



, объединяет все виды качественной")









 - разнородная группа заболеваний, обусловленных патологией сосудистой стенки и не относящихся")

")









")





 medicine
medicineSimilar presentations:



. Геморрагические диатезы")






Патология системы гемостаза
1. ПАТОЛОГИЯ
СИСТЕМЫ ГЕМОСТАЗА2.
В клинической практике наиболее часто встречаютсятри разновидности нарушений формирования
коагуляционного потенциала крови :
1. геморрагические диатезы, обусловленные
недостаточностью тромбоцитарно-сосудистого и
коагуляционного механизмов гемостаза;
2. тромбофилии, связанные с преобладанием
прокоагулянтных механизмов, недостаточностью
антикоагулянтных механизмов или системы фибринолиза;
3. синдром диссеминированного внутрисосудистого
свертывания крови (ДВС-синдром, или
тромбо-геморрагический синдром),
который характеризуется фазными системными или
локальными нарушениями коагуляционного потенциала
крови.
3. ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ
Геморрагические диатезы - группа наследственных илиприобретенных заболеваний, основным клиническим
признаком которых является повышенная кровоточивость,
возникающая самопроизвольно или после незначительных
травм.
4.
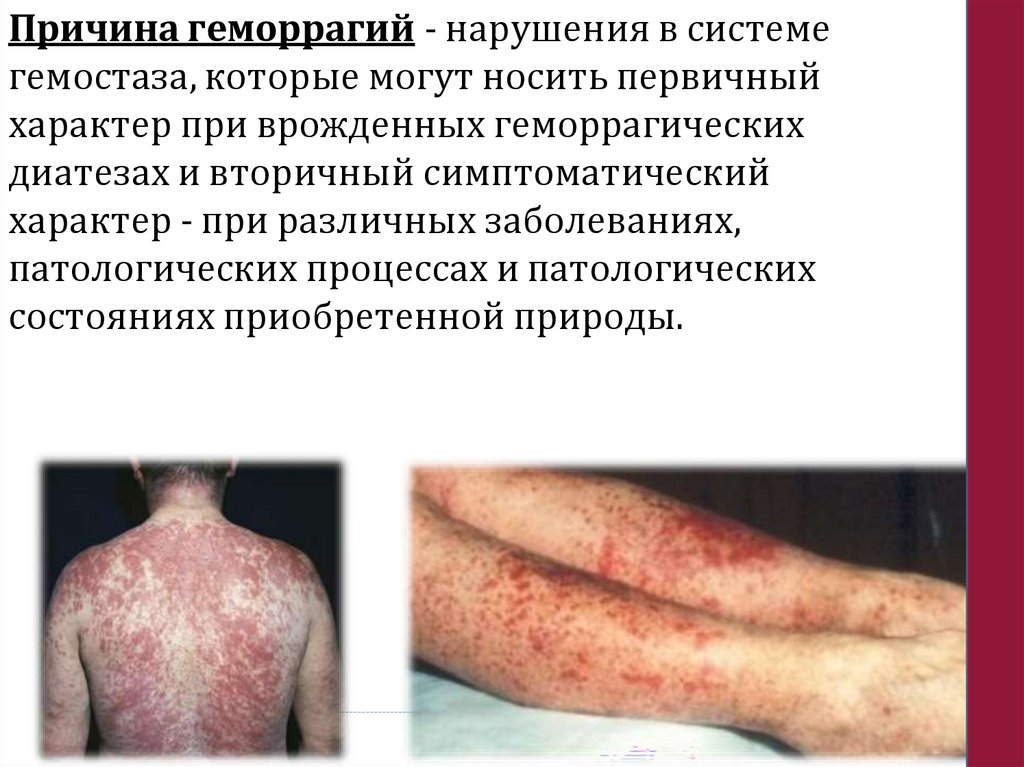
Причина геморрагий - нарушения в системегемостаза, которые могут носить первичный
характер при врожденных геморрагических
диатезах и вторичный симптоматический
характер - при различных заболеваниях,
патологических процессах и патологических
состояниях приобретенной природы.
5.
Развитие различных форм геморрагическихдиатезов связано с патологией определенных
компонентов системы, регулирующей
агрегатное состояние крови (РАСК), что
позволяет классифицировать данные
заболевания и синдромы следующим образом:
6.
1. Тромбоцитопатии - состояния, в основе которыхлежит качественная неполноценность тромбоцитов.
2. Тромбоцитопении - формы геморрагических
диатезов, обусловленные уменьшением количества
тромбоцитов в единице объема крови.
3. Коагулопатии - заболевания или синдромы,
связанные со снижением активности плазменных
факторов свертывания крови.
4. Вазопатии - заболевания или синдромы,
обусловленные поражением сосудов в виде
повышенной проницаемости их или аномалий
сосудистой стенки.
7. Термин "тромбоцитопатии", согласно рекомендациям Комитета экспертов ВОЗ (1969), объединяет все виды качественной
Термин "тромбоцитопатии", согласнорекомендациям Комитета экспертов ВОЗ (1969),
объединяет все виды качественной
неполноценности тромбоцитов.
