























 medicine
medicineSimilar presentations:

")



")




Семиотика нарушения обмена веществ
1.
Доцент каф. пропедевтики педиатрии,к.м.н. Г.А. Зубова
2.
1. новообразованияанаболизма
или
2.распада или
катаболизма
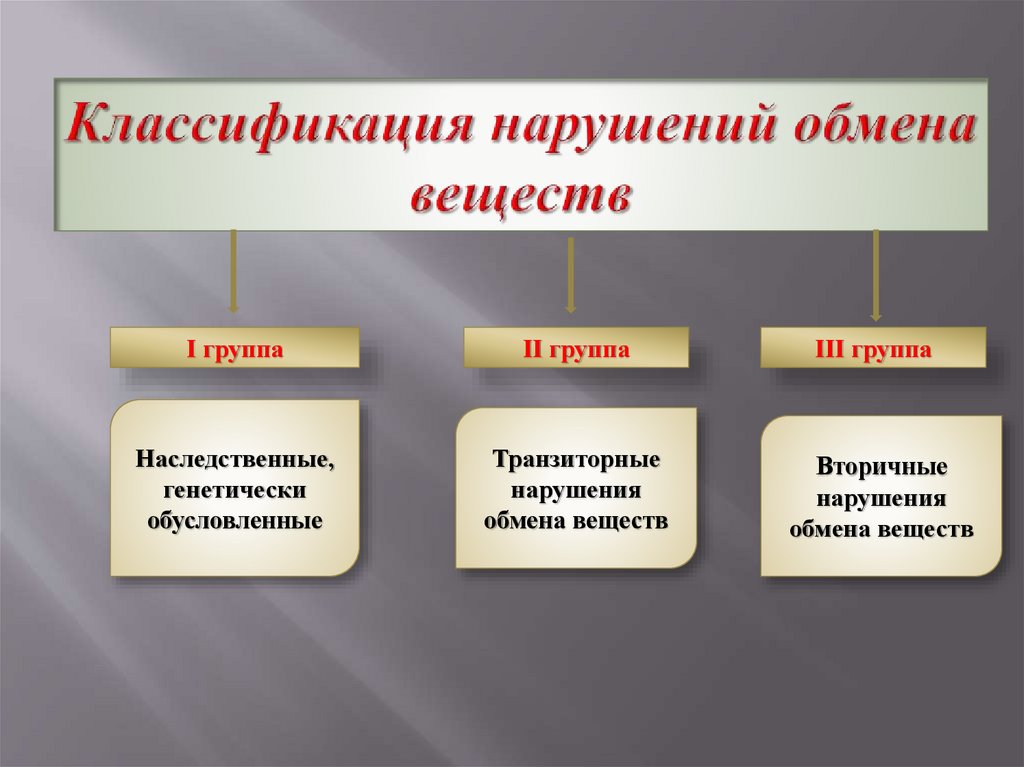
3.
І группаІІ группа
Наследственные,
генетически
обусловленные
Транзиторные
нарушения
обмена веществ
ІІІ группа
Вторичные
нарушения
обмена веществ
4.
Хотя отдельные наследственные нарушения обмена веществвстречаются редко, их общая распространенность превышает
1:500.
Наследственные нарушения обмена веществ обусловлены
мутациями — изменениями последовательности нуклеотидов
ДНК. Мутации приводят к синтезу дефектных белков (в том числе
— структурных белков, ферментов, гормонов, факторов роста,
белков-рецепторов). В частности, если мутация затрагивает ген,
кодирующий фермент, то последний частично или полностью
утрачивает каталитическую активность.
Подавляющее
большинство
наследственных
нарушений
метаболизма обусловлено генетическими дефектами ферментов,
участвующих в обмене аминокислот, углеводов и липидов.
Патогенез и клинические проявления определяются отсутствием
промежуточных или конечных нормальных метаболитов и
накоплением токсических метаболитов.
Клиническая картина в значительной мере зависит от степени
проявления (экспрессивности) мутантного гена, а также от других
генетических факторов и условий окружающей среды.
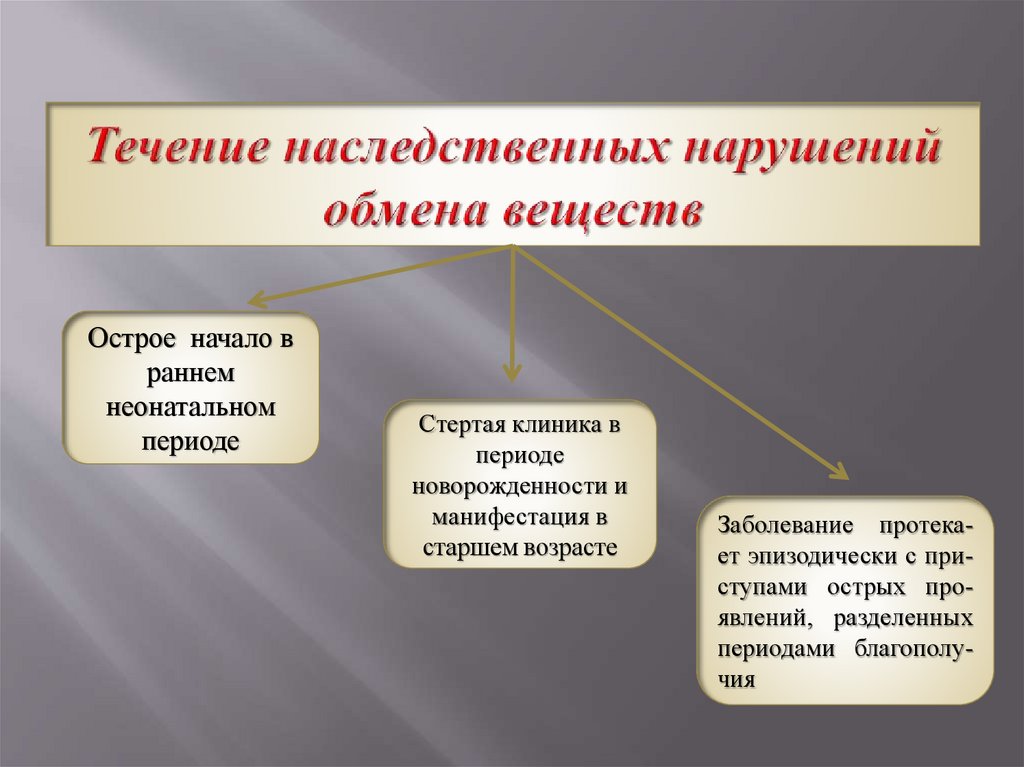
5.
Острое начало враннем
неонатальном
периоде
Стертая клиника в
периоде
новорожденности и
манифестация в
старшем возрасте
Заболевание протекает эпизодически с приступами острых проявлений, разделенных
периодами благополучия
6.
ЗаболеваниеДефицит ацил-СоА-дегидрогеназы
жирных кислот
Частота
1/9000 – 1/13000
Тирозинемия І
1/30000
Метилмалоновая ацидемия
1/48000
Синдром Цельвегера
1/50000
Дефицит биотинидазы
1/35000 – 1/54000
Болезнь с запахом кленового сиропа
1/200000
Пропионовая ацидемия и др.
1/350000
7.
