3 C
CH3
N
O
N
N
NaSO 3 - H2 C
CH3
N
H3 C
O
CH3
N
N
O
H9 C4
CH3
O
N
N
C6 H5
C6 H5
C6 H5
амидопирин
анальгин
бутадион
C6 H5
Все эти препараты проявляют жаропонижающее, болеутоляющее и противовоспалительное действие. Амидопирин применяется в первую очередь как жаропонижающее средство, анальгин – как болеутоляющее, а бутадион – как противовоспалительный препарат.
Тиазол проявляет основные свойства и образует соли с минеральными кислотами:
+
NH
N
+ HCl
Cl
-
S
S
тиазол
тиазолия хлорид
Тиазол способен к реакциям гидрирования (восстановления)
с образованием тиазолидина:
N
NH
4 [H]
S
S
тиазол
тиазолидин
Тиазолидин является структурным фрагментом молекул антибиотиков пенициллинов.
R-CO-NH
S
CH3
CH3
N
O
COOH
общая формула пенициллинов
Ядро тиазола входит в структуру лекарственного препарата
норсульфазола (стр. 129), витамина B1 (тиамина).
NH2
N
CH2
H3 C
N
кольцо пиримидина
Тиамин
+
N
CH3
S
CH2 -CH2 -OH
кольцо тиазола
143
144.
Недостаток витамина B1 в организме приводит к тяжеломузаболеванию «бери-бери». Потребность в витамине B1 связана с
тем, что он входит в структуру кофермента кокарбоксилазы, принимающего участие в декарбоксилировании α-кетокислот и синтезе ацетилкофермента А. Кокарбоксилаза – сложный эфир тиамина и пирофосфорной кислоты (тиаминдифосфат):
NH2
N
H3 C
N
CH2
+
N
CH3
O
O
CH2 -CH 2 -O-P-O-P-OH
S
OH O-
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ
СОЕДИНЕНИЯ
Представителями шестичленных гетероциклов с одним
гетероатомом азота являются пиридин и хинолин (бензопиридин).
Пиридин и хинолин являются аро4
4
5
5
3
3
6
матическими соединениями. Это
2
1
7
замкнутые π,π-сопряженные систе6
1
2
N
N
..
..
8
мы. Неподеленная электронная пара
пиридин
хинолин
атома азота в сопряжении не участвует. В ароматическом облаке пиридина делокализовано 6 электронов, в хинолине – 10 электронов.
Пиридин и хинолин относят к π-недостаточным системам:
электронная плотность ароматических колец понижена в сравнении с бензолом. Это связано с электроноакцепторным влиянием
пиридинового атома азота. В результате пиридин труднее, чем
бензол, вступает в реакции электрофильного замещения. Замещение протекает по β-положению цикла, т.к. в нем электронная
плотность выше, чем в α- и γ-положениях:
β
β
N
Как и бензол, пиридин вступает в реакции нитрования, бромирования, сульфирования, но в гораздо более жестких условиях:
144
145.
NO2KNO3,H2SO4
300o
γ
β
β
α
α
N
β-нитропиридин
N
SO3H β-пиридинсульфокислота
H2SO4
350o
N
Br
Br2, 300o
β-бромпиридин
FeBr3
N
Реакции электрофильного замещения в хинолине протекают
по положениям 5 и 8 бензольного кольца, на которое электроноакцепторный гетероатом азота влияет в меньшей степени, чем на
собственное – пиридиновое. Реакции идут труднее, чем в бензоле, но легче, чем в пиридине:
5
6
7
1
8
NO 2
4
N
3
HNO3
2
H2 SO4
+
N
N
5-нитрохинолин
NO 2
8-нитрохинолин
Из-за пониженной электронной плотности в ароматическом
кольце для пиридина и хинолина характерны реакции нуклеофильного замешения. Они протекают по 2 (α-) положению в пиридине и положению 2 в хинолине. Примерами нуклеофильного
замещения являются реакции аминирования и гидроксилирования.
Реакцию аминирования (реакцию Чичибабина) проводят
при действии амида натрия в жидком аммиаке. Вначале образуется натриевая соль α-аминопиридина, из которой действием аммиака выделяется свободный α-аминопиридин:
N
α
+ NaNH2
to
-H2
NH3
N
NHNa
натриевая соль
2-аминопиридина
+ NaNH2
NH2
N
2- аминопиридин
( α-аминопиридин)
145
146.
Гидроксилирование протекает при нагревании пиридина ствердым гидроксидом калия. Вначале также образуется соль (калиевая соль α-гидроксипиридина), из которой затем действием
минеральной кислоты выделяют свободный α-гидроксипиридин:
+ KOH
300o
HCl
N
OK
N
калиевая соль
2-гидроксипиридина
+ KCl
OH
N
2-гидроксипиридин
(α-гидроксипиридин)
Аналогичным образом в реакции нуклеофильного замещения вступает хинолин.
Неподеленная электронная пара атома азота, не участвующая в сопряжении, обусловливает основные свойства пиридина и
хинолина. Например, пиридин образует соли с минеральными
кислотами:
+ HCl
+
N
N
..
H
Cl пиридиния хлорид
Пиридин является достаточно сильным основанием, чтобы
взаимодействовать с водой:
+ H2 O
+
N
N
..
H
OH пиридиния гидроксид
Поэтому водные растворы пиридина имеют щелочную реакцию.
С кислотами Льюиса пиридин образует комплексы по донорно-акцепторному механизму. Например, при взаимодействии
пиридина с триоксидом серы образуется пиридинсульфотриоксид, который применяют для сульфирования ацидофобных гетероциклов (см. стр. 134).
O
δ+
+ S O
N
..
146
O
N . SO3
пиридинсульфотриоксид
147.
За счет неподеленной электронной пары азота пиридин проявляет также нуклеофильные свойства, например, в реакциях алкилирования галогеналканами:+
N
..
δ+
CH3
δ−
I
+
N
CH3
I-
N-метилпиридиния
йодид
Катион N-метилпиридиния является ароматической структурой, но еще более π-недостаточной, чем сам пиридин (из-за положительного заряда на атоме азота). Ароматическое кольцо
становится еще более способным к взаимодействию с нуклеофилами. Например, при действии гидрид-аниона протекает реакция
нуклеофильного присоединения и N-метилпиридиний-катион
восстанавливается в 1,4-дигидро-N-метилпиридин.
H
δ+
δ+
+
N
δ+
CH3
N-метилпиридинийкатион
H
H[O]
N
CH3
1,4-дигидро-Nметилпиридин
1,4-дигидро-N-метилпиридин не ароматичен (атом углерода в 4
положении sp3-гибридизован и не принимает участия в сопряжении), его молекула нестабильна и стремится за счет обратной реакции окисления вернуться в ароматическое состояние. Эти реакции окисления-восстановления моделируют действие важного
кофермента НАД+, в структуру которого входит замещенный катион пиридиния.
147
148.
никотинамидаденин
O
C
NH2
NH2
+
N
O
N
O
CH2 -O-P-O-P-O-CH 2
OH
HO
O
N
N
N
O
OH OH
OH
OH
Никотинамидадениндинуклеотид (НАД + )
HO
H
H
C
NH2
N
NH2
O
N
O
CH2 -O-P-O-P-O-CH 2
OH
HO
O
N
N
N
O
OH OH
OH
OH
Никотинамидадениндинуклеотид (НАДH )
В ходе реакции дегидрирования in vivo, которая может рассматриваться как особый случай окисления, субстрат теряет два
атома водорода, т.е. протон и гидрид-анион (H+ и H-). Кофермент
НАД+ принимает гидрид-анион, и пиридиниевое кольцо переходит в восстановленную форму – 1,4-дигидропиридиновый фрагмент. Этот процесс обратим.
C
+
N
R
НАД +
148
H
H
O
NH2
фермент
+ H-Субстрат-H
Восстановленная
форма
N
R
НАДН
O
C
NH2
+ H+ + Субстрат
Окисленная
форма
149.
Типичный пример биохимических реакций с участиемНАД – окисление спиртовых групп в альдегидные (превращение
ретинола в ретиналь). НАДН, наоборот, участвует в восстановлении карбонильных групп в спиртовые (например, при превращении пировиноградной кислоты в молочную).
Многие производные пиридина являются биологически важными соединениями, используются в медицине. Например, витамин B6 – пиридоксаль. В виде сложного эфира с фосфорной кислотой (пиридоксальфосфата) он участвует в реакции переаминирования, ведущей к получению α-аминокислот.
+
H
C
H
O
CH2 OH
HO
O
C
O
CH2 O-P-OH
HO
OH
H3 C
H3 C
N
Пиридоксаль
N
Пиридоксаль фосфат
Никотиновая (β-пиридинкарбоновая) кислота и никотинамид являются формами витамина PP, при недостатке которого
развивается пеллагра. Никотинамид является также структурным
фрагментом кофермента НАДH. Диэтиламид никотиновой кислоты – кордиамин – применяется как стимулятор центральной
нервной системы. Все эти соединения могут быть получены из βметилпиридина (β-пиколина):
O
O
COOH
SOCl2
CH3
[O]
C
C
Cl NH
3
N
N
N
N
β-пиколин
никотиновая
кислота
хлорангидрид
никотиновой кислоты
никотинамид
NH2
O
(C2 H5 )2 NH
C
N(C 2 H5 )2
N
диэтиламид никотиновой
кислоты (кордиамин)
Изоникотиновая (γ-пиридинкарбоновая) кислота образуется
при окислении γ-пиколина. Гидразид изоникотиновой кислоты –
тубазид – используется при лечении туберкулеза. Для снижения
149
150.
токсичности был получен гидразон тубазида с ароматическимальдегидом ванилином – препарат фтивазид:
O
COOH
CH3
[O]
C
N
γ-пиколин
Cl
SOCl2
N
изоникотиновая
кислота
O
C
NH2 -NH2
N
хлорангидрид
изоникотиновой
кислоты
N
гидразид
изоникотиновой
кислоты (тубазид)
O
C
NH-NH2
O
C
NH-NH2
+
N
O
H
OH
C
- H2 O
OCH3
ванилин
тубазид
NH-N=C
OH
OCH3
N
фтивазид
Некоторые производные хинолина также используются в
медицине. Например, 8-гидроксихинолин (оксин) и его производные применяются как антисептические средства.
Антибактериальное действие оксина связывают с его способностью образовывать токN
сичные для микроорганизмов хелатные комOH
плексы с ионами металлов (Fe2+, Cu2+). При8-гидроксихинолин
чем доказано, что антибактериальным дейст(Оксин)
вием обладают только комплексы оксина с
металлами (1:1), при повышении концентрации оксина его биологический эффект снижается.
Fe2+
(1:1)
N
O
проявляет антимикробное
действие
Fe+
N
OH
оксин
Fe2+
(2:1)
N
O
N
150
Fe
O
не проявляет антимикробного
действия
151.
Бактерицидным действием обладает продукт нитрования8-гидроксихинолина – 8-гидрокси-5-нитрохинолин (5-НОК, нитроксолин):
NO
2
HNO3
N
N
OH
OH
8-гидроксихинолин
8-гидрокси5-нитрохинолин
Нитроксолин применяется для лечения инфекций мочевыводящих путей.
Cl
При кишечных инфекциях применяется
другое производное 8-гидроксихинолина – 8-гидрокси-7-иод-5-хлорхинолин
I
N
(энтеросептол).
OH
энтеросептол
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ДВУМЯ
ГЕТЕРОАТОМАМИ
Пиридазин, пиримидин и пиразин являются изомерными
шестичленными гетероциклами с двумя гетероатомами азота.
Они носят общее название азины:
4
4
4
5
3
5
6
1 N2
6
N
пиридазин
1
N3
5
2
6
N
пиримидин
N
1
3
N
пиразин
2
Диазины являются ароматическими соединениями (это
замкнутые π,π-сопряженные системы, в которых делокализовано
6 электронов).
Подробнее остановимся на свойствах пиримидина.
Реакционная способность пиримидина в реакциях электрофильного замещения понижена из-за электроноакцепторного
влияния двух пиридиновых атомов азота, поэтому реакции SE
практически не протекают.
151
152.
В связи с тем, что пиримидин является ещё более πнедостаточной системой, чем пиридин, для него характерны реакции нуклеофильного замещения, например, реакция Чичибабина. Аминирование протекает в положениях 2, 4 или 6, в которыхэлектронная плотность особенно понижена:
NH2
N
N
NaNH2
N
4-аминопиримидин
N
пиримидин
Основные свойства пиримидина ниже, чем пиридина, также
из-за электроноакцепторного влияния второго атома азота. В отличие от пиридина пиримидин не образует соли с водой, он реагирует с сильными минеральными кислотами:
N:
N
+ H2 SO4
+
N
N
..
пиримидин
HSO4 -
H
пиримидиния
гидросульфат
Особенно важны гидрокси- и амино-производные пиримидина – урацил, тимин и цитозин, являющиеся компонентами нуклеиновых кислот (см. стр.204).
К производным пиримидина относится также барбитуро вая кислота и барбитураты .
Барбитуровая кислота является циклическим уреидом малоновой кислоты. Она может быть получена при взаимодействии
мочевины с диэтиловым эфиром малоновой кислоты:
O
C OC 2 H5
H2 C
C=O
+
C OC 2 H5
O
диэтилмалонат
O
H2 N
H2 N
мочевина
C2 H5 ONa
NH
O
O
NH
барбитуровая
кислота
Для барбитуровой кислоты характерны два вида таутомерии – лактам - лактимная и кето - енольная :
152
153.
OH N3
лактамные
фрагменты
2
4
1 6
H
кето-енольная таутомерия H N
OH
N
O
O
N
O
O
кето-фрагмент
H
5 H
енольный
фрагмент
H
H
триоксоформа
лактам-лактимная
таутомерия
лактам-лактимная
таутомерия
OH
OH
лактимные
фрагменты
N
H
H
N
HO
кето-енольная таутомерия
N
HO
O
OH
N
тригидроксиформа
Наиболее стабильной является триоксоформа, т.е. лактамная
кето-форма. За счет существования в енольной таутомерной
форме барбитуровая кислота проявляет сильные кислотные свойства (она сильнее, чем уксусная).
O
Многие 5,5-дизамещенные производные барR
битуровой кислоты, так называемые барбиту4
H N3
5
R'
раты, применяются в медицине как снотвор2
1 6
O
N
O
ные и противосудорожные препараты.
H
Для барбитуратов возможна только лак общая формула
там - лактимная таутомерия . Они проявбарбитуратов
ляют кислотные свойства за счет лактимной
формы, образуя соли с одним эквивалентом щелочи:
O
O
O
H N3
4
R
O
N
O
5
R'
2
1 6
H
лактамная
форма
H N
R
R'
NaOH
O
N
HO
лактимная форма
кислотный
центр
R
R'
H N
Na O
N
O
натриевая соль
153
154.
Примерами барбитуратов являются барбитал (5,5диэтилбарбитуроваякислота),
фенобарбитал
(5-фенил-5этилбарбитуровая кислота).
Пиримидиновое кольцо входит также в структуру витамина
В1 (тиамина) и кокарбоксилазы (см. стр. 143,144).
Из конденсированных гетероциклических систем в природе наиболее распространены соединения пуринового ряда.
6
7
Пурин образован конденсированными яд1
5 N
N
рами пиримидина и имидазола. Пурин яв8
9
3
2
ляется ароматическим соединением. Это
N 4 N
H
замкнутая p,π-сопряженная система, в которой делокализовано 10 электронов (пиридиновые атомы азота в
положениях 1, 3 и 7 отдают в сопряжение по одному электрону,
пиррольный азот в положении 9 – неподеленную электронную
пару).
Пурин проявляет амфотерные свойства: пиридиновый азот в
положении 7 отвечает за основные свойства, а пиррольный азот –
..
за кислотные:
N
основный центр
N
N
N
кислотный центр
H
Пурин образует соли с минеральными кислотами и со щелочными металлами:
N
N
Na
N
N
N
N
N
H
HCl
натриевая соль
пурина
N
Na
+
NH
N
Cl - пуриния хлорид
N
N
H
Для пурина характерна протропная таутомерия за счет переноса протона водорода от кислотного центра к основному, поэтому положения 7 и 9 в молекуле равноценны:
1
154
6
5
2
N 4 N
H
3
H
N
7
N
N
9
8
N
7
9
N
N
155.
Гидроксипурины. Гипоксантин (6-гидроксипурин), ксантин (2,6-дигидроксипурин) и мочевая кислота (2,6,8тригидроксипурин) образуются в организме в процессе метаболизма нуклеиновых кислот.O
O
H
N
HN 6
HN 6
2
N
H
ксантин
O
N
N
гипоксантин
O
H
N
H
N
6
HN
8
2
N
O
N
N
H
H
мочевая кислота
O
Для гидроксипуринов характерна лактам - лактимная
таутомерия , за счет лактимной формы они проявляют кислотные свойства. Более стабильной является лактамная форма.
O
HN
O
OH
H
N
H
N
N
N
N
H
лактамная форма
ксантина
HO
N
N
лактимная форма
ксантина
Важными производными ксантина являются алкалоиды
(так называемые метилированные ксантины) – теофилин (1,3диметилксантин), теобромин (3,7-диметилксантин) и кофеин
(1,3,7-триметилксантин):
O
1
CH3 N
3
O
N
H
N
N
O
HN
O
7
O
CH3
1
N
CH3 N
N
O
3
N
7
CH3
N
3
N
CH3
CH3
CH3
теофиллин
теобромин
кофеин
N
Их природными источниками являются листья чая, зерна
кофе, какао-бобы. Кофеин возбуждает центральную нервную
систему. Теофиллин и теобромин также стимулируют ЦНС, но в
меньшей степени. Они проявляют диуретический эффект.
Мочевая кислота – конечный продукт метаболизма пуриновых соединений в организме. Мочевая кислота образует два ряда
солей, т.е. является двух-, а не трехосновной кислотой, как можно
было бы предположить.
155
156.
"закрепленный"лактамный фрагмент
N
HN
O
NaOH
O
N
H
O : ..... H
O : ..... H
O : ..... H
N
H
мочевая кислота
N
HN
O
NaOH
N
N
ONa
ONa
N
H
N
мононатриевая соль
мочевой кислоты
NaO
N
N
динатриевая соль
мочевой кислоты
Дело в том, что один из лактамных фрагментов мочевой кислоты является «закрепленным», т.е. не может перейти в лактимную форму из-за наличия водородной связи.
Соли мочевой кислоты называют уратами. Кислые ураты
(кроме литиевых) нерастворимы в воде, средние ураты растворимы. Нерастворимые ураты могут откладываться в суставах при
подагре, а также в виде почечных камней.
Производными пурина являются также аденин и гуанин –
нуклеиновые основания (см. стр. 204).
УГЛЕВОДЫ. МОНОСАХАРИДЫ
Углеводы (сахара) составляют основную массу органического вещества на Земле. Они входят в состав клеток и тканей
всех растительных и животных организмов. Углеводы являются
источниками энергии (у растений – крахмал, у животных – гликоген), структурными компонентами клеточных стенок растений
(целлюлоза), входят в структуру жизненно важных веществ –
нуклеиновых кислот, витаминов, коферментов. Некоторые углеводы и их производные используются в медицине (глюкоза, лактоза, ксилит, сорбит, глюконат кальция).