Тромбоцитопатии
Наследственные
Приобретенные
8. В настоящее время выделены следующие варианты наследственных тромбоцитопатий:
1. Формы с преимущественным нарушением агрегационной функции(дизагрегационные):
1. Формы с сохраненной "реакцей освобождения":
а) с развернутым нарушением агрегационной функции: - тромбоцитастения
Гланцмана I и II типов; - эссенциальная атромбия I типа; - другие формы;
б) парциальные тромбоцитопатии: - с изолированным нарушением коллагенагрегации; - аномалия Мей-Хегглина; - с изолированным нарушением АДФ- и
(или) тромбин-агрегации (аномалия Пирсон-Стоба, наследственная
афибриногенемия и др.).
2. Формы с нарушением "реакции освобождения" и второй фазы агрегации
(аспириноподобный синдром, эссенциальная атромбия II типа и др.).
3. Болезни недостаточного пула накопления (недостаточного хранения
гранул и их компонентов)
а) недостаток плотных телец I типа и их компонентов (АДФ, серотонина,
адреналина);
б) недостаток плотных телец II типа (белковых) и их компонентов (фактора 4 и
его носителя, - тромбоглобулина, ростового фактора);
в) нарушения лизосом и кислых гидролаз.
9.
2. Формы с преимущественным нарушением адгезии тромбоцитов кколлагену и стеклу.
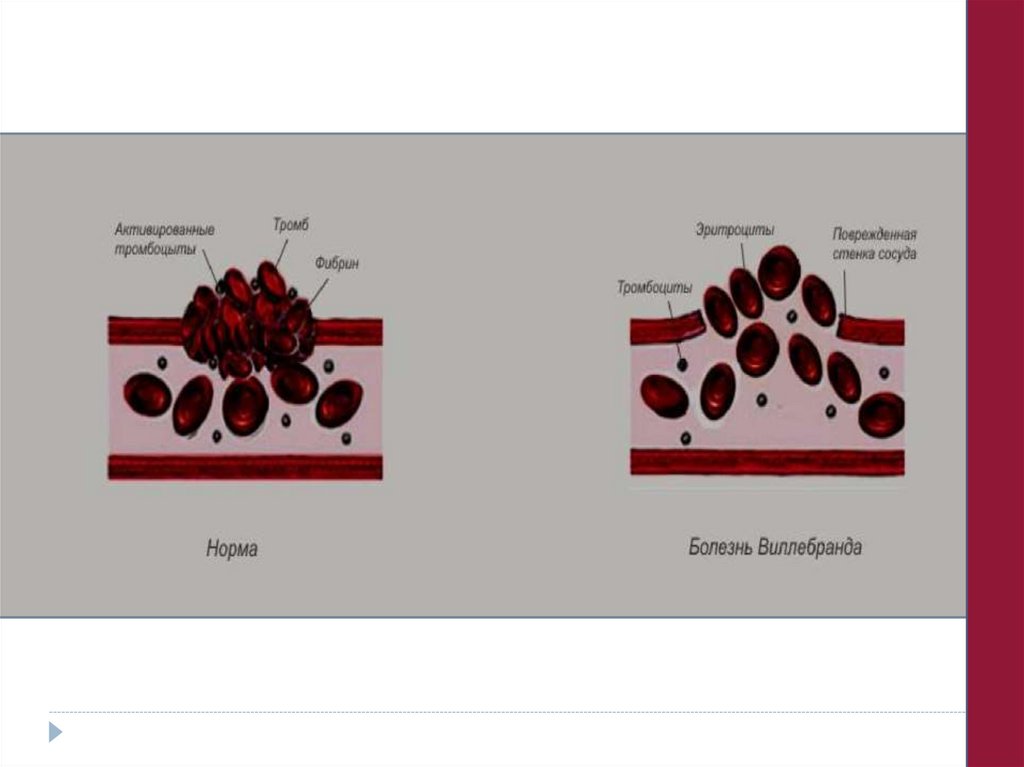
1. Формы с нарушенной ристомицинагрегацией:
а) плазменного генеза (болезнь Виллебранда);
б) тромбоцитарного генеза (макроцитарная тромбоцитодистрофия БернараСулье);
в) плазменно-пластиночного генеза (синдром Виллебранда-Юргенса
2. Формы с нормальной ристомицинагрегацией:
а) молекулярные варианты болезни Виллебранда;
б) изолированное нарушение агрегации тромбоцитов к коллагену.
10.
11. 3. Формы с дефицитом и снижением доступности фактора 3:
1. С генетически обусловленным дефицитом фактора 3 (врожденныедефицитные тромбопатии по Bowie и Owen).
2. С нарушением освобождения фактора 3 при адгезии и агрегации
(функциональные тромбопатии по Bowie и Owen).