Анамнез• Близкородственные браки
• Неврологические симптомы у одного из родителей
• Наличие сибса, страдающего неврологическим
заболеванием или умершего в неонатальном
периоде или младенчестве
• Несоответствие тяжести состояния новорожденного
благоприятному течению беременности
• Наличие «светлого промежутка»
продолжительностью в несколько дней от
Начало
рождения до клинических первых проявлений
заболевания заболевания
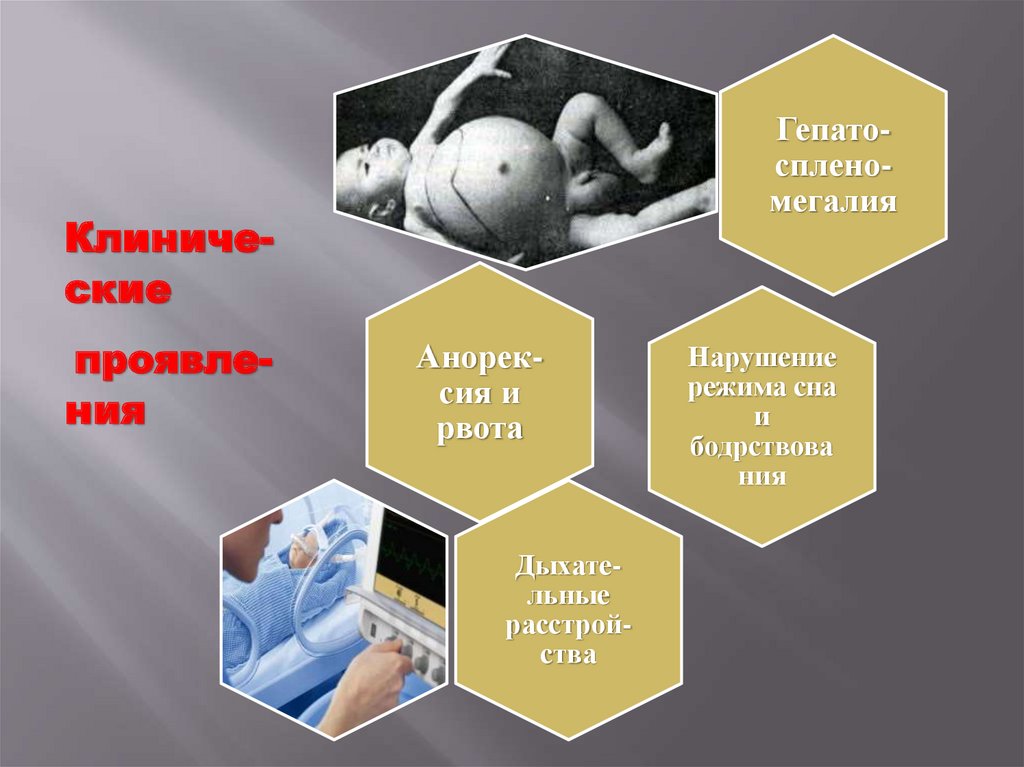
8.
ГепатоспленомегалияКлинические
проявления
Анорексия и
рвота
Дыхательные
расстройства
Нарушение
режима сна
и
бодрствова
ния
9.
Специфический запахмочи
Глутаровая ацидемия
Фенилкетонурия
Болезнь кленового сиропа
Метилкротонилглицинурия
Мальабсорбция метионина
Триметиламинурия
Тирозенемия
Болезнь хмелесушилки
Хокинсинурия
Запах потных ног
Мышиный запах
Запах кленового сиропа
Запах кошачей мочи
Капустный запах
Запах гниющей рыбы
Прогорклый рыбный запах
Запах хмеля
Запах плавательного
бассейна
10.
Симптомокомплекс«вялого ребенка»:
поза «лягушки»,
снижение сопротивления к
пассивным движениям,
гипермобильость суставов,
снижение
общей
двигательной активности,
задержка
моторного
и
психоневрологического
развития.
11.
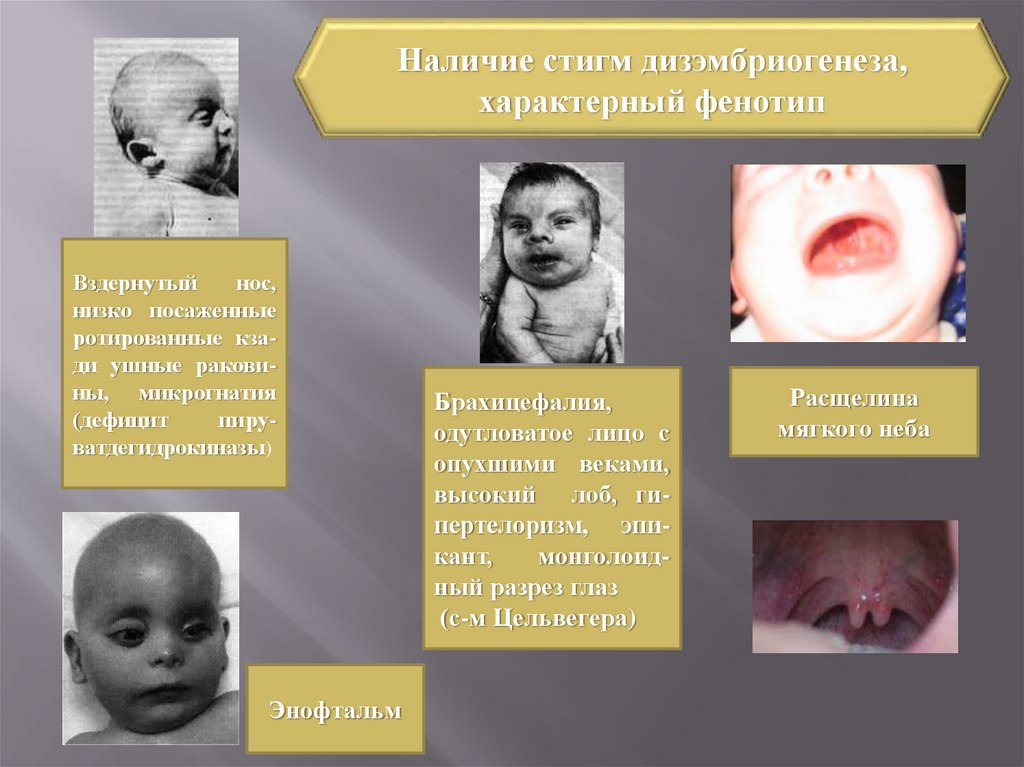
Наличие стигм дизэмбриогенеза,характерный фенотип
Вздернутый
нос,
низко посаженные
ротированные кзади ушные раковины, микрогнатия
(дефицит
пируватдегидрокиназы)
Энофтальм
Брахицефалия,
одутловатое лицо с
опухшими веками,
высокий лоб, гипертелоризм, эпикант,