По способности к гидролизу углеводы классифицируют на
моносахариды (не способны к гидролизу до более простых углеводов) и полисахариды (способны к гидролизу). Среди полисахаридов выделяют группу олигосахаридов (гидролизуются с образованием 2-10 молекул моносахаридов) и собственно полисахаридов (состоят из более 10 моносахаридных остатков).
156
157.
МОНОСАХАРИДЫМоносахариды являются гетерофункциональными соединениями – многоатомными альдегидо- или кетоноспиртами.
Моносахариды классифицируют по характеру оксо-группы
(на альдозы и кетозы) и по числу атомов углерода в цепи (на
триозы, тетрозы, пентозы, гексозы и т.д.). Суффикс –оза характерен для названий всех моносахаридов. Строение альдоз и кетоз в
общем виде можно представить следующим образом:
O
C
CH2 OH
C=O
H
(CH-OH)n
(CH-OH)n
CH2 OH
альдозы
CH2 OH
кетозы
Наиболее распространены в природе пентозы и гексозы.
Стереоизомерия моносахаридов. В молекулах моносахаридов присутствуют несколько центров хиральности, что служит
причиной существования большого числа стереоизомеров, соответствующих одной и той же структурной формуле.
Например, в альдогексозе имеется четыре хиральных атома углерода, и она может существовать в виде
O
* * * *
4
CH2 -CH-CH-CH-CH-C
2
=16 стереоизомеров, т.е. 8 пар энанH
тиомеров. Стереоизомеры изображают
OH OH OH OH OH
с использованием проекционных фор1 O
мул Фишера.
C
Относительную конфигурацию стереоизоH
2
OH
H
меров моносахаридов принято определять
HO 3 H
по последнему асимметрическому центру (в
H 4 OH
5
глюкозе это C-5). Его конфигурацию сравH 6 OH
CH2 OH
нивают со стандартом – стереоизомерами
D-глюкоза
глицеринового альдегида и относят стереоизомер моносахарида к D- или L-ряду. Большинство природных
моносахаридов имеет D-конфигурацию.
Каждому моносахариду D-ряда соответствует энантиомер
L-ряда с противоположной конфигурацией всех хиральных центров.
157
158.
Остальные стереоизомеры являются по отношению к нимдиастереоизомерами:
O
C
O
O
C
H
H
HO
H
OH
HO
H
HO
H
CH2 OH
L-глюкоза
O
C
C
H
OH
H
HO
H
OH
H
H
OH
CH2 OH
H
HO
H
HO
H
OH
H
H
OH
CH2 OH
H
HO
HO
H
D-глюкоза
D-манноза
D-галактоза
H
OH
H
H
OH
CH2 OH
энантиомеры
эпимеры
диастереомеры
Альдогексозы существуют в виде 16 стереоизомеров, 8 из
которых относятся к D-ряду, а 8 – к L-ряду, т.е. существует 8 пар
энантиомеров. Энантиомеры имеют одно название (D-глюкоза и
L-глюкоза). По отношению к ним остальные 14 стереоизомеров
являются диастереомерами. Они обладают различными свойствами и имеют различные названия (глюкоза, галактоза, манноза и
т.д.).
Диастереомеры моносахаридов, отличающиеся конфигурацией только одного асимметрического атома углерода, называются эпимерами. Например, D-глюкоза и D-манноза – эпимеры
по C-2; D-глюкоза и D-галактоза – эпимеры по C-4.
Другими представителями моносахаридов являются:
C
C
C
H
OH
OH
CH2 OH
CH2 OH
CH2 OH
D-рибоза
D-дезоксирибоза
D-ксилоза
HO
H
CH2 OH
H
OH
H
OH
OH
OH
альдопентозы
158
O
O
O
C=O
HO
OH
OH
OH
CH2 OH
D-фруктоза
кетогексоза
159.
Цикло-оксо-таутомерия моносахаридовВпервые предположение о циклическом строении глюкозы
было высказано российским ученым А.А. Колли (1870), а затем
развито немецким ученым Б. Толленсом (1883).
CH2OH
..
CH
OH O
OH
H
C
C
C C
H
OH
H
H
OH
Моносахариды, являясь многоатомными альдегидо- или
кетоноспиртами, образуют циклические полуацетали при
взаимодействии
пространственно
сближенных
оксои
гидроксигрупп. В альдопентозах и альдогексозах сближенными
оказываются альдегидная группа и спиртовые гидроксилы при
C-4 или C-5.
При взаимодействии альдегидов со спиртами образуются
полуацетали. В случае, если обе функциональные группы
принадлежат одной молекуле, эти полуацетали будут
циклическими:
полуацетальный
δ+
R C
O:
H
+ HO-R'
R
гидроксил
кислородный
OH
"мостик"
C O R'
H
При образовании полуацеталя в структуре молекулы появляется новый, полуацетальный гидроксил и кислородный «мостик» между C-1 и C-4 или C-1 и C-5.
Рассмотрим цикло-оксо-таутомерию на примере глюкозы.
При взаимодействии альдегидной группы с гидроксилом
при C-5 образуется шестичленный, пиранозный цикл; при взаимодействии с C-4 – пятичленный, фуранозный.
159
160.
полуацетальныйгидроксил
H
O
C
HO
H
OH
OH
OH
CH2 OH
5
D-глюкоза
OH
*
1C
2
3
HO
4
5
6
CH2 OH
O
4
1
OH
2
OH
HO 3
OH
5
OH
OH
O
CH2 OH
α-D-глюкопираноза
(формула Толленса)
HO
полуацетальный
гидроксил
6
*
1C
2
HO 3
4
5
6
H
6
OH
OH
α-D-глюкопираноза
(формула Хеуорса)
O
CH2 OH
β-D-глюкопираноза
CH2 OH
5
O OH
4
1
OH
2
HO 3
OH
β-D-глюкопираноза
Внутримолекулярное образование полуацеталя приводит к
тому, что атом C-1 становится асимметрическим. В результате
образуются стереоизомеры, которые отличаются лишь пространственным расположением полуацетального гидроксила. Их называют α- и β-аномерами. В проекционных формулах моносахаридов (формулах Толленса) полуацетальный гидроксил α-аномера
расположен для моносахаридов D-ряда справа. Полуацетальный
гидроксил β-аномера – слева.
Проекционные формулы Толленса неудобны для изображения циклических структур. Хеуорс предложил так называемые
перспективные формулы. В этих формуO
O
лах пиранозные и фуранозные циклы
пираноза
фураноза изображают в виде плоских колец. Заместители, расположенные в формуле
Толленса справа, в формулах Хеуорса располагаются под плоскостью кольца; слева – над плоскостью.
160
161.
При взаимодействии альдегидной группы глюкозы с гидроксилом при C-4 образуются пятичленные, фуранозные циклы:полуацетальный
гидроксил
H
2
3
HO
4
5
6
O
C
HO
OH
*
1C
OH
O
3
OH
CH2 OH
H
OH
α-D-глюкофураноза
(формула Толленса)
4
OH
OH
CH2 OH
HO
D-глюкоза
*
1C
2
3
HO
4
5
6
полуацетальный
гидроксил
6
CH2 OH
5
O
HO
4
OH
H
OH
1
OH
2
OH
α-D-глюкофураноза
(формула Хеуорса)
6
CH2 OH
5
O OH
HO
1
4
OH
O
2
3
OH
CH2 OH
OH
β-D-глюкофураноза
β-D-глюкофураноза
В кристаллическом виде моносахариды имеют циклическое
строение (например, глюкоза существует в виде β-Dглюкопиранозы), в растворах – в нескольких таутомерных формах. Так как циклические формы моносахаридов представляют
собой внутренние полуацетали, то в водной среде их циклы легко
раскрываются с образованием ациклической (оксо-) формы. Между таутомерными формами устанавливается равновесие:
CH 2 OH
CH 2 OH
O
HO
OH
O
OH
OH
α-D-глюкофураноза
C
H
OH
O
OH
OH
OH
α−D-глюкопираноза (~36%)
HO
HO
CH 2 OH
O
HO
OH
OH
OH
CH 2 OH
OH
CH 2 OH
O
OH
D-глюкоза
OH
HO
OH
β-D-глюкофураноза
OH
β−D-глюкопираноза (64%)
< 0.5%
161
162.
С таутомерными превращениями моносахаридов в водномрастворе связано явление мутаротации, т.е. изменение величины
оптической активности свежеприготовленных растворов моносахаридов. Каждая из таутомерных форм обладает определенной
оптической активностью. Пока между таутомерными формами не
установится равновесие, оптическая активность будет изменяться.
В равновесной смеси присутствуют пять таутомеров
D-глюкозы, однако преобладают β-D-глюкопираноза (64%) и
α-D-глюкопираноза (~36%). На ациклическую и две фуранозные
формы приходится менее 0,5%.
Пиранозные формы являются более устойчивыми, т.к. шестичленный цикл более энергетически выгоден. Почему в равновесии преобладает β-таутомер?
Пиранозные циклы не плоские, для них предпочтительнее
конформация кресла. Из двух кресловидных конформаций более
стабильной является та, в которой объемные заместители (OH- и
-CH2OH группы) находятся в экваториальном положении. Именно у β-аномера глюкозы все оксигруппы и группа –CH2OH занимают экваториальные положения.
HO
HO
CH2 OH
O
OH (экв.)
HO
HO
CH2 OH
O
HO
HO
OH (акс.)
α-D-глюкопираноза
β-D-глюкопираноза
Таутомерные превращения фруктозы представлены следующей схемой:
6
CH2 OH 1
CH2 OH
O
2
5
HO
4
3 OH
OH
α-D-фруктофураноза
CH2 OH
O OH
HO
CH2 OH
OH
β-D-фруктофураноза
162
6
5
1
CH2 OH
2
C=O
HO
4
OH
α-D-фруктопираноза
3
HO
4
5
6
OH
OH
CH2 OH
D-фруктоза
1
O CH2 OH
HO 2
3
OH
HO
OH
O OH
HO
CH2 OH
β-D-фруктопираноза
163.
Обратите внимание, что полуацетальный гидроксил в циклических таутомерах образуется при C-2, т.к. оксо-группа в ациклической форме фруктозы находится в положении 2.Наиболее стабильной таутомерной формой D-фруктозы является β-D-фруктофуранозная.
Итак, моносахариды – циклические полуацетали многоатомных альдегидо- (кетоно-) спиртов, находящиеся в равновесии со своими ациклическими формами.
Так как в растворах моносахаридов устанавливается динамическое равновесие между ациклическим и циклическими таутомерами, то в зависимости от природы реагента они могут реагировать и как многоатомные спирты, и как альдегиды или кетоны, и как циклические полуацетали. При этом равновесие будет
смещаться в сторону образования той формы, которая вступает
во взаимодействие и выводится из сферы реакции.
Производные моносахаридов
К производным моносахаридов относят соединения, в
структуре которых одна или несколько гидроксильных групп отсутствуют или замещены другими функциональными группами
(чаще всего амино- или карбоксильной группой). Наиболее важными из них являются следующие дезокси- и аминосахара:
O
O
C
C
C
C
CH2OH
H
CH2
OH
OH
OH
CH3
H
NH2
H
HO
H
OH
H
H
OH
CH2OH
D-дезоксирибоза
D-дигитоксоза
D-глюкозамин
H
CH2
OH
OH
O
O
H
HO
HO
H
H
NH2
H
H
OH
CH2OH
D-галактозамин
D-дезоксирибоза является структурным компонентом дезоксирибонуклеиновых кислот (ДНК), остаток D-дигитоксозы входит в структуру сердечных гликозидов – природных соединений,
использующихся в кардиологии. Аминосахара являются структурными фрагментами полисахаридов – хитона, хондроитинсульфата, гиалуроновой кислоты, гепарина. Аминогруппы в этих
соединениях обычно ацилированы или сульфатированы.
163
164.
Аскорбиновая кислота (витамин С) по структуре близка к моносахаридам. Она присутствует в овощах, фруктах и ягодах. При недостаткевитамина C в пище вызывает различные заболевания, например, цингу. В
промышленности аскорбиновая кислота производится из D-глюкозы. Аскорбиновая кислота – γ-лактон 2-оксо-L-гулоновой кислоты.
COOH
C=O
HO
CO
-H2O
C=O
HO
OH
HO
CO
таутомерия
O
C-OH
HO
CH2OH
O
HO
CH2OH
2-оксо-L-гулоновая
кислота
C-OH
CH2OH
γ-лактон 2-оксо-L-гулоновой
кислоты
O
O
ендиольный
фрагмент
CHOH-CH2OH
HO
OH
Аскорбиновая кислота является довольно сильной кислотой (pKa
4,2). Её кислотные свойства обусловлены наличием енольного гидроксила.
Аскорбиновая кислота легко окисляется в дегидроаскорбиновую кислоту:
O
HO
O
CHOH-CH2OH окисление
OH
аскорбиновая кислота
восстановление
O
O
O
CHOH-CH2OH
O
дегидроаскорбиновая кислота
Это обратимый процесс, который обеспечивает некоторые окислительно-восстановительные реакции в организме.
РЕАКЦИИ ЦИКЛИЧЕСКИХ ФОРМ МОНОСАХАРИДОВ
Образование гликозидов. O-Гликозиды образуются при
взаимодействии моносахаридов со спиртами в присутствии газообразного хлороводорода. Реакция протекает только за счет полуацетального (гликозидного) гидроксила:
164
165.
CH2 OHO OH
OH
CH3 OH, HCl газ
HO
HO
OH
β−D-глюкопираноза
CH2 OH
O OCH3
OH
+ H2 O
OH
O-метил - β-D-глюкопиранозид
В названиях гликозидов указывается вначале наименование
введенного радикала, затем конфигурация аномерного центра и
название углеводного остатка с заменой суффикса –оза на –озид.
Гликозиды являются полными ацеталями. Водные растворы
гликозидов не мутаротируют, т.к. для них не характерна циклооксо-таутомерия.
Как и все ацетали, гликозиды легко подвергаются гидролизу
в кислой среде и устойчивы к щелочному гидролизу. При гидролизе гликозидов образуются соответствующие моносахариды и
спирты:
CH2 OH
O OC 2 H5
HO
OH
H2 O, H +
OH
O- этил - β −D-галактопиранозид
CH2 OH
O OH
HO
OH
+ C2 H5 OH
OH
β-D- галактопираноза
этанол
При взаимодействии моносахаридов с аминами образуются
N-гликозиды:
CH2 OH
O OH
OH
OH
β-D-рибофураноза
C2 H5 NH2
CH2 OH
O NH-C 2 H5
+ H2 O
OH
OH
Ν−этил-β-D-рибофуранозид
N-гликозидами являются нуклеозиды – структурные компоненты нуклеиновых кислот.
Реакции моносахаридов по гидроксильным группам. Как
многоатомные спирты моносахариды образуют простые и сложные эфиры. В реакциях участвует и полуацетальный гидроксил.
165
166.
CH2 OHO OH
CH3 I , NaOH
OH
HO
избыток
OH
простая эфирная
связь
гликозидная связь
CH2 OCH3
O OCH3
OCH3
H3 CO
OCH3
β-D- глюкопираноза
O- этил - β-D-2,3,4,6- тетраметилглюкопиранозид
Простые эфиры получают с использованием алкилгалогенидов в щелочной среде. Одновременно реакция идет и по полуацетальному гидроксилу с образованием гликозида. Простые
эфиры не гидролизуются, гликозиды легко подвергаются гидролизу в кислой среде:
H3 CO
CH2 OCH3
O OCH3
OCH3
H2 O, H+
H3 CO
OCH3
CH2 OCH3
O OH
OCH 3
+ CH3 OH
OCH3
2,3,4,6-тетераметилβ -D-глюкопираноза
Моносахариды легко ацилируются ангидридами карбоновых кислот, образуя сложные эфиры.
CH2 OH
O
OH
OH
HO
O
CH2 OAц
O O-C-CH 3
OAц
(CH3 CO) 2 O
избыток
OH
β−D-глюкопираноза
AцO
OCOCH 3
Aц
пентаацетилглюкоза
Сложные эфиры моносахаридов гидролизуются и в кислой,
и в щелочной среде:
NaOH, H2 O
AцO
O
CH2 OAц
O O-C-CH 3
OAц
OCOCH 3
пентаацетилглюкоза
166
H2 O, H+
CH2 OH
O OH
OH
+ 5 CH3 COONa
HO
ацетат натрия
OH
глюкоза
CH2 OH
O OH
OH
+ 5 CH3 COOH
HO
уксусная кислота
OH
167.
Среди сложных эфиров моносахаридов особое место занимают эфиры фосфорной кислоты. Они содержатся во всех растительных и животных организмах, являясь основными веществамиметаболизма углеводов, структурными элементами нуклеиновых
кислот и коферментов.
HO
6
O=P-O-CH 2
HO
5
O=P-O-CH 2
O
HO
OH
HO
OH
1
CH2 -O-P
O
OH
HO
O
OH
OH
1,6-дифосфат
α-D-фруктофуранозы
OH OH
5-фосфат
β-D-рибофуранозы
РЕАКЦИИ АЦИКЛИЧЕСКИХ ФОРМ МОНОСАХАРИДОВ
Эпимеризация и изомеризация. Одно из характерных
свойств моносахаридов – способность к взаимопревращению
изомерных альдоз под влиянием щелочи (в мягких условиях).
Например, D-глюкоза в присутствии известковой воды превращается в D-маннозу (эпимер глюкозы) и D-фруктозу (изомер
глюкозы):
H
H
O:
C
H
HO
H
H
OH
H
OH
OH
CH2 OH
D-глюкоза
H
OH
HO
HO
H
H
C
C OH
HO
H
OH
H
OH
H
CH2 OH
ендиольная форма
O
C
H
H
OH
OH
CH2 OH
D-манноза
CH2 OH
C=O
HO
OH
OH
CH2 OH
D-фруктоза
167
168.
Атом водорода при C-2 подвижен из-за электроноакцепторного влияния двух соседних групп (оксо-группы и спиртовогогидроксила). В щелочной среде происходит отщепление протона
и присоединение его к оксониевому основному центру – атому
кислорода оксо-группы. Образуется ендиольная форма. Этот
процесс схож с кето-енольной таутомерией (см. стр.113). Ендиольная форма не стабильна и превращается в смесь альдоз (Dглюкозы и D-маннозы) и кетозы (D-фруктозы).
Возможностью взаимных превращений альдоз и кетоз в
слабощелочной среде объясняется способность фруктозы давать
положительные пробы Толленса и Троммера (см. стр.169).
Реакции восстановления. При восстановлении моносахаридов образуются многоатомные спирты (альдиты), например, глюкоза восстанавливается в глюцит (сорбит), манноза – в маннит,
ксилоза – в ксилит. Восстановление проводят водородом в присутствии палладия или никеля.
C
O
H
OH
HO
OH
OH
CH2 OH
D-глюкоза
CH2 OH
[H]
C
OH
HO
OH
OH
CH2 OH
глюцит (сорбит)
O
H
OH
HO
CH2 OH
[H]
OH
HO
OH
OH
CH2 OH
D-ксилоза
CH2 OH
ксилит
Ксилит и сорбит применяются как заменители сахара при
сахарном диабете, т.к. они обладают сладким вкусом, но по химической структуре не являются сахарами и не участвуют в процессах метаболизма моносахаридов, которые при сахарном диабете нарушены. Ксилит и сорбит используют также вместо сахара
при изготовлении жевательных резинок, т.к. в отличие от глюкозы эти многоатомные спирты не участвуют в процессах молочнокислого брожения (см. стр.173), и молочная кислота не оказывает
негативного действия на зубную эмаль.