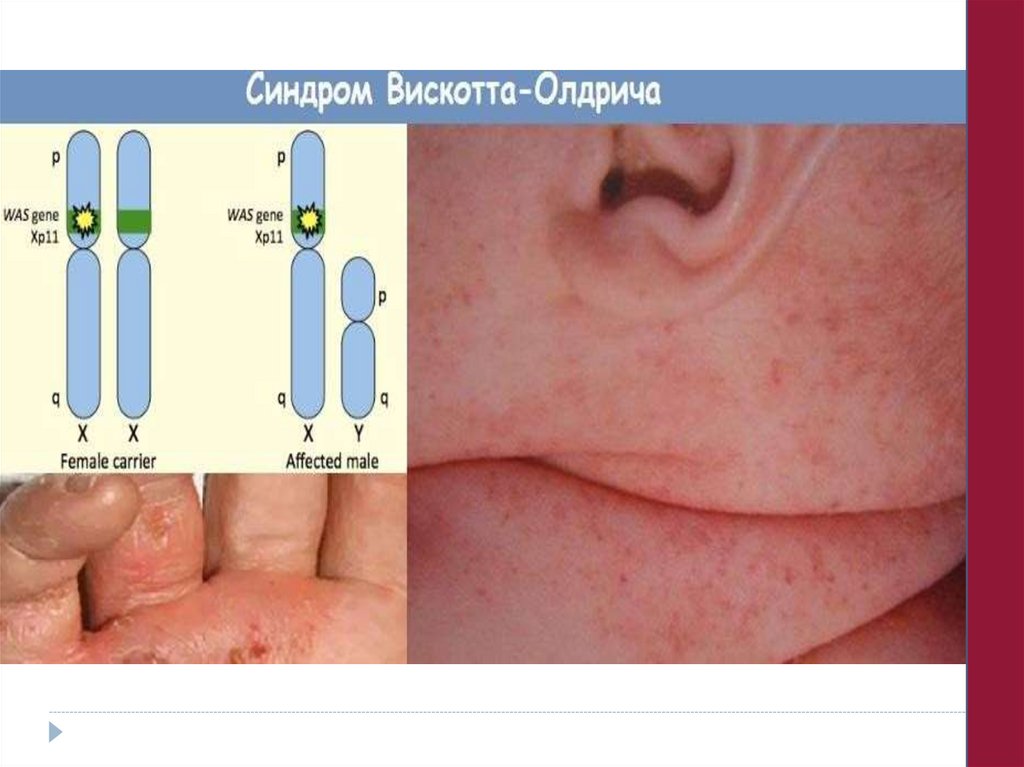
4. Сложные аномалии и дисфункции тромбоцитов,
сочетающиеся с другими генетическими дефектами:
1. При иммунных нарушениях (синдром Вискотта-Олдрича).
2. При ферментопатиях (гликогенозы I и II типов и др.).
3. При дисплазиях соединительной ткани (синдромы Элерса-Данлоса,
Марфана и др.). 4.4. При врожденных пороках сердца.
5. Недостаточно идентифицированные формы.
12.
13. При приобретенных тромбоцитопатиях кровоточивость относится к сосудисто- тромбоцитарному типу и протекает обычно нетяжело.
Виды приобретенных (симптоматических)тромбоцитопатий:
1. При гемобластозах:
а) дизагрегационные гипорегенераторные;
б) формы потребления (при ДВС-синдроме);
в) смешанного типа.
2. При миелопролиферативных заболеваниях и эссенциальной
тромбоцитемии.
3. При В12-дефицитной анемии.
4. При уремии.
5. При циррозах, опухолях и паразитарных заболеваниях печени.
6. При других формах ДВС-синдрома (быстрое потребление
тромбоцитов, блокада их функции продуктами расщепления
фибриногена).
14.
7. Блокада тромбоцитов макро- и парапротеинами (миеломнаяболезнь, болезнь Вальденстрема и др.).
8. При цинге (нарушения АДФ-агрегации).
9. При гормональных нарушениях (гипо- и дистиреозы,
гипоэстрогенемии и др.).
10. Лекарственные и токсические (при лечении ацетилсалициловой
кислотой, пиразолоновыми производными, бруфеном, бетаадреноблокаторами, дипиридомолом, большими дозами папаверина,
антибиотиками - карбенициллином, пенициллином,
транквилизаторами, мочегонными, нитрофуранами,
антигистаминами, цитостатиками, бутазолидинами и др.).
11. При лучевой болезни.
12. При массивных гемотрансфузиях.
13. При больших тромбозах и гигантских ангиомах
15. Тромбоцитопении - это группа заболеваний, при которых количество тромбоцитов ниже существующей нормы – 150-10*9 /л. Снижение
количества тромбоцитов может бытьобусловлено повышенным их разрушением, повышенным
потреблением или недостаточным образованием. Наиболее
частой причиной является повышенное разрушение
тромбоцитов.
Наследственные
Приобретенные
16.