монголоидный разрез глаз
(с-м Цельвегера)
Расщелина
мягкого неба
12.
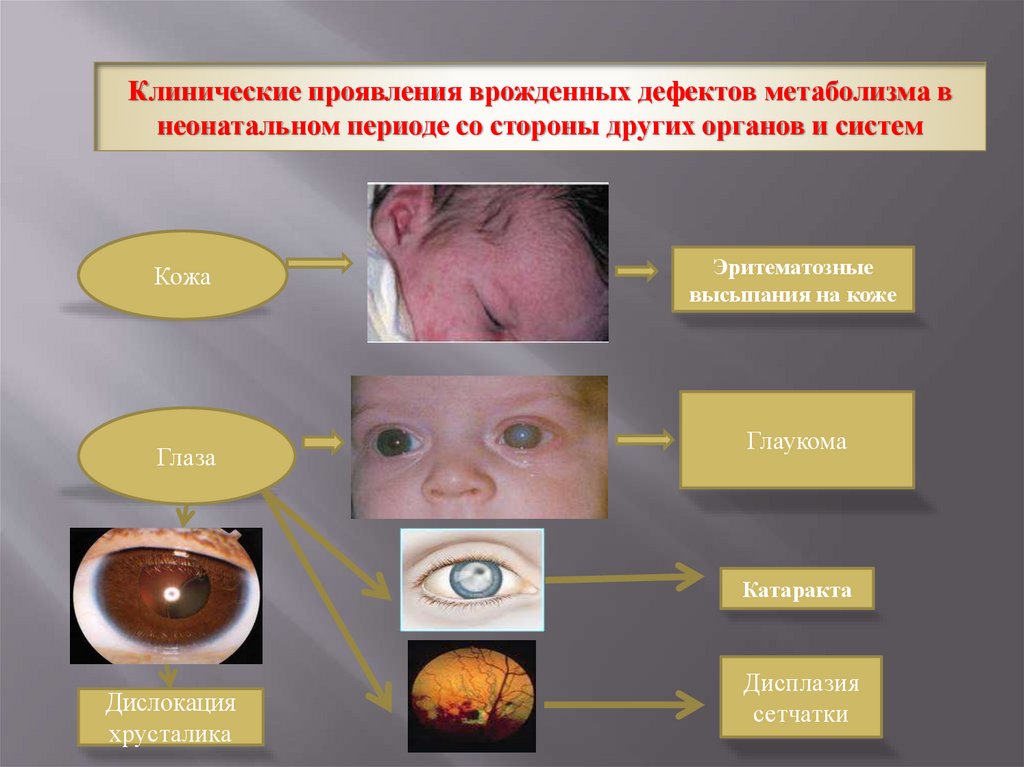
Клинические проявления врожденных дефектов метаболизма внеонатальном периоде со стороны других органов и систем
Кожа
Глаза
Эритематозные
высыпания на коже
Глаукома
Катаракта
Дислокация
хрусталика
Дисплазия
сетчатки
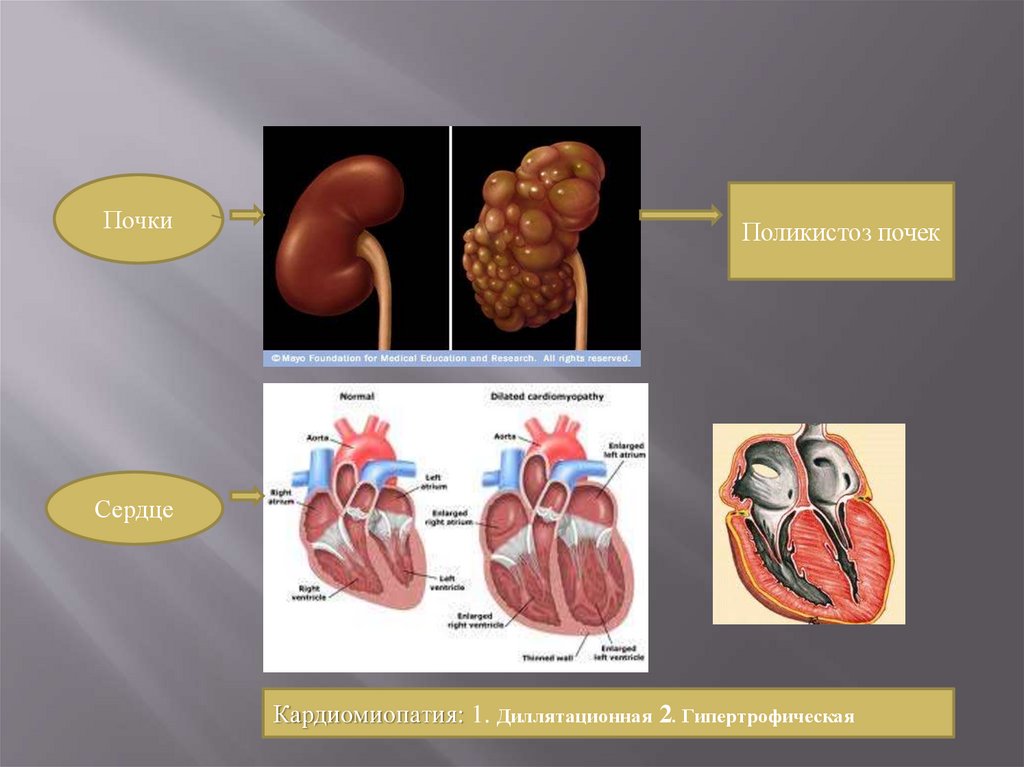
13.
ПочкиПоликистоз почек
Сердце
Кардиомиопатия: 1. Диллятационная 2. Гипертрофическая
14.
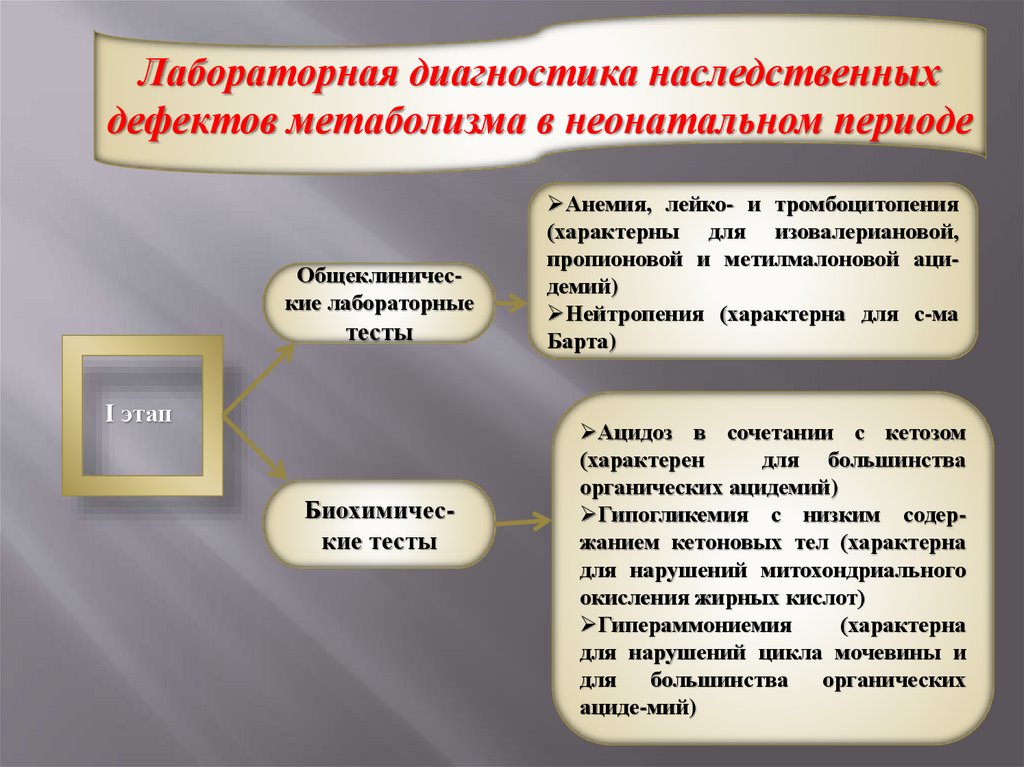
Лабораторная диагностика наследственныхдефектов метаболизма в неонатальном периоде
Общеклинические лабораторные
тесты
І этап
Биохимические тесты
Анемия, лейко- и тромбоцитопения
(характерны для изовалериановой,
пропионовой и метилмалоновой ацидемий)
Нейтропения (характерна для с-ма
Барта)
Ацидоз в сочетании с кетозом
(характерен
для большинства
органических ацидемий)
Гипогликемия с низким содержанием кетоновых тел (характерна
для нарушений митохондриального
окисления жирных кислот)
Гипераммониемия
(характерна
для нарушений цикла мочевины и
для большинства
органических
ациде-мий)
15.
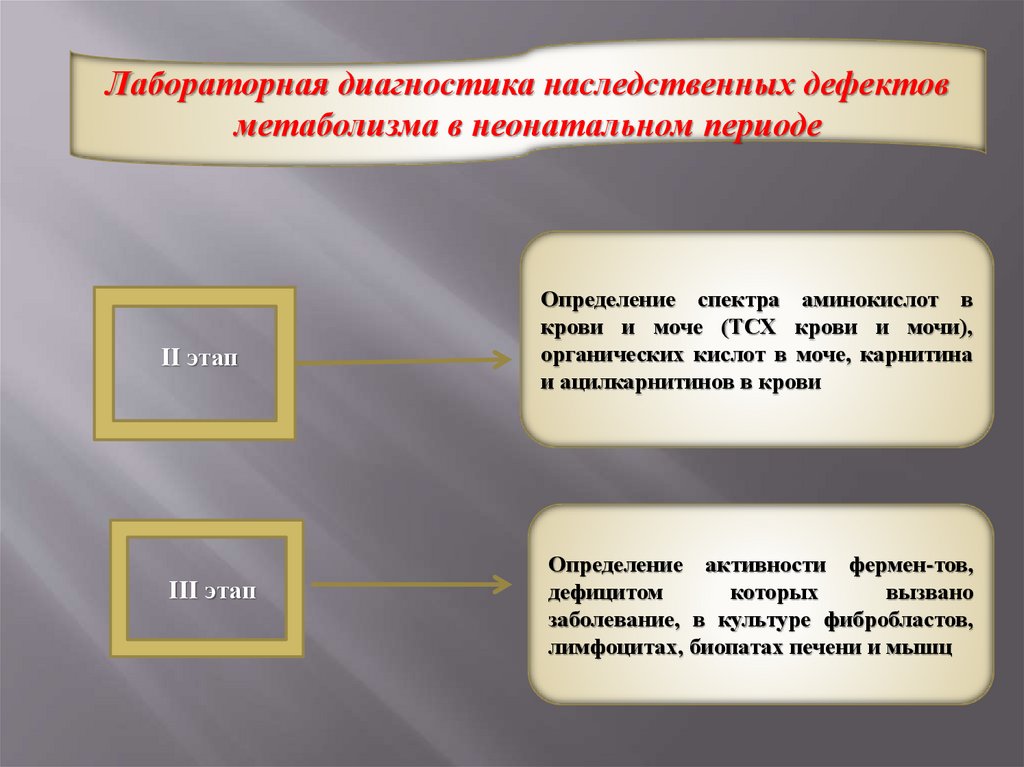
Лабораторная диагностика наследственных дефектовметаболизма в неонатальном периоде
ІІ этап
ІІІ этап
Определение спектра аминокислот в
крови и моче (ТСХ крови и мочи),
органических кислот в моче, карнитина
и ацилкарнитинов в крови
Определение активности фермен-тов,
дефицитом
которых
вызвано
заболевание, в культуре фибробластов,
лимфоцитах, биопатах печени и мышц
16.