Восстановление глюкозы в сорбит является одной из стадий
промышленного синтеза аскорбиновой кислоты. Гипертонические растворы маннита используют как диуретическое средство
для снижения внутричерепного давления и уменьшения отека
мозга.
168
169.
Реакции моносахаридов как многоатомных спиртов. Какмногоатомные спирты, моносахариды взаимодействуют с гидроксидом меди (II) при комнатной температуре с образованием растворимых хелатных комплексов ярко-синего цвета:
C
O
C
H
OH
CH2 OH
HO
OH + Cu(OH) HO
2
OH
HO
CH2 OH
HO
D-глюкоза
OH
O
H
OH
HO
O
Cu
OH
CH2 OH
CH2 OH
HO
O
HO
C
H
OH
O
H
C
O
Реакции окисления моносахаридов протекают по-разному, в
зависимости от условий.
Окисление слабыми окислителями в щелочной среде. Как
альдегиды, моносахариды окисляются такими слабыми окислителями, как гидроксид меди (II) (проба Троммера) или аммиачный раствор оксида серебра Ag(NH3)2OH (реакция Толленса).
Моносахариды в щелочной среде неустойчивы, поэтому при
окислении образуется смесь продуктов.
C
O
H
OH
Ag(NH3 )2 OH , to
HO
OH
OH
CH2 OH
D-глюкоза
Cu(OH)2 , to
продукты окисления + Ag + NH3 + H2 O
"серебряное
глюкозы
зеркало"
продукты окисления + Cu2 O + H2 O
глюкозы
кирпичнокрасный осадок
В этих реакциях моносахариды проявляют восстанавливающие свойства.
Гликозиды не обладают восстанавливающими свойствами,
т.к. для них невозможен переход в открытую таутомерную форму.
Фруктоза (кетоза) также окисляется слабыми окислителями,
т.к. в щелочной среде изомеризуется в глюкозу и маннозу.
169
170.
Качественной реакцией на фруктозу является реакция Селиванова: при нагревании фруктозы с резорцином в присутствииконцентрированной соляной или серной кислоты наблюдается
винно-красное окрашивание. Эта реакция основана на том, что
при нагревании с минеральными кислотами фруктоза подвергается дегидратации с образованием 5-гидроксиметилфурфурола:
to
фруктоза
- 3 H2 O
O
HOCH2
O
C
H
5-гидроксиметилфурфурол
5-гидроксиметилфурфурол вступает затем в реакцию конденсации с резорцином, образуя окрашенный продукт.
OH
OH
O
HOCH2
C
- H2O
+ 2
H
O
5-гидроксиметилфурфурол
OH
HOCH2
O
CH
O
резорцин
продукт конденсации
OH
Окисление в нейтральной и слабокислой среде. При окислении альдоз в мягких условиях (например, бромной водой) окисляется только альдегидная группа и образуются гликоновые
(глюконовая, манноновая, галактоновая) кислоты.
C
O
H
OH
HO
OH
OH
CH2 OH
D-глюкоза
C
Br2 , H2 O
(HOBr)
O
OH
OH
HO
OH
OH
CH2 OH
D-глюконовая кислота
Кальциевая соль глюконовой кислоты – глюконат кальция –
применяется в медицине как источник ионов кальция (см. стр.
115):
170
171.
CO
C
OH
OH
Ca(OH)2
HO
O
OOH
HO
OH
OH
CH2OH
OH
OH
CH2OH
Ca
2
глюконат кальция
D-глюконовая кислота
При окислении сильным окислителем в сильнокислой среде
(например, разбавленной азотной кислотой) окисляются и альдегидная, и первичная спиртовая группы. При этом образуются дикарбоновые, гликаровые кислоты (глюкаровая, галактаровая, риO
баровая):
C
COOH
H
OH
HO
HNO3
OH
HO
OH
OH
CH2 OH
OH
OH
COOH
D-глюкоза
D-глюкаровая кислота
При окислении первичной спиртовой группы при сохранении альдегидной образуются уроновые кислоты. Для получения
уроновых кислот альдегидную группу нужно предварительно защитить от окисления, образовав гликозид:
CH2 OH
O OH
OH
HO
OH
β−D-глюкопираноза
[O]
CH2 OH
O OCH3
OH
CH3 OH, HCl газ
- H2 O
HO
OH
O-метил - β-D-глюкопиранозид
O
C
COOH
COOH
H
O OH
O OCH3 H2O, H+
OH
OH
OH
HO
HO
- CH3OH
HO
OH
OH
OH
OH
D-глюкуроновая
O-метилгликозид
COOH
кислота (циклическая
D-глюкуроновой кислоты
форма)
D-глюкуроновая
кислота (ациклическая
форма)
171
172.
Уроновые кислоты выполняют в организме важную функцию: они образуют растворимые гликозиды с токсическими веществами (продукты метаболизма лекарственных веществ и др.)и выводятся с мочой. Образование гликозидов глюкуроновой кислоты – глюкуронидов – является частным случаем процесса
конъюгации, т.е. процесса взаимодействия лекарственных веществ или продуктов их метаболизма с биогенными веществами.
Например, парацетамол выводится из организма в виде глюкуронида:
NHCOCH
3
NHCOCH3
COOH
OH
COOH
O O
O OH +
OH
HO
OH
D-глюкуроновая
кислота
HO
OH
глюкуронид
парацетамола
OH
парацетамол
При нагревании солей уроновых кислот с никелем или магнием происходит реакция декарбоксилирования, при этом из гексуроновых кислот образуются соответствующие пентозы. В организме сходным образом в присутствии ферментов протекает
декарбоксилирование D-глюкуроновой кислоты с образованием
D-ксилозы:
O
C
COOH
OH
HO
O OH - СO
2
OH
D-глюкуроновая
кислота
OH
HO
O OH
OH
D-ксилопираноза
HO
H
H
OH
OH
CH2OH
D-ксилоза
Реакции брожения глюкозы. Брожение – это сложный процесс расщепления моносахаридов под влиянием ферментов микроорганизмов. Для глюкозы характерно спиртовое, молочнокислое, лимоннокислое и маслянокислое брожение с образованием
соответствующих продуктов:
172
173.
C2 H5 OH + CO 2этанол
CH3 -CH-COOH
OH
молочная кислота
C6 H12 O6
глюкоза
COOH
HOOC-CH 2 -C-CH 2 -COOH
OH
лимонная кислота
CH3 -CH2 -CH2 -COOH
масляная кислота
В организме человека и высших животных непрерывно протекают процессы биохимического расщепления и синтеза моносахаридов. При мышечных сокращениях в результате расщепления углеводов образуется молочная кислота. Этот процесс называется гликолизом.
УГЛЕВОДЫ. ДИ- И ПОЛИСАХАРИДЫ
Природные дисахариды (биозы) состоят из двух остатков
одинаковых или разных моносахаридов, связанных Oгликозидной связью, т.е. они являются O-гликозидами, или полными ацеталями.
Моносахаридные остатки могут быть связаны по-разному:
если гликозидная связь образуется за счет полуацетального гидроксила одной молекулы и спиртового гидроксила другой молекулы, образуется восстанавливающий дисахарид; если гликозидная связь образуется за счет взаимодействия двух полуацетальных гидроксилов, образуется невосстанавливающий дисахарид.
Примерами восстанавливающих дисахаридов являются
мальтоза, целлобиоза, лактоза.
Молекула мальтозы образуется при взаимодействии двух
молекул α-D-глюкопиранозы.
173
174.
Полуацетальный гидроксил первой молекулы взаимодействует со спиртовым гидроксилом у C-4 второго остатка:CH2 OH
O
OH
1
CH2 OH
O
4
OH
OH
HO
OH
α-D-глюкопираноза
+
OH
OH
α-D-глюкопираноза
HO
CH2 OH
O
OH
HO
1
O
CH2 OH
O
4
OH
OH α-1,4-глико-
OH
полуацетальный
гидроксил
OH
зидная связь
α-мальтоза
(α-D-глюкопиранозил-1,4-α-D-глюкопираноза)
Мальтоза – солодовый сахар, основной продукт расщепления крахмала под действием фермента амилазы, выделяемой
слюнной железой.
В номенклатуре дисахаридов в основном используют тривиальные названия. Систематические названия восстанавливающих
дисахаридов составляют так: название гликозильного остатка
(первый моносахаридный остаток) получает суффикс «озил», а
название аликона (второй моносахаридный остаток) сохраняет
суффикс «оза». Между названиями гликозильного остатка и
агликона указывают порядковые номера атомов углерода, связанных гликозидной связью.
CH2 OH
O 1
OH
HO
OH
O
β-1,4связь
CH2 OH
O
4 OH
OH
OH
β-целлобиоза
(β-D-глюкопиранозил-1,4-β-D-глюкопираноза)
174
Молекула целлобиозы тоже построена из остатков
глюкозы, но они связаны
β-1,4-гликозидной связью.
175.
CH2 OHHO
O 1
OH
CH2 OH
O
4 OH
O
β-1,4связь
OH
Молекула лактозы построена из остатков галактозы и глюкозы, связанных
β-1,4-гликозидной связью.
OH
OH
β-лактоза
(β-D-галактопиранозил-1,4- β-D-глюкопираноза)
Лактоза (молочный сахар) содержится в молоке (4-5%). Применяется в фармацевтической практике при изготовлении порошков
и таблеток, т.к. она менее гигроскопична, чем сахар, как питательное средство для грудных детей. Из женского молока выделено более 10 олигосахаридов, структурным фрагментом которых
является лактоза. Эти олигосахариды играют важную роль для
формирования кишечной микрофлоры новорожденных.
В структуре всех восстанавливающих дисахаридов есть
свободный полуацетальный гидроксил.
Наличие свободного полуацетального гидроксила ведет к
возможности цикло-оксо-таутомерии, следовательно, свежеприготовленные растворы восстанавливающих дисахаридов мутаротируют. Например, мальтоза существует в растворе в виде α- и βциклических таутомеров, а также в открытой (оксо-) таутомерной
форме:
CH2 OH
O
OH
HO
OH
O
α-мальтоза
OH
восстанавливающий
фрагмент
O
OH
OH
O
OH
оксо-таутомер
CH2 OH
O
OH
HO
OH
CH2 OH
CH2 OH
O
OH
HO
полуацетальный
гидроксил
CH2 OH
O
OH
O
C
H
OH
CH2 OH
O OH
OH
полуацетальный
гидроксил
OH
OH
β-мальтоза
175
176.
Из-за наличия альдегидной группы в оксо-форме восстанавливающие дисахариды обладают восстанавливающими свойствами, они окисляются слабыми окислителями (аммиачный раствор оксида серебра, гидроксид меди (II)) в щелочной среде. Также, как моносахариды, дисахариды неустойчивы при нагревании
в щелочной среде, поэтому в реакции образуются различные продукты окисления:
CH2 OH
O
OH
HO
CH2 OH
OH
OH
O
OH
Ag(NH3 )2 OH
to
O
C
продукты
окисления
H
Cu(OH)2
to
OH
открытый таутомер
целлобиозы
+ Ag + NH3 +H2 O
серебряное
зеркало
продукты + Cu O + H O
2
2
окисления
кирпичнокрасный
осадок
При окислении слабыми окислителями в слабокислой или
нейтральной среде (например, бромной водой) образуются бионовые кислоты (мальтобионовая, лактобионовая и т.п.):
CH2 OH
CH2 OH
O
OH
HO
OH
OH
O
OH
O
C
H
Br2 , H2 O
OH
мальтоза
CH2 OH
O
OH
HO
CH2 OH
OH
OH
O
OH
O
C
OH
OH
мальтобионовая кислота
Полуацетальный гидроксил восстанавливающих дисахаридов участвует в образовании гликозидов:
CH2 OH
O
OH
HO
176
O
OH
β-целлобиоза
CH2 OH
O OH CH3 OH
OH
HCl газ
OH
CH2 OH
O
OH
HO
O
CH2 OH
O OCH3
OH
OH
OH
O-метил- β-целлобиозид
177.
Все дисахариды являются O-гликозидами, поэтому для ниххарактерны реакции гидролиза в кислой среде:
CH2 OH
CH2 OH
O
HO
O 1
OH
O 4 OH
CH2 OH
CH2 OH
O OH
O OH
+
OH
OH
HO
OH
OH
β-D-галактопираноза β-D-глюкопираноза
OH H O,H+ HO
2
OH
OH
Как и моносахариды, восстанавливающие дисахариды алкилируются и ацилируются по всем гидроксильным группам. Например, при исчерпывающем метилировании мальтозы образуется продукт октаметилмальтоза, в структуре которой присутствуют как O-гликозидные, так и простые эфирные связи:
CH2 OH
O
OH
HO
O
CH2 OH
O OH
OH
(CH3 )2 SO4
NaOH
OH
OH
мальтоза
H3 CO
О-гликозидные
связи
CH2 OCH3
O
OCH3
O
CH2 OCH3
O OCH3
OCH3
OCH3
октаметилмальтоза
OCH3
H2 O, H+
H3 CO
CH2 OCH3
O
OCH3
OH
OCH3
2,3,4,6-тетраметилα-D-глюкопираноза
CH2 OCH3
O OH
+ CH3 OH
OCH3
+
HO
OCH3
2,3,6-триметилβ-D-глюкопираноза
177
178.
Кислотному гидролизу подвергаются только гликозидныесвязи.
В реакциях ацилирования также участвуют и спиртовые, и
полуацетальные гидроксильные группы. Все вновь образующиеся связи – сложноэфирные, связь между двумя моносахаридными
остатками – O-гликозидная. Сложные эфиры подвергаются гидролизу как в кислой, так и в щелочной среде, гликозиды – только
в кислой. Поэтому в зависимости от условий гидролиза ацилированного дисахарида можно получить различные продукты:
CH2 OH
O
OH
HO
O
CH2 OH
O OH
OH
(CH3 CO) 2 O
OH
OH
мальтоза
CH2 OAc
O OCOCH 3
OAc
Ac
O
OAc
OAc
октаацетилмальтоза
CH2 OAc
O
OAc
AcO
H2 O, H+
2
CH2 OH
O
OH
HO
OH
глюкоза
CH2 OH
O
H2 O, NaOH
HO
OH + 8 CH3 COOH
CH2 OH
O OH
+ 8 CH3 COONa
O
OH
OH
OH
мальтоза
OH
К невосстанавливающим дисахаридам относится сахароза, молекула которой построена из остатков α-D-глюкопиранозы
и β-D-фруктофуранозы, связанных 1,2-гликозидной связью (α-Dглюкопиранозил-1,2-β-D-фруктофуранозид):
178
179.
CH2 OHO
OH
HO
CH2 OH
O
OH
1
OH
OH
α-D-глюкопираноза
+
CH2 OH
O OH
HO 2
CH2 OH
1
OH
β-D-фруктофураноза
HO
1
OH
O 1,2-гликозидная связь
CH2 OH
O
HO 2
CH2 OH
1
OH
сахароза
Гликозидная связь образовалась за счет взаимодействия
двух полуацетальных гидроксилов, свободная полуацетальная
группа отсутствует, следовательно, невозможна цикло-цепная
таутомерия. Отсюда отсутствие восстанавливающих свойств у
сахарозы.
Как гликозид сахароза гидролизуется в кислой среде до
глюкозы и фруктозы. Образующуюся смесь моносахаридов называют искусственным медом, или инвертным сахаром («инверсия» – обращение). Это название появилось потому, что сахароза
обладает правым вращением, а образовавшаяся смесь моносахаридов – левым, т.к. фруктоза значительно сильнее вращает плоскость поляризованного луча влево, чем глюкоза – вправо.
ПОЛИСАХАРИДЫ
Полисахариды (полиозы) – высокомолекулярные соединения. Их молекулы состоят из нескольких сотен моносахаридных
звеньев, связанных O-гликозидными связями, образующих разветвленные или неразветвленные цепи. По химической природе
все полисахариды являются полигликозидами. В полисахаридах
растительного происхождения чаще всего встречаются 1,4- и 1,6гликозидные связи. В полисахаридах животного и бактериального происхождения – также 1,2- и 1,3-гликозидные связи.
На концах цепей имеются свободные полуацетальные гидроксильные группы, но восстанавливающие свойства практически не проявляются, т.к. доля восстанавливающего фрагмента относительно всей молекулы очень мала.
179
180.
Полисахариды, состоящие из остатков одного моносахарида, называются гомополисахаридами, из остатков разных моносахаридов – гетерополисахаридами.Гомополисахариды. Гомополисахариды, построенные из
остатков пентоз, называются пентозанами, из остатков гексоз –
гексозанами. Большинство природных гомополисахаридов – гексозаны. К ним относятся крахмал, целлюлоза, гликоген, декстраны.
Крахмал является основным источником резервной энергии
в растениях. Крахмал на 10-20% состоит из растворимой в горячей воде амилозы и на 80-90% – из нерастворимого амилопектина.
При постепенном кислотном или ферментативном гидролизе амилоза и амилопектин расщепляются до декстринов (смесь
полисахаридов с меньшей молекулярной массой), затем до мальтозы и глюкозы:
(C6 H10 O5 )n
(C6 H10 O5 )m
C12 H22 O11
C6 H12 O 6
крахмал
декстрины
(m<n)
мальтоза
глюкоза
Мономером обеих фракций крахмала является D-глюкоза.
Различия в строении амилозы и амилопектина связаны с характером гликозидных связей.
В
амилозе
D-глюкопиранозные
остатки
связаны
α-1,4-гликозидными связями. Цепь амилозы неразветвленная,
включает до 1000 глюкозных остатков (молекулярная масса до
160 000).
CH2OH
O
OH
HO
OH
O
CH2OH
O
OH
OH
O
α-1,4
CH2OH
O
OH
OH
O
n
CH2OH
O
OH
OH
OH
амилоза
По данным рентгеноструктурного анализа, молекула амилозы свернута в спираль (рис. 3), на каждый виток которой приходится 6 моносахаридных звеньев. Возможность такой пространственной структуры связана с конфигурацией аномерного угле180
181.
рода у α-D-глюкопиранозы – полуацетальный гидроксил находится в аксиальном положении (см. стр. 162).Рис. 3. Фрагмент спирали амилозы.
Во внутреннюю полость этой спирали могут входить подходящие по размеру молекулы, например, молекулы иода, образуя
комплексы, так называемые соединения включения. Комплекс
амилозы с иодом имеет синюю окраску. Эта реакция используется и для открытия крахмала, и для обнаружения иода.
Амилопектин имеет разветвленное строение. В цепи
D-глюкопиранозные остатки связаны α-1,4-гликозидными связями, а в точках ветвления – α-1,6-гликозидными связями. Между
точками ветвления расположены 20-25 моносахаридных остатков. Молекула амилопектина состоит из нескольких тысяч остатков D-глюкозы (молекулярная масса – 1-6 млн.).
