




























































































 medicine
medicineSimilar presentations:










Моногенные наследственные болезни обмена веществ
1.
Мустафин Рустам Наилевич,доцент кафедры МГиФМ
2.
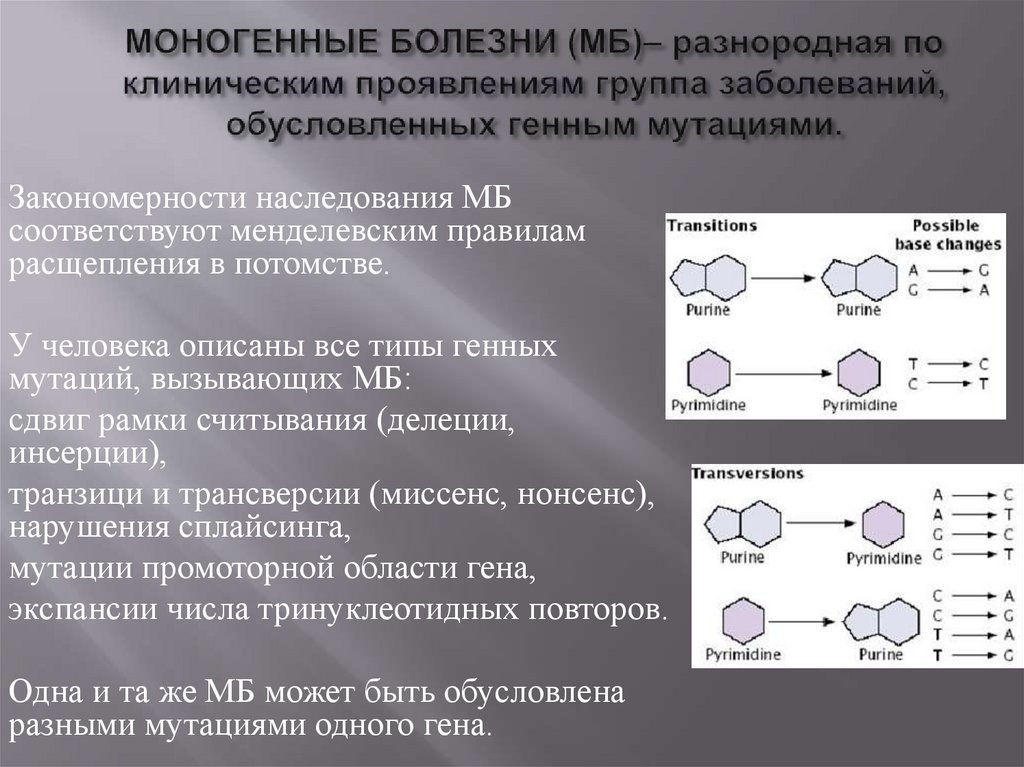
Закономерности наследования МБсоответствуют менделевским правилам
расщепления в потомстве.
У человека описаны все типы генных
мутаций, вызывающих МБ:
сдвиг рамки считывания (делеции,
инсерции),
транзици и трансверсии (миссенс, нонсенс),
нарушения сплайсинга,
мутации промоторной области гена,
экспансии числа тринуклеотидных повторов.
Одна и та же МБ может быть обусловлена
разными мутациями одного гена.
3.
Ген–
структурно-функциональная
единица
наследственности,
участок
молекулы
ДНК,
кодирующий
определенный
белок
или
функциональную РНК.
Гены, которые занимают идентичные локусы в
гомологичных
хромосомах,
называются
аллельными. У каждого человека есть по два
аллельных гена.
Аллели (греч. allenon — различные формы) — это
альтернативные
формы
гена,
определяющие
альтернативные формы одного и того же признака.
Формы взаимодействия между аллельными генами:
полное доминирование,
неполное доминирование,
кодоминированием и
сверхдоминирование.
4.
Основная форма взаимодействия - полное доминирование,которое впервые описано Г. Менделем. В гетерозиготном
организме проявление одной из аллелей доминирует над
проявлением другой.
Неполное доминирование - форма взаимодействия, при
которой у гетерозиготного организма (Аа) доминантный ген
(А) не полностью подавляет рецессивный ген (а), вследствие
чего проявляется промежуточный между родительскими
признак.
При кодоминировании в гетерозиготных организмах
каждый из аллельных генов вызывает формирование
зависимого от него продукта, то есть оказываются
продукты обеих аллелей. Классическим примером такого
проявления является система групп крови, в частности
система АBО, когда эритроциты человека несут на
поверхности антигены, контролируемые обеими аллелями
(IV группа).
Сверхдоминирование - когда доминантный ген в
гетерозиготном состоянии проявляется сильнее, чем в
гомозиготном. Так, у дрозофилы при генотипе ААнормальная продолжительность жизни; Аа - удлиненная
жизнь; аа - летальный исход.
5.
Плейотропное действие генов - этозависимость нескольких признаков от одного
гена, то есть множественное действие одного
гена.
Пенетрантность - это частота проявления
гена, появления или отсутствия признака у
организмов, одинаковых по генотипу (%
людей с проявлением болезни).
Экспрессивность (лат. ехргеssio выражение) - это изменение
количественного проявления признака
(степень выраженности болезни) в разных
особях-носителях соответствующего аллеля.
6.
Online Mendelian Inheritance in Man – OMIMwww. Omim.org (Менделевское наследование у человека)
Для каждой болезни суммированы клинические и молекулярногенетические данные (о картировании, идентификации гена, практических
возможностях генодиагностики). База находится в Национальном центре
биотехнологической информации (США). Адрес в Интернете:
www.ncbi.nlm.nih.gov/omim/
Gene Clinics
National Newborn Screening and Genetics Resource
Center web site: NNSGRC –
www.geneclinics.org
www.genes-r-us.uthscsa.edu/
Alliance of Genetic Support Groups
www.medhlp.netusa.net/www/agsg.htm
7.
Классификация наследственных болезней обмена веществ:1) Болезни углеводного обмена:
Галактоземия (мутации гена галактозо-1-фосфатуридилтрансферазы, 9р13, ЧВ=1:15 000)
Фруктоземия (мутации гена альдолазы В, 9q31, ЧВ=1:20000)
Гликогенозы
2) Болезни обмена аминокислот:
фенилкетонурия (мутации гена фенилаланин-4-гидроксилазы, 12q22-24.2, ЧВ=1:10 000)
тирозинемия (мутации гена FAH, 15q23-25, ЧВ=1:100 000).
лейциноз (мутации гена ферментов окислительного декарбоксилирования лейцина, изолейцина, валина; ЧВ=120 000).
гомоцистинурия (мутации гена цистатоинин-бета-синтазы, 21q22.1, ЧВ=1:200 000).
3) Болезни обмена органических кислот:
алкаптонурия (мутации гена оксидазы гомогентезиновой кислоты, 3q21-23, АР т/н, ЧВ=100 000 ).
4) Болезни обмена жирных кислот:
дефицит ацетил-КоА-дегидрогеназы коротких цепей (мутации гена VLCAD, 17p13, ЧВ=1:50 000).
5) Болезни обмена пуринов и пиримидинов:
болезнь Леша-Нихена (мутации гена гипоксантин-гуанинфосфорибозил-трансферазы, Хq26, ЧВ=1:300 000).
6) Болезни обмена порфиринов:
острая перемежающая порфирия (мутация гена синтазы уропорфириногена-1, 11q23.2, ЧВ=1:100 000).
7) Болезни обмена стероидов:
врожденная гиперплазия надпочечников (мутации гена стероид-21-гидроксилазы, 6р21.13, 1:15 000).
врожденный гипотиреоз (мутации гена тиреоидного фактора транскрипции TITF1, 14q13-21, ЧВ=1:5000).
8) Митохондриальные болезни: синдром Кернса-Сэйра; синдром MELAS; синдром MERRF
8.
9) Пероксисомные болезни:синдром Цельвегера (мутации в генах пероксинов, ЧВ=1:50 000)
синдром Рефсума (мутации в гене фитаноил-КоА-гидроксилазы, 6q22-24, ЧВ=1:25000)
10) Лизосомальные болезни:
Мукополисахаридозы: синдромы 1-Гурлер 4p16.2, 2-Хантера Xq28, 3-Санфилиппо 17q25.3, 4Моркио 16q24.3, 6-Марото-Лами 5q14.1, 7-Слая 7q11.21, 9-Натовича 3p21.31.
Сфинголипидозы: болезни Ниманна-Пика (11р15,1:40 000), Фабри (Хq22.1, 1:40 000) , Тея-Сакса
(15q23, 1:320 000), Гоше (1q22, 1:50 000), Краббе (14q31, 1:100 000).
11) Болезни обмена липидов:
наследственные гиперлипидемии (тип I - гиперхиломикронемия, IIa - гиперхолестеринемия, III дис-бета-липопротеинемия, V – гипертриглицеридемия)
12) Болезни обмена циркулирующих белков:
гемоглобинопатии (серповидноклеточная анемия, талассемии).
13) Нарушения обмена металлов: болезнь Вильсона-Коновалова (13q14.3, 1:30 000).
14) Болезни обмена факторов свертывания: гемофилия А (Xq28, ген фактора VIII), гемофилия В
(Xq27, ген фактора IX), гемофилия С (АР т/н, ген фактора XI)
15) Нарушения транспорта ионов: муковисцедоз (7q31, 1:5000).
16) Компонентов соединительной ткани (ахондроплазия, синдром Марфана и Элерса-Данло).
9.
В России проводится массовый скрининг новорожденных (на 4 сутки послерождения (у недоношенных на 7) натощак берется кровь из пяточной области и
наносится на 5 отдельных бланков фильтровальной бумаги) на 5
наследственных болезней обмена веществ:
Фенилкетонурия (ЧВ=1:10 000).
Муковисцедоз (ЧВ= 1:5 000).
Врожденный гипотиреоз
(ЧВ=1:5 000).
Адреногенитальный синдром
(ЧВ=1:15 000).
Галактоземия (ЧВ=1:15 000).
10.
В США – ОБ – с частотой встречаемости реже, чем1:1500 населения.
В Японии – реже, чем 1:2500 населения.
В России – реже, чем 1:10000 человек.
В мировой медицинской литературе – ОБ – с
частотой встречаемости от 1:1000 до 1:200000
населения.
11.
Углеводами называют органические вещества, которые содержат в своем составекарбонильную (=С=О) группу и несколько гидроксильных (ОН) групп. Различают
моно-, ди-, олиго- и полисахариды.
Моносахариды могут состоять из углеродной цепи, содержащей от 3 до 10 атомов
углерода. В соответствии с этим их называют триозам (3 атома С), тетрозами (4),
пентозами (5), гексозами (6) и т.д.
Углеводы могут входить в структуру сложных белков – если основную часть
молекулы составляют углеводы (около 90 – 95%), эти соединения называют
протеогликанами, если же углеводы занимают МЕНЬШЕ 40% общей массы
молекулы, они называются гликопротеинами.