Дифференциальный диагноз в неонатальном периодеСходные с наследственными дефектами метаболизма
клинические
симптомы
(неврологические
симптомы,
нарушение дыхания, сознания, судороги, мышечная гипотония,
рвота) часто встречаются при:
1. гипоксически-ишемических энцефалопатиях
2. пороках развития головного мозга
3. внутричерепных кровоизлияниях
4. внутриутробных инфекциях
17.
Дифференциальный диагноз наследственных заболеваний обменавеществ и гипоксически-ишемичкой энцефалопатией в неонатальном
периоде
Гипоксическиишемическая
энцефалопатия
Симптомы
Отягощенность родословной по неврологическим заболеваниям
Гипоксия плода
Отягощенное течение беременности и родов
Заболевания матери, вызывающие нарушения плацентарного кровотока
Мекониальное прокрашивание околоплодных вод
Недоношенность
Проявление неврологических нарушений сразу после
рождения
Черепно-лицевая дисморфия
Гепатомегалия,кардиомиопатия, поликистоз почек
НСГ изменения, характерные
ишемической энцефалопатии
для
гипоксически-
Наследственная
болезнь метаболизма
18.
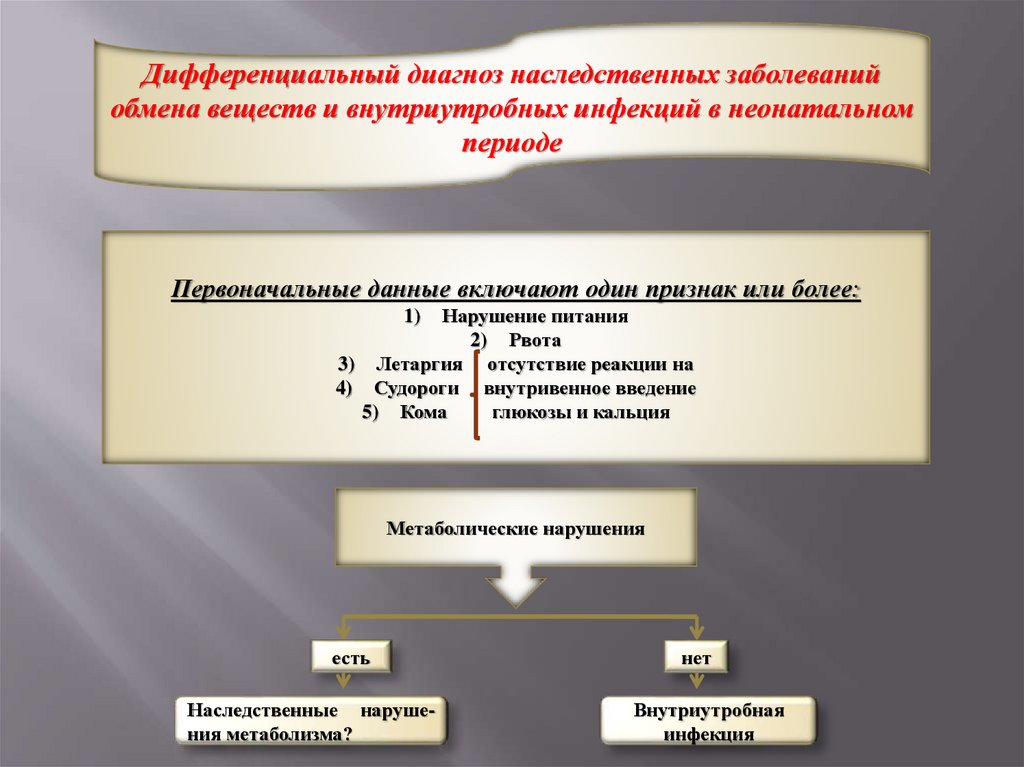
Дифференциальный диагноз наследственных заболеванийобмена веществ и внутриутробных инфекций в неонатальном
периоде
Первоначальные данные включают один признак или более:
Нарушение питания
2) Рвота
3) Летаргия отсутствие реакции на
4) Судороги внутривенное введение
5) Кома
глюкозы и кальция
1)
Метаболические нарушения
есть
Наследственные нарушения метаболизма?
нет
Внутриутробная
инфекция
19.
Врожденные нарушения метаболизма в некоторых случаях в неонатальномпериоде представляют собой более умеренно выраженные формы со стертым началом.
Поэтому они могут оставаться нераспознанными в периоде новорожденности, и диагноз
может быть поставлен только через несколько месяцев или лет. Ранние клинические
проявления обычно носят неспецифический характер и могут быть отнесены к
перинатальной патологии.
Кроме того, течение заболеваний, обусловленных врожденными нарушениями
метаболизма, может быть эпизодическим или интермиттирующим. Эпизоды заболевания
обычно провоцируются стрессовыми ситуациями или неспецифическими повреждающими
факторами.
В связи с этим врожденное нарушение метаболизма должно рассматриваться как
возможное состояние у любого ребенка с одним или более из указанных клинических
проявлений:
Необъяснимое отставание умственного и двигательного развития
Наличие необъяснимых судорог
Необычайный запах, особенно во время острого течения заболевания
Интермиттирующие эпизоды необъяснимой рвоты, ацидоза, нарушений
психики, комы
5) Гепатомегалия
6) уролитиаз
1)
2)
3)
4)
20.
Общим в проявлении нарушений обменавеществ среди детей ІІ и ІІІ групп является
то, что в основном наблюдаются соматические
нарушения,
нет
тяжелых
проявлений
поражения ЦНС, и при правильном ведении
пациентов
клиническая
симптоматика
полностью купируется без каких-либо грубых
последствий.
21.
22.
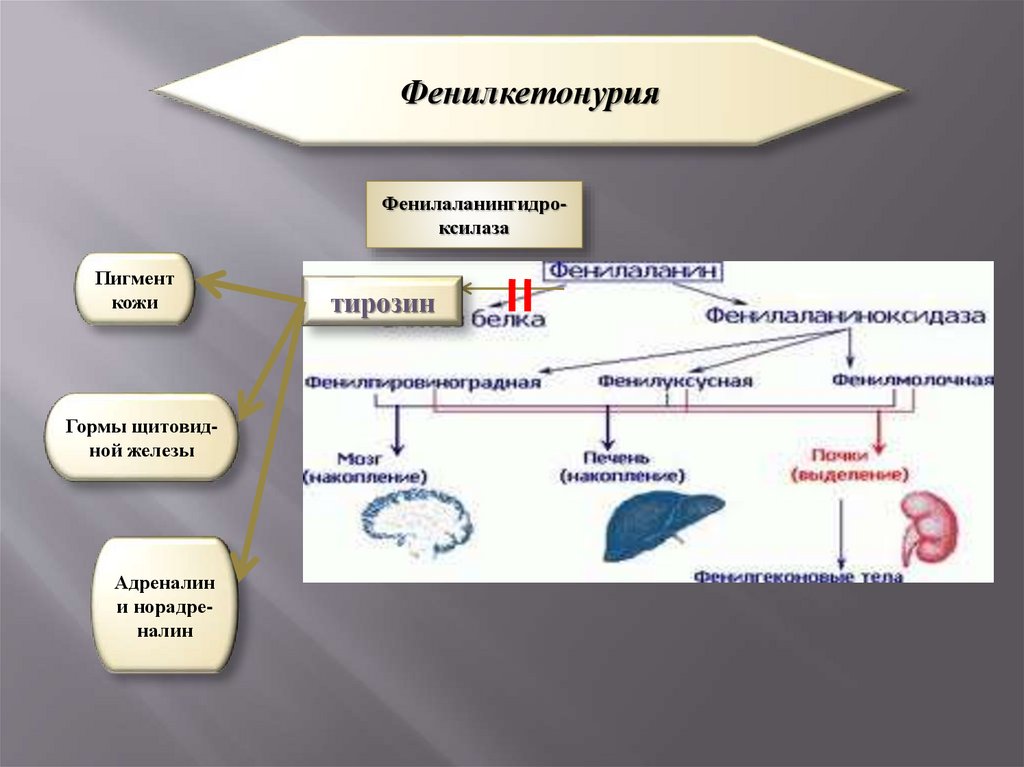
ФенилкетонурияФенилаланингидроксилаза
Пигмент
кожи
Гормы щитовидной железы
Адреналин
и норадреналин
тирозин
23.