CH2OH
O
OH
OH
O α-1,6
CH2OH
O
OH
OH
CH2
O
OH
O
OH
O
α-1,4
CH2OH
O
OH
OH
O
CH2OH
O
OH
O
OH
амилопектин
В связи с наличием разветвлений молекула амилопектина не
способна принимать форму спирали и связывает иод в незначительных количествах.
181
182.
Гидролиз крахмала в пищеварительном тракте происходит сучастием фермента амилазы, расщепляющего α-1,4- и
α-1,6-гликозидные связи. Конечными продуктами гидролиза являются мальтоза и глюкоза.
В животных организмах функцию запасного питательного
вещества выполняет гликоген (животный крахмал). Строение
гликогена сходно со строением амилопектина, но его молекула
еще более разветвлена. Между точками ветвления находится
10-12 глюкозных остатков (молекулярная масса гликогена может
достигать многих млн.). Сильная разветвленность цепей гликогена способствует атаке его ферментами сразу со многих концов.
Это приводит к чрезвычайно высокой скорости расщепления полисахарида и, следовательно, к почти мгновенной мобилизации
заключенных в гликогене энергетических запасов. Наиболее богаты гликогеном печень и мышцы.
Декстраны – полисахариды бактериального происхождения. Они построены из остатков α-D-глюкопиранозы. Молекулы
декстранов сильно разветвлены. Основной тип связи –
α-1,6-гликозидная, в местах ветвления – α-1,3 и α-1,4:
O
CH2
O
α-1,6
O
CH2
O
OH
O
CH2
OH
O
α-1,3
O
CH2
OH
CH2
O
O
OH
HO
HO
HO
O
OH
OH
O
CH2
OH
O
OH
O
α-1,4
O
HO
HO
OH
OH
Структура декстрана
Молекулярная масса декстранов достигает нескольких млн.
Они плохо растворимы в воде. Путем частичного гидролиза получают так называемые «клинические декстраны» с молекулярной массой 50-100 тыс. Их используют в качестве заменителя
плазмы крови (препарат «Полиглюкин»).
Наиболее широко распространенный растительный гомополисахарид – целлюлоза (клетчатка). Молекулы целлюлозы обла182
183.
дают большой механической прочностью, поэтому выполняютроль опорного материала растений.
Молекула целлюлозы – длинная линейная цепь, состоящая
из остатков β-D-глюкопиранозы, тип связи между моносахаридными остатками – β-1,4-гликозидная. Молекулярная масса целлюлозы от 400 000 до 2 млн.
CH2 OH
O
OH
HO
OH
O
CH2 OH
O
OH
OH
O
β-1,4
CH2 OH
O
OH
OH
O
n
CH2 OH
O OH
OH
OH
Целлюлоза
β-Конфигурация аномерного атома углерода приводит к линейному строению молекулы (β-полуацетальный гидроксил занимает экваториальное положение) (рис. 4).
Внутри цепи и между параллельными цепями образуются
Рис. 4. Строение фрагмента молекулы целлюлозы.
Внутри цепи и между параллельными цепями образуются
водородные связи, что обеспечивает высокую механическую
прочность, волокнистость, нерастворимость целлюлозы в воде.
Как O-гликозид целлюлоза способна к кислотному гидролизу с образованием глюкозы.
В человеческом организме нет фермента, расщепляющего
β-гликозидные связи целлюлозы, поэтому она не усваивается организмом, но является необходимым компонентом пищи, т.к.
улучшает пищеварение.
Целлюлоза является важным сырьем в бумажной и текстильной промышленности. Большое практическое применение
183
184.
находят эфиры целлюлозы – ацетаты (искусственный шелк),ксантогенаты (вискоза, целлофан), нитраты (смесь моно- и динитратов – коллоксилин в виде спирто-эфирного раствора используется для заклеивания мелких ранок и ссадин; тринитрат
является взрывчатым веществом).
В животных организмах опорные и механические функции
выполняет хитин (роговые оболочки насекомых, панцири ракообразных). Хитин построен из остатков N-ацетилглюкозамина,
связанных β-1,4-гликозидными связями:
CH2 OH
O
OH
β-1,4
O
CH2 OH
O
OH
O
CH2 OH
O
OH
NHCOCH3
NHCOCH3
O
NHCOCH3
Хитин
К гомополисахаридам относятся также пектины. В основе
их структуры лежит пектовая (полигалактуроновая) кислота. Остатки галактуроновой кислоты связаны α-1,4-гликозидными свяCOOH
зями:
COOH
COOH
OH
O
O
OH
OH
O
O
OH
OH
O
O
OH
Пектовая кислота
В состав пектинов полигалактуроновая кислота может входить в виде сложных эфиров по карбоксильной группе и солей.
Пектины содержатся в овощах и плодах, для них характерно
желеобразование под действием органических кислот, что используется в пищевой промышленности (желе, мармелад). Пектины обладают противоязвенным действием (препарат «Плантаглюцид» содержит пектины подорожника).
Гетерополисахариды. К гетерополисахаридам принадлежат полисахариды соединительной ткани – хондроитинсульфаты
(содержатся в коже, хрящах), гиалуроновая кислота (в стекловидном теле, суставной жидкости, хрящах) гепарин (в печени).
В строении гетерополисахаридов есть общие черты – их молекулы линейны и состоят из повторяющихся дисахаридных
фрагментов, в состав которых входят остатки уроновых кислот
184
185.
(глюкуроновая,галактуроновая) и N-ацетилгексозаминов
(N-ацетилглюкозамин, N-ацетилгалактозамин).
Хондроитинсульфаты состоят из дисахаридных остатков
N-ацетилированного хондрозина, соединенных β-1,4-гликозидными связями. Сам хондрозин состоит из остатков
D-глюкуроновой кислоты и D-галактозамина, связанных
β-1,3-гликозидной связью:
COOH
OH
O
HO
1
CH2 OH
O OH
O
HO
OH
D-глюкуроновая
кислота
3
NHCOCH3
N-ацетил-Dгалактозамин
β-1,3
N-ацетилхондрозин
Хондроитинсульфаты являются эфирами серной кислоты.
Сульфатная группа присутствует в 4-м или 6-м положении
N-ацетилгалактозамина. Соответственно различают хондроитин4-сульфат и хондроитин-6-сульфат.
COOH
HO3 SO
O
OH
OH
CH2 OH
O
COOH
β-1,4
OH
O
O
β-1,3
NHCOCH 3
HO3 SO
O
OH
CH2 OH
O
O
O
β-1,3
NHCOCH 3
Хондроитин-4-сульфат
Хондроитинсульфатные цепи соединены O-гликозидными
связями с гидроксильными группами аминокислотных остатков
белков.
Гиалуроновая кислота построена из дисахаридных фрагментов, связанных β-1,4-гликозидными связями. Дисахаридный
фрагмент состоит из остатков D-глюкуроновой кислоты и Nацетил-D-глюкозамина, связанных β-1,3-гликозидной связью.
COOH
OH
O
CH2 OH
O
1
O HO
OH
D-глюкуроновая β-1,3
кислота
3
COOH
β-1,4
O
NHCOCH3
N-ацетил-Dглюкозамин
OH
O
CH2 OH
O
1
O HO
OH
β-1,3
3
O
NHCOCH3
Гиалуроновая кислота
185
186.
Гиалуроновая кислота имеет высокую молекулярную массу(2-7 млн.). Ее растворы обладают высокой вязкостью, с чем связывают ее функции по обеспечению непроницаемости соединительной ткани для патогенных микроорганизмов.
Хондроитинсульфаты и гиалуроновая кислота содержатся в
организме в связанном виде с полипептидными цепями. Углеводсодержащие смешанные биополимеры составляют основу клеток и жидкостей животных организмов. Если в молекуле биополимера преобладает полисахаридная часть, его относят к протеогликанам, если полипептидная часть – к гликопротеинам.
ПРИРОДНЫЕ α-АМИНОКИСЛОТЫ. ПЕПТИДЫ, БЕЛКИ
α-Аминокислоты являются мономерными единицами биополимеров – пептидов и белков. Белки – основа всего живого.
Функции белков в природе многообразны. Это ферменты и гормоны, выполняющие регуляторную функцию, структурные белки
(кератин, коллаген), транспортные (гемоглобин), защитные (иммуноглобулины). Некоторые аминокислоты используются как
лекарственные средства, например, глутаминовая кислота – при
заболеваниях ЦНС, метионин – для лечения заболеваний печени,
цистеин – как радиопротектор, а также в глазной практике.
В природе в свободном и связанном виде обнаружены сотни
аминокислот, 20-25 из них постоянно встречаются во всех белках.
Строение и классификация природных α-аминокислот
Общая формула α-аминокислот (исключение – пролин и оксипролин):
R-CH-COOH
NH2
В основном используют тривиальные названия αаминокислот. В биохимии часто пользуются сокращенными
трехбуквенными названиями.
Согласно общей формуле, аминокислоты отличаются лишь
строением радикала, в соответствии с чем они классифицируются
на алифатические, ароматические и гетероциклические. Среди
алифатических аминокислот в зависимости от наличия в радика186
187.
ле функциональной группы выделяют подгруппы гидрокси- и серосодержащих аминокислот.Примеры алифатических аминокислот:
CH2 -COOH
NH2
CH3
Глицин
CH3
CH3 -CH-COOH
Аланин
NH2
CH3
CH-CH2 -CH-COOH
Лейцин *
NH
2
CH3 -CH2 -CH-CH-COOH
CH3 NH2
CH-CH-COOH
CH3
Валин *
NH2
CH2 -CH-COOH
OH NH2
Серин
CH3 -CH-CH-COOH
OH NH2
Треонин *
CH2 -CH-COOH
CH2 -CH-COOH
SH NH2
Изолейцин *
Цистеин
CH3 -S-CH 2 -CH2 -CH-COOH
NH2
Мет ионин
HOOC-CH 2 -CH-COOH
S
NH2
S
NH2
Цистин (Цис-S-S-Цис)
CH2 -CH-COOH
HOOC-CH 2 -CH 2 -CH-COOH
NH2
NH2
Аспарагиновая кислота
Глютаминовая кислота
H2 NOC-CH 2 -CH-COOH
H2 NOC-CH 2 -CH 2 -CH-COOH
NH2
NH2
Аспарагин (Асн )
Глютамин (Глн )
H2 N-CH 2 -CH2 -CH2 -CH-COOH
H2 N-C-NH-CH 2 -CH 2 -CH 2 -CH-COOH
Орнитин
NH2
NH
Аргинин
NH2
H2 N-CH2 -CH2 -CH2 -CH2 -CH-COOH
Лизин *
NH2
187
188.
Примерами ароматических аминокислот являются фенилаланин и тирозин:CH2 -CH-COOH
CH2 -CH-COOH
HO
NH2
NH2
Тирозин
Фен илаланин *
К гетероциклическим аминокислотам относятся:
CH2 -CH-COOH
N
H
N
NH2
N
H
Триптофан*
CH2 -CH-COOH
NH2 Гистидин
HO
N
H
COOH
Пролин
N
H
COOH
Оксипролин ( Про-OH )
В зависимости от соотношения количества карбоксильных и
аминогрупп в молекулах различают нейтральные (моноаминомонокарбоновые) – глицин, аланин и др., кислые (моноаминодикарбоновые) – аспарагиновая, глютаминовая кислоты, основные
(диаминомонокарбоновые) аминокислоты – лизин, орнитин, аргинин.
Природа радикала в молекулах аминокислот значительно
влияет на свойства белков. Например, наличие полярных групп
(-SH, NH2, -OH, COOH, фенольного гидроксила) увеличивает
растворимость белков. Кроме того, эти группы принимают участие в образовании связей, формирующих пространственную
структуру белка. За счет карбоксильной и аминогруппы многие
ферменты обеспечивают кислотный или основный катализ ряда
биохимических процессов.
Большинство α-аминокислот синтезируется в организме, но
некоторые организм не способен синтезировать, они должны поступать с пищей. Эти аминокислоты называют незаменимыми, в
норме их восемь: валин, лейцин, изолейцин, треонин, метилнин,
фенилаланин, триптофан, лизин. При некоторых заболеваниях
незаменимых аминокислот больше. Так, фенилкетонурия (пиро188
189.
виноградная олигофрения) представляет собой врожденное нарушение обмена веществ, каким-то образом связанное с некоторыми умственными расстройствами. Люди, страдающие этим заболеванием, нуждаются еще в одной незаменимой для них аминокислоте – тирозине, т.к. в отличие от здоровых людей их организм не способен превращать фенилаланин в тирозин.Стереоизомерия α-аминокислот
Все α-аминокислоты, за исключением глицина, имеют в
своей структуре хотя бы один хиральный центр, значит, для них
характерна оптическая изомерия. Например, аланин существует
в виде пары энантиомеров:
*
CH3 -CH-COOH
NH2
аланин
COOH
COOH
H
NH2
H
H2 N
CH3
CH3
D-аланин
L-аланин
Относительная конфигурация стереоизомеров определяется
в сравнении с конфигурационным стандартом – стереоизомерами
глицеринового альдегида.
Все α-аминокислоты, участвующие в построении молекул
белков животных и человека, имеют L-конфигурацию. D-аминокислоты встречаются в некоторых грибах и микроорганизмах.
α-Аминокислоты представляют собой кристаллические вещества с высокой температурой плавления, растворимые в воде,
но не растворимые в неполярных растворителях. Эти свойства
результат того, что и в кристаллическом состоянии, и в водных
растворах аминокислоты существуют в виде биполярных ионов
(цвиттер-ионов). Возможность образования биполярных ионов
связана с амфотерностью аминокислот, в молекулах которых
присутствуют и кислотные COOH-группы, и основные
NH2-группы. Близость этих групп у α-аминокислот облегчает переход протона H+:
R-CH-COOH
R-CH-COO NH2
+
NH3
189
190.
Практически α-аминокислоты в водном растворе существуют в виде равновесной смеси из цвиттер-ионов, катионной и анионной форм:+
OH +
H2N-CH-COO R
анион
+
H3N-CH-COO -
H+
R
биполя рный ион
H
OH -
H3N-CH-COOH
R
катион
Положение такого равновесия существенно зависит от pH
среды: в сильнокислой среде (pH=1-2) преобладает катионная
форма, в сильнощелочной (pH=13-14) – анионная.
Если поместить раствор аминокислоты в электрическое поле, то в кислых растворах она будет мигрировать к катоду, а в
щелочных – к аноду. При некотором значении pH, характерном
для данной аминокислоты, она не будет перемещаться в электрическом поле. При этом значении pH, называемом изоэлектрической точкой (ИЭТ, pI), аминокислота существует в виде биполярного иона и в целом электронейтральна. Итак, изоэлектрическая точка аминокислоты – это то значение pH раствора, при
котором большинство молекул данной аминокислоты существуют в виде биполярных ионов, а концентрации анионной
и катионной форм минимальны и равны. Изоэлектрическая
точка зависит от соотношения кислотных и основных групп в
молекуле: pI кислых аминокислот имеет значение <7, pI основных аминокислот >7.
При пропускании электрического тока через раствор, содержащий смесь аминокислот, каждая из них будет двигаться к
катоду или аноду со скоростью, зависящей от природы аминокислоты и pH среды. Это явление используют для разделения и
анализа смеси аминокислот методом электрофореза.
Химические свойства
Являясь амфотерными соединениями, аминокислоты реагируют и с кислотами, и с щелочами: HCl R-CH-COOH
R-CH-COOH
NH2
+
NH3 Cl гидрохлорид α−аминокислоты
NaOH
R-CH-COONa + H 2 O
NH2
натриевая соль α−аминокислоты
190
191.
Характерной особенностью α-аминокислот является способность образовывать комплексные соли с ионами тяжелых металлов:O
O
C
H2 N
OH
R-CH
CH-R
+ Cu 2+ + HO
NH2
C
O
C
R-CH
O
H2 N
Cu
NH2
O
CH-R
C
O
Медные соли аминокислот нерастворимы в воде, имеют интенсивное синее окрашивание. Эта реакция используется для обнаружения α-аминокислот.
Другой общей качественной реакцией α-аминокислот является их взаимодействие с нингидрином с образованием продукта
сине-фиолетового цвета:
O
O
OH
OH
N
+ H2 N-CH-COOH
O
нингидрин
R
O
OH
O
продукт сине-фиолетового цвета
O
+ R-C
+ CO 2
H
Как карбоновые кислоты , α-аминокислоты образуют
сложные эфиры при взаимодействии со спиртами и хлорангидриды при взаимодействии с тионилхлоридом или хлоридами фосфора:
CH3 OH
HCl газ
R-CH-COOH
NH2
SOCl2
R-CH-COOCH 3 + H2 O
NH2
метиловый эфир
R-CH-COCl + SO 2 + HCl
NH2
хлорангидрид
Сложные эфиры α-аминокислот летучи, они имеют сравнительно низкие температуры кипения. Это их свойство используется для разделения смеси аминокислот в белковых гидролизатах
(эфирный метод Фишера). С этой целью аминокислоты сначала
этерифицируют, а потом подвергают перегонке.
191
192.
За счет аминогруппы α-аминокислоты подвергаются реакциям ацилирования и алкилирования, взаимодействуют с оксосоединениями. Так, при ацилировании аминокислоты уксуснымангидридом образуется N-ацетильное производное. При взаимодействии с формальдегидом образуется продукт нуклеофильного
присоединения – N-метилольное производное, которое достаточно устойчиво (с другими оксосоединениями протекает реакция
нуклеофильного присоединения-отщепления).
(CH3 CO) 2 O
R-CH-COOH
NH2
R-CH-COOH
+ CH 3 COOH
NH-CO-CH 3
N-ацетилпроизводное
O
H-C-H
R-CH-COOH
NH-CH2 OH
N-метилольное производное
Реакцию ацилирования раньше использовали для защиты
аминогруппы в синтезе пептидов. Реакция с формальдегидом лежит в основе метода количественного анализа аминокислот (метод формольного титрования, метод Зеренсена). Сущность метода формольного титрования заключается в следующем: до взаимодействия с формальдегидом растворы большинства аминокислот имеют реакцию, близкую к нейтральной; N-метилольное
производное проявляет кислотные свойства и может быть оттитровано раствором щелочи с известной концентрацией.
Как первичные алифатические амины α-аминокислоты подвергаются действию азотистой кислоты с образованием соответствующих α-оксикислот и выделением азота:
R-CH-COOH
NH2
HNO2
R-CH-COOH + N 2 + H2 O
OH
α -оксикислота
Эту реакцию называют реакцией дезаминирования in vitro.
Ее используют и как качественную реакцию для доказательства
наличия первичной алифатической аминогруппы (наблюдают
выделение пузырьков газа), и для количественного анализа (ме192
193.
тод Ван-Слайка) – по объему выделившегося азота рассчитываютколичество аминокислоты, вступившей в реакцию.
Специфическим свойством α-аминокислот является их
способность к декарбоксилированию при нагревании в присутствии гидроксида бария:
to
Ba(OH)2
R-CH-COOH
NH2
R-CH2 -NH 2 + BaCO3 + H2 O
амин
Рассмотренные выше реакции характерны для всех αаминокислот. Существуют также реакции на определенные группы аминокислот.