12.
ГАЛАКТОЗЕМИЯ (E74.2).Впервые описана в 1908 г. А. Рессом.
АР тип наследования, частота1:15 000.
Дефицит ферментов, превращающих галактозу в глюкозу:
I тип (классическая галактоземия) обусловлен дефектом фермента галактозо-1фосфатуридилтрансферазы (ген GALT, 9р13).
II тип вызван дефектом фермента галактокиназы (ген GALK1, 17q25).
III тип – фермента уридинфосфат-галакто-4-эпимеразы (ген GALE, 1р36).
13.
ПАТОГЕНЕЗТоксическое действие накапливающейся в результате метаболического блока галактозы и галактозо-1фосфата на клетки ЦНС, печени, кишечника, почек.
Ингибируется бактерицидная активность лейкоцитов – септические проявления.
КЛИНИКА
На 3-4 сутки после рождения: многократная рвота, диарея, желтуха.
При отсутствии лечения – гепатомегалия, гемолитические изменения. Поражение ЦНС (судорожный
синдром, нарушение мышечного тонуса, задержка ПМР, внутричерепная гипертензия). В дальнейшем
гипотрофия, катаракты, поражение почек.
14.
ДИАГНОСТИКАПо результатам неонатального скрининга
при повышении галактозы и снижении
активности фермента галактозо-1фосфатуридилтрансферазы.
Если содержание галактозы повышено, а
активность данного фермента в норме, то
рекомендуется проверить активность
уридинфосфат-галакто-4-эпимеразы или
галактокиназы.
Для подтверждения используется ДНКдиагностика.
ЛЕЧЕНИЕ ограничении поступления
субстрата (галактозы) пожизненно.
15.
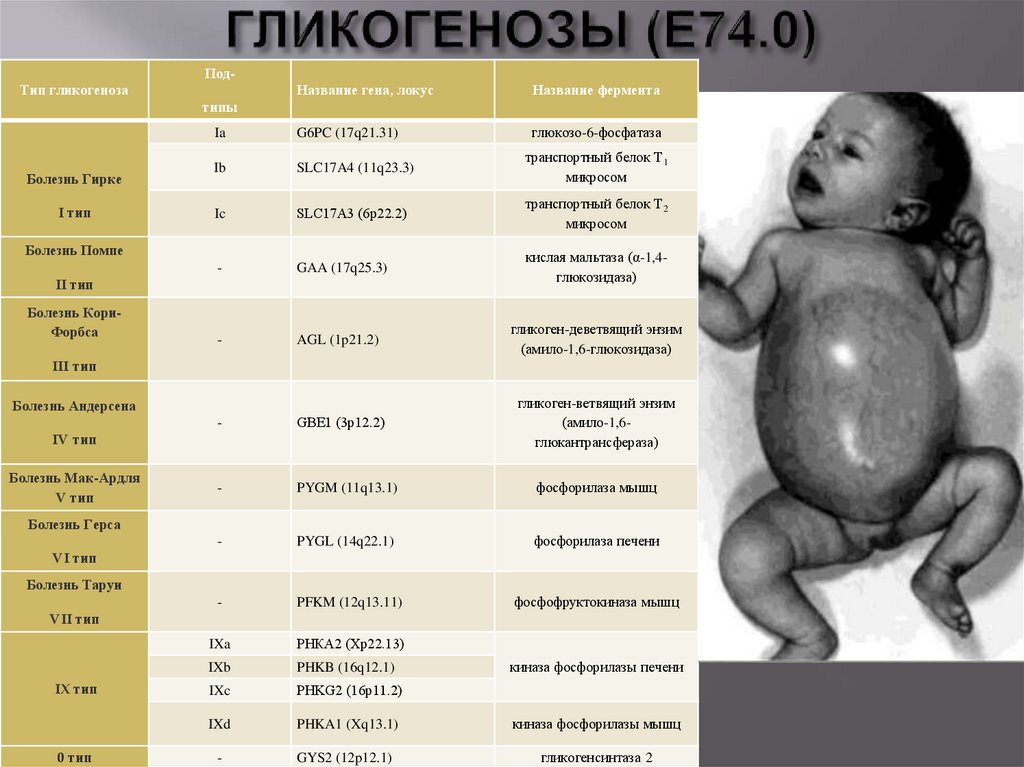
ПодТип гликогенозаНазвание гена, локус
Название фермента
Ia
G6PC (17q21.31)
глюкозо-6-фосфатаза
Ib
SLC17A4 (11q23.3)
транспортный белок Т1
микросом
Ic
SLC17A3 (6р22.2)
транспортный белок Т2
микросом
-
GAA (17q25.3)
кислая мальтаза (α-1,4глюкозидаза)
-
AGL (1p21.2)
гликоген-деветвящий энзим
(амило-1,6-глюкозидаза)
-
GBE1 (3р12.2)
гликоген-ветвящий энзим
(амило-1,6глюкантрансфераза)
-
PYGM (11q13.1)
фосфорилаза мышц
-
PYGL (14q22.1)
фосфорилаза печени
-
PFKM (12q13.11)
IXa
РНКА2 (Хр22.13)
IXb
PHKB (16q12.1)
IXc
PHKG2 (16р11.2)
IXd
PHKA1 (Xq13.1)
киназа фосфорилазы мышц
-
GYS2 (12p12.1)
гликогенсинтаза 2
типы
Болезнь Гирке
I тип
Болезнь Помпе
II тип
Болезнь КориФорбса
III тип
Болезнь Андерсена
IV тип
Болезнь Мак-Ардля
V тип
Болезнь Герса
VI тип
Болезнь Таруи
фосфофруктокиназа мышц
VII тип
IX тип
0 тип
киназа фосфорилазы печени
16.
Возраст манифестации гликогенозов варьирует в зависимостиот типа, но даже внутри одного типа бывают варианты с разным
возрастом начала болезни (от инфантильного до зрелого).
По клинике с преимущественным поражением
печени: гипогликемия, гепатомегалия, задержка роста (типы I,
VI, IX),
мышц: непереносимость физических упражнений, мышечные
спазмы (типы V, VII) и
генерализованные/смешанные типы: кардиомипатия, поражение
печени, мышц, ЦНС (типы II, III, IV).
Диагностика :
Биохимический метод (активность фермента),
Биопсия печени/мышц,
молекулярно-генетический анализ
Для лечения гликогенозов используется диета, направленная на
предупреждение и борьбу с гипогликемией, метаболическим
ацидозом, и кетозом, гиперлипидемией. Обязательно
соблюдение режима питания для предотвращения
гипогликемии – дробный прием пищи до 8 – 10 раз в сутки с
использованием полимеров глюкозы (крахмал, мальдекстрин).
Для лечения метаболического ацидоза внутривенно вводится
раствор гидрокарбоната натрия.
17.
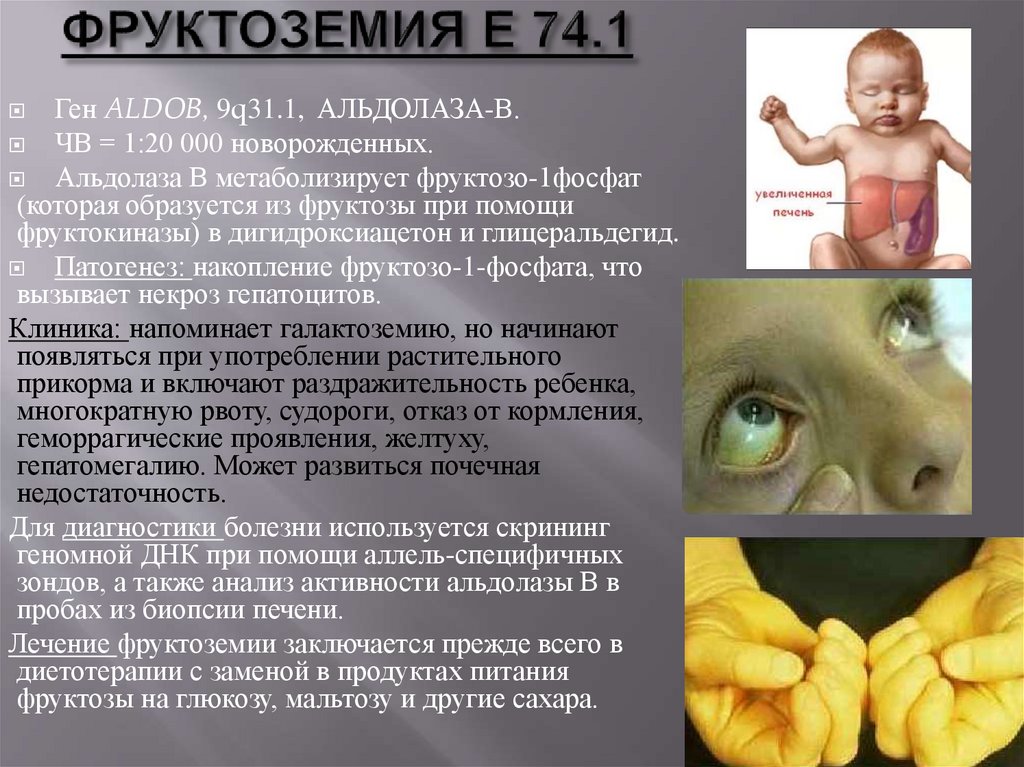
Ген ALDOB, 9q31.1, АЛЬДОЛАЗА-В.ЧВ = 1:20 000 новорожденных.
Альдолаза В метаболизирует фруктозо-1фосфат
(которая образуется из фруктозы при помощи
фруктокиназы) в дигидроксиацетон и глицеральдегид.
Патогенез: накопление фруктозо-1-фосфата, что
вызывает некроз гепатоцитов.
Клиника: напоминает галактоземию, но начинают
появляться при употреблении растительного
прикорма и включают раздражительность ребенка,
многократную рвоту, судороги, отказ от кормления,
геморрагические проявления, желтуху,
гепатомегалию. Может развиться почечная
недостаточность.
Для диагностики болезни используется скрининг
геномной ДНК при помощи аллель-специфичных
зондов, а также анализ активности альдолазы В в
пробах из биопсии печени.
Лечение фруктоземии заключается прежде всего в
диетотерапии с заменой в продуктах питания
фруктозы на глюкозу, мальтозу и другие сахара.
18.