Клиническая картина24.
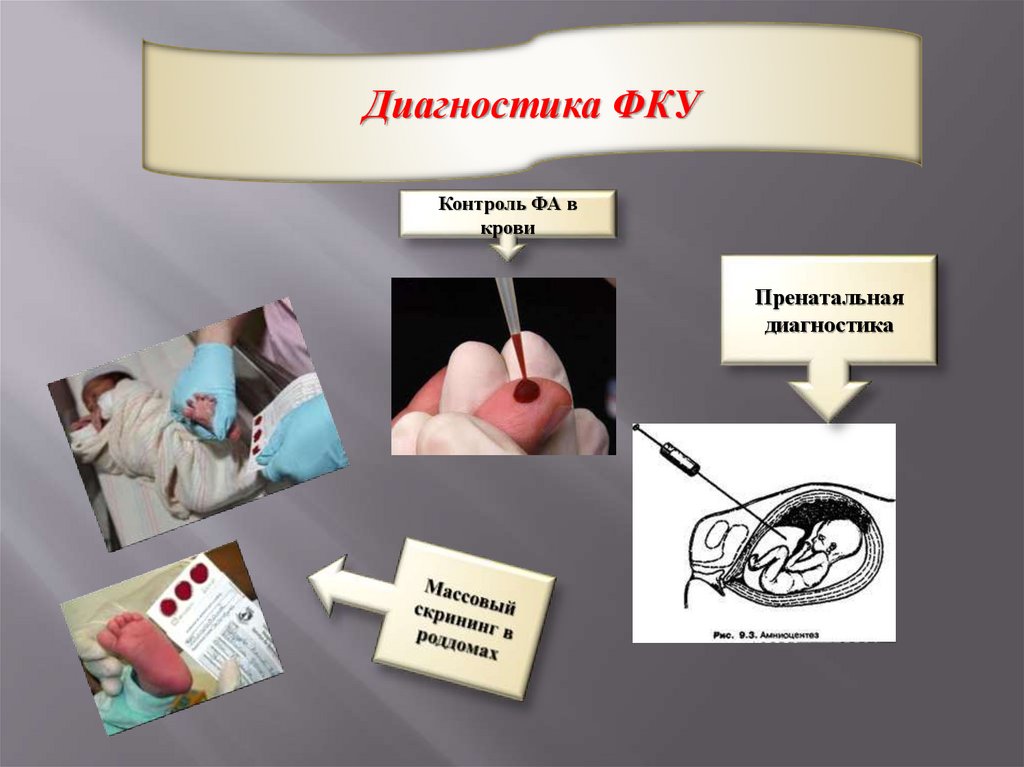
Диагностика ФКУКонтроль ФА в
крови
Пренатальная
диагностика
25.
Вскармливание детей с ФКУ26.
27.
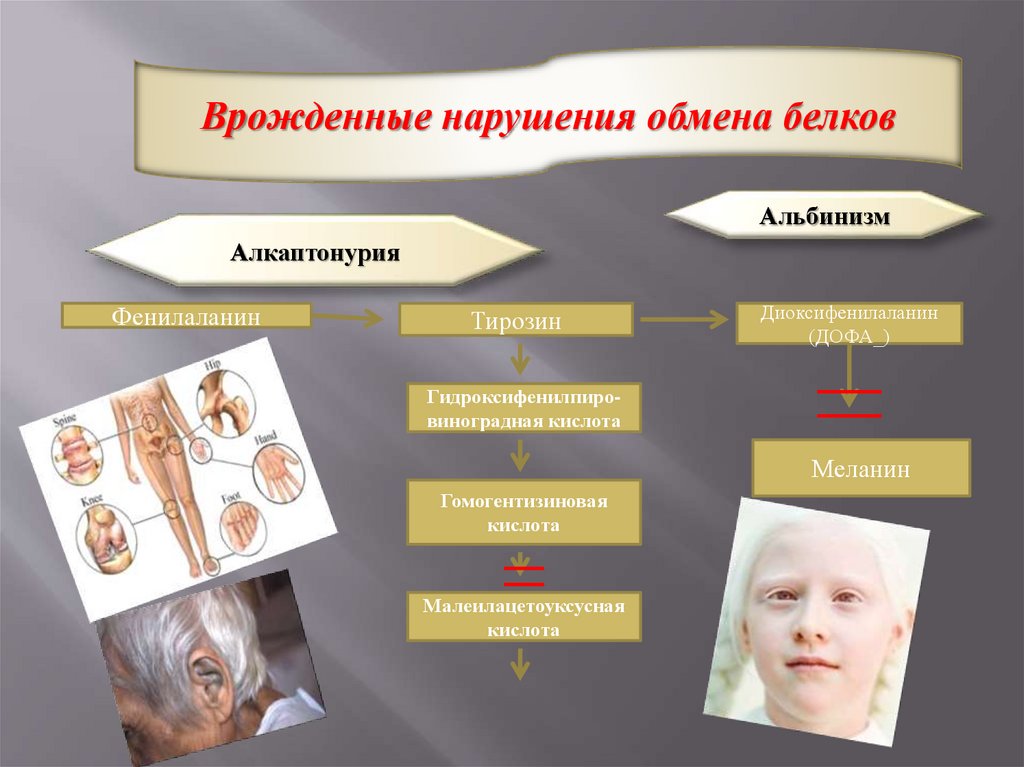
Врожденные нарушения обмена белковАльбинизм
Алкаптонурия
Фенилаланин
Тирозин
Диоксифенилаланин
(ДОФА_)
Гидроксифенилпировиноградная кислота
Меланин
Гомогентизиновая
кислота
Малеилацетоуксусная
кислота
28.
Общие клинические признаки дляаминоацидопатий
1. Наличие специфического запаха
2. Нервно-психические нарушения
3. Судорожный синдром
4. Изменения со стороны кожи и ее придатков
5. Поражение ЖКТ с вовлечением печени
6. Поражение почек
29.
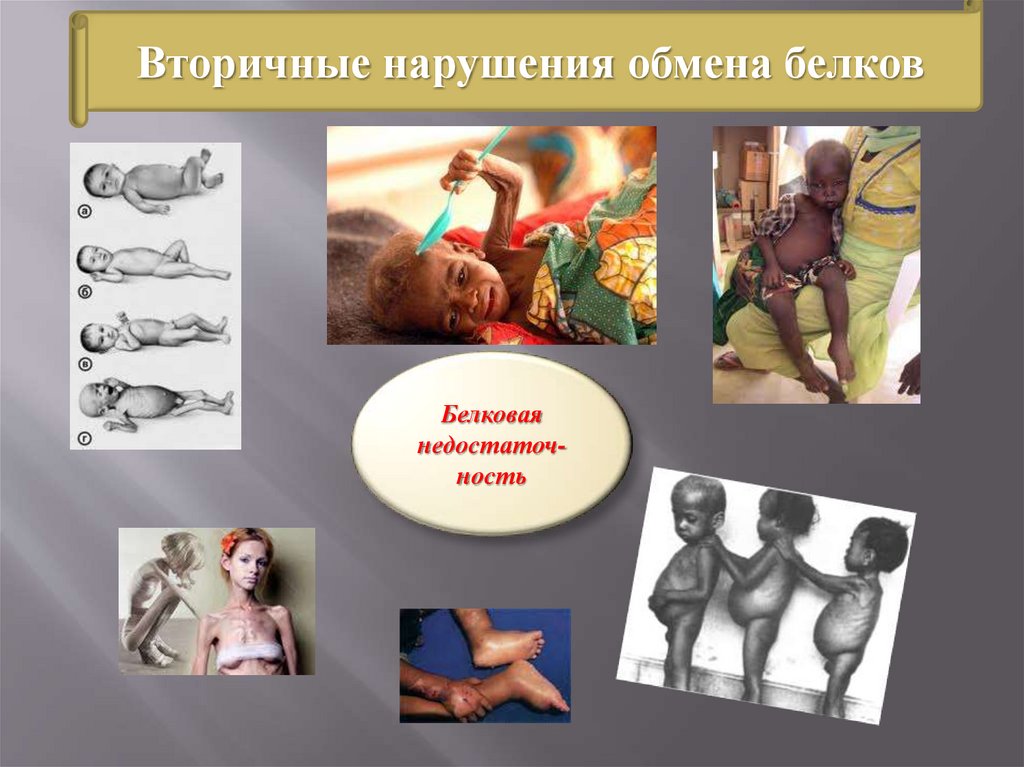
Вторичные нарушения обмена белковБелковая
недостаточность
30.