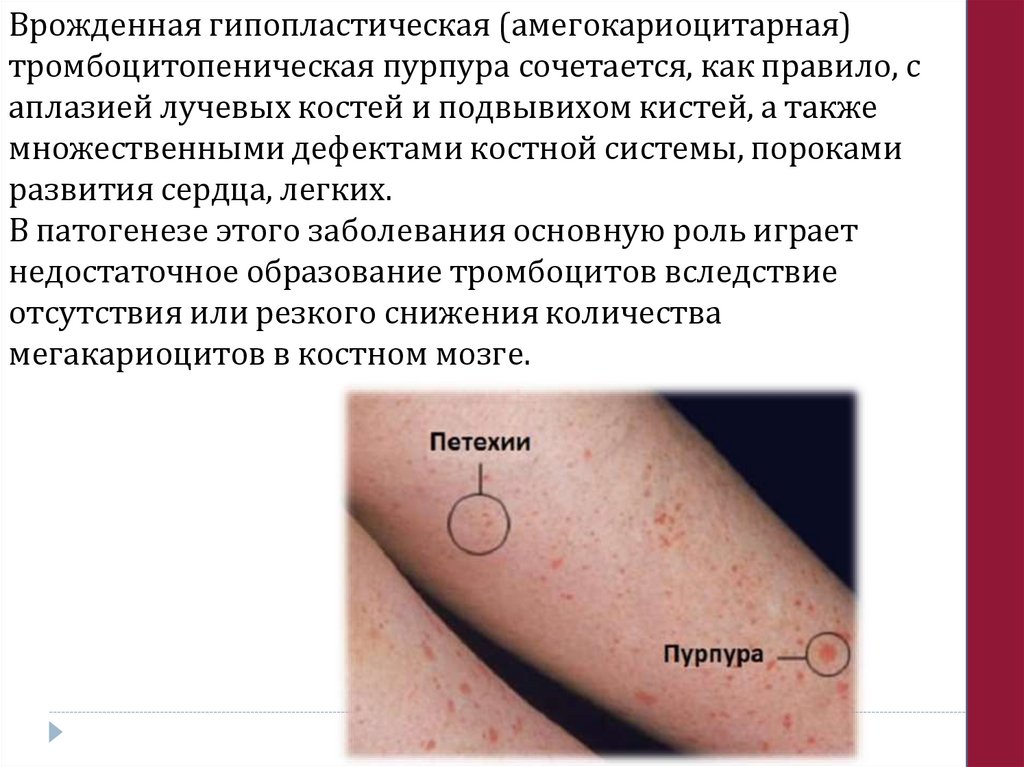
Врожденная гипопластическая (амегокариоцитарная)тромбоцитопеническая пурпура сочетается, как правило, с
аплазией лучевых костей и подвывихом кистей, а также
множественными дефектами костной системы, пороками
развития сердца, легких.
В патогенезе этого заболевания основную роль играет
недостаточное образование тромбоцитов вследствие
отсутствия или резкого снижения количества
мегакариоцитов в костном мозге.
17. Приобретенные формы тромбоцитопений подразделяются на несколько групп:
1. Иммунные формы.2. Формы, обусловленные механической травматизацией тромбоцитов
(при гемангиомах, спленомегалии и др.).
3. Тромбоцитопении, связанные с угнетением пролиферации клеток
костного мозга (при апластической анемии, химическом и радиационном
повреждении костного мозга).
4. Формы, обусловленные замещением костного мозга опухолевой
тканью.
5. Тромбоцитопении вследствие соматических мутаций (болезнь
Маркиафы-Микели).
6. Повышенное потребление тромбоцитов (тромбозы, ДВС-синдром и
др.).
7. Формы, связанные с недостатком витамина В12 и фолиевой кислоты.
18. Среди иммунных тромбоцитопений выделяют следующие группы:
а) Аллоиммунные, при которых разрушение тромбоцитов связано снесовместимостью по одной из групповых систем крови при
гемотрансфузии, либо с трансплацентарным проникновением из организма
матери антител к антигенам тромбоцитов плода, полученных от отца и
отсутствующих у матери.
б) Трансиммунные, при которых аутоантитела матери, страдающей
аутоиммуннойтромбоцитопенией, проникают через плаценту и
вызывают разрушение тромбоцитов у ребенка. Тромбоцитопения
развивается у 30-50% новорожденных детей, рожденных от матерей,
больных иммунными тромбоцитопениями. Тромбоцитопения у детей
наблюдается, как правило, в течение 2-12 недель после рождения.
в) Гетероиммунные, связанные с нарушением антигенной структуры
мембран тромбоцитов под влиянием вирусов, гаптенов и др.
г) Аутоиммунные, при которых антитела вырабатываются против собственных
неизмененных антигенов тромбоцитов.
19. Выделяют несколько форм гетероиммунных тромбоцитопений:
- алиментарно-аллергическую, вызываемую некоторыми пищевымипродуктами (коровье молоко, лимоны и др.);
-медикаментозную (хинин, хинидин, антибиотики - пенициллин,
тетрациклин, рифампицин, стрептомицин, левомицетин и др.,
сульфаниламиды - бактрим, диакарб, гипотиазид, дигитоксин, ПАСК,
фенобарбитал, калия йодид и др.;
-- пара- и постинфекционную (краснуха, корь, цитомегалия,
инфекционный мононуклеоз и др.);
-- аллергические тромбоцитопении после прививок.