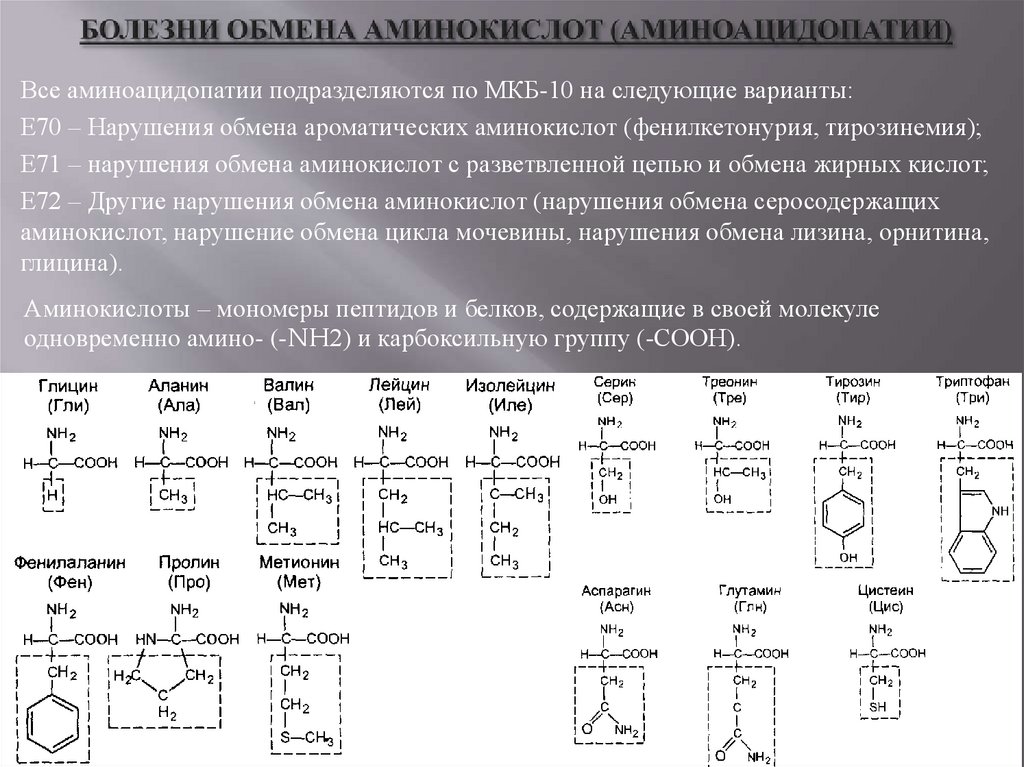
Все аминоацидопатии подразделяются по МКБ-10 на следующие варианты:Е70 – Нарушения обмена ароматических аминокислот (фенилкетонурия, тирозинемия);
Е71 – нарушения обмена аминокислот с разветвленной цепью и обмена жирных кислот;
Е72 – Другие нарушения обмена аминокислот (нарушения обмена серосодержащих
аминокислот, нарушение обмена цикла мочевины, нарушения обмена лизина, орнитина,
глицина).
Аминокислоты – мономеры пептидов и белков, содержащие в своей молекуле
одновременно амино- (-NH2) и карбоксильную группу (-СООН).
19.
ФКУ – АР тип наследования. Мутация гена РАН (фенилаланингидроксилаза), локализованного на 12q22-24. ЧВ=1:10 000.ФКУ впервые описана Фелингом в 1934 г., который разработал
метод диагностики патологии путем обнаружения фенилпирувата в
моче у новорожденных.
Для проведения пробы Фелинга к 2 мл мочи добавляют несколько
капель 10% раствора FeCl3 и уксусной кислоты. При окраске мочи в
оливково-зеленый цвет проба считается положительной.
20.
В основе патогенеза ФКУлежат аминоацидурия и ацидоз тканей,
причинами которых являются следующие
метаболические нарушения:
• внутриклеточное накопление (до
метаболического блока) ФА и его
производных (фенилпировиноградная,
фенилмолочная и фенилуксусная
кислоты, фенилэтиламин,
фенилацетилглютамин);
• высокая концентрация ФА и его
производных в крови и моче;
• нарушение реабсорбции и
транспорта ФА и тирозина.
В результате этих нарушений
наблюдается прямое токсическое
действие продуктов метаболизма на
ЦНС, нарушаются функции печени,
обмен белков, липо- и гликопротеидов,
метаболизм гормонов.
21.
Схема превращений фенилаланина и тирозина и основныеметаболические блоки на их пути
1 - фенилкетонурия
4 – врожденный гипотиреоз
2 - альбинизм
3- алкаптонурия
22.
КЛИНИКАПервые проявления болезни:
вялость, отсутствие интереса
к окружающему, повышенная
возбудимость, беспокойство,
частое срыгивание,
гипотонус мышц,
судорожный синдром,
признаки аллергического
дерматита.
Появляется специфический
«мышиный» запах.
Характерны
гипопигментация кожи,
волос и радужной оболочки
глаз.
Задержка психомоторного
развития.
При отсутствии лечения
развивается необратимая УО.
23.
ДиагностикаБХ (превышение уровня ФА в
крови более 900-1200 мкмоль/л);
положительная проба Феллинга.
Прямая диагностика ФКУ идентификация наиболее частой
мутации R408W (в Китае и Японии
не встречается) – ПЦР-ПДРФ.
Лечение ограничение субстрата с
пищей до уровня минимальной
возрастной потребности.
Для коррекции питания назначают
белковые гидролизаты, лишенные
ФА, но имеющие все другие
необходимые аминокислоты
(фенил-фри, берлофен, нофелан).
24.
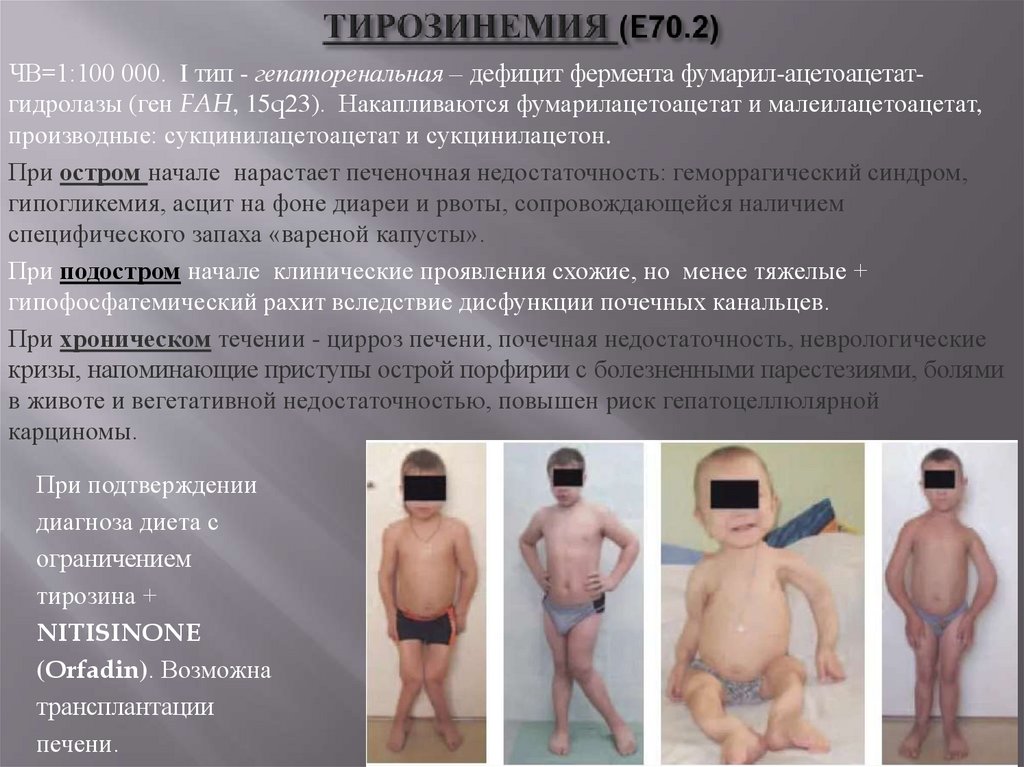
ЧВ=1:100 000. I тип - гепаторенальная – дефицит фермента фумарил-ацетоацетатгидролазы (ген FAH, 15q23). Накапливаются фумарилацетоацетат и малеилацетоацетат,производные: сукцинилацетоацетат и сукцинилацетон.
При остром начале нарастает печеночная недостаточность: геморрагический синдром,
гипогликемия, асцит на фоне диареи и рвоты, сопровождающейся наличием
специфического запаха «вареной капусты».
При подостром начале клинические проявления схожие, но менее тяжелые +
гипофосфатемический рахит вследствие дисфункции почечных канальцев.
При хроническом течении - цирроз печени, почечная недостаточность, неврологические
кризы, напоминающие приступы острой порфирии с болезненными парестезиями, болями
в животе и вегетативной недостаточностью, повышен риск гепатоцеллюлярной
карциномы.
При подтверждении
диагноза диета с
ограничением
тирозина +
NITISINONE
(Orfadin). Возможна
трансплантации
печени.
25.
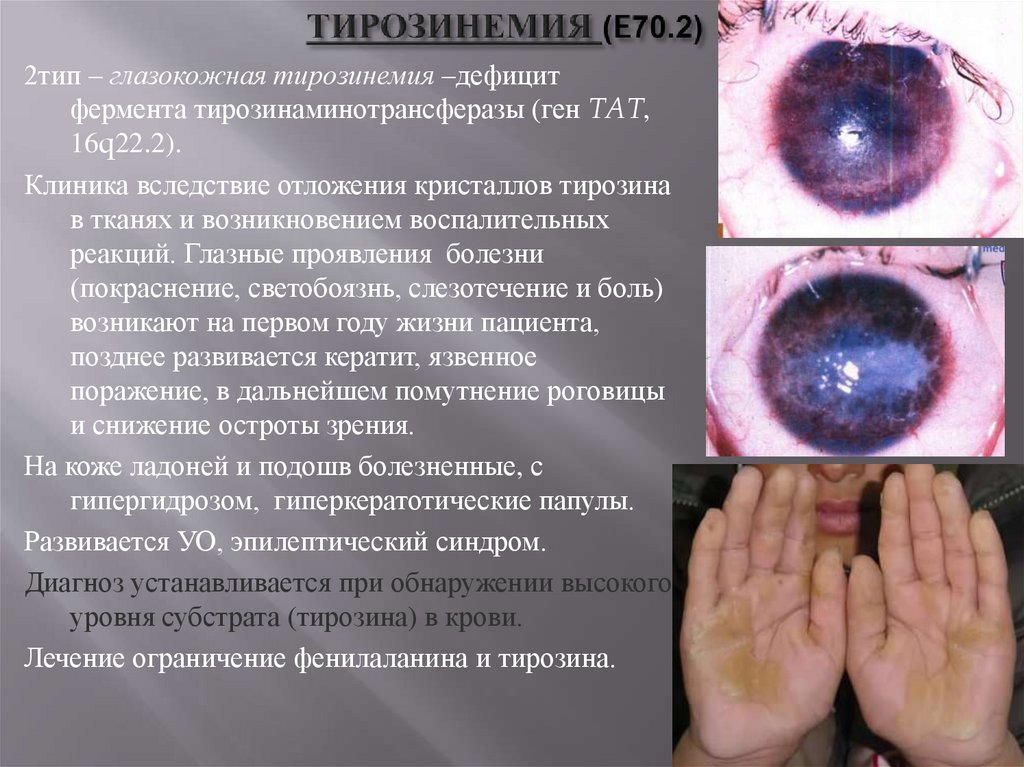
2тип – глазокожная тирозинемия –дефицитфермента тирозинаминотрансферазы (ген TAT,
16q22.2).