Для обнаружения ароматических аминокислот используют так называемую ксантопротеиновую реакцию. При нагревании этих аминокислот (или белков, в структуре которых присутствуют их остатки) с концентрированной азотной кислотой
образуются продукты нитрования желтого цвета, которые в щелочной среде приобретают оранжевую окраску:
CH2 -CH-COOH
HO
NH2
HNO3 , to
HO
CH2 -CH-COOH
O2 N
NH2
продукт нитрования
(желтая окраска)
тирозин
NaOH
оранжевая
окраска
Серосодержащие аминокислоты (цистеин, цистин, метионин) обнаруживают по реакции с ацетатом свинца (реакция
Фоля).
CH2 -CH-COOH
SH NH2
цистеин
NaOH, to
продукты разложения (Na2 S)
(CH3 COO) 2 Pb
PbS
сульфид
свинца
(серо-черный
осадок)
При нагревании с щелочью серосодержащие аминокислоты
разлагаются, одним из продуктов разложения является сульфид
натрия. При дальнейшем добавлении ацетата свинца образуется
осадок сульфида свинца серо-черного цвета.
193
194.
Реакции α-аминокислот in vivoАминокислоты не накапливаются в клетке: как правило, их
избыток разрушается при помощи реакций, которые снабжают
живую систему энергией. Аминокислоты принимают участие в
метаболических процессах, в результате которых в клетке синтезируются необходимые ей соединения. Основные реакции, за
счет которых осуществляется превращение аминокислот в клетке, – это декарбоксилирование, дезаминирование, переаминирование. Эти реакции катализируются соответствующими ферментами.
В результате реакций декарбоксилирования образуются биогенные амины.
N
N
N
H
CH2 -CH-COOH
декарбоксилазы
N
H
NH2
гистидин
CH2 -CH-COOH
декарбоксилазы
CH2 -CH2 -NH2 + CO2
гистамин
CH2 -CH2 -OH + CO 2
NH2
OH NH2
коламин
серин
Реакции дезаминирования могут осуществляться как по
окислительному, так и по неокислительному пути.
Неокислительное дезаминирование встречается в основном
у растений и грибов. Так, например, с участием фермента аспартазы протекает дезаминирование аспарагиновой кислоты с образованием транс-бутендиовой (фумаровой) кислоты:
HOOC-CH 2 -CH-COOH
NH2
аспарагиновая кислота
аспартаза
HOOC-CH=CH-COOH + NH 3
фумаровая кислота
Окислительное дезаминирование осуществляется через образование промежуточной иминокислоты, которая затем подвергается гидролизу:
CH3 -CH-COOH
NH2
аланин
194
[O]
фермент
CH3 -C-COOH
H2 O
фермент
NH
α- иминопропионовая
кислота
CH3 -C-COOH + NH 3
O
пировиноградная
кислота
195.
Переаминирование α-аминокислот сводится к взаимопревращению их аминогрупп и карбонильных групп α-кетокислотпод действием ферментов трансаминаз (аминотрансфераз). Это
основной путь биосинтеза α-аминокислот из α-кетокислот. Донором аминогруппы служит аминокислота, имеющаяся в клетке в
достаточном количестве, а ее акцептором – кетокислота. Например, таким образом, из аспарагиновой кислоты в клетке может
быть синтезирован аланин или глютаминовая кислота:
HOOC-CH2-CH-COOH +
NH2
аспарагиновая кислота
CH3-C-COOH
фермент
O
пировиноградная
кислота
HOOC-CH2-C-COOH +
CH3-CH-COOH
O
щавелевоуксусная
кислота
NH2
аланин
HOOC-CH2-CH-COOH +
HOOC-CH2-CH2-C-COOH
NH2
аспарагиновая кислота
HOOC-CH2-C-COOH +
O
щавелевоуксусная
кислота
фермент
O
α-кетоглютаровая
кислота
HOOC-CH2-CH2-CH-COOH
NH2
глютаминовая
кислота
Реакция переаминирования является связующим звеном
между процессами метаболизма белков (аминокислоты) и углеводов (кетокислоты).
ПЕПТИДЫ И БЕЛКИ
Пептиды и белки – соединения, построенные из остатков αаминокислот. Деление между ними условное: принято считать,
что пептиды построены из остатков менее 100 аминокислот, белки – более 100 аминокислот.
Можно рассматривать полипептидную цепь как продукт поликонденсации α-аминокислот, а пептидную связь – как амид195
196.
ную, т.е. пептиды и белки являются полиамидами . Схематичнообразование трипептида можно показать следующим образом:
H2 N-CH-C
R
O
O
O
+
H2 N-CH-C
OH
R'
+
OH
N-конец
O
H2 N-CH-C
R"
O
OH
- 2 H2 O
C-конец
H2 N-CH-C-NH-CH-C-NH-CH-COOH
R'
R
R"
пептидные связи
трипептид
Пептидная цепь имеет неразветвленное строение и состоит
из чередующихся пептидных (амидных) групп и CH-групп с соответствующими радикалами. Принято записывать структуру
пептида, начиная со свободной аминогруппы (N-конца молекулы)
и заканчивая свободной карбоксильной группой (C-конец).
Названия пептидов строятся путем последовательного перечисления аминокислотных остатков, начиная с N-конца. Названия аминокислот, вступивших в образование пептидной связи
своей карбоксильной группой, заканчиваются суффиксом «-ил»,
например:
O
O
H2N-CH-C-NH-CH2-C-NH-CH-COOH
CH3
CH2
аланилглицилфенилаланин (Ала-Гли-Фен)
Амидная связь между двумя α-аминокислотными остатками
называется пептидной связью. Атомы углерода, азота и кислорода пептидной связи находятся в sp2-гибридизации, все 4 атома
CONH-группы расположены в одной плоскости под углом 120о.
Неподеленная пара электронов азота
O
H H
R
вступает в p,π-сопряжение с электрон..
N
C
C
ным облаком π-связи карбонильной
C ..
N
группы, вследствие чего связь C=O неH
O
сколько удлиняется (0,124 нм вместо
196
197.
0,121 нм), а связь C-N несколько укорачивается (0,132 нм вместо0,147 нм). Вследствие делокализации электронной плотности в
p,π-сопряженной системе связи C-O и C-N становятся
O
как бы «полуторными», поэтому вращение вокруг них
C
N
затруднено. Такое электронное строение обусловливает жесткую плоскостную структуру пептидной связи:
H
Качественной реакцией на пептидную связь является биуретовая реакция: при взаимодействии пептида с Cu(OH)2 наблюдается красновато-фиолетовое окрашивание. Обратите внимание,
что эта реакция не характерна для дипептидов.
При единообразно построенной цепи пептиды и белки отличаются аминокислотным составом и аминокислотной последовательностью. Аминокислотный состав показывает, какие аминокислоты и в каких соотношениях входят в структуру пептидов и
белков. Для определения аминокислотного состава пептид или
белок подвергают гидролизу и гидролизат анализируют (чаще
всего хроматографическими методами).
Пептидные связи гидролизуются и в кислой, и в щелочной
среде. При кислотном гидролизе пептидов образуются соли аминокислот по аминогруппе, при щелочном гидролизе – соли по
карбоксильной группе:
H2 O,HCl
H2 N-CH 2 -CO-NH-CH-COOH
глицилаланин
CH3
H2 O,NaOH
+
+
H3 N-CH 2 -COOH + H3 N-CH-COOH
ClClCH3
гидрохлорид
гидрохлорид
глицина
аланина
H2 N-CH 2 -COONa + H2 N-CH-COONa
натриевая соль
глицина
CH3
натриевая соль
аланина
197
198.
Практически для гидролиза пептидов и белков используюткислотный катализ, т.к. многие аминокислоты в щелочной среде
неустойчивы. Полный кислотный гидролиз любого белка протекает при его нагревании с 20% раствором соляной кислоты при
110оС в течение 24 час. Некоторые аминокислоты (например,
триптофан) разрушаются и в кислой среде.
Более удобным является ферментативный гидролиз пептидов и белков. Ферментативный гидролиз протекает под действием ферментов – пептидаз. Определенные пептидазы расщепляют
пептидные связи между остатками определенных аминокислот.
Например, трипсин гидролизует связи, образованные лизином
или аргинином с другими аминокислотами, пепсин – связи между
аминокислотами с гидрофобными радикалами (аланин, валин,
лейцин, изолейцин), химотрипсин – связи, образованные ароматическими аминокислотами. Чтобы осуществить полный ферментативный гидролиз, нужно использовать набор различных ферментов.
При ферментативном гидролизе образуются свободные
аминокислоты.
Первичная структура белков – это определенная аминокислотная последовательность, т.е. порядок чередования аминокислотных остатков в полипептидной цепи. Определение аминокислотной последовательности заключается в последовательном отщеплении аминокислотных остатков с N- или C-конца и их
идентификации. Существуют различные методы определения
аминокислотной последовательности, один из них – фенилтиогидантоиновый, или метод Эдмана.
Этот метод заключается во взаимодействии N-концевой
аминокислоты с фенилизотиоцианатом в щелочной среде:
O
..
δ+
C6 H5 -N=C=S + H2 N-CH-C-NH-CH-CONHR
R'
O
..
δ+
C6 H5 -NH-C-NH-CH-C-NH-CH-CONHS
R
R'
меченый полипептид
Образуется так называемый меченый полипептид, в структуре которого появился остаток фенилизотиоцианата (это реак198
199.
ция AN). При обработке меченого пептида слабой кислотой прикомнатной температуре происходит внутримолекулярная циклизация в фенилтиогидантоиновое производное (ФТГ-производное)
с одновременным гидролизом пептидной связи:
S
C6 H5 - N
C
C CH
O
+ H2 N-CH-CO-
NH
R
R'
ФТГ-производное
ФТГ-производное идентифицируется методом тонкослойной хроматографии. Преимущество метода Эдмана в том, что остальная часть пептидной молекулы не нарушается, и можно повторять операции по отщеплению следующей N-концевой аминокислоты. Сейчас этот метод воспроизводится в автоматическом
приборе, который осуществляет до 50 стадий отщепления и автоматически анализирует ФТГ-производные.
Для белков, наряду с первичной структурой, характерны более высокие уровни организации – вторичная, третичная и четвертичная структуры.
Под вторичной структурой белка понимают пространственное расположение атомов полипептидной цепи. Существует
два типа вторичной структуры – α-спираль и β-складчатая
структура.
На основании теоретических расчетов в 1950 г. Полинг и
Кори доказали, что для полипептидной цепи наиболее выгодной
конформацией является α-спираль – структура, сходная с правозакрученной винтовой лестницей (рис. 5). Поскольку α-спираль
построена из повторяющихся единиц –NH-CHR-CO-, то размеры
ее относительно постоянны. На один виток спирали приходится
3,6 аминокислотных остатка, шаг спирали 0,54 нм, диаметр – 0,5
нм. Плоскости двух соседних пептидных групп находятся под углом ~108о, боковые радикалы находятся на наружной стороне
спирали (направлены как бы от цилиндра).
Фиксируют такую спиральную конформацию водородные
связи, которые возникают между атомом кислорода карбониль199
200.
ной группы каждого первого аминокислотного остатка и атомомводорода NH-группы каждого пятого остатка:
O
H
H
O
O
H
-N-CH-C-N-CH-C-N-CH-C-N-CH-C-N-CH-C-N-CH-CHR
I
O
..
R'
H R" O
R'"
H R"" O
R'""
II
III
IV
V
VI
Водородные связи почти параллельны оси спирали, а так
как каждая C=O и NH-группы
α-спирали участвуют в образовании водородной связи, это
делает вторичную структуру
весьма устойчивой.
Рис. 5. Фрагмент белковой α-спирали.
Другим видом вторичной структуры является β-складчатая
структура. В складчатые листы укладываются вытянутые полипептидные цепи, связываемые множеством водородных связей
между пептидными группами этих цепей. Складчатый лист
включает обычно не более шести полипептидных цепей. Если
полипептидные цепи имеют одинаковую направленность от N- к
C-концу, то образуется параллельная β-структура, если противоположную – то антипараллельная:
200
201.
N-конецN-конец
R-HC
C =O:
H-N
CH-R
O= C
N-H
R-HC
N-конец
R-HC
C =O
R-HC
C H-R
H-N
N -H
O= C
CH-R
:O= C
R-HC
C-конец
C-конец
параллельная β-структура
H-N
R-HC
C =O
N-H
N-H
R-HC
C-конец
CH-R
C =O:
H- N
C-конец
:O= C
CH-R
N-конец
антипараллельная β-структура
Многие белки содержат одновременно и α-спиральные, и βскладчатые участки.
Полипептидная цепь, имеющая ту или иную вторичную
структуру, способна определенным образом
закручиваться в пространстве, что и определяет третичную структуру белка, то есть
общую форму полипептидной цепи (рис. 6).
Рис. 6. Третичная
структура белка.
Третичная структура, наряду с водородными связями, стабилизируется ионными связями (между остатками кислых и основных аминокислот), ковалентными связями (дисульфидные
мостики в цистине), гидрофобными взаимодействиями (силы
притяжения между неполярными радикалами).
H-N
O= C
CH-CH2 -COOH + H2 N-(CH 2 )4 -H C
Асп
Лиз
C =O
N-H
H-N
C =O
+
CH-CH2 -COO - H3 N-(CH 2 )4 -HC
O= C
N-H
ионная связь
201
202.
H-NO= C
CH-CH2 -SH + HS-CH2 -H C
Цис
Цис
C =O
N-H
[O]
H-N
O= C
CH-CH2 -S- S-CH 2 -H C
дисульфидный
мостик
C =O
N-H
+ H2 O
По пространственной структуре белки делятся на глобулярные и
фибриллярные. Для глобулярных белков более характерна α-спиральная
структура, а цепи их изогнуты в пространстве так, что макромолекула приобретает сферическую форму. Глобулярные белки растворяются в воде и
солевых растворах с образованием коллоидных растворов. Это альбумин
(яичный белок), глобин (белковая часть гемоглобина), большинство ферментов. Фибриллярные белки имеют β-складчатую структуру, волокнистое
строение. Они нерастворимы в воде. Это β-кератин (волосы, роговая
ткань), коллаген (соединительная ткань), миоинозин (мускульная ткань).
Несколько отдельных полипептидных цепей способны укладываться
в более сложные образования. При этом каждая цепь, сохраняя свою первичную, вторичную и третичную структуру, выступает в роли субъединицы комплекса с более высокой пространственной организацией – четвертичной структуры. Четвертичная структура закрепляется за счет водородных связей и гидрофобных взаимодействий между полипептидными цепями. Четвертичная структура характерна лишь для некоторых белков, например гемоглобина.
Пространственная структура белков способна нарушаться под влиянием некоторых факторов – высокая или низкая температура, изменение
pH среды, УФ- или рентгеновское облучение, механические воздействия.
Нарушение природной пространственной структуры белка называется денатурацией. Денатурация может быть обратимой и необратимой. При денатурации белков снижается их растворимость и изменяется биологическая активность.
Сложные белки. По составу белки подразделяются на простые и
сложные (конъюгированные). При гидролизе простых белков образуются
только α-аминокислоты. Сложные белки, наряду с белковой частью, содержат так называемую простетическую группу непептидной структуры.
По характеру простетической группы сложные белки делят на фосфопротеины (содержат остатки фосфорной кислоты), металлопротеины (ионы
202
203.
металлов), гликопротеины (углеводы), липопротеины (липидная часть),нуклеопротеины (нуклеиновые кислоты).
НУКЛЕИНОВЫЕ КИСЛОТЫ
Нуклеиновые кислоты играют основную роль в передаче генетической информации, отвечают за процессы биосинтеза белка.
Нуклеиновые кислоты – биополимеры, молекулярная масса
которых может достигать 1 млн. и более. Их полимерные цепи
построены из мономерных звеньев – нуклеотидов, т.е. нуклеиновые кислоты являются полинуклеотидами. Особенность нуклеотидов в том, что они в свою очередь состоят из остатка гетероциклического основания (нуклеинового основания), моносахарида и фосфорной кислоты.
Углеводным компонентом нуклеотидов служат пентозы –
рибоза и дезоксирибоза. В зависимости от этого нуклеиновые кислоты подразделяют на рибонуклеиновые (РНК) и дезоксирибонуклеиновые (ДНК).
В общем виде структуру нуклеиновых кислот можно представить так:
гетероциклическое
основание
гетероциклическое
основание
сахар
O-P-O
O OH
сахар
O-P-O
O
OH
нуклеотид
Нуклеиновые основания
Нуклеиновые основания – гетероциклические соединения,
являющиеся производными пурина или пиримидина. Все нуклеиновые основания являются амино- или гидроксипроизводными.
Пиримидиновыми основаниями являются урацил, тимин и
цитозин; пуриновыми – аденин и гуанин.
203
204.
ONH2
O
H3 C
NH
O
N
H
Урацил (У)
N
NH
O
N
H
Цитозин (Ц)
O
N
H
Тимин (Т)
O
NH2
N
N
N
NH
NH2
N
N
H
Гуанин (Г)
N
N
H
Аденин (А)
Для нуклеиновых оснований (кроме аденина) характерна
лактам - лактимная таутомерия :
O
O
OH
NH
5
4
6
1
N
N3
2
O
N
OH
N
H
лактамная форма
лактимная форма
Урацил (2,4-дигидроксипиримидин)
OH
NH
N
H
N
5
N7
NH2
8
9
N
H
4
6
N1
3 2
N
NH2
лактамная форма
лактимная форма
Гуанин (2-амино-6-гидроксипурин)
Лактамная форма более устойчива. В структуру нуклеиновых кислот остатки нуклеиновых оснований входят в лактамной
форме.
Нуклеиновые кислоты отличаются входящими в них нуклеиновыми основаниями: урацил присутствует только в структуре РНК, тимин – только в ДНК. Остальные нуклеиновые основания участвуют в построении молекул и РНК, и ДНК.
Кроме перечисленных нуклеиновых оснований, в нуклеиновых кислотах в небольших количествах присутствуют так называемые минорные основания, например:
O
NH
N
H
O
дигидроурацил
204
O
NH2
H3 C
N
O
N
H
5-метилцитозин
N
NH
N
H
N
гипоксантин
205.
В медицинской практике в качестве противоопухолевыхсредств используются производные пурина и пиримидина, «похожие» на нуклеиновые основания, но отличающиеся от них какой-то группой. Эта схожесть структур позволяет им встраиваться в молекулы нуклеиновых кислот и выполнять антагонистическую роль, нарушая информацию о биосинтезе белка опухолевой
клетки. Так, 5-фторурацил является антагонистом урацила, 6меркаптопурин – антагонистом аденина:
SH
O
F
4
3NH
5
6 1 2
8
O
N
H
5
N7
5-фторурацил
9
6 N1
3
N 4 N
H
2
6-меркаптопурин
Нуклеозиды
Нуклеозиды – соединения, молекулы которых состоят из остатка нуклеинового основания, связанного N-гликозидной связью
с остатком моносахарида. Таким образом, нуклеозиды – это Nгликозиды рибозы или дезоксирибозы и нуклеинового основания.
Гликозидная связь в нуклеозидах осуществляется за счет полуацетального гидроксила моносахарида и атома водорода при N-1
у пиримидиновых оснований и N-9 у пуриновых. Рибоза и дезоксирибоза входят в структуру нуклеозидов в β-фуранозной форме.
Атомы углерода фуранозного цикла нумеруют цифрой со штрихом.