20. Вазопатии (ангиодисплазии) - разнородная группа заболеваний, обусловленных патологией сосудистой стенки и не относящихся
непосредственно к патологиисистемы крови
В группе врожденных ангиодисплазий выделяют следующие
варианты патологии:
1. Собственно геморрагические ангиодисплазии:
-телеангиэктазия (болезнь Рандю-Ослера);
- телеангиэктатическая атаксия (синдром Луи-Бар);
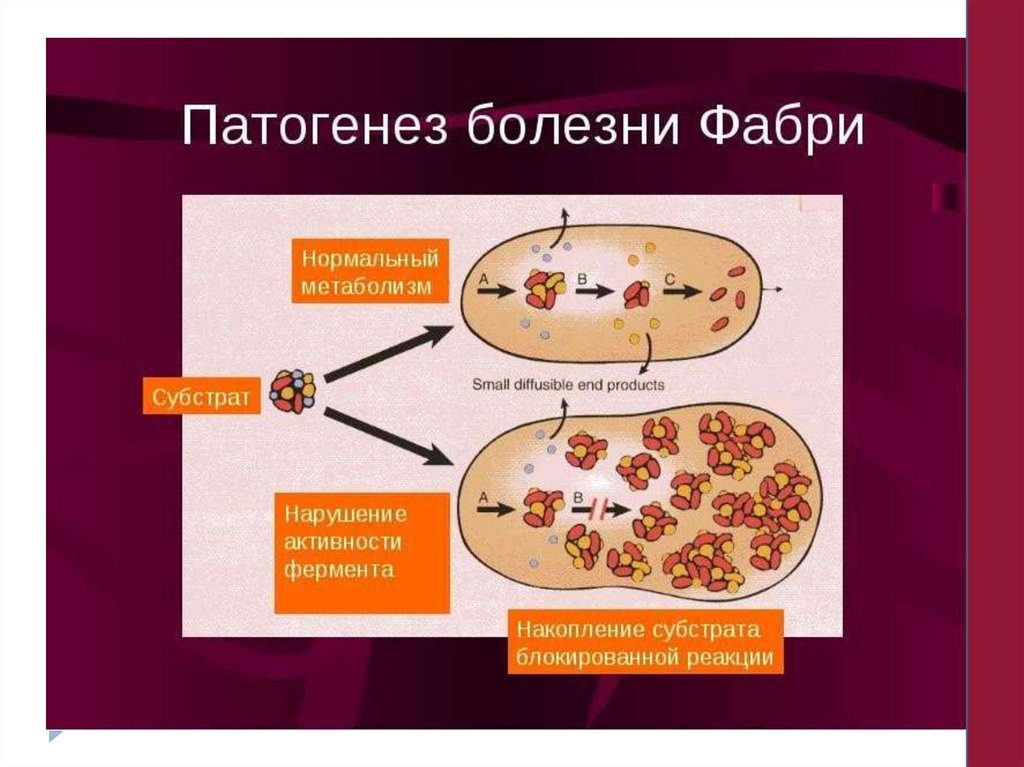
-диффузная ангикератома туловища (болезнь Фабри);
- синдром Богарта-Диври и др.
21.
22. 2. Гемангиомы, протекающие с тромбоцитарными и коагуляционными нарушениями:
- синдромы Казабаха-Мерритта;- микроангиоматозы с тромбоцитопенией и др.
3. Формы с наследственной неполноценностью
соединительной ткани, которые могут сочетаться с
дисфункцией тромбоцитов, дефицитом фактора
Виллебранда и другими нарушениями гемостаза:
- синдром Черногубова-Элерса-Данло;
-синдром Марфана и др.
- 4. Комбинированные аномалии вышеперечисленных
групп.
23. Синдром Казабаха-Мерритта (солитарная гемангиома с тромбоцитопенией и нарушениями коагуляции)
Гемангиома - доброкачественная сосудистая опухоль, которая можетбыть локализована в разных участках тела.
Гемангиомы подразделяются на:
- капиллярные;
- кавернозные;
-капиллярно-кавернозные.
Кавернозный тип часто сопровождается тромбоцитопенией и
гемокоагуляционным нарушениями. Коагуляционный дефект связан с
внутрисосудистым свертыванием крови в гемангиоме. Клинически
гемангиомы обнаруживаются сразу же после рождения, а также спустя
несколько недель или месяцев.
Характерен быстрый рост опухоли.
24. Синдром Черногубова-Элерса-Данло
Патогенез кровоточивости связан с недоразвитием коллагеновыхструктур стенок сосудов, вследствие чего легко происходит их разрыв.
Проявления геморрагического диатеза сочетаются с истончением,
растяжимостью кожи, разболтанностью суставов, плохим
заживлением ран, что обусловлено недоразвитием соединительной
ткани.
В зависимости от клинических проявлений болезни, от
ультраструктурных и биохимических особенностей дефектов
коллагена, различают несколько типов синдрома (тяжелый,
среднетяжелый, доброкачественный, глазной, периодонтитный,
экхимозный и др.).
25. Приобретенные вазопатии
Эта группа геморрагических васкулитов весьма неоднороднапо этиологии и патогенезу и недостаточно изучена.
Среди приобретенных вазопатий выделяют
идиопатические, застойные и ортостатические,
атрофические и дистрофические, неврогенные,
механические и другие формы патологии.
С приобретенными вазопатиями связывают
преимущественно кожные формы кровоточивости, которые
обусловлены экзо- и эндогенными поражениями
кровеносных сосудов без существенных нарушений
тромбоцитарно-сосудистого и коагуляционного звеньев
гемостаза и фибринолиза.
26. Наиболее частой разновидностью приобретенных пурпур является геморрагический васкулит Шенлейна- Геноха (анафилактоидная,
Наиболее частой разновидностью приобретенныхпурпур является геморрагический васкулит ШенлейнаГеноха (анафилактоидная, аллергическая пурпура),
он обусловлен генерализованным гиперергическим воспалением
мелких сосудов и характеризуется полиморфизмом клинических
проявлений.