Клиника вследствие отложения кристаллов тирозина
в тканях и возникновением воспалительных
реакций. Глазные проявления болезни
(покраснение, светобоязнь, слезотечение и боль)
возникают на первом году жизни пациента,
позднее развивается кератит, язвенное
поражение, в дальнейшем помутнение роговицы
и снижение остроты зрения.
На коже ладоней и подошв болезненные, с
гипергидрозом, гиперкератотические папулы.
Развивается УО, эпилептический синдром.
Диагноз устанавливается при обнаружении высокого
уровня субстрата (тирозина) в крови.
Лечение ограничение фенилаланина и тирозина.
26.
3 типдефицит фермента 4гидроксифенилпируватдиоксигеназы
(HPD, 12q24.31).
Характеризуется мягкой
гипертирозинемией.
Описаны бессимптомные формы и
формы с выраженной
неврологической симптоматикой
(двигательные нарушения и
интеллектуальный дефицит),
поражение кожи не характерно.
Пациентам с тирозинемией 3 типа
также рекомендуется ограничение
фенилаланина и тирозина.
27.
АР тип наследования, ЧВ=1:120 000.ЭТИОЛОГИЯ: Мутации в генах мультиферментного комплекса (дегидрогеназа
кетокислот с разветвленной цепью – BCKAD окислительного
декарбоксилирования лейцина, изолейцина, валина). В состав комплекса входят 4
субъединицы:
E1a (α-субъединица 2-оксоизовалерат дегидрогеназы )- ген BCKDHA, 19q13.2;
Е1b (β- субъединицa 2-оксоизовалерат дегидрогеназы)- ген BCKDHB, 6q14.1;
E2 (ацетилтрансфераза липоамида), ген DBT, 1р21.2;
E3 (дегидрогеназа дигидролипоамида), ген DLD, 7q31.
ПАТОГЕНЕЗ: Накопление в биологических жидкостях и тканях токсичных
метаболитов – органических кетокислот.
28.
Клиника: 4 варианта течения (при всех будет УО):1) Классический – на 3-5 день рождения сладкий запах ушной серы и мочи, снижение
аппетита, вялость, сонливость. Без лечения на 7-10 день развивается кома.
2) Промежуточный – позднее и менее тяжелые проявления.
3) Интермиттирующий – эпизоды острой метаболической декомпенсации (повторная
рвота, сонливость, атаксия и дистония, судороги).
4) Тиамин-чувствительный – эффективность витамина В1 при 3 варианте.
Диагностика – БХ анализ крови при помощи масс-спектрометрии и газо-жидкостной
хроматографии повышенного уровня лейцина, изолейцина и валина,
аллоизолейцина. МГ-диагностика (мутации в генах BCKDHA, BCKDHB, DBT, DLD)
Лечение – ограничение субстрата с пищей (лейцина, изолейцина и валина). При острых
кризах – гемодиализ.
29.
Нарушение обмена аминокислоты метионина.АР тип наследования, ген 21q22.1. ЧВ=1:200 000.
Типы:
I (цистатионин-бета-синтаза, ген CBS, 21q22.3),
II (10-метилентетрагидрофолат-редуктаза),
III (5-метилентетрагидрофолат),
IV (гомоцистеин трансметилаза).
Патогенез: Токсическое действие на нервную
и ОДС, поражение интимы сосудов.
Клиника развивается в первые 10 лет жизни.
Глазные симптомы: близорукость, подвывих
хрусталика, дрожание радужки, глаукома,
катаракта, атрофия зрительных нервов,
отслойка сетчатки.
Судорожный синдром, гиперкинезы.
Поражение ОДС: килевидная грудная клетка,
арахнодактилия, кифосколиоз, остеопороз,
искривление голеней и стоп.
Артериальные и венозные тромбозы.
Высокий рост, длинные конечности и укороченное
тело, голубые глаза, редкие светлые волосы.
Телеангиэктазии и
эритематозные пятна на
скулах.
Снижение интеллекта.
30.
Диагностика: БХ анализ крови и мочи(высокое содержание гомоцистина и
метионина при низком уровне
цистина).
Лечение: При В6-резистентной
(независимой) – низкобелковая диета
с ограничением метионина.
Употребление главным образом
растительной пищи.
При В6-зависимой форме –
активность ферментов удается
нормализовать большими дозами
витамина В6.
При всех формах назначается бетаин,
фолиевая кислота.
Для снижения уровня гомоцистеина
рекомендуется препарат
CYSTADANE (betaine) –
альтернативный донор метильных
групп для превращения
гомоцистеина в метионин.
31.
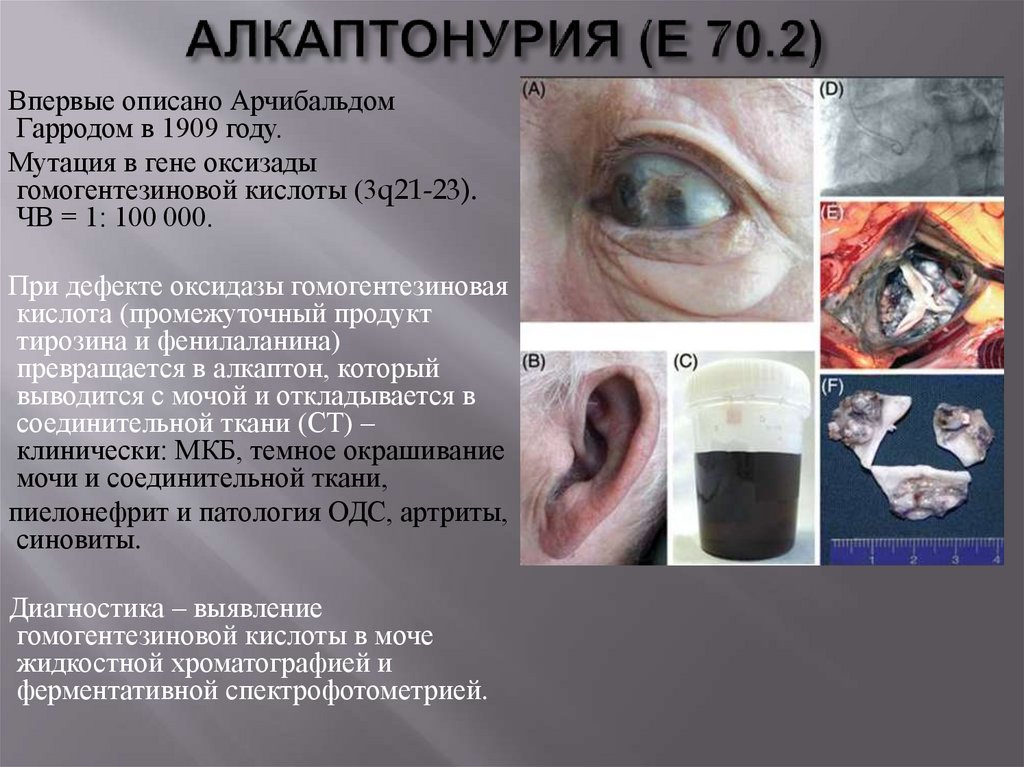
Впервые описано АрчибальдомГарродом в 1909 году.
Мутация в гене оксизады
гомогентезиновой кислоты (3q21-23).
ЧВ = 1: 100 000.
При дефекте оксидазы гомогентезиновая
кислота (промежуточный продукт
тирозина и фенилаланина)
превращается в алкаптон, который
выводится с мочой и откладывается в
соединительной ткани (СТ) –
клинически: МКБ, темное окрашивание
мочи и соединительной ткани,
пиелонефрит и патология ОДС, артриты,
синовиты.
Диагностика – выявление
гомогентезиновой кислоты в моче
жидкостной хроматографией и
ферментативной спектрофотометрией.
32.
ЧВ=1:50 000.Мутации в гене VLCAD (17p.13). Синоним гена ACADVL.
Клинические формы:
1)
Системная (ранняя неонатальная) – кардиомиопатия,
печеночная энцефалопатия, гипокетонемическая
гипогликемия.
2)
Печеночная (манифестация в первые годы жизни) гипокетонемическая гипогликемия при стрессе,
голодании, гепатомегалия вследствие жировой дистрофии
печени.
3)
Миопатическая (поздняя манифестация) - эпизоды
мышечной слабости, боли в мышцах.
Диагностика – биохимическая масс-спектрометрия
(повышение концентрации тетрадеценоилкарнитина выше
0,7 мкмоль/л, низкий уровень свободного карнитина ниже
20 мкмоль/л).
Лечение – снижение потребления жиров, применение
Карнитина (элькар 50 мг/кг массы в сутки) и глицина (200
мг в сутки), которые конъюгируют дериваты жирных
кислот.
33.
ЧВ=1:300 000. (В 1964 г. – Майкл Лёш и Билл Нихен).Мутации гена гипоксантин-гуанин-фосфорибозилтрансферазы (HPRT – Xq26).
Данный фермент участвует в метаболизме гипоксантина и гуанина.
При мутации накапливается мочевая кислота.
Х-сцепленный рецессивный тип наследования.
34.
Клиника: хореоатетоз (хорея – быстрые прерывистые движения, атетоз – медленныесудорожные движения), задержка психомоторного развития, аутоагрессия.
Диагностика: клиника + гиперурикемия + МГА.
Лечение: аллопуринол (синтетический аналог гипоксантина, который конкурентно
ингибирует ксантиноксидазу).
35.
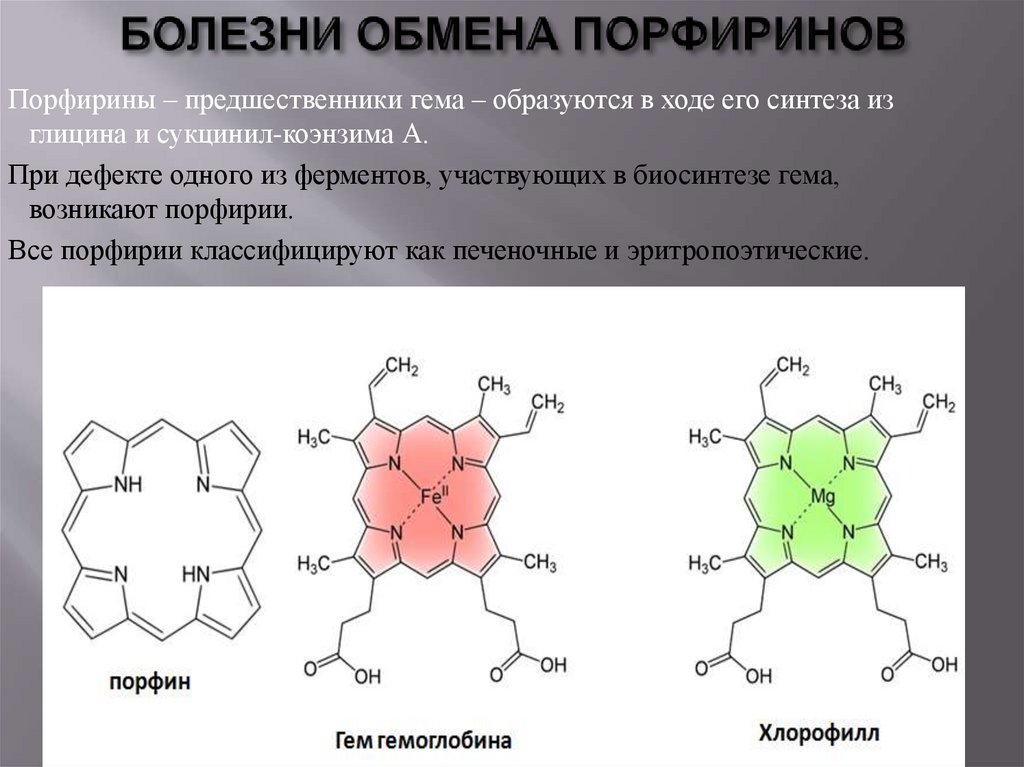
Порфирины – предшественники гема – образуются в ходе его синтеза изглицина и сукцинил-коэнзима А.