O
4
3NH
6 1 2
5
урацил
N
H
O
O
N
HO-CH2
OH
OH OH
β-D-рибофураноза
4
3NH
6 1 2
5
5'
+
HO-CH2
O
O
4'
1'
3'
2'
OH
OH
O
+ H2 O
N-гликозидная
связь
Уридин
205
206.
Названия пиримидиновых нуклеозидов строятся путем замещения суффикса в тривиальных названиях нуклеиновых оснований на –идин (например, уридин, цитидин); названия пуриновых нуклеозидов – с использованием суффикса –озин (аденозин,гуанозин). Нуклеозиды ДНК называют аналогично, добавляя
приставку дезокси- (дезоксицитидин, дезоксиаденозин).
NH2
N
N
NH2
аденин
N
H
7
N
5'
HO-CH2
+
HO-CH2
O
5 6
1N
2
8 9
3
4
N
N
N
OH
O
4'
3'
OH
β-D-дезоксирибофураноза
1'
2'
N-гликозидная
связь
OH
Дезоксиаденозин
Таблица 5
Названия нуклеозидов
Нуклеиновое
основание
Моносахарид
Название нуклеозида
Урацил
Тимин
Цитозин
Цитозин
Аденин
Аденин
Гуанин
Гуанин
рибоза
дезоксирибоза
рибоза
дезоксирибоза
рибоза
дезоксирибоза
рибоза
дезоксирибоза
уридин
тимидин
цитидин
дезоксицитидин
аденозин
дезоксиаденозин
гуанозин
дезоксиаденозин
Как для N-гликозидов, для нуклеозидов характерен кислотный гидролиз, который протекает сравнительно легко, особенно
для пуриновых нуклеозидов.
206
207.
Например, при кислотном гидролизе тимидина образуетсятимин и дезоксирибоза (тимидин – нуклеозид ДНК):
O
H3 C
NH
N
HO-CH2
O
OH
O
O
H3 C
HO-CH2
OH
O
NH
H2 O, H+
+
O
N
H
тимин
OH
дезоксирибоза
Тимидин
Некоторые нуклеозиды не входят в структуру нуклеиновых
кислот, а присутствуют в растительных и животных клетках в
свободном виде. Они обладают антибиотической и противоопухолевой активностью. Некоторые из них применяются в медицине.
Нуклезиды-антибиотики отличаются от обычных нуклеозидов природой пентозы или строением гетероциклического основания. Например, кордицепин отличается от аденозина только
отсутствием гидроксильной группы в положении 3’ моносахаридного остатка.
Такое подобие кордицепина аденозину,
NH2
по-видимому, делает его антагонистом
N
N
аденозина, что и приводит к антибиотиN
N
HO-CH 2
ческой активности.
O
3'
OH
Кордицепин
Метод аналогии был использован в синтезе некоторых новых антивирусных препаратов, например,
арабинозида аденина (ингибирует
синтез ДНК вируса):
NH2
N
N
N
HO-CH 2
N
O
HO
OH
остаток
арабинозы
207
208.
Одним из первых препаратов, применяемых для леченияВИЧ-инфекцированных больных, был азидотимидин (антагонист
тимидина).
Азидотимидин снижает скорость размноO
жения вируса СПИД.
H3 C
NH
O
N
HO-CH2
O
N3
Азидотимидин
Нуклеотиды
Нуклеотиды – это сложные эфиры нуклеозидов и фосфорной кислоты (фосфаты нуклеозидов). В зависимости от природы
нуклеозидов выделяют рибонуклеотиды и дезоксирибонуклеотиды.
Фосфатная группа связана с пентозным циклом обычно в
положениях 3’ или 5’.
O
сложноэфирная
связь
4 NH
3
5
6 1 2
5'
N
HO-CH 2
H3 PO 4
O
4'
HO
O
1'
3'
2'
OH
OH
уридин
HO
5'
O
4 NH
3
5
6 1 2
N
P-O-CH 2
O
O
4'
O
N-гликозидная
связь
1'
3'
2'
OH
OH
уридин-5'-фосфат,
или 5'-уридиловая кислота
Нуклеотиды называют как сложные эфиры фосфорной кислоты – фосфаты, например, уридин-5’-фосфат, дезоксигуанозин3’-фосфат. С другой стороны, нуклеотиды можно рассматривать
как двухосновные кислоты. Тогда их называют 5’-уридиловая кислота, 3’-дезоксигуаниловая кислота и т.д.
208
209.
O7
5 6
N
8 9
3
N 4 N
5'
HO-CH2
O
4'
1NH
2
NH2
1'
2'
3'
O
OH
O= P
дезоксигуанозин-3'-фосфат,
или 3'-дезоксигуаниловая кислота
OH
Фосфорная кислота может этерифицировать одновременно
две гидроксильные группы углеводного остатка, образуя циклофосфаты. Практически во всех клетках присутствуют аденозин3’,5’-циклофосфат (3’,5’-циклоадениловая кислота) и гуанозин3’,5’-циклофосфат (3’,5’-циклогуаниловая кислота):
NH2
7
5
N
8 9
5'
HO
P
O
6 N
1
2
3
7
O
1'
3'
2'
O
OH
аденозин-3',5'-циклофосфат
5
N
N 4 N
O-CH 2
4'
O
8 9
5'
N 4 N
O-CH 2
O
4'
HO
P
O
6 NH
1
2
3
NH2
1'
3'
2'
O
OH
гуанозин-3',5'-циклофосфат
Циклические нуклеотиды играют в клетке важную роль: они
являются посредниками между некоторыми гормонами (например, адреналин) и клеточными процессами.
Так как в молекулах нуклеотидов присутствуют и
N-гликозидные, и сложноэфирные связи, в зависимости от условий гидролиза образуются различные продукты. N-гликозидная
связь гидролизуется только в кислой среде, а сложноэфирная – и
в кислой, и в щелочной:
209
210.
OH3 C
O
H3 C
HO
HO
H2 O, H+
NH
N
P-O-CH 2
OH
+ H3 PO 4
O
OH
дезоксирибоза
O
O
O
O
+
N
H
тимин
O
HOCH2
NH
NaOH, H2 O
OH
5'-тимидиловая кислота
H3 C
NH
N
HOCH2
O
O
+ H3 PO 4
OH
тимидин
Нуклеотиды имеют большое значение не только как мономеры для строительства нуклеиновых кислот. Они участвуют в
биохимических процессах и особенно важны в роли коферментов, т.е. веществ, необходимых для проявления активности ферментов.
Во всех тканях организма содержатся моно-, ди- и трифосфаты нуклеотидов. Особенно широко известны аденозинмонофосфат (АМФ), аденозиндифосфат (АДФ) и аденозинтрифосфат
(АТФ). Нуклеотиды, фосфорилированные в разной степени, способны к взаимным преврашениям путем отщепления остатка
фосфорной кислоты или наращиванием его:
NH2
N
OH
N
N
O
N
H3 PO4
H2 O
OH OH
АМФ (аденозинмонофосфат)
210
N
OH OH
HO-P-O-CH 2
O
NH2
N
HO-P-O- P-O-CH 2
O
O
N
N
O
OH OH
АДФ (аденозиндифосфат)
H3 PO4
H2 O
211.
NH2сложноэфирная
связь
N
OH OH OH
N
HO-P- O-P-O- P-O-CH 2
O
O
N
N
N-гликозидная
связь
O
O
ангидридные
связи
OH
OH
АТФ (аденозинтрифосфат)
Ангидридные связи между остатками фосфорной кислоты
обладают большим запасом энергии (~32 кДж/моль), поэтому их
называют макроэргическими. При гидролизе АТФ до АДФ и
АМФ эта энергия выделяется. Важнейшая роль АТФ – поставщик
энергии во всех живых клетках.
С участием АТФ и АДФ в организме осуществляется важнейший биохимический процесс – перенос фосфатных групп.
Образование фосфатов – типичная реакция в метаболизме углеводов:
HO
O=P-O-CH 2
HO
OH
HO
CH2 OH
O OH
+ АТФ
OH
HO
OH
глюкоза
O OH
+ АДФ
OH
глюкозо-6-фосфат
АТФ активирует α-аминокислоты в биосинтезе белков:
NH2
N
OH OH OH
N
CH3 -CH-COOH + HO-P- O-P-O- P-O-CH 2
NH2
аланин
O
O O
АТФ
N
N
O
OH
OH
211
212.
NH2ангидридная
связь
N
OH
N
N
CH3 -CH-C-O- P-O-CH 2
NH2 O
+ HO-P-O-P-OH
O
O
OH
OH OH
N
O O
пирофосфорная кислота
OH
аминоациладенилат
При взаимодействии α-аминокислоты с молекулой АТФ образуется смешанный ангидрид аминокислоты и АМФ – аминоациладенилат. Ангидриды кислот являются более активными ацилирующими средствами, чем сами кислоты. Активированная
аминокислота затем взаимодействует с аминогруппой следующей
аминокислоты в синтезе пептидов.
Нуклеотидную природу имеет никотинамидадениндинуклеотид (НАД+) – кофермент дегидрогеназ, который является участником окислительно-восстановительных реакций (см. стр. 148). С
участием НАД+ протекают реакции окисления спиртов в альдегиды (ретинол – ретиналь и др.).
Флавинадениндинуклеотид (ФАД) является метаболически активной
формой рибофлавина (витамина B2).
нуклеотидный фрагмент
NH2
N
OH OH
N
CH2- O-P-O- P-O-CH2
фрагмент
рибитола
HO
HO
HO
H
H
H
H
H
H3C
N
H3C
N
O
O
O
OH
O
N
NH
O
фрагмент изоаллоксазина
212
N
OH
N
213.
В структуре рибофлавина присутствует остаток D-рибитола(многоатомный спирт – продукт восстановления рибозы) и гетероциклическая система изоаллоксазина (флавина). В структуре
же ФАД присутствует ещё и остаток адениндинуклеотида.
Флавинадениндинуклеотид является коферментом окислительно-восстановительных процессов с участием ферментов оксидаз и дегидрогеназ. С участием ФАД происходит, например,
окислительное дезаминирование α-аминокислот в организме.
Ответственной за окислительно-восстановительный процесс
является остаток изоаллоксазина, способный присоединять два
атома водорода с образованием восстановленной формы ФАДH2:
R
R
H3C
N
H3C
N
O
N
NH
O
ФАД (окисленная форма)
+2H
H3C
N
-2H
H3C
N
H
H
N
O
NH
O
ФАДН (восстановленная форма)
Нуклеиновые кислоты
ДНК содержатся в основном в ядрах клеток, РНК – в рибосомах и протоплазме. Основная роль РНК – участие в биосинтезе
белка.
Под первичной структурой нуклеиновых кислот понимают
нуклеотидную последовательность в полинуклеотидной цепи.
Нуклеотидные звенья связываются через фосфатную группу. Например, структура фрагмента РНК с нуклеотидной последовательностью У-А-Ц:
213
214.
O4 NH
5
3
6 1 2
У
O
N
-O-CH 2
O
NH2
3'
O
N
OH
5'
N
O
O
NH2
3'
O
A
N
N
HO-P-O-CH 2
N
OH
5'
N
HO-P-O-CH 2
O
O
O
O
Ц
OH
HO-P-OO
Вторичная структура нуклеиновых кислот – пространственная организация полинуклеотидных цепей, т.е. определенное
расположение полинуклеотидной цепи в пространстве.
В 1953 г. Уотсон и Крик описали вторичную структуру ДНК
в виде двойной спирали (рис. 7). Согласно этой модели молекула
ДНК состоит из двух полинуклеотидных цепей, правозакрученных вокруг общей оси. Диаметр спирали 1,8-2,0 нм. Две полинуклеотидные цепи антипараллельны, т.е. направления образования фосфодиэфирных связей у них противоположны – в одной
3’→5’, в другой 5’→3’. Остатки пуриновых и пиримидиновых
оснований направлены внутрь спирали. Вторичная структура
стабилизируется водородными связями между нуклеиновыми основаниями двух цепей. Каждый виток спирали содержит 10 пар
оснований.
214
215.
Рис. 7. Вторичная структура ДНК.Водородные связи возникают между пуриновым основанием одной цепи и пиримидиновым – другой. Такие пары оснований называются комплементарными . В молекуле ДНК аденину комплементарен тимин, гуанину – цитозин:
N
N:
N
H
N
A
CH3
HN
O
H2 N
O:
:O
NH2
N
N
H
N
H
T
:N
NH
N
Г
NH2
:O
N
H
Ц
Комплементарность цепей составляет химическую основу
важнейшей функции ДНК – хранения и передачи наследственной
информации. Сохранность нуклеотидной последовательности является залогом безошибочной передачи генетической информации. Однако нуклеотидная последовательность ДНК под действием некоторых факторов может изменяться – происходят мутации.
215
216.
Наиболее распространенный вид мутаций – замена однойпары нуклеиновых оснований на другую (А-Т на Г-Ц). Мутации
могут вызывать действие излучения, химические факторы. Например, мутагенным действием обладают нитриты – соли азотистой кислоты:
O
OH
NH2
N
N
N
N
HNO2
N
N
N
аденозин
N
N
NH
N
N
инозин (нуклеозид
гипоксантина)
Под действием азотистой кислоты происходит дезаминирование остатка аденина с образованием гипоксантина, т.е. остаток
аденозина превращается в остаток инозина. Гипоксантину комплементарен не тимин, а цитозин. Таким образом, под действием
химического фактора произошло нарушение первичной структуры ДНК, а значит и генетической информации, заложенной в
этом фрагменте ДНК.
Генетическая информация, заложенная в ДНК, передается
от родительских к дочерним клеткам путем синтеза новой спирали ДНК. При делении клеток двойная спираль ДНК раскручивается с образованием двух односпиральных цепей, являющихся
матрицами для синтеза двух новых спиралей, комплементарных
матричным (рис. 8).
Рис. 8. Схема удвоения цепи ДНК.
216
217.
Информация, заложенная в ДНК, переписывается на матричнуюРНК (м-РНК) – происходит транскрипция. м-РНК становится
матрицей для биосинтеза белка в цитоплазме. Каждая аминокислота кодируется трехнуклеотидной последовательностью (триплетом).
Аминокислоты для биосинтеза белка доставляются с помощью транспортной РНК.
ОМЫЛЯЕМЫЕ ЛИПИДЫ
К липидам относятся разнородные в химическом отношении
природные соединения. Их объединяют в один класс по признаку
растворимости: липиды нерастворимы в воде и растворимы в неполярных органических растворителях (бензол, диэтиловый
эфир).
В зависимости от способности к гидролизу липиды делят на
омыляемые (подвергаются гидролизу) и неомыляемые (не подвергаются гидролизу).
К омыляемым липидам относятся жиры, фосфолипиды и
воски.
Основу омыляемых липидов составляют высшие карбоновые кислоты и спирты (глицерин в случае жиров и фосфолипидов
и высшие спирты – в случае восков).
Жиры
В организме жиры играют роль структурного компонента
клетки и роль запасного питательного вещества.
Жиры являются сложными эфирами глицерина и
O
высших карбоновых кислот, т.е. триацилглицеCH2 -O-C-R
ринами .
O
CH -O-C-R' Природные жиры являются смесью различных
O
триацилглицеринов.
CH2 -O-C-R''
В структуре жиров присутствуют остатки
насыщенных высших карбоновых кислот (стеариновой и пальмитиновой) и ненасыщенных кислот (олеиновой,
линолевой, линоленовой, арахидоновой).
ацил
217
218.
Углеводородные радикалы высших карбоновых кислот нелинейны. Они существуют в зигзагообразной конформации:16
O
C15 H31 -C
CH3
1
HOOC
OH
пальмитиновая
кислота
18
O
CH3
1
HOOC
C17 H35 -C
OH
стеариновая
кислота
Высшие ненасыщенные кислоты способствуют снижению
содержания в крови холестерина – фактора развития атеросклероза. Смесь сложных эфиров ненасыщенных жирных кислот под
названием «Линетол» применяется в медицине.
C17 H33 -COOH
10
9
13
12
CH3 -(CH2 )7 -CH=CH-(CH2 )7 -COOH
олеиновая кислота
C17 H31 -COOH
10
9
CH3 -(CH2 )4 -CH=CH-CH 2 -CH=CH-(CH2 )7 -COOH
ленолевая кислота
16
C17 H29 -COOH
13
15
12
10
9
CH3 -CH 2 -CH=CH-CH 2 -CH=CH-CH 2 -CH=CH-(CH2 )7 -COOH
леноленовая кислота
Для ненасыщенных карбоновых кислот характерна цистранс-изомерия. В структуру жиров и фосфолипидов они входят
в цис-конфигурации.
1
HOOC
9
CH3
10
CH3
1
HOOC
9
10
12
9
10
12
13
13 15
1
HOOC
9
8
6
5
20
218
14
15
CH3
линолевая кислота
линоленовая кислота
COOH
CH3
11 12
16
олеиновая кислота
арахидоновая кислота
219.
Если в структуре жира преобладают остатки насыщенныхкарбоновых кислот, то жир твердый, если преобладают остатки
ненасыщенных кислот, то жир жидкий (масло). Как правило, животные жиры твердые, растительные – жидкие.
Жиры, включающие остатки одинаковых кислот, называют
простыми, остатки разных жирных кислот – смешанными.
Жиры называют по тривиальной номенклатуре, заменяя
суффикс «-овая» в названии кислоты на «-ин» (тристеарин, трипальмитин), и по систематической номенклатуре как сложные
эфиры глицерина, используя суффикс «-оил» (тристеароилглицерин, трипальмитоилглицерин). Например:
CH2 -O-CO-C 17 H35
CH2 -O-CO-C 15 H31
CH -O-CO-C 17 H35
CH -O-CO-C 17 H33
CH2 -O-CO-C 17 H35
CH2 -O-CO-C 17 H31
тристеарин
(тристеароилглицерин)
α-пальмито-β-олео-α'-линолеин
(1-пальмитоил-2-олеоил3-линолеоилглицерин)
Так как жиры являются сложными эфирами, то для них характерны реакции гидролиза как в кислой, так и в щелочной среде.
Кислотный гидролиз обратим, в результате образуются глицерин и соответствующие карбоновые кислоты:
CH2 -O-CO-C 17 H35
CH -O-CO-C 17 H35
CH2 -O-CO-C 17 H35
тристеарин
H2 O, H+
CH2 -OH
CH -OH + 3 C17 H35 -COOH
CH2 -OH
глицерин
стеариновая кислота
При щелочном гидролизе жиров образуются глицерин и соли высших карбоновых кислот:
CH2 -O-CO-C 17 H33
CH -O-CO-C 17 H33
CH2 -O-CO-C 17 H33
триолеин
H2 O, NaOH
CH2 -OH
CH -OH + 3 C17 H35 -COONa
олеат натрия
CH2 -OH
глицерин
Соли высших карбоновых кислот нзываются мылами. Вот
почему жиры относят к омыляемым липидам. Натриевые и кали219
220.
вые мыла растворимы в воде, кальциевые, магниевые – нет.Моющее действие мыл основано на дифильности их структуры.
Молекулы мыл включают полярную часть – карбоксилатную
группу с гидрофильными свойствами и неполярный углеводородный радикал – с гидрофобными (липофильными).