Согласно современным представлениям, болезнь Шенлейна-Геноха
относится к болезням иммунных комплексов.
В основе поражения капилляров и артериол при этом заболевании
лежит повреждающее действие на сосудистый эндотелий комплексов
антиген-антитело, которые накапливаются в крови при различных
инфекционных процессах, чаще стрептококковой этиологии.
Нередко обнаруживаются иммунные комплексы в виде смешанных
криоглобулинов как проявление аллергии замедленного типа.
27. КОАГУЛОПАТИИ
Коагулопатии - заболевания или синдромы, причиной которыхявляется дефицит плазменных факторов свертывания крови; они
могут носить наследственный и приобретенный характер. Четкие
представления о патогенезе коагулопатий и характере нарушений
коагуляционного потенциала крови при указанных формах патологии
могут быть сформулированы лишь на основе современных концепций
формирования коагуляционного гемостаза в условиях нормы.
28. Наследственные коагулопатии классифицируются следующим образом:
Группа I. С изолированным нарушением внутреннего механизмаформирования протромбиназной активности:
- гемофилия А (дефицит антигемофильного глобулина - VIII:K);
- кофакторная гемофилия и другие аутосомные формы (дефицит
антигемофильного глобулина - VIII:C);
-болезнь Виллебранда (дефицит фактора Виллебранда - VIII:ФВ);
-- гемофилия В
- - болезнь Кристмаса (дефицит плазменного компонента
тромбопластина - IX);
-- гемофилия С - РТА-недостаточность (дефицит предшественника
плазменного тромбопластина - XI);
- - дефект Хагемана (дефицит фактора Хагемана - XII);
- - дефект Флетчера (дефицит плазменного прекалликреина, фактора
Флетчера);
-- дефект Вильямса, Фитцжеральда, Фложак (дефицит
высокомолекулярного кининогена).
29. -Группа II. С изолированным нарушением внешнего механизма формированиия протромбиназной активности:
- - гипопроконвертинемия (дефицит проконвертина - VII).-Группа III. С нарушениием внешнего и внутреннего механизмов
формирования протромбиназной активности:
- - гипоакцелеринемия
- - парагемофилия (дефицит Ас-глобулина, проакцелерина - V);
- - болезнь Стюарта-Прауэра (дефицит фактора Стюарта-Прауэра - Х);
- - гипо(дис)протромбинемия (дефицит протромбина - II);
-- комплексный дефицит факторов II, VII, X, IX.
-Группа IV. С нарушением конечного этапа свертывания крови:
-- а(гипо)фибриногенемия (дефицит фибриногена - I).
-Группа V. Нарушения стабилизации фибрина:
-- дефицит фибринстабилизирующего фактора (XIII).
-Группа VI. Смешанные формы дефицитных факторов (чаще
VIII+V).
- Группа VII. Дефицит физиологических антикоагулянтов:
- - тромбофилия (дефицит антитромбина III);
-- дефицит 2-макроглобулина;
- - дефицит протеина С и его кофакторов.
30. По частоте встречаемости эти формы патологии подразделяются следующим образом:
1) часто встречающиеся (примитивные формы, характеризующиесяизолированной недостаточностью какого-либо одного фактора гемофилия А (68-78%), болезнь Виллебранда (9- 18%), гемофилия В (613%) и др.);
2) редкие формы, на долю которых приходится около 2-3% коагулопатий
(дефицит XI, VII, V факторов);
3) крайне редкие (казуистические) формы (комплексные,
разнонаправленные нарушения коагуляционного гемостаза).
31. ПРИОБРЕТЕННЫЕ КОАГУЛОПАТИИ
Причинами приобретенных коагулопатий могут быть следующие заболевания,патологическиепроцессы и состояния:
1. Патологическое течение беременности и родов, прием лекарств матерью (ДВСсиндром, трансплацентарная передача ингибиторов факторов свертывания крови
и т.д.).
2. 2. Инфекционные заболевания, сепсис, риккетсиозы, протозойные заболевания.
3. Все виды шока, терминальные состояния, тяжелые травмы, ожоговая болезнь.
4. Холестаз вследствие внутри- или внепеченочного блока.
5. Заболевания печени (острые и хронические, инфекционные, паразитарные,
токсические, аутоиммунные и т.д.).
6. Заболевания почек (нефротический синдром, нефриты, почечная
недостаточность).
7. Все виды острого внутрисосудистого гемолиза.
8. Системный амилоидоз.
9. Гемобластозы, злокачественные новообразования различной локализации.
10.Коллагенозы (системная красная волчанка, ревматоидный артрит и др.).
11. Лекарственная терапия (антикоагулянты непрямого действия, актиномицин D,
гепарин, аспарагиназа, кислота аминокапроновая, амбен, антипротеазы и др.).
12. "Синдром массивных трансфузий".
13. Хирургические вмешательства.
14. Парапротеинемия (миеломная болезнь, макроглобулия Вальденстрема и др.).
15.Асфиксия, респираторный дистресс-синдром.
32. ТРОМБОФИЛИИ
Тромбофилия - состояние, характеризующееся предрасположенностьюк тромбозу.
Инициирующими этиопатогенетическими факторами
тромбофилий могут быть следующие:
1.
Повышение функциональной активности тромбоцитов и
увеличение количества тромбоцитов в единице объема крови.