При дефекте одного из ферментов, участвующих в биосинтезе гема,
возникают порфирии.
Все порфирии классифицируют как печеночные и эритропоэтические.
36.
ЧВ=1:100 000 населения. АД тип наследования, неполная пенетрантность.Мутация гена третьего фермента биосинтезе гема – порфобилиноген-дезаминазы
(HMBS, 11q23.3).
Патогенез: в нервных клетках накапливается аминолевулиновая кислота. Это
приводит к нарушению транспорта ионов через мембраны – нейропатия,
демиелинизация.
Клинически: после полового созревания, чаще у женщин, боли в различных
отделах живота,
полиневрит, тетрапарез, паралич дыхательной мускулатуры,
эпилептиформные припадки, галлюцинации и бред.
Провокация приступов лекарствами , алкоголем, менструациями.
Диагностика: Окраска мочи в красный цвет под действием света. Обнаружение в
моче повышенного содержания порфобилиногена и аминолевулиновой
кислоты (предшественники порфиринов). МГА.
Лечение: 1) Исключить провоцирующие приступы препараты (ТиопенталНатрия, барбитураты, сульфаниламиды).
2) Глюкоза до 200 г в сутки внутривенно
или фосфаден (аденил) до 250 мг в сутки в/м.
3) Плазмаферез.
4) Гематин в тяжелых случаях.
5) При болях – НПВП.
37.
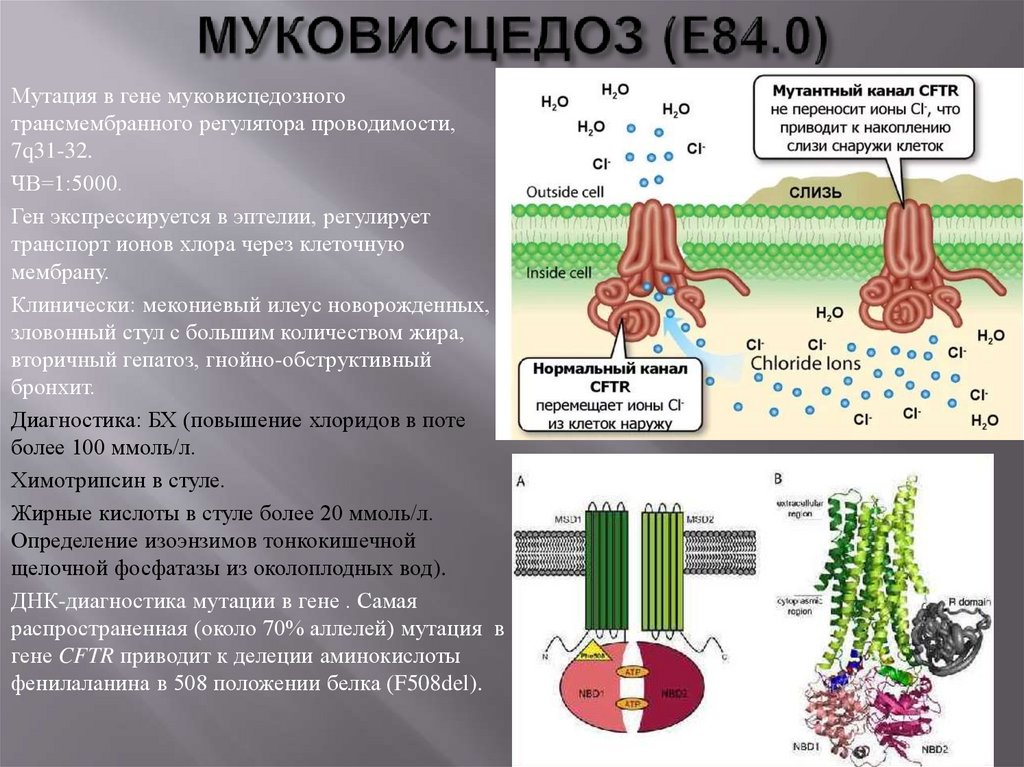
Мутация в гене муковисцедозноготрансмембранного регулятора проводимости,
7q31-32.
ЧВ=1:5000.
Ген экспрессируется в эптелии, регулирует
транспорт ионов хлора через клеточную
мембрану.
Клинически: мекониевый илеус новорожденных,
зловонный стул с большим количеством жира,
вторичный гепатоз, гнойно-обструктивный
бронхит.
Диагностика: БХ (повышение хлоридов в поте
более 100 ммоль/л.
Химотрипсин в стуле.
Жирные кислоты в стуле более 20 ммоль/л.
Определение изоэнзимов тонкокишечной
щелочной фосфатазы из околоплодных вод).
ДНК-диагностика мутации в гене . Самая
распространенная (около 70% аллелей) мутация в
гене CFTR приводит к делеции аминокислоты
фенилаланина в 508 положении белка (F508del).
38.
Строение гонана (циклопентанпергидрофенантрена), основыстроения стероидов
39.
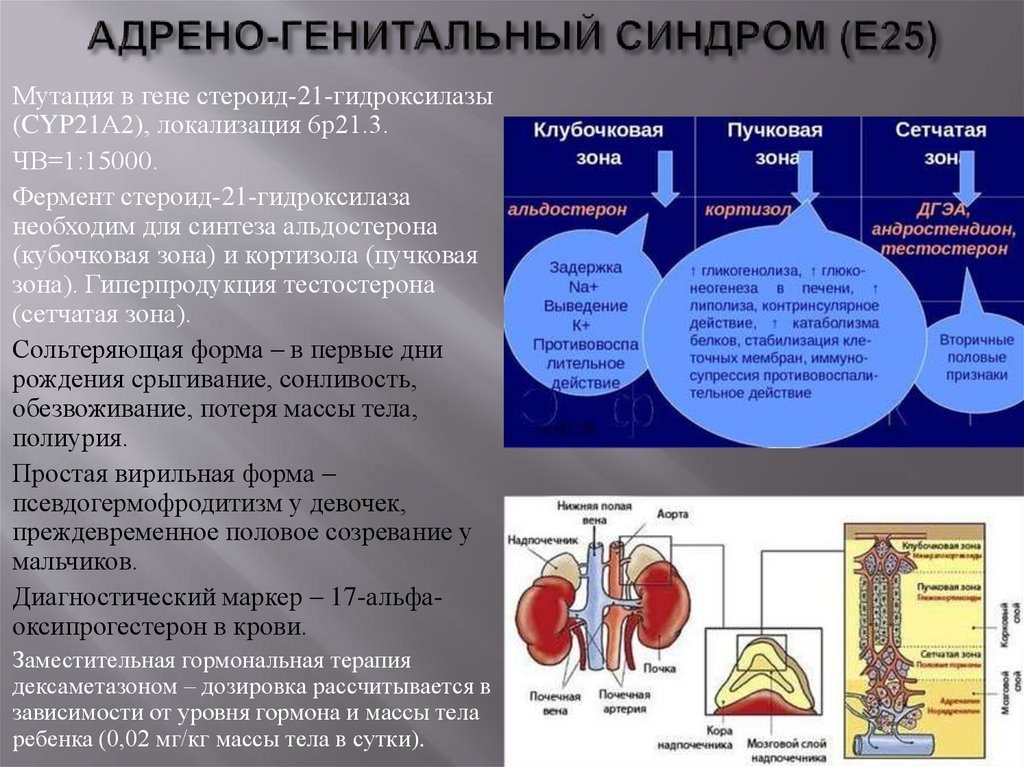
Мутация в гене стероид-21-гидроксилазы(CYP21А2), локализация 6р21.3.
ЧВ=1:15000.
Фермент стероид-21-гидроксилаза
необходим для синтеза альдостерона
(кубочковая зона) и кортизола (пучковая
зона). Гиперпродукция тестостерона
(сетчатая зона).
Сольтеряющая форма – в первые дни
рождения срыгивание, сонливость,
обезвоживание, потеря массы тела,
полиурия.
Простая вирильная форма –
псевдогермофродитизм у девочек,
преждевременное половое созревание у
мальчиков.
Диагностический маркер – 17-альфаоксипрогестерон в крови.
Заместительная гормональная терапия
дексаметазоном – дозировка рассчитывается в
зависимости от уровня гормона и массы тела
ребенка (0,02 мг/кг массы тела в сутки).
40.
41.
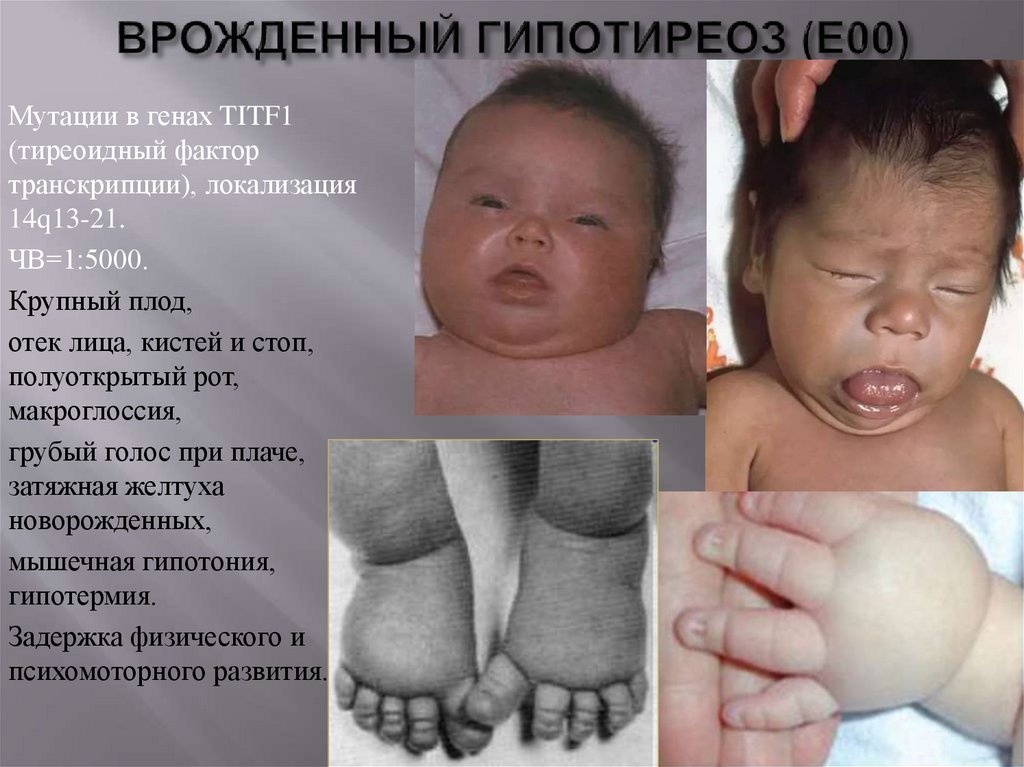
Мутации в генах TITF1(тиреоидный фактор
транскрипции), локализация
14q13-21.