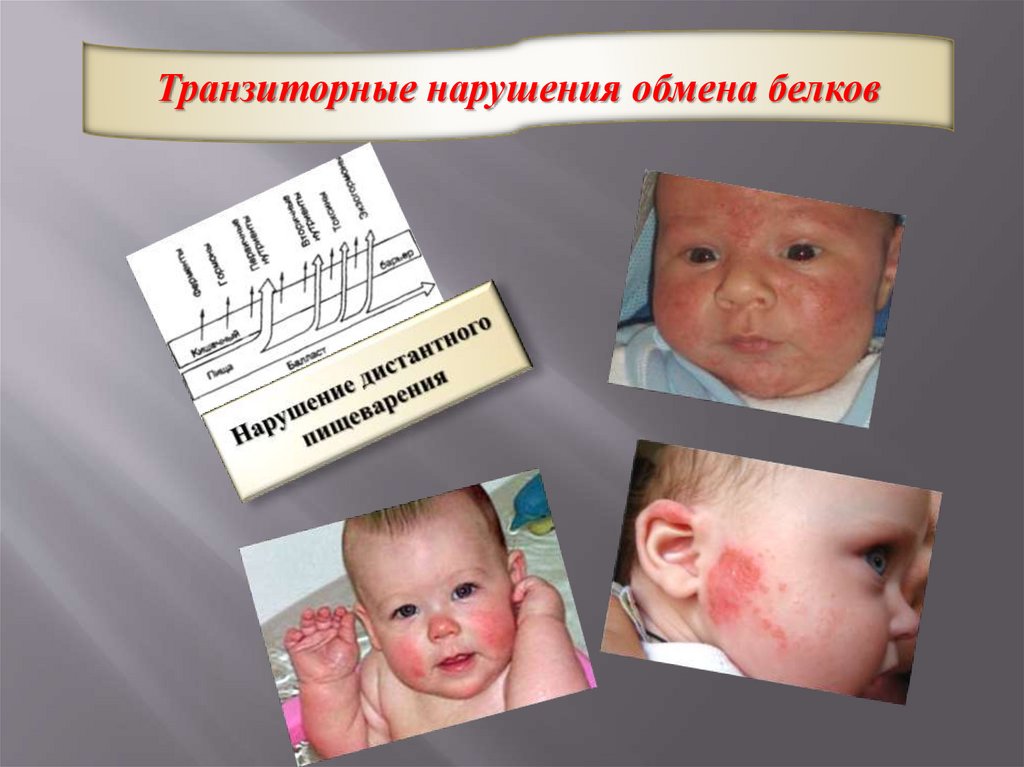
Транзиторные нарушения обмена белков31.
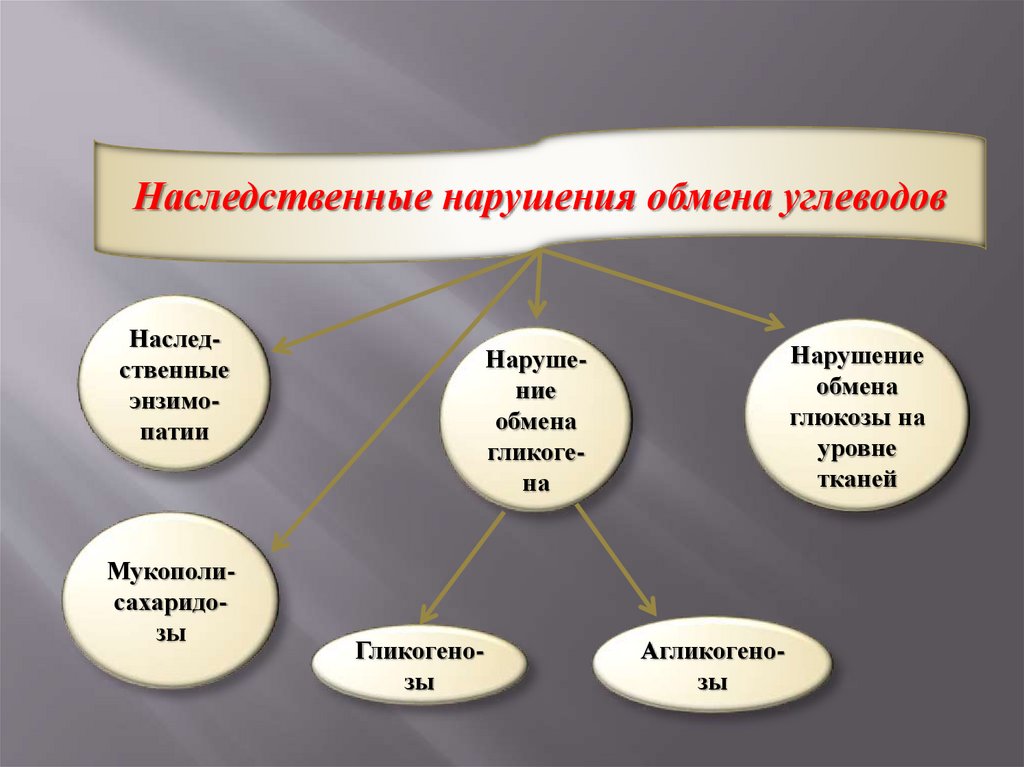
Наследственные нарушения обмена углеводовНаследственные
энзимопатии
Мукополисахаридозы
Нарушение
обмена
глюкозы на
уровне
тканей
Нарушение
обмена
гликогена
Гликогенозы
Агликогенозы
32.
Наследственные энзимопатииДефект
Клиническая картина
Дефицит сахаразы
При
употреблении
в
пищу
крахмалсодержащих продуктов или
сахарозы в виде столового сахара у
пациентов появляются боли в
животе, метеоризм, диарея. Молоко
переносится хорошо.
Непереносимость лактозы
Глюкозогалактозная
недостаточность
Метеоризм, диарея, боли в животе,
рвота после приема молока.
Метеоризм, диарея, дегидротация и
ацидоз при приеме молока и
глюкозы.
Фруктозу
переносят
хорошо.
33.
Нарушение обмена углеводов на уровне тканейГалактоземия
5.
1. Желтуха
2. Гепатомегалия
3. Рвота
4. Гипогликемия
Задержка физического развития
6. Катаракта
7. Цирроз печени
8. Асцит
9. Спленомегалия
10. Умственная отсталость
34.