2. Изменение тромбогенной активности и тромборезистентности
сосудов.
3. Повреждение сосудистой стенки.
4. Увеличение содержания активных коагулянтов в крови.
5. Уменьшение антикоагулянтной активности крови.
6. Угнетение фибринолиза.
33. ДИССЕМИНИРОВАННОЕ ВНУТРИСОСУДИСТОЕ СВЕРТЫВАНИЕ КРОВИ (ДВС-синдром)
ДВС-синдром - неспецифический патологический процесс,характеризующийся распространенным свертыванием
крови и агрегацией клеток крови в микроциркуляции,
ведущим к блокаде микроциркуляции, гипоксии, ацидозу,
дистрофии органов, развитию полиорганной
недостаточности.
34.
ДВС-синдром осложняет самые разнообразные формы патологии:инфаркт миокарда, кардиогенный шок, различные виды
злокачественных новообразований, обширные оперативные
вмешательства, тяжелую гипоксию, акушерскую патологию
(преждевременная отслойка плаценты, эмболия околоплодными
водами, внутриутробная гибель плода), переливание несовместимой
крови, системную красную волчанку, иммунокомплексные
заболевания, цирроз печени.
Диссеминированное внутрисосудистое свертывание крови динамический патологический процесс, характеризующийся
последовательной сменой генерализованной гиперкоагуляции с
внутрисосудистым свертыванием крови, агрегацией тромбоцитов,
блокадой микроциркуляции и гипокоагуляции с
гипофибриногенемией и тромбоцитопенией потребления
35. Общие закономерности его развития, включающего следующие инициирующие механизмы:
1. Первичное поражение сосудистой стенки, десквамация эндотелия,обнажение субэндотелиальных белков: коллагена, тромбоспондина,
фибронектина, фактора Виллебранда, обладающих способностью
активировать процессы адгезии и агрегации тромбоцитов, активировать XII
фактор Хагемана с последующей активацией внутреннего механизма
протромбиназной активности, системы комплемента, фибринолиза,
калликреин-кининовой систем.
2. Первичное преимущественное воздействие патогенного фактора на
тромбоциты, индукция процессов адгезии, агрегации тромбоцитов,
высвобождение из тромбоцитов биогенных аминов, тромбоцитарных
факторов свертывания крови, в частности, III и IV, инициирующих
образование тромбина с возможной последующей активацией под
влиянием тромбина ряда факторов формирования протромбиназы,
развития каскада реакций вторичной активации
коагуляционногогемостаза
36.
3. Сочетанное одномоментное воздействие бактериальных, токсических,иммуноаллергических факторов на тромбоцитарно-сосудистое и
коагуляционное звенья системы гемостаза.
4. Развитие альтернативных механизмов гемокоагуляции за счет
активации моноцитарно- макрофагального и эритроцитарного звеньев
системы гемостаза.
37. В развитии ДВС-синдрома следует выделять следующие фазы:
1. Гиперкоагуляция и агрегация клеток крови.2. Переход гиперкоагуляции в гипокоагуляцию.
3. Стадия глубокой гипокоагуляции, вплоть до полной
несвертываемости крови, котораяобусловлена
потреблением, расщеплением и блокадой ряда факторов
свертывания крови; накоплением и циркуляцией
продуктов их распада, обладающих антикоагулянтной
активностью, а также тромбоцитопенией потребления.
4. Восстановительная стадия при благоприятном течении
заболевания или формирование полиорганной
недостаточности.
38.
При остром ДВС возможно быстрое развитие тромбоцитопении игипофибриногенемии. Истинной афибриногенемии при ДВС-синдроме
практически не бывает. Часто наблюдается усиление связывания
фибриногена с фибринмономерами с образованием растворимого
фибрина. При затяжных и хронических формах ДВС
гипофибриногенемия встречается крайне редко.
При медленно протекающем ДВС-синдроме число тромбоцитов может
быть нормальным или даже повышенным.
.
При острых и подострых формах ДВС протромбиновое время
увеличено из-за снижения уровня V фактора и фибриногена;
тромбиновое время обычно увеличено, что обусловлено
гипофибриногенемией и ингибирующим воздействием продуктов
деградации фибриногена.
39.