COO - Na+
H3 C
неполярный "хвост"
(липофильная часть)
полярная "голова"
(гидрофильная часть)
Рассмотрим механизм моющего действия мыл. Загрязнения
имеют жироподобную природу. По принципу
- - - «подобное растворяется в подобном» липофиль- ная часть молекулы мыла растворяется в капельке
- жира, а гидрофильная остается на поверхности.
- - Образуется так называемая мицелла. В целом она
гидрофильна (на ее поверхности гидрофильная пленка) и может
быть вымыта водой. Кроме того, на поверхности мицеллы сосредоточен отрицательный заряд, значит, отдельные капельки жира
отталкиваются друг от друга и не сливаются. Таким образом, мыло способствует образованию эмульсии жира в воде, обладает
эмульгирующим действием. Таким действием обладают и молекулы других соединений, имеющие дифильную структуру (фосфолипиды, желчные кислоты).
Мыла как моющие средства имеют ряд недостатков. Они не
эффективны в жесткой воде, т.к. кальциевые и магниевые соли
высших жирных кислот не растворимы в воде. Растворы натриевых и калиевых мыл имеют щелочную реакцию (они образованы
сильными основаниями и слабыми кислотами), поэтому сушат и
раздражают кожу.
В настоящее время используются также синтетические
моющие средства – детергенты. В их структурах также имеются
гидрофильная и липофильная части, т.е. они являются поверхностно-активными соединениями.
220
221.
- +CH3 (CH2 )n -O-SO 3 Na
анионный тип
липофильная гидрофильная
часть
часть
+
CH3 (CH2 )n -N(CH 3 )3 Br
липофильная гидрофильная
часть
часть
катионный тип
CH3 (CH2 )n -O-(CH 2 -CH2 -O) m -CH2 -CH2 -OH
липофильная гидрофильная часть
часть
неионный тип
Синтетические моющие средства имеют нейтральную реакцию, эффективны в жесткой воде, но плохо подвергаются биологическому распаду и загрязняют окружающую среду.
Для жиров, содержащих остатки непредельных карбоновых
кислот, характерны реакции гидрирования:
CH2 -O-CO-C 17 H33
CH -O-CO-C 17 H33
CH2 -O-CO-C 17 H35
H2 , Ni
p, to
CH -O-CO-C 17 H35
CH2 -O-CO-C 17 H33
CH2 -O-CO-C 17 H35
триолеин
тристеарин
В результате этой реакции можно получить из более дешевых жидких растительных масел твердые жиры. Это свойство используют в производстве маргарина.
Наличие остатков непредельных карбоновых кислот в
структуре жиров можно подтвердить реакцией с бромной водой:
CH2 -O-CO-(CH 2 )7 -CH=CH-(CH2 )7 -CH3
H2 O
CH -O-CO-C 17 H35
+ Br2
CH2 -O-CO-(CH 2 )7 -CH-CH-(CH 2 )7 -CH3
CH2 -O-CO-C 17 H35
CH2 -O-CO-C 17 H35
CH -O-CO-C 17 H35 Br Br
α-олео-α',β-дистеарин
Бромная вода обесцвечивается.
Ненасыщенные жиры способны к реакциям окисления в
различных условиях. Окисление раствором перманганата калия в
мягких условиях приводит к образованию диольного фрагмента
по месту разрыва двойной связи, качественным признаком этой
221
222.
реакции является исчезновение розовой окраски перманганатакалия и выпадение бурого осадка диоксида марганца:
CH2 -O-CO-(CH 2 )7 -CH=CH-(CH 2 )7 -CH 3
CH -O-CO-C 17 H35
H2 O
+ KMnO4
CH2 -O-CO-(CH 2 )7 -CH-CH-(CH 2 )7 -CH 3
CH -O-CO-C 17 H35 OH OH
CH2 -O-CO-C 17 H35
CH2 -O-CO-C 17 H35
α-олео-α',β-дистеарин
+ MnO2
+ KOH
При окислении в жестких условиях происходит расщепление молекулы по месту двойной связи:
CH2 -O-CO-(CH 2 )7 -CH=CH-(CH2 )7 -CH 3
CH -O-CO-C 17 H35
H+
to
+ KMnO4
CH2 -O-CO-C 17 H35
CH2 -O-CO-(CH 2 )7 -COOH
CH -O-CO-C 17 H35 + HOOC-(CH 2 )7 -CH3
CH2 -O-CO-C 17 H35
+ MnSO4 + K2 SO4
α-олео- α',β-дистеарин
Сходным образом (но медленно) протекает реакция окисления жиров и под действием кислорода воздуха (так называемое
автоокисление). В результате автоокисления образуются низшие
карбоновые кислоты, имеющие неприятный запах и вкус, – жир
«прогоркает».
Некоторые лекарственные препараты используются в медицине в виде масляных растворов (камфора, эстрадиола дипропионат, прогестерон и т.д.), поэтому важно уметь контролировать
качество этих масел. С этой целью используют специальную характеристику – иодное число жира. Иодное число – это количество граммов иода, способное присоединиться к 100 граммам жира.
CH2 -O-CO-(CH 2 )7 -CH=CH-(CH2 )7 -CH3
CH2 -O-CO-(CH 2 )7 -CH-CH-(CH 2 )7 -CH3
CH -O-CO-C 17 H35
CH -O-CO-C 17 H35 I
CH2 -O-CO-C 17 H35
+ I2
I
CH2 -O-CO-C 17 H35
α-олео-α',β-дистеарин
Чем более ненасыщенный жир, тем выше его иодное число.
В результате автоокисления иодное число жира снижается по
сравнению со стандартом, т.к. уменьшается количество двойных
связей в молекуле.
222
223.
Еще один вид окисления жиров (а также фосфолипидов) –пероксидное окисление, протекающее с участием свободных радикалов, которые постоянно образуются в организме:
α
R-CH2 -CH=CH-R'
HO
.
- H2 O
R-CH-CH=CH-R'
O2
R-CH-CH=CH-R'
.
R-C
O
+
H
O OH
гидропероксид
H2 O
R-CH-CH=CH-R'
- HO.
.
O-O
пероксид
CH=CH-R'
OH
O2
O
H
C-CH 2 -R'
O2
R-COOH
+
R'-CH 2 -COOH
Реакция пероксидного окисления начинается по αположению по отношению к двойной связи, т.к. радикалы аллильного типа устойчивы (см. стр. 32), далее образуются пероксид и гидропероксид, который расщепляется с образованием соответствующих альдегидов, которые в свою очередь окисляются
в карбоновые кислоты. В результате пероксидного окисления нарушается структура жиров и фосфолипидов, которые являются
составляющими клеточных мембран, т.е. нарушаются функции
мембран. С этим явлением связывают и процессы старения, и
вредное воздействие радиации на организм (один из механизмов
возникновения лучевой болезни). Кроме того предполагают, что
промежуточные продукты пероксидного окисления липидов обладают мутагенным действием.
Воски
Воски – сложные эфиры высших жирных кислот и одноили двухатомных высших спиртов. В восках содержатся также
свободные карбоновые кислоты, высшие спирты, углеводороды.
Воски делятся на животные (спермацет, пчелиный воск, ланолин)
223
224.
и растительные (карнаубский воск). Они образуют защитнуюсмазку на коже человека и животных и предотвращают растения
от высыхания. Воски используют при приготовлении косметических средств, мазей, водоотталкивающих пропиток для тканей.
Главным компонентом спермацета (содержится в спермацетовом масле, получаемом из головы кашалота) является цетиловый эфир пальмитиновой кислоты:
O
C15H31-C
O-CH2-(CH2)14-CH3
цетилпальмитат
Другой эфир пальмитиновой кислоты – мирицилпальмитат – содержится в пчелином воске:
O
C15H31-C
O-CH2-(CH2)29-CH3
мирицилпальмитат
Как сложные эфиры, воски способны к гидролизу. Их относят к омыляемым липидам.
Фосфолипиды
Фосфолипиды являются главным компонентом клеточных
мембран, они сопутствуют жирам в пище и служат источником
фосфорной кислоты, необходимой организму человека.
Фосфолипиды относят к сложным липидам. Они являются
сложными эфирами фосфатидовых кислот и аминоспиртов. В
свою очередь фосфатидовые кислоты –
CH2 OH
сложные эфиры глицерофосфата и высHO
H
ших карбоновых кислот.
O
Глицерофосфат содержит асимметричеCH2 O-P-OH
ский атом углерода, поэтому может суOH
ществовать в виде двух стереоизомеров.
L-глицерофосфат
Все природные фосфолипиды являются
производными L-глицерофосфата.
Фосфатидовые кислоты – ацильные производные
l-глицерофосфата. Как правило, в положении 1 находится остаток
224
225.
насыщенной карбоновой кислоты, в положении 2 – ненасыщенной:O
1
CH2 O-C-R
O
2
R'-C-O
H
O
CH2 O-P-OH
OH
L-фосфатидовые кислоты
Примерами фосфолипидов являются фосфатидилколамины
(коламинкефалины), фосфатидилхолины (лецитины), фосфатидилсерины (серинкефалины), в которых фосфатидовые кислоты
этерифицированы по фосфатному гидроксилу коламином, холином и серином соответственно.
CH2 -O-CO-R
CH2 -O-CO-R
CH -O-CO-R'
CH -O-CO-R'
CH -O-CO-R'
CH2 -O-P-OH + HO-CH2 -CH 2
CH2 -O-P-O -CH2 -CH 2
CH2 -O-P-O -CH 2 -CH 2
+ NH
O O3
CH2 -O-CO-R
O
OH
фосфатидовая
кислота
- H2 O
NH2
коламин
O
OH
NH2
фосфатидилколамин
(кефалин)
диполярный ион кефалина
В условиях организма фосфолипиды существуют в виде биполярных ионов.
CH2 -O-CO-R
- H2 O
CH -O-CO-R'
CH2 -O-P-OH
O
OH
фосфатидовая
кислота
+ HO-CH2 -CH 2
+N(CH )
3 3
холин
CH2 -O-P-OH
O
OH
фосфатидовая
кислота
- H2 O
+ HO-CH2 -CH-COOH
NH2
серин
CH -O-CO-R'
CH2 -O-P-O -CH 2 -CH 2
+N(CH )
O O-
3 3
фосфатидилхолин
(лецитин)
CH2 -O-CO-R
CH -O-CO-R'
CH2 -O-CO-R
CH2 -O-CO-R
CH -O-CO-R'
CH2 -O-P-O -CH 2 -CH-COOH
+ NH
O O3
фосфатидилсерин
(серинкефалин)
225
226.
Молекулы фосфолипидов дифильны: полярная часть (ионизированная) гидрофильна, неполярные остатки карбоновых кислот – липофильны.липофильная часть
O
O
C-O
CH2 O-C
H
O
CH2 O-P
-O
O
+
CH2
H3 N
CH2
схематичное
изображение
гидрофильная часть
На границе раздела фаз за счет дифильности структуры
фосфолипиды действут как эмульгаторы. Липидные компоненты,
входящие в структуру биомембран, обеспечивают их высокое
электрическое сопротивление, непроницаемость для полярных
молекул и ионов и проницаемость для неполярных веществ.
Фосфолипиды составляют до 90% от общего количества липидов
в мембране.
Существует несколько моделей клеточной мембраны. В одной из них мембрана рассматривается как липидный бислой
(двойной липидный слой).
Неполярные углеводородные хвосты липидов удерживаются друг возле друга в вытянутом состоянии во внутренней полости за счет гидрофобных взаимодействий. Полярные группы липидов располагаются на внешней поверхности бислоя.
При использовании данной модели биомембран нельзя объяснить, как сквозь нее могут транспортироваться полярные молекулы воды, углеводов, ионы Na+ и K+. Эти факты объясняет жид226
227.
ко-мозаичная модель. В структуру бислоя встроены молекулыбелков.
Молекулы белков свернуты таким образом, что их гидрофобные остатки погружены в липидный бислой, а гидрофильные – находятся внутри, поэтому молекула белка является «каналом» для транспорта полярных молекул и ионов.
К менее распространенным фосфолипидам относятся плазмалогены,
в молекулах которых присутствует простая эфирная связь с остатком непредельного спирта:
простая эфирная связь
CH2 -O-CH=CH-(CH 2 )n -CH3
O
R-C-O
H
O
CH2 O-P-O-CH 2 -CH2 -NH 2
OH
остаток этаноламина
Плазмалогены
Плазмалогены составляют до 10% от общего количества липидов
ЦНС.
Кроме фосфолипидов, к сложным липидам относят сфинголипиды и
гликолипиды.
Сфинголипиды являются производными не глицерина, а сфингозина – ненасыщенного длинноцепочечного двухатомного аминоспирта с
транс-конфигурацией двойной связи.
1
Примером сфинголипидов явCH2 -OH
CH2 -OH
ляются
церамиды
–
N2
H
NH-C-R
H
NH2
ацильные производные сфин3
O
H
H
OH
OH
гозина, аминогруппа которого
4
ацилирована жирной кислотой.
5
(CH2 )12
(CH2 )12
CH3
CH3
Сфингозин
Церамид
227
228.
Гликолипиды включают углеводные остатки, чаще всего D-галактозы. Представителями гликолипидов являются цереброзиды (входят в состав оболочек нервных клеток) и ганглиозиды (выделены из серого вещества мозга). В цереброзидах остаток церамида связан с D-галактозой илиD-глюкозой β-гликозидной связью:
остаток галактозы
HO
CH2 OH
O
OH
O-CH 2
OH
H
NH-C-R
H
OH O
остаток церамида
(CH2 )12
CH3
Галактоцереброзид
Ганглиозиды отличаются от цереброзидов тем, что вместо остатка
моносахарида они содержат сложный олигосахарид.
НЕОМЫЛЯЕМЫЕ ЛИПИДЫ
Неомыляемые липиды – группа негидролизующихся природных веществ, растворимых в неполярных органических растворителях (бензол, хлороформ) и не растворимых в воде. К ним
относятся терпеноиды и стероиды. Терпеноиды имеют в основном растительное происхождение, а стероиды – животное. И терпеноиды, и стероиды построены из фрагментов изопрена, поэтому их общее название – изопреноиды.
ТЕРПЕНОИДЫ
Терпеноиды – обширный класс природных кислородсодержащих соединений, производных терпенов. Терпены – это углеводороды общей формулы (C5H8)n, где n≥ 2. Углеводородный
228
229.
скелет всех терпеноидов построен из остатков изопрена(2-метилбутадиена-1,3).
CH2=C-CH=CH2
CH3
изопрен
Терпеноиды широко растпространены в природе. Они выделены из цветковых растений семейств Amarantaceae, Lamiaceae,
Apiaceae, Asteraceae и др., а также некоторых мхов и грибов.
Терпеноиды в больших количествах содержатся в эфирных маслах мяты перечной, эвкалипта, герани, розы, лимона, ромашки
аптечной, смоле хвойных деревьев.
К терпеноидам относятся растительные пигменты, смолы,
фитогормоны, сапонины, жирорастворимые витамины.
В большинстве терпеноидов изопреновые фрагменты соединены по принципу «голова к хвосту» (т.н. «изопреновое правило», впервые сформулированное О. Валлахом и подтвержденное Л. Ружичкой). Например:
"голова"
"голова"
"хвост"
"хвост"
изопрен
"голова"
.
.
лимонен
"хвост"
.
.
CH2 OH
гераниол
(В химии терпеноидов принято пользоваться краткими формулами, без обозначения символов углерода). Наряду с таким построением, но гораздо реже, наблюдается порядок соединения
«голова к голове». Известны также природные вещества терпенового типа, структура которых не отвечает изопреновому правилу,
но эти исключения немногочисленны.
Терпеновые углеводороды общей формулы (C5H8)n классифицируют по количеству изопреновых звеньев в молекуле на монотерпены (n=2), сесквитерпены (n=3), дитерпены (n=4), тритерпены (n=6), тетратерпены (n=8). Другой вид классификации – по
количеству циклов в молекуле. Терпены и терпеноиды могут
229
230.
быть ациклическими (цикл отсутствует), моноциклическими, бициклическими и полициклическими.Примером ациклических терпеноидов является спирт геранил и продукт его окисления – альдегид гераниаль (цитраль). Они
содержатся в эфирных маслах герани, лимона и розы.
Цитраль используется в
O
C
CH2 OH
глазной практике как протиH
вовоспалительное средство.
[O]
Примером моноциклических терпенов является лимонен – компонент эфирного масла
лимона и скипидара. При гидрировании лимонена образуется
ментан, производным которого является ментол.
гераниол
гераниаль
H2 , Ni
лимонен
ментан
Ментол присутствует в эфирном масле перечной мяты. Ментол обладает антисептическим,
болеутоляющим и успокаивающим действием.
OH
Он входит в состав валидола, мазей, применяемых при лечении ревматизма и при насморке.
ментол
В промышленности ментол получают из
м-крезола. Вначале проводят реакцию алкилирования по Фриделю-Крафтсу с получением тимола, который затем гидрируют:
Cl
CH3
CH3
CH3-CH-CH3
OH
м-крезол
[H]
AlCl 3
OH
OH
CH3-CH-CH3
тимол
ментол
Как непредельное соединение лимонен способен к реакции
гидратации. При полной гидратации в кислой среде, которая про-
230
231.
текает по правилу Марковникова, образуется двухатомный спирттерпин:
OH
OH
2 H2O, H
+
H2O
OH
OH
терпин
лимонен
. H2O
терпингидрат
Терпин применяется в медицине в виде гидрата как отхаркивающее средство.
Представителями бициклических терпенов являются пинан
и камфан:
пинан
камфан
Ненасыщенным производным пинана является α-пинен –
составная часть скипидара. Как непредельный углеводород αпинен вступает в реакции присоединения (например, с бромной
водой) и окисления:
Br
Br2 , H2 O
α- пинен
O
камфора
KMnO4 , H2 O
Br
HO
HO
+ MnO 2 + KOH
Производным камфана является кетон камфора,
которую применяют в медицине как стимулятор
сердечной деятельности.
При бромировании камфоры образуется α-бромкамфора, которая используется как успокаивающее средство:
O
камфора
Br2
O
Br
α-бромкамфора
231
232.
Особую группу терпенов составляют растительные пигменты каротиноиды. Они широко распространены в природе, играютроль витаминов или предшественников витаминов, участвуют в
процессах фотосинтеза. Большинство каротиноидов являются
тетратерпенами. В их молекулах присутствуют длинные сопряженные системы, поэтому они окрашены. Каротиноиды окрашивают морковь в оранжево-красный цвет (carrot – морковь), придают различную окраску плодам и ягодам, присутствуют во всех
зеленых частях растений. Для каротиноидов характерна трансконфигурация двойных связей.
β-Каротин – растительный пигмент оранжевого цвета, содержащийся в моркови, томатах:
β-каротин
Многие каротиноиды являются провитамином А, то есть соединениями, которые в организме человека и животных способны превращаться в витамин А.
CH2 OH
β-каротин
2
витамин A
Витамин А относится к жирорастворимым витаминам.
И каротиноиды, и витамин А неустойчивы и легко разрушаются при нагревании, под действием кислорода воздуха и света.