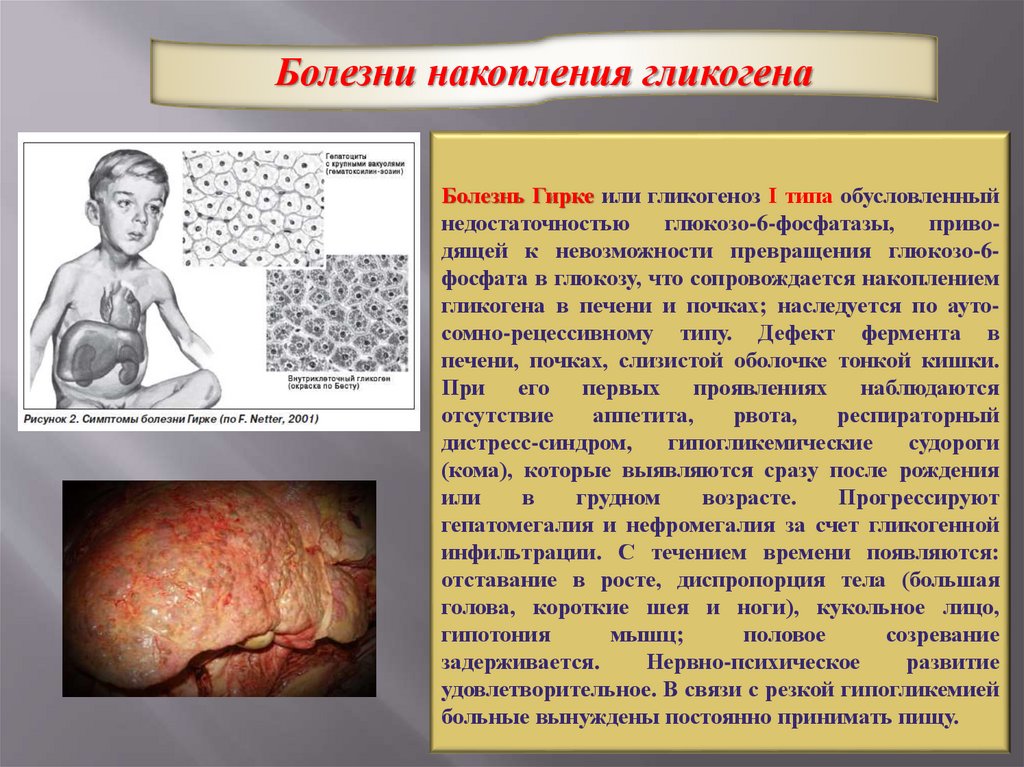
Болезни накопления гликогенаБолезнь Гирке или гликогеноз І типа обусловленный
недостаточностью
глюкозо-6-фосфатазы,
приводящей к невозможности превращения глюкозо-6фосфата в глюкозу, что сопровождается накоплением
гликогена в печени и почках; наследуется по аутосомно-рецессивному типу. Дефект фермента в
печени, почках, слизистой оболочке тонкой кишки.
При его первых проявлениях наблюдаются
отсутствие
аппетита,
рвота,
респираторный
дистресс-синдром,
гипогликемические
судороги
(кома), которые выявляются сразу после рождения
или
в
грудном
возрасте.
Прогрессируют
гепатомегалия и нефромегалия за счет гликогенной
инфильтрации. С течением времени появляются:
отставание в росте, диспропорция тела (большая
голова, короткие шея и ноги), кукольное лицо,
гипотония
мышц;
половое
созревание
задерживается.
Нервно-психическое
развитие
удовлетворительное. В связи с резкой гипогликемией
больные вынуждены постоянно принимать пищу.
35.
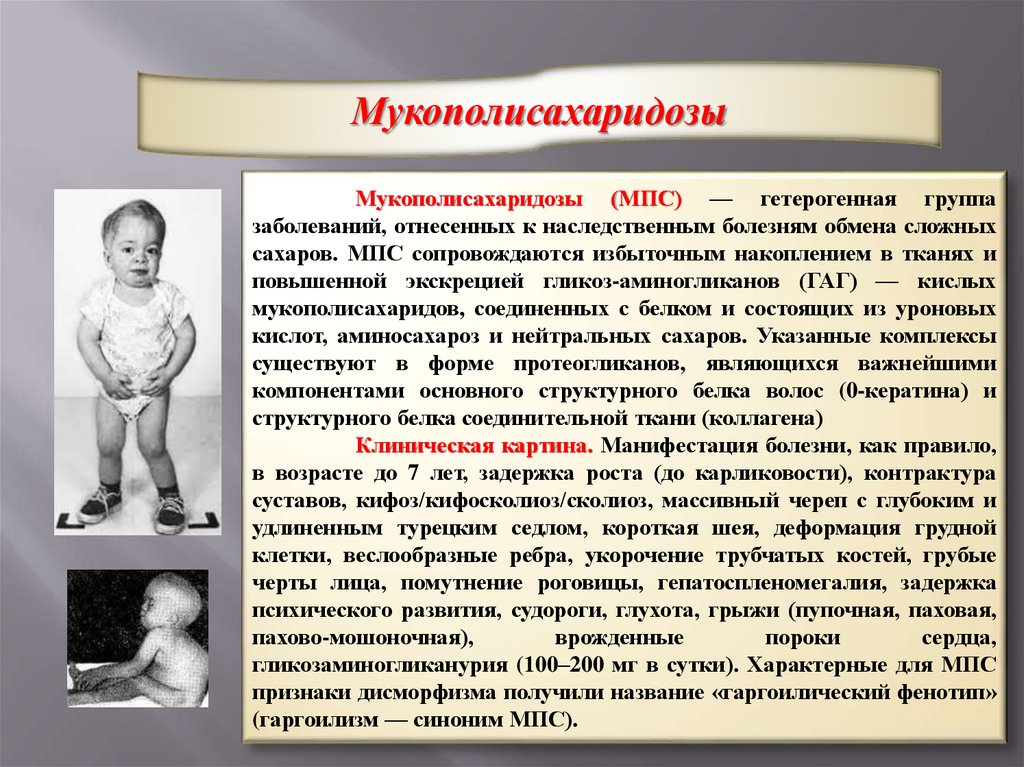
МукополисахаридозыМукополисахаридозы (МПС) — гетерогенная группа
заболеваний, отнесенных к наследственным болезням обмена сложных
сахаров. МПС сопровождаются избыточным накоплением в тканях и
повышенной экскрецией гликоз-аминогликанов (ГАГ) — кислых
мукополисахаридов, соединенных с белком и состоящих из уроновых
кислот, аминосахароз и нейтральных сахаров. Указанные комплексы
существуют в форме протеогликанов, являющихся важнейшими
компонентами основного структурного белка волос (0-кератина) и
структурного белка соединительной ткани (коллагена)
Клиническая картина. Манифестация болезни, как правило,
в возрасте до 7 лет, задержка роста (до карликовости), контрактура
суставов, кифоз/кифосколиоз/сколиоз, массивный череп с глубоким и
удлиненным турецким седлом, короткая шея, деформация грудной
клетки, веслообразные ребра, укорочение трубчатых костей, грубые
черты лица, помутнение роговицы, гепатоспленомегалия, задержка
психического развития, судороги, глухота, грыжи (пупочная, паховая,
пахово-мошоночная),
врожденные
пороки
сердца,
гликозаминогликанурия (100–200 мг в сутки). Характерные для МПС
признаки дисморфизма получили название «гаргоилический фенотип»
(гаргоилизм — синоним МПС).
36.
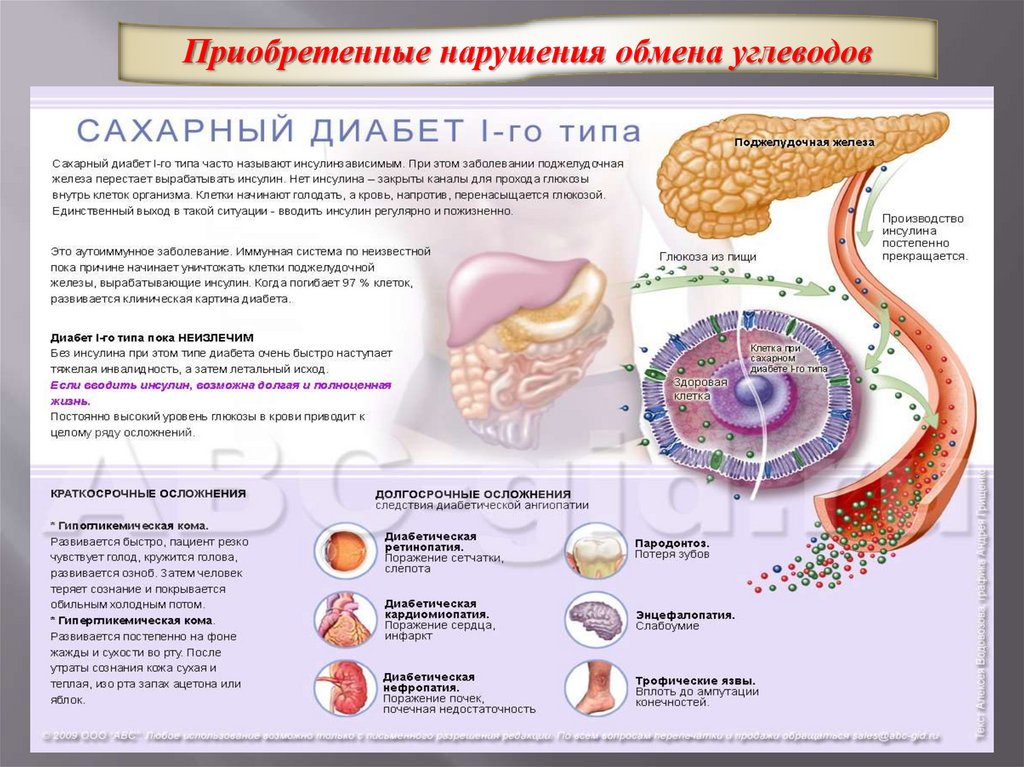
Приобретенные нарушения обмена углеводов37.