ЧВ=1:5000.
Крупный плод,
отек лица, кистей и стоп,
полуоткрытый рот,
макроглоссия,
грубый голос при плаче,
затяжная желтуха
новорожденных,
мышечная гипотония,
гипотермия.
Задержка физического и
психомоторного развития.
42.
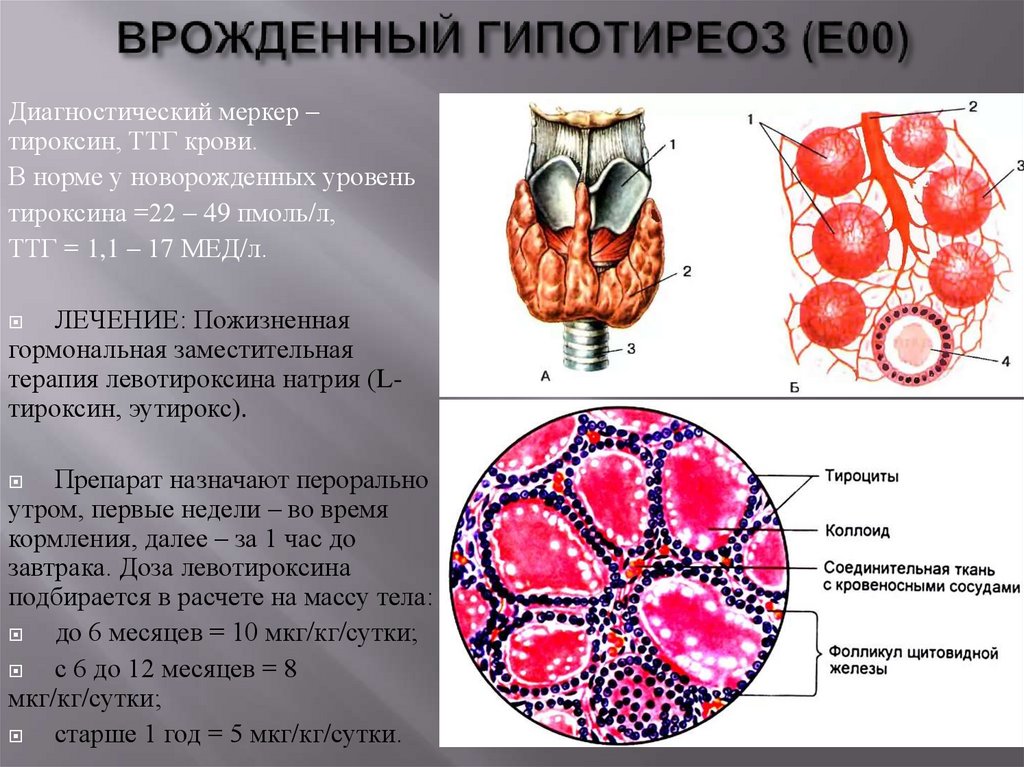
Диагностический меркер –тироксин, ТТГ крови.
В норме у новорожденных уровень
тироксина =22 – 49 пмоль/л,
ТТГ = 1,1 – 17 МЕД/л.
ЛЕЧЕНИЕ: Пожизненная
гормональная заместительная
терапия левотироксина натрия (Lтироксин, эутирокс).
Препарат назначают перорально
утром, первые недели – во время
кормления, далее – за 1 час до
завтрака. Доза левотироксина
подбирается в расчете на массу тела:
до 6 месяцев = 10 мкг/кг/сутки;
с 6 до 12 месяцев = 8
мкг/кг/сутки;
старше 1 год = 5 мкг/кг/сутки.
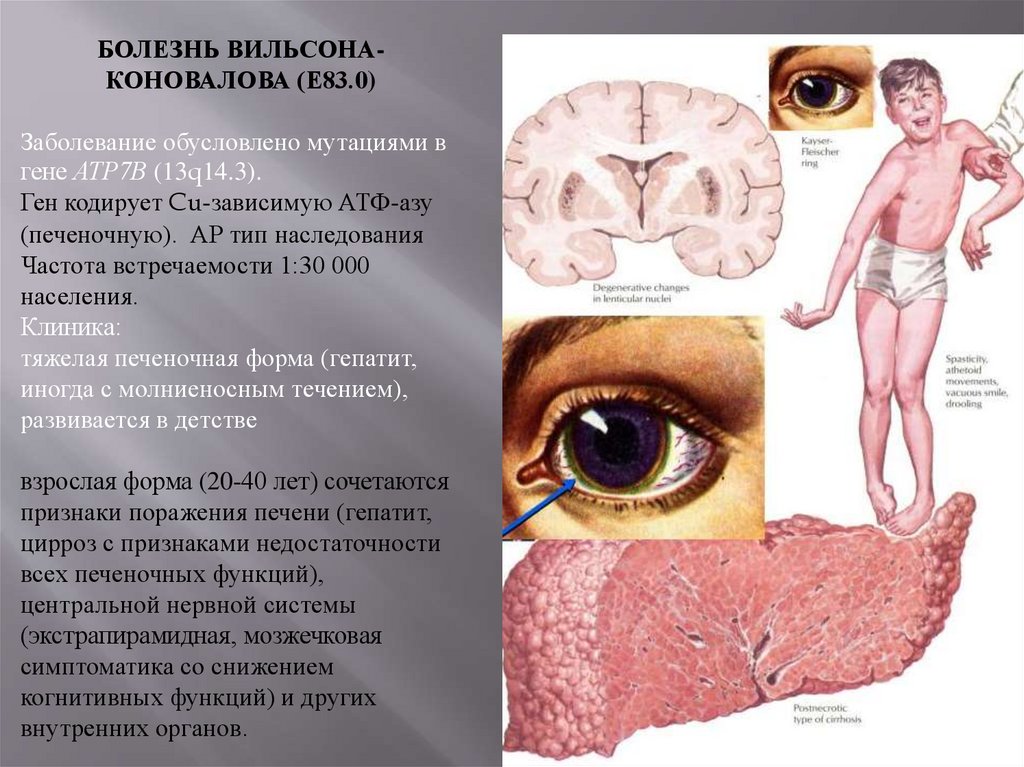
43.
БОЛЕЗНЬ ВИЛЬСОНАКОНОВАЛОВА (Е83.0)Заболевание обусловлено мутациями в
гене АТР7В (13q14.3).
Ген кодирует Cu-зависимую АТФ-азу
(печеночную). АР тип наследования
Частота встречаемости 1:30 000
населения.
Клиника:
тяжелая печеночная форма (гепатит,
иногда с молниеносным течением),
развивается в детстве
взрослая форма (20-40 лет) сочетаются
признаки поражения печени (гепатит,
цирроз с признаками недостаточности
всех печеночных функций),
центральной нервной системы
(экстрапирамидная, мозжечковая
симптоматика со снижением
когнитивных функций) и других
внутренних органов.
44.
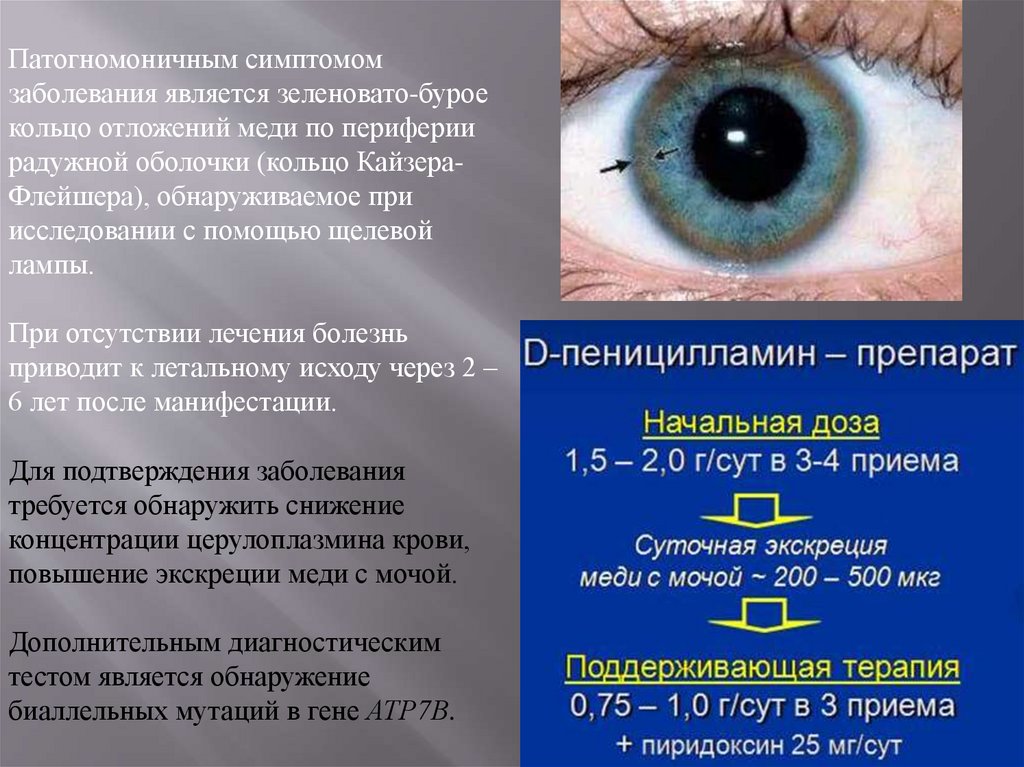
Патогномоничным симптомомзаболевания является зеленовато-бурое
кольцо отложений меди по периферии
радужной оболочки (кольцо КайзераФлейшера), обнаруживаемое при
исследовании с помощью щелевой
лампы.
При отсутствии лечения болезнь
приводит к летальному исходу через 2 –
6 лет после манифестации.
Для подтверждения заболевания
требуется обнаружить снижение
концентрации церулоплазмина крови,
повышение экскреции меди с мочой.
Дополнительным диагностическим
тестом является обнаружение
биаллельных мутаций в гене АТР7В.
45.
БОЛЕЗНЬ МЕНКЕСА (Е83.0)Ген АТР7А (Xq21.1). Ген кодирует Cu-зависимую АТФ-азу (кишечную), в результате в
кишечнике не всасывается медь. ЧВ=1:300 000.
Клинические проявления заболевания представлены в следующих формах: классическая
(наиболее часто), синдром «затылочного рога», дистальная моторная нейропатия.