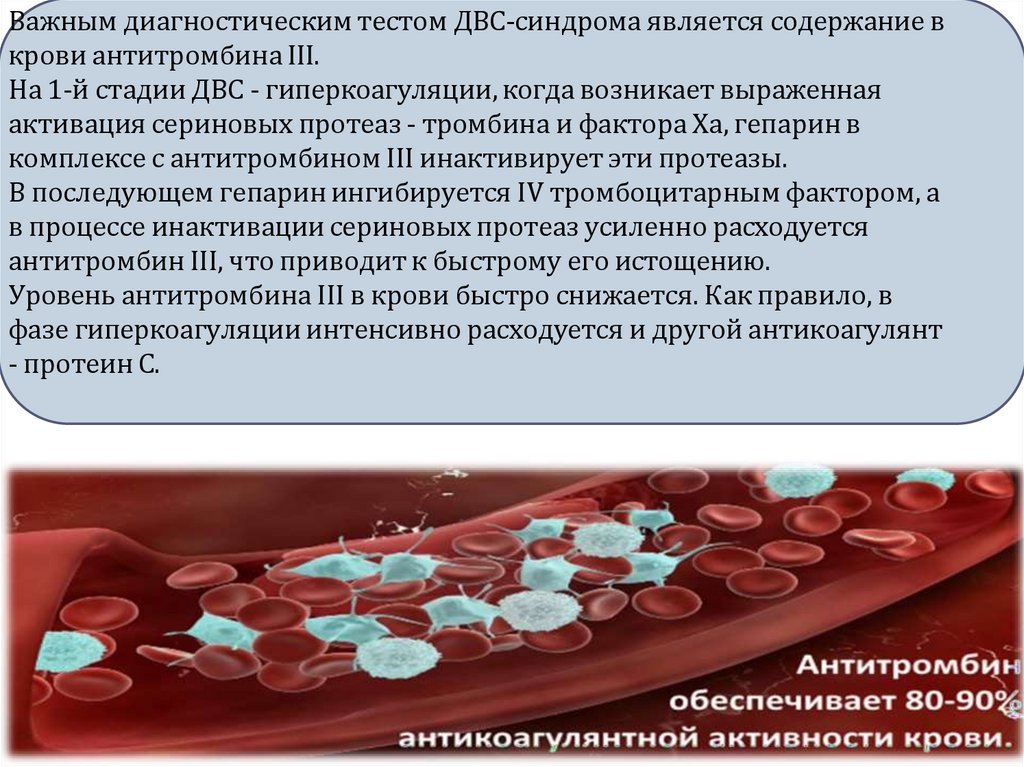
Важным диагностическим тестом ДВС-синдрома является содержание вкрови антитромбина III.
На 1-й стадии ДВС - гиперкоагуляции, когда возникает выраженная
активация сериновых протеаз - тромбина и фактора Ха, гепарин в
комплексе с антитромбином III инактивирует эти протеазы.
В последующем гепарин ингибируется IV тромбоцитарным фактором, а
в процессе инактивации сериновых протеаз усиленно расходуется
антитромбин III, что приводит к быстрому его истощению.
Уровень антитромбина III в крови быстро снижается. Как правило, в
фазе гиперкоагуляции интенсивно расходуется и другой антикоагулянт
- протеин С.
40.
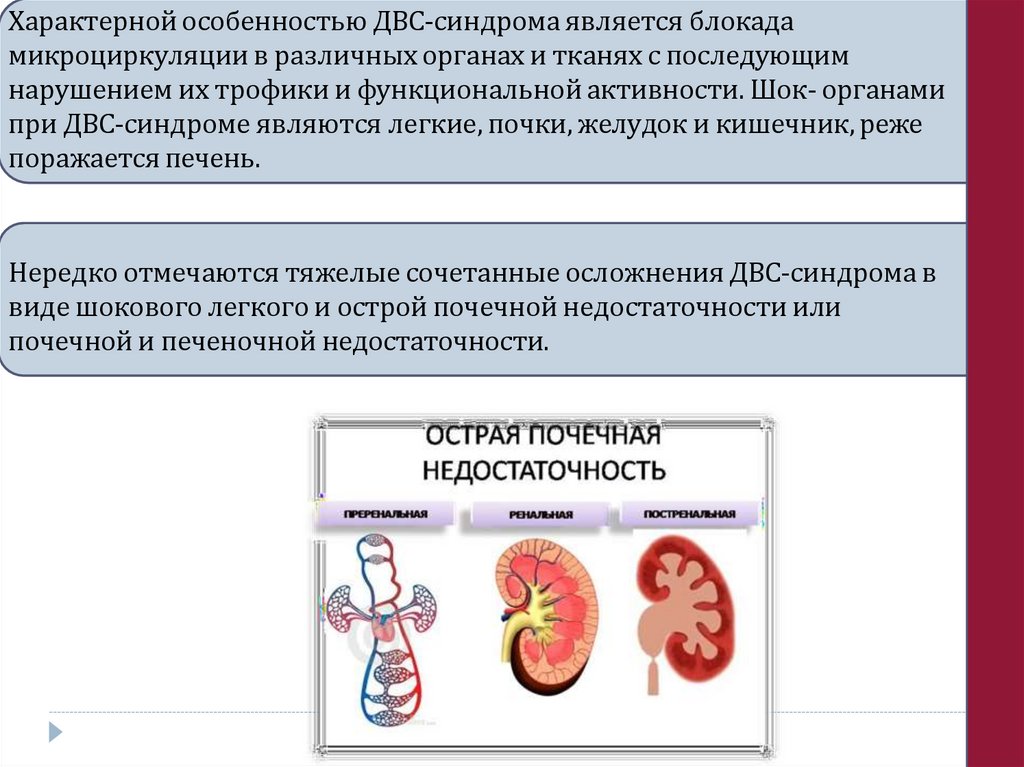
Характерной особенностью ДВС-синдрома является блокадамикроциркуляции в различных органах и тканях с последующим
нарушением их трофики и функциональной активности. Шок- органами
при ДВС-синдроме являются легкие, почки, желудок и кишечник, реже
поражается печень.
Нередко отмечаются тяжелые сочетанные осложнения ДВС-синдрома в
виде шокового легкого и острой почечной недостаточности или
почечной и печеночной недостаточности.
.