Наследственные нарушения обмена жировЛипидозы
К генетическим заболеваниям, особенно распространенным среди
ашкеназов, относятся болезнь Тея-Сакса, истощающая организм ребенка и обычно
приводящая к летальному исходу в возрасте четырех лет, а также синдром Канавана. Это
заболевание, как правило, проявляется у детей в возрасте от 2 до 4 месяцев, и они
начинают забывать ранее полученные навыки. Большинство детей умирает в возрасте до
5 лет. Носителем гена этого заболевания является один из 40 евреев-ашкеназов.
Такого рода заболевания приводят к повреждению молекул жирных кислот, именуемых
сфинголипидами, которые играют важную роль в передаче клеточного сигнала и в
клеточном распознавании. Известно только 108 генов (из более 20 тысяч, наследуемых
человеком), которые могут привести к сфинголипидному метаболизму. Кохран рассчитал,
что вероятность рождения больного ребенка, при условии, что у одного из родителей
была генетическая предрасположенность к определенной болезни, составляет
приблизительно 1:100 000.
38.
ЛипидозыБолезнь Нимана-Пика. (синоним: сфингомиелолипидоз) Описана в 1914 г. A. Niemann
и в 1926 г. L. Pick.
Минимальные диагностические признаки:
гепатоспленомегалия, «пенистые»
клетки в костном мозге, печени и селезенке, накопление сфингомиелина в
ретикулоэндотелиальных клетках и клетках других органов.
Клиническая характеристика
Выделяют несколько форм заболевания, различающихся клинически
(временем начала, течением и тяжестью неврологических и висцеральных проявлений) и
имеющих, по-видимому, различную генетическую природу. Общими для всех форм
симптомами являются увеличение печени и селезенки, генерализованное увеличение
лимфатических узлов. Обычно отмечаются побочные признаки гиперспленизма.
Характерна инфильтрация легких, выявляемая рентгенологически.
Неврологические симптомы (отсутствующие при висцеральной форме
заболевания, тип В) включают задержку психомоторного развития, атаксию, судороги,
снижение мышечного тонуса и угнетение сухожильных рефлексов. У некоторых больных
при исследовании глазного дна обнаруживают симптом «вишневой косточки». Иногда
отмечаются небольшие или нодулярные ксантомы на коже.
39.
ЛипидозыБолезнь Гоше - редкое наследственное системное заболевание обмена
липидов, характеризующееся накоплением цереброзидов в клетках селезенки, печени,
костного мозга, лимфатических узлов. Впервые описана в 1882 г. Gaucher. Различают острую
форму (детский тип), передающуюся аутосомно-рецессивно, и хроническую форму
(ювенильный тип), наследуемую по доминантному типу с неполной пенетрантностью гена.
Симптомы болезни Гоше. Острой формой болеют только дети грудного возраста.
Начинаясь уже в первые месяцы жизни, заболевание характеризуется задержкой
физического и нервно-психического развития ребенка. Отмечаются лихорадка,
значительное увеличение объема живота (вследствие спленогепатомегалии), симптомы
дыхательной недостаточности (цианоз, кашель), отечность суставов и болезненность в
области трубчатых костей, усиливающаяся при движении, остеопороз, декальцификация.
Возможны спонтанные переломы костей. Увеличение лимфатических узлов встречается
редко. Характерны своеобразная коричневая окраска кожи, а также петехиальные
высыпания в области лица и кистей, иногда других участков тела. У всех больных
обнаруживают анемию, лейкопению, тромбоцитопению. Содержание липидов и холестерина
в крови не увеличено. Наблюдаются многочисленные неврологические симптомы гипертония мышц, тризм, опистотонус, затруднения глотания, косоглазие, слепота,
тонические и клонические судороги, параличи различной локализации. Диагноз ставят на
основании результатов клинико-рентгенологического обследования и обнаружения в
стернальном пунктате характерных клеток Гоше - округлой формы и крупных размеров,
наполненных цереброзидами.
40.
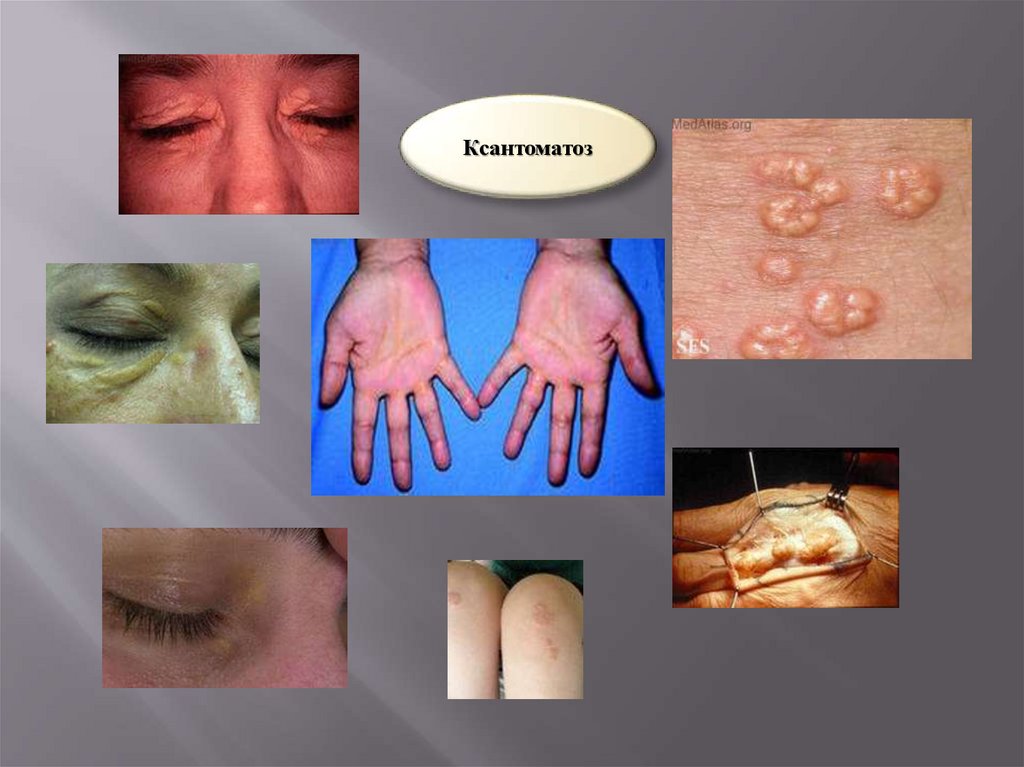
Нарушение обмена липидовГиперлипидемии
Тип
Липидограмма
Клинические симптомы
І
ХМ , холестерин-N, αи β-липопротеиды
Носит семейный характер. Наблюдаются
частые
«абдоминальные
кризы», ксантоматоз
ІІ
β-липротеиды и преβ-липопротеиды
Ксантоматоз, атеросклероз, ИБС
ІІІ
Холестерин
и
триглецириды
Атеросклероз, ИБС, сахарный диабет,
перемежающаяся
хромота,
ксантоматоз
ІУ
пре-β-липопротеиды
и триглицириды
Сахарный диабет, ожирение, рано
начинается ИБС
У
пре-β-липопротеиды
и ХМ
Абдоминальные колики, спленомегалия, ксантоматоз, сахарный диабет
41.
Ксантоматоз42.
43.
Анамнез• Близкородственные браки
• Неврологические симптомы у одного из родителей
• Наличие сибса, страдающего неврологическим
заболеванием или умершего в неонатальном периоде или
младенчестве
• Несоответствие тяжести состояния новорожденного
благоприятному течению беременности
Начало
заболевания
• Наличие «светлого промежутка»
продолжительностью в несколько дней
от рождения до клинических первых
проявлений заболевания
Клинические
симптомы
Нарушение режима сна и бодрствования
Неонатальные судороги
Гепатоспленомегалия
Анорексия и рвота
Дыхательные нарушения
Специфический запах мочи