Классическая форма начинается в инфантильном периоде (обычно в 3-6 месяцев) с
неврологических проявлений (задержка психомоторного развития, эпилепсия,
двигательные расстройства), сочетается со своеобразным дефектом волос («перекрученные
волосы») и повышенной растяжимостью кожи. Заболевание носит прогрессирующий
характер.
46.
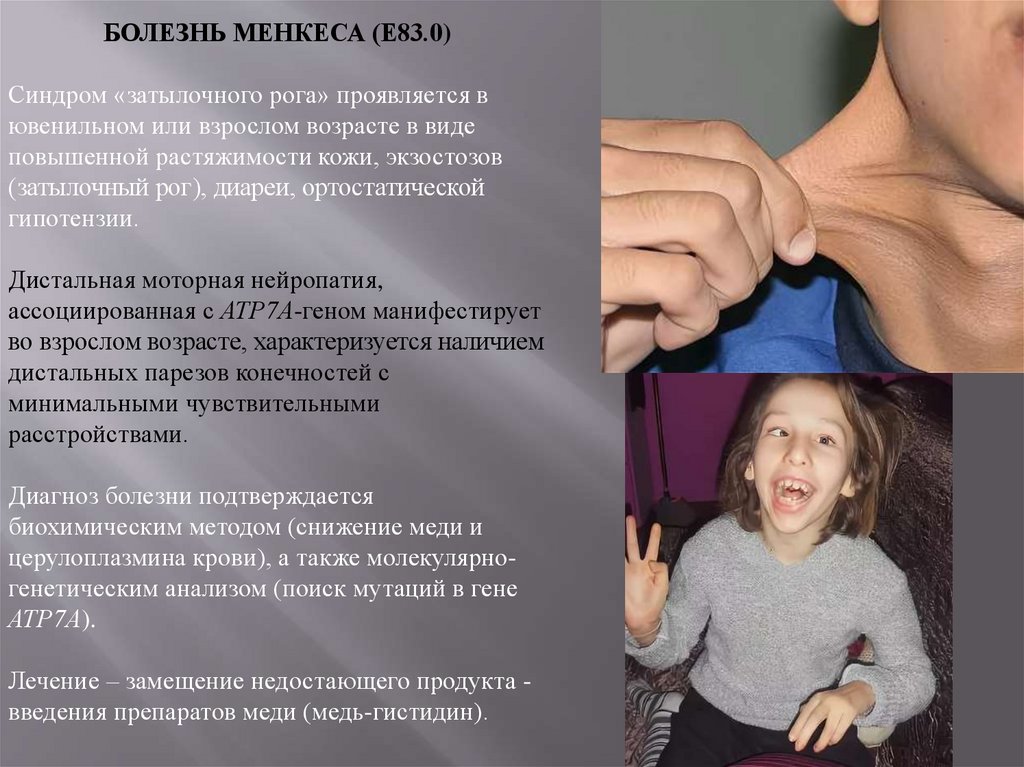
БОЛЕЗНЬ МЕНКЕСА (Е83.0)Синдром «затылочного рога» проявляется в
ювенильном или взрослом возрасте в виде
повышенной растяжимости кожи, экзостозов
(затылочный рог), диареи, ортостатической
гипотензии.
Дистальная моторная нейропатия,
ассоциированная с АТР7А-геном манифестирует
во взрослом возрасте, характеризуется наличием
дистальных парезов конечностей с
минимальными чувствительными
расстройствами.
Диагноз болезни подтверждается
биохимическим методом (снижение меди и
церулоплазмина крови), а также молекулярногенетическим анализом (поиск мутаций в гене
АТР7А).
Лечение – замещение недостающего продукта введения препаратов меди (медь-гистидин).
47.
ГЕМОХРОМАТОЗ (Е83.1)Накопление избытка Fe в органах, c их
тяжелым поражением (цирроз, диабет,
кардиомиопатия, артрит). ЧВ=1:300.
Мутации в гене HFE (16р22.2), C282Y
(замещение цистеина на тирозин).
Заболевание манифестирует в возрасте
40 - 50 лет, мужчины болеют чаще.
Клиника: гепатоспленомегалия, цирроз
печени, кардиомиопатия, гипогонадизм,
сахарный диабет, желтуха, поражение
суставов, гиперпигментация.
Диагностика БХ: в крови содержание
железа, трансферрина, ферритина, МГА.
Лечение : ограничение Fe с пищей,
коррекции выведения железа
(дефероксамин), регулярная флеботомии
(удаление Fe c кровью).
48.
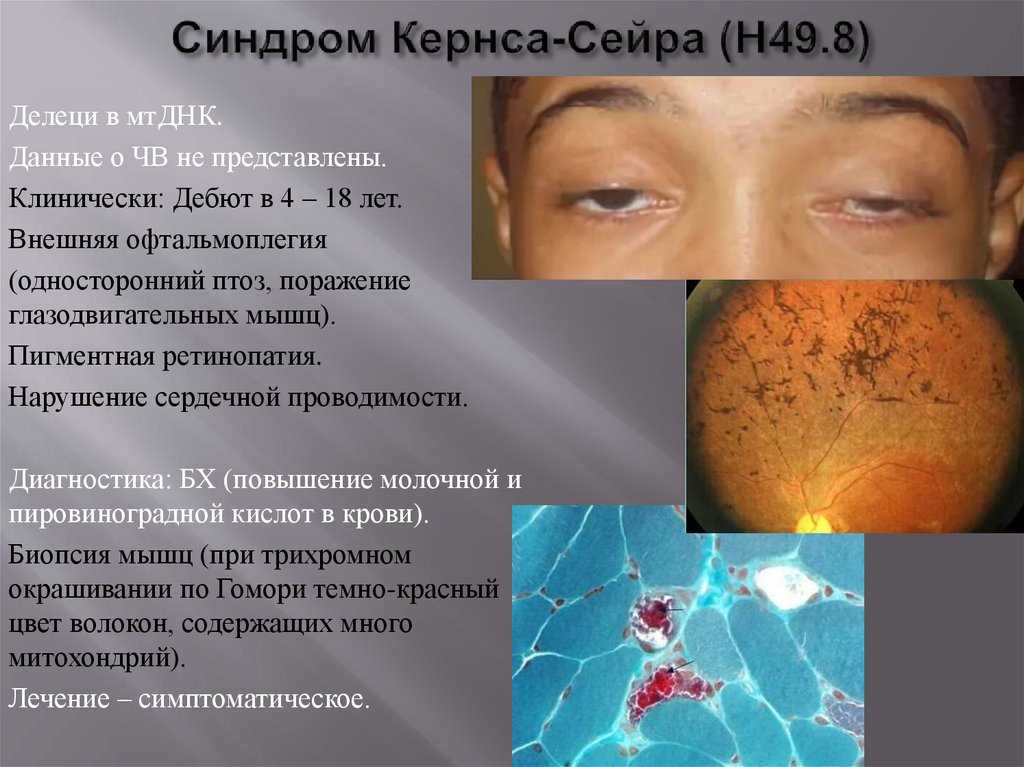
Делеци в мтДНК.Данные о ЧВ не представлены.
Клинически: Дебют в 4 – 18 лет.
Внешняя офтальмоплегия
(односторонний птоз, поражение
глазодвигательных мышц).
Пигментная ретинопатия.
Нарушение сердечной проводимости.
Диагностика: БХ (повышение молочной и
пировиноградной кислот в крови).
Биопсия мышц (при трихромном
окрашивании по Гомори темно-красный
цвет волокон, содержащих много
митохондрий).
Лечение – симптоматическое.
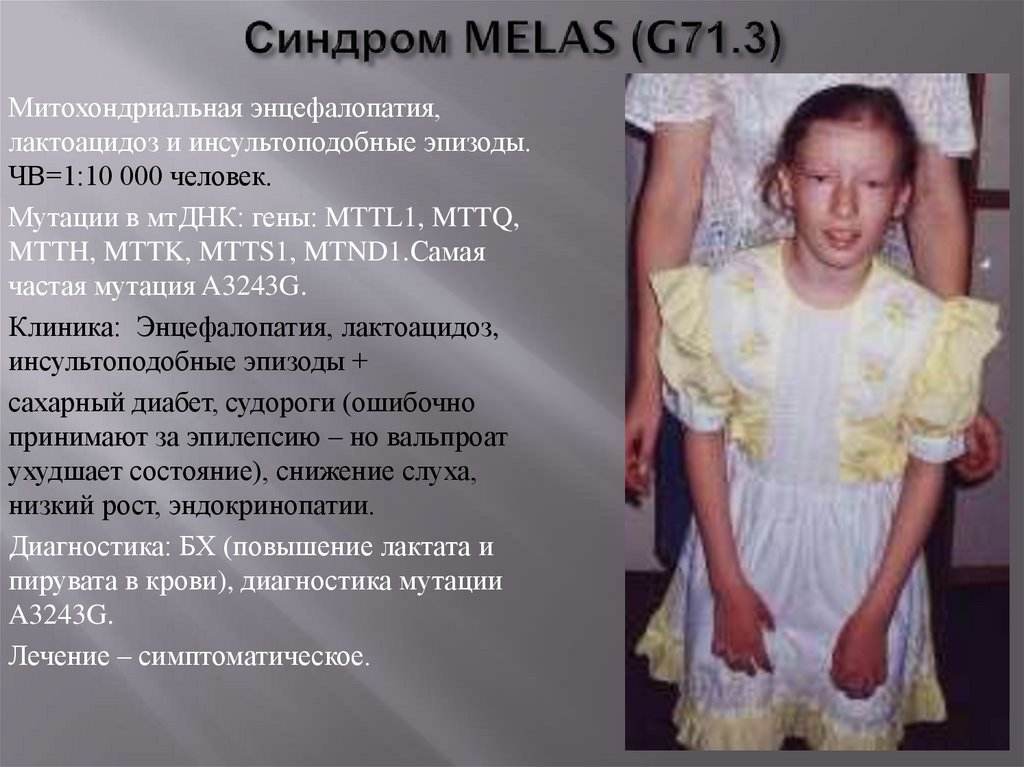
49.
Митохондриальная энцефалопатия,лактоацидоз и инсультоподобные эпизоды.
ЧВ=1:10 000 человек.
Мутации в мтДНК: гены: MTTL1, MTTQ,
MTTH, MTTK, MTTS1, MTND1.Самая
частая мутация A3243G.
Клиника: Энцефалопатия, лактоацидоз,
инсультоподобные эпизоды +
сахарный диабет, судороги (ошибочно
принимают за эпилепсию – но вальпроат
ухудшает состояние), снижение слуха,
низкий рост, эндокринопатии.
Диагностика: БХ (повышение лактата и
пирувата в крови), диагностика мутации
A3243G.
Лечение – симптоматическое.
50.
Миоклоническая эпилепсия с рванымимышечными волокнами.
Мутации в мтДНК: MTTK, MTTL1, MTTH,
MTTS1, MTTS2, MTTF.
ЧВ=1:100 000 населения.