Витмин А (ретинол) – важнейший витамин, влияющий на
рост человека, животных и птиц. Главными признаками авитаминоза А являются заболевание глаз (куриная слепота), исхудание,
понижение сопротивляемости организма инфекциям. Перерождение и ороговение эпителия в различных органах вследствие недостатка витамина А приводит к заболеванию дыхательных путей, к желудочно-кишечным и инфекционным заболеваниям, к
нарушению деятельности ЦНС, образованию камней в почках и
232
233.
мочевом пузыре и другим патологиям. К жирорастворимым относятся также витамины группы Е и К.Витамины группы Е – токоферолы – присутствуют в растительных маслах. Витамины группы Е можно рассматривать и как
производные гетероциклической системы хромана, и как производные двухатомного фенола гидрохинона. Они выполняют роль
антиоксидантов по отношению к ненасыщенным липидам, предохраняя их от пероксидного окисления, участвуют в синтезе
белков, тканевом дыхании, в регуляции развития зародыша и
функций эпителия половых желез.
CH3
HO
CH3
H3C
O
CH3
CH3
CH3
CH3
CH3
Витамин Е (α-токоферол)
Витамины группы К являются антигеморрагическим фактором, они нормализуют процесс свертываемости крови. Витамины группы К – производные 2-метил-1,4-нафтохинона. В природе
данная группа витаминов представлена несколькими соединениями. Витамин К1 встречается в высших растениях, витамин
К2 – в организмах животных и бактерий.
O
CH3
CH3
O
CH3
CH3
CH3
CH3
Витамин К 1
В медицине применяется синтетический водорастворимый
аналог витаминов группы К – викасол, который повышает свертываемость крови:
O
CH3
SO 3 Na
O
Викасол
233
234.
СТЕРОИДЫСтероиды – большая группа природных соединений как животного, так и растительного происхождения, объединенная
общностью углеродного скелета и путями биогенеза.
Соединения стероидной структуры широко распространены
в природе. Они найдены практически во всех организмах – от одноклеточных до млекопитающих. Стероидами выполняются самые разнообразные функции (регуляция углеводного обмена –
глюкокортикоиды, обмена минеральных солей – минералокортикоиды, процессов размножения – половые гормоны и т.д.). Стероиды появились в организмах на самых ранних стадиях их эволюции.
Почему же природа выбрала именно эти соединения в качестве химических регуляторов биологических процессов? Возможно, из-за высокой устойчивости их молекул и из-за высокой
информационной емкости, которая обусловлена многообразием
производных и стереоизомеров.
В настоящее время известно около 20 тыс. различных стероидов и свыше 100 из них применяются в медицине.
12
11
1
2
10 9
A
3
4
17
D
C
8
B
5
13
14
7
16
15
Все стероиды являются производными
циклопентанпергидрофенантрена, или
стерана, или гонана. Кольца принято
обозначать как A, B, C и D.
6
стеран
Стереоизомерия стерана. Все циклогексановые кольца в
структуре стерана находятся в конформации кресла. Сочленены
они могут быть по-разному. Рассмотрим типы сочленения колец
на более простом примере – декалине:
H
H
234
Это цис-сочленение колец.
Атомы водорода находя тся
по одну сторону от плоскости
сочленения .
H
H
цис-декалин
235.
HH
H
Это транс-сочленение колец.
Атомы водорода находя тся
по разные стороны от плоскости сочленения .
H
транс-декалин
Более энергетически выгодным является транссочленение
колец.
В структуре стероидов кольца B и C и C и D всегда транссочленены (за исключением сердечных гликозидов и ядов жаб – в
них C и D цис-сочленены). Кольца A и B могут иметь как цис-,
так и транссочленение:
H
H
5
5
H
H
5-β-стеран (A/B-цис).
β-связи выходят из плоскости
бумаги. Мы обозначаем их
сплошными линиями.
5-α-стеран (A/B-транс).
α-связи находятся за плоскостью
бумаги. Мы обозначаем их пунктиром.
Классификация стероидов. Выделяют следующие группы
стероидов:
- стерины
- желчные кислоты
- гормоны коры надпочечников (кортикостероиды)
- половые гормоны (мужские и женские)
- агликоны сердечных гликозидов.
Для родоначальных структур каждой группы стероидов
приняты тривиальные названия, т.к. использование международной номенклатуры привело бы к очень сложным названиям.
235
236.
СтериныВ основе структуры всех стеринов лежит углеводород холестан.
В молекуле холестана присут22 24
21
26
20
25
18
ствуют две так называемые ангу23
12
17 16 27
11
лярные (угловые) метильные груп13
19
1
15
пы в положениях 10 и 13 и углево9 8 14
2
10
дородный радикал из восьми ато3
7
5
мов углерода в положении 17.
6
4
Холестан
Наиболее широко распространенным стерином является холестерин. Он присутст1
9 8
вует в нервной ткани и надпо2
10
3
чечниках, в крови, желчи. В
7
5
HO
6
4
организме присутствует и в
свободном виде, и в виде
Холестерин (холестен-5-ол-3)
сложных эфиров с высшими
карбоновыми кислотами (по спиртовому гидроксилу), например,
холестерина пальмитат.
Только 20% от общего количества холестерина поступает в
организм с пищей, основное количество холестерина синтезируется в печени и кишечнике из уксусной кислоты (синтез включает более 20 стадий). Нарушение уровня холестерина (нормальная
концентрация в крови ~2г/л) ведет к различным нарушениям. Повышение концентрации холестерина ведет к отложению его на
стенках сосудов, к снижению их эластичности и развитию атеросклероза (как следствие – ишемическая болезнь сердца, нарушение мозгового кровообращения). При пересыщении желчи холестерином развивается желчнокаменная болезнь. Значительное
падение концентрации холестерина в плазме крови тоже может
вести к заболеваниям: гипертиреозу, аддисоновой болезни (поражению коры надпочечников), истощению.
На уровень холестерина влияет состав пищевых жиров.
Употребление животных жиров ведет к повышению концентрации холестерина. На 1 г насыщенных жиров должно приходиться
2 г ненасыщенных.
236
237.
2118
12
11
1 19
2
10
3
HO
Эргостерин – 24-метилхолестатриен-5,7,22-ол-3
(содержится в дрожжах) является провитамином D2, т.к.
при его облучении образуется этот витамин.
24 26
25
23
16
27
15
14
7
5
4
8
22
17
13
9
20
6
Эргостерин
CH2
UV
HO
HO
Витамин D2
Эргостерин
Витамины группы D регулируют обмен кальция и фосфора.
Их недостаток ведет к рахиту.
Желчные кислоты
21
18
12
11
1 19
2
10
3
5
4
9
13
8
14
20
22
23
17
16
24
В основе структуры желчных кислот лежит углеводород холан.
15
7
6
Холан
Желчные кислоты вырабатываются печенью при окислении
холестерина и выделяются с желчью в кишечник. Особенностью
структуры желчных кислот является цис-сочленение колец A и B.
Наиболее распространены холевая кислота и ее производные.
237
238.
OH12
11
1 19
2
10
3
HO
4
5
9
21
18
Холевая кислота является
3,7,12-тригидроксихолановой
кислотой.
COOH
23
17
13
8
20
24
22
16
15
14
7
OH
H 6
Холевая кислота
В желчи содержится не свободная холевая кислота, а ее
производные – амиды с глицином или таурином:
COOH
OH
O
C
OH
NH
+ H2 N-CH 2 -COOH
CH2
глицин
COOH
OH
HO
HO
Холевая кислота
OH
Гликохолевая кислота
COOH
OH
OH
C
O
NH
CH2
CH2
+ H2 N-CH 2 -CH2 -SO 3 H
таурин
OH
HO
HO
OH
SO3 H
Таурохолевая кислота
Холевая кислота
NaOH
OH
C
O
NH
гидрофильная часть
CH2
CH2
HO
OH
SO3 Na
липофильная часть
В кишечнике и желчи гликохолевая и таурохолевая кислоты
присутствуют в виде солей. Они являются дифильными соедине238
239.
ниями, т.к. имеют в структуре гидрофильную и гидрофобнуючасти. Желчные кислоты обладают поверхностно-активными
свойствами, действуют как эмульгаторы.
Сами желчные кислоты плохо растворимы в воде, могут откладываться в виде камней в желчном пузыре.
Кортикостероиды
18
12
11
1
2
3
4
9
17
13
19
10
20
8
14
21
16
Кортикостероиды являются производными углеводорода прегнана.
15
7
5
6
Прегнан
Кортикостероиды синтезируются в коре надпочечников из
холестерина. В чрезвычайно малых концентрациях влияют на
процессы жизнедеятельности. Удаление коры надпочечников ведет к смерти.
Гормоны коры надпочечников регулируют водно-солевой
обмен (минералокортикоиды) и углеводный обмен (глюкокорти21
коиды).
CH
2 OH
20
O
Кортикостерон – 11,21-дигидC
18
12
рок-сипрегнен-4-дион-3,20. ЯвHO 11
17 16
13
ляется глюкокортикоидом, анта1 19
15
9 8 14
гонистом инсулина (повышает
2
10
3
уровень сахара).
7
5
O
6
4
Кортикостерон
21
O 20
C
18
11 12
1 19
2
10
3
O
4
5
13
9
CH2 OH
8
14
17
16
Дезоксикортикостерон – 21-гидроксипрегнен-4-дион-3,20 является
минералокортикоидом.
15
7
6
Дезоксикортикостерон
239
240.
21O 20
C
1 19
2
3
O
8
17
O 20
C
OH
HO 11
16
1 19
15
14
2
10
3
7
5
4
13
9
10
18
12
HO 11
21
CH2 OH
O
6
5
4
Гидрокортизон
9
18
12
13
8
CH2 OH
OH
17
16
15
14
7
6
Преднизолон
Глюкокортикоид
гидрокортизон
(11,17,21тригидроксипрегнен-4-дион-3,20) и синтетический аналог глюкокортикоидов преднизолон (11,17,21-тригидроксипрегнадиен-1,4дион-3,20) используются как противовоспалительные и антиаллергические средства при лечении ревматоидного артрита, бронхиальной астмы и т.д. Используются в медицине в виде ацетатов
по первичному спиртовому гидроксилу в положении 21.
Андрогенные гормоны
18
12
11
1 19
2
10
3
5
4
9
13
8
14
17
Мужские половые гормоны являются производными андростана.
16
15
7
6
Андростан
Главными андрогенными гормонами являются андростерон
и тестостерон. Они влияют на развитие вторичных половых признаков, выработку спермы, оказывают активизирующее действие
на синтез ДНК и биосинтез белка, потенцируют сгорание углеводов и жирных кислот с образованием энергии.
O
OH
17
17
3
HO
240
Андростерон
(3-гидроксиандростанон-17)
3
O
4
Тестостерон
(17-гидроксиандростен-4-он-3)
241.
В медицинской практике тестостерон применяется в видепропионата (сложные эфиры обладают более длительным действием в организме):
O-CO-CH 2 -CH 3
OH
+ CH3 -CH 2 -C
O
O
Cl
O
Тестостерона пропионат
Тестостерон
Реакция ацилирования протекает по спиртовому гидроксилу. В качестве ацилирующего агента можно использовать хлорангидрид или ангидрид пропионовой кислоты.
Женские половые гормоны
12 18
11
1
10
2
3
8
9
16
15
14
7
5
4
17
13
Основой структуры эстрогенных гормонов является эстран (обратите внимание
на отсутствие ангулярной метильной
группы в положении 10).
6
Эстран
Эстрогены контролируют менструальный цикл у женщин.
Представителями эстрогенных гормонов являются эстрадиол и
эстрон:
18
12
11
1
2
HO
10
3
4
5
9
13
8
14
O
18
12
17
16
11
1
15
7
6
эстрон
(3-гидроксиэстратриен-1,3,5(10)-он-17)
2
HO
10
3
4
5
9
13
8
14
OH
17
16
15
7
6
эстрадиол
(эстратриен-1,3,5(10)-диол-3,17)
Эстрадиол применяется в медицинской практике в виде дипропионата.
241
242.
OOH
O
2 CH3 -CH2 -C
HO
O-C-CH 2 -CH 3
Cl
CH3 -CH2 -C-O
эстрадиол
O
эстрадиола дипропионат
Для ацилирования можно использовать также пропионовый
ангидрид.
К женским половым гормонам относятся также гестагены
(гормоны желтого тела яичников, гормоны беременности). Гестагены являются производными прегнана. Наиболее активным гестагеном является прогестерон:
21
O 20
C
18
11 12
1 19
2
10
3
O
4
5
13
9
CH3
8
14
17
16
15
7
6
Прогестерон
(прегнен-4-дион-3,20)
Сердечные гликозиды
Сердечные гликозиды – это соединения, в которых стероидная часть молекулы является агликоном (несахарной частью)
гликозидов, образованных моно- или олигосахаридами. В небольших дозах сердечные гликозиды используются в кардиологии. Они увеличивают силу и уменьшают частоту сердечных сокращений, улучшают тканевой обмен сердечной мышцы. В больших дозах сердечные гликозиды являются ядами. В мировой
медицинской практике широко используют препараты, получаемые из наперстянки (дигиталиса), строфанта, ландыша, горицвета.
Например, агликоном ланатозида А, выделяемого из наперстянки шерстистой, является дигитоксигенин:
242
243.
OO
Характерной особенностью
агликонов сердечных глико18
зидов
является
цис11 12
17 16
13
сочленение колец A и B и C
14
1 19
15
и D, а также наличие нена9 8
2
10
OH
сыщенного пяти- или шес3
7
5
HO
тичленного
лактонного
4 H 6
кольца в положении 17. УгДигитоксигенин
леводная часть молекулы
содержит от одного до пяти моносахаридных остатков. Огликозидная связь с углеводным остатком осуществляется за
счет спиртового гидроксила в положении 3.
АЛКАЛОИДЫ
Алкалоиды – большая группа органических азотсодержащих соединений основного характера, встречающихся в растительных организмах и обладающих сильным физиологическим
действием.
В растениях алкалоиды содержатся в виде оснований и солей органических кислот – лимонной, яблочной, янтарной, щавелевой и др. Алкалоиды-основания практически нерастворимы в
воде, но растворимы в органических растворителях – диэтиловом
эфире, бензоле, хлороформе. Алкалоиды-соли, наоборот, растворимы в воде и нерастворимы в органических растворителях.
Большинство алкалоидов оптически активны. Многие алкалоиды
применяются в медицине (морфин, кофеин, хинин и др.).
Алкалоиды принято классифицировать по характеру гетероциклов, составляющих основу их структуры. Рассмотрим некоторые из них.
Никотин является производным пиридина и
пирролидина. Воздействует на вегетативную
нервную систему, сужает кровеносные сосуN
ды. Содержание никотина в листьях табака –
CH3
N
до 8%.
никотин
243
244.
В жестких условиях никотин окисляется до никотиновойкислоты:
COOH
[O]
N
N
CH3
N
никотин
никотиновая кислота
Хинин является производным хинолина (второе
гетероциклическое ядро в
его структуре – хинуклидин). Применяется для лечения малярии.
CH=CH2
HO
CH
N
CH3 O
N
хинин
CH3 O
Производным изохинолина
является алкалоид папаверин,
применяемый в медицине в виде
гидрохлорида как противосудорожное средство и при лечении
гипертонии.
N
CH3 O
CH2
OCH3
OCH3
папаверин
Синтетическим аналогом папаверина, применяемым как
спазмолитик, является но-шпа:
C2 H5 O
N
C2 H5 O
H
C
H
OC 2 H5
OC 2 H5
но-шпа
Другим алкалоидом – производным изохинолина является
морфин. В молекуле морфина две гидроксильные группы –
спиртовая и фенольная. Морфин проявляет как основные (за счет
244
245.
гетероциклического атома азота), так и кислотные свойства (засчет фенольного гидроксила), поэтому он
HO
растворим и в кислотах, и в щелочах.
Морфин обладает сильным обезболиO
вающим действием, но при длительном
N-CH 3
употреблении вызывает привыкание –
наркоманию.
HO
Диацетильное производное морфиморфин
на – героин является еще более сильным
наркотиком.
Метиловый эфир морфина – кодеин применяется в медицине как противокашлевое средство.
CH3 O
O
N-CH 3
HO
кодеин
Алкалоид резерпин является производным индола.
CH3 O
N
N
H
O-CO
CH3 OOC
CH3 O
резерпин
OCH3
OCH3
OCH3
Резерпин применяют при лечении гипертонии.
Кофеин, теофиллин и теобромин (см. стр.155) являются
производными пурина.
245
246.
ЛИТЕРАТУРА1. Тюкавкина Н.А., Бауков Ю.И. Биоорганическая химия.
М.: Медицина, 2006.
2. Органическая химия (т. 1, 2) / В.Л. Белобородов, С.Э. Зурабян, А.П. Лузин, Н.А. Тюкавкина / Под ред. Н.А. Тюкавкиной. М.: Дрофа, 2002.
3. Овчинников Ю.А. Биоорганическая химия. М.: Просвещение, 1987.
4. Райлс А., Смит Л., Уорд Р. Основы органической химии.
М.: Мир, 1983.
5. Терней А. Современная органическая химия (т. 1, 2). М.:
Мир, 1981.
6. Робертс Дж., Кассерио М. Основы органической химии
(т. 1, 2). - М.: Мир, 1978.
7. Березин Б.Д., Березин Д.Б. Курс современной органической химии. - М.: Высшая школа, 2001.
8. Чалый Г.А., Зубкова И.В., Лазарев А.И. Метаболизм ксенобиотиков в организме. - Курск, 2000.
9. Альберт А. Избирательная токсичность (т. 1, 2). - М.: Медицина, 1989.
246
247.
ОГЛАВЛЕНИЕВведение……………………………………………………...….
3
Электронное строение элементов-органогенов. Химическая 4
связь в органических молекулах……………………………….
Классификация химических реакций. Химические свойства 13
алканов, алкенов и алкадиенов………………………………….
Сопряжённые системы. Ароматичность. Электронные 30
эффекты. Реакции электрофильного замещения в бензоле
и его производных……………………………………………….
Химические свойства галогеналканов, спиртов и фено- 48
лов………………………………………………………………..
Кислотные и основные свойства органических соединений. 64
Реакционная способность аминов………………………………
Реакционная способность оксосоединений……………………. 75
Химические свойства карбоновых кислот и их функциональ- 87
ных производных…………………………………………………
Гетерофункциональные соединения алифатического ряда – 103
метаболиты и биорегуляторы……………………………………
Оптическая изомерия……………………………………………. 117
Гетерофункциональные производные бензольного ряда…….. 124
Гетероциклические соединения. Производные пятичленных 132
гетероциклов……………………………………………………..
Шестичленные гетероциклические соединения………………. 144
Углеводы. Моносахариды………………………………………. 156
Углеводы. Ди- и полисахариды…………………………………. 173
Природные α-аминокислоты. Пептиды, белки………………… 186
Нуклеиновые кислоты…………………………………………… 203
Омыляемые липиды……………………………………………… 217
Неомыляемые липиды…………………………………………… 228
Алкалоиды……………………………………………………….. 243
247
248.
Издательство Курского государственного медицинского университета.305041, г. Курск, ул. К. Маркса, 3.
Лицензия ЛР № 020862 от 30.04.99 г.
Тираж
экз.
Отпечатано в типографии КГМУ.
305041, г. Курск, ул. К. Маркса, 3.
Заказ № 313.
248