Клиника: дебют в любом возрасте
(чем раньше, тем тяжелее),
Эпилепсия,
Мозжечковая атаксия,
Прогрессирующая мышечная слабость
Снижение интеллекта, нейросенсорная
тугоухость, полинейропатия.
Диагностика: БХ (высокие уровни лактата и
пирувата в крови и ЦСЖ),
биопсия (разорванные мышечные волокна
(трихромном окрашивании по Гомори темнокрасный цвет волокон, содержащих много
митохондрий).
Лечение – симптоматическое.
51.
Мукополисахаридозы – группа заболеваний, обусловленных генетическимдефектом ферментного расщепления углеводной части молекулы
мукополисахаридов (кислые ГАГ, составляющих основу межклеточного вещества
соединительной ткани), при этом наблюдается накопление мукополисахаридов в
тканях и органах.
I – Hurler syndrome = синдром Гурлер (АР, IDUA, 4р16.3, идуронидаза).
II – Hunter syndrome = с.Хантера (X-сц.Р, IDS, Xq28, идуронатсульфатаза).
III – Sanfilippo syndrome = A (SGSH, 17q25.3, гепарансульфатаза),
B (NAGLU – 17q21.2, ацетилглюкозаминидаза),
C (HGNAT, 8p11.21, ацетил-КоА-глюкозаминидин-N-ацетилтрансфераза),
D (GNS, 12q14.3, ацетилглюкозамин-6-сульфатаза).
IV – Morquio syndrome = А (GALNS, 16q24.3, галактозаминсульфатаза),
В (GLB1, 3p22.3, бета-галактозидаза).
VI – Maroteaux-Lamy syndrome = (ARSB, 5q14.1, арилсульфатаза).
VII – Sly syndrome = с. Слая (АР, GUSB, 7q11.21, бета-глюкуронидаза).
IX – Natowicz s. = с. Натовича (АР, HYAL1, 3p21.31).
52.
МПС53.
ЧВ= 1:100 000 населения. АР т/н.Ген IDUA, 4p16.3.
АЛЬФА-L-ИДУРОНИДАЗЫ
КП: когнитивные расстройства,
микрогнатия,
грубые черты лица,
макроглоссия,
дегенерация сетчатки,
непрозрачность роговицы,
кардиомиопатия,
гепатоспленомегалия.
Тугоподвижность суставов,
тугоухость, шумное дыхание,
кардиальная патология (поражение
клапанов, миокарда, эндокарда,
магистральных сосудов).
54.
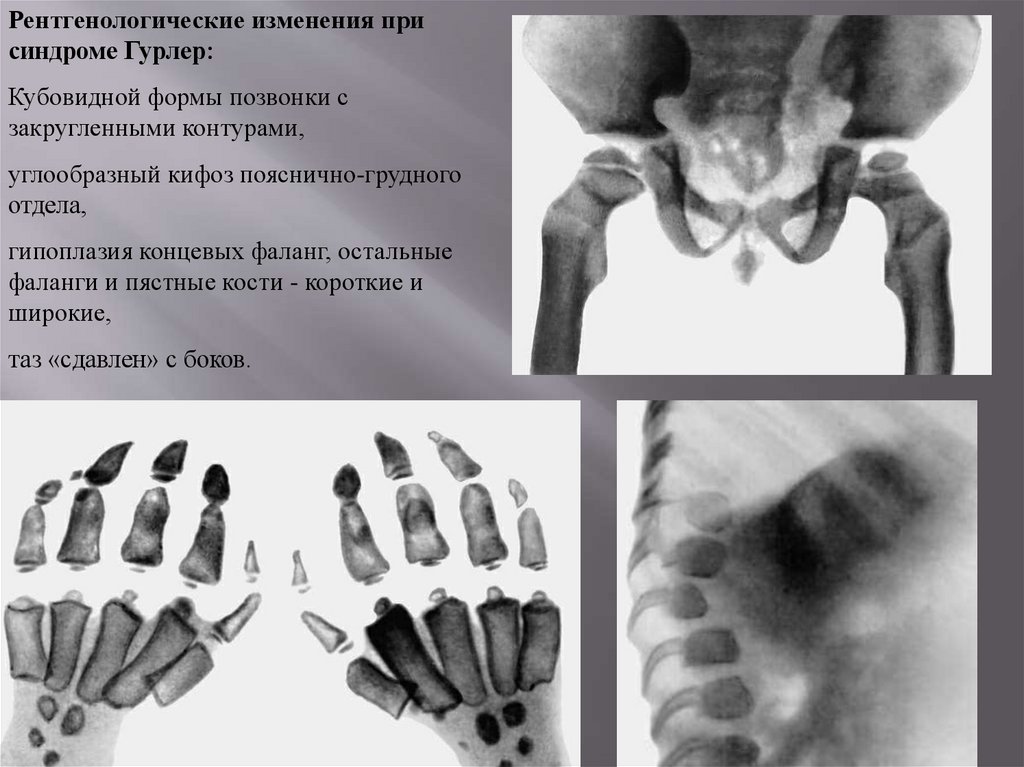
Рентгенологические изменения присиндроме Гурлер:
Кубовидной формы позвонки с
закругленными контурами,
углообразный кифоз пояснично-грудного
отдела,
гипоплазия концевых фаланг, остальные
фаланги и пястные кости - короткие и
широкие,
таз «сдавлен» с боков.
55.
Пациенты с вариантом Шейе имеют нормальный или почтинормальный рост и интеллект, из признаков болезни присутствуют
помутнение роговицы, тугоподвижность суставов, синдром
запястного канала, поражение аортального клапана. Сужение
позвоночного канала в шейном отделе может привести к развитию у
больных спастического тетрапареза.
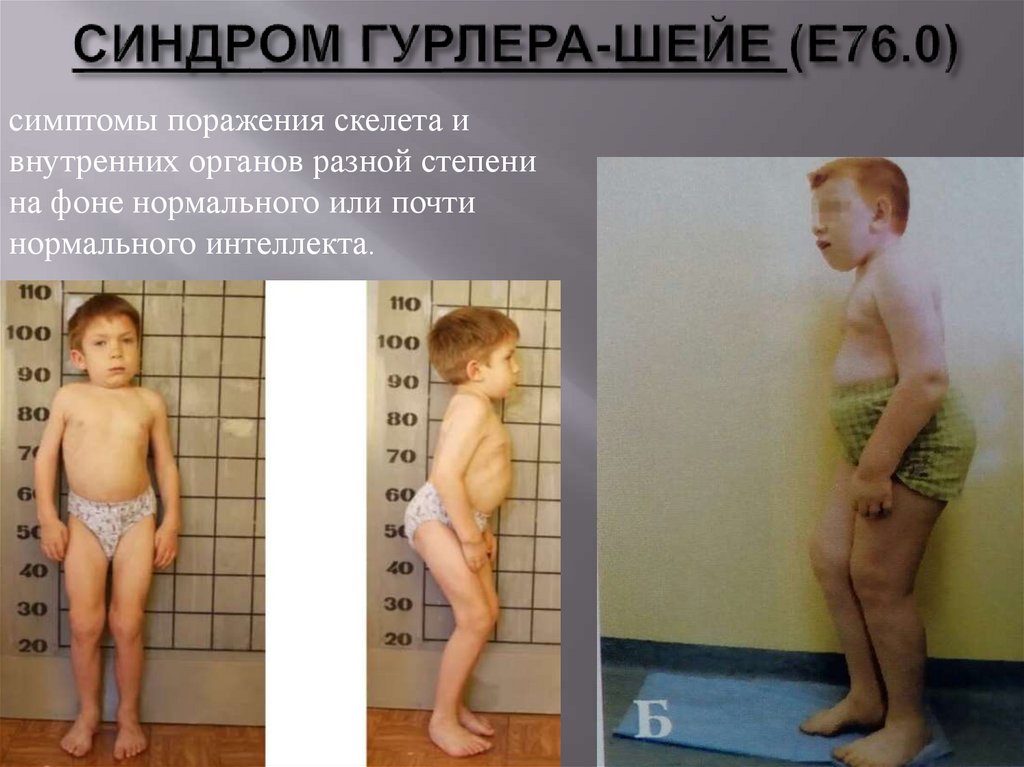
56.
симптомы поражения скелета ивнутренних органов разной степени
на фоне нормального или почти
нормального интеллекта.
57.
Диагностика:биохимический метод (повышение в моче уровня
дерматансульфатов и гепарансульфатов и определение
активности фермента α–L–идуронидазы в лейкоцитах
крови), а также МГА.
Патогенетическая терапия – замещение функции
фермента при помощи препарата Aldurazyme LARONIDASE (Genzyme);
в случае ранней диагностики эффективна трансплантация
гемопоэтических клеток.
58.
ЧВ= 1:132 000 населения. Ген IDS, Xq28.ИДУРОНАТ-L-СУЛЬФАТАЗА
КП: когнитивные расстройства,
микрогнатия, грубые черты лица,
макроглоссия, дегенерация сетчатки,
непрозрачность роговицы,
кардиомиопатия,
гепатоспленомегалия, светлы жесткие
волосы, мутная роговица.
Узелково-папулезное поражение кожи,
умственная отсталость,
расторможенность поведения.
Элапраза разработана британско-американской компанией Шайер (SHIRE)
для лечения МПСII
Ферменты вводятся внутривенно капельно
в течение нескольких часов пожизненно
один раз в неделю.
59.
Тип А (1:100 000, SGSH, 17q25.3,СУЛЬФОГИДРОЛАЗА NСУЛЬФОГЛЮКОЗАМИНА),
тип B (1:200 000, NAGLU, 17q21.2 α-Nацетилглюкозаминидаза,),
тип С (1:500 000, HGSNAT, 8p11.21, αглюкозаминид-N-ацетилтрансфераза),
тип D (1:1млн, GNS, 12q14.3, Nацетилглюкозамин-6-сульфат-сульфатазы).
Манифестация в 2 года: незначительное
отставание в росте, аномалииОДС, далее
прогрессирует распад приобретенных
моторных и психических навыков,
ригидность суставов, гепатоспленомегалия,
УО, судороги.
Д/ка: гепарансульфат в суточной моче,
активность ферментов в лейкоцитах.
Лечение симптоматическое.
60.
Тип А (ЧВ=1:40 000, АР т/н, ген GALNS,16q24.3, ГАЛАКТОЗАМИН-6-СУЛЬФАТ
СУЛЬФАТАЗА).
Тип В (ЧВ=1:40 000, АР т/н, ген ген GLB1,
3p22.3, БЕТА-ГАЛАКТОЗИДАЗА).
Клиника – на 2 году жизни – отставание
роста, выраженные деформации ОДС,
компрессия корешков спинного мозга,
нарушение двигательной функции.
Гепатомегалия, помутнение роговицы,
нарушение дыхания, клапанные пороки
сердца. Потеря слуха. Интеллект сохранен.
Диагностика: повышенный уровень
кератансульфата в суточной моче, снижение
активности ферментов в лейкоцитах. МГА.
Ферментзаместетильная терапия препарат
Elosulfase alfa - VIMIZIM (BioMarin
Pharmaceutical Inc
61.
ЧВ=1:215 000. АР т/н.Ген ARSB, 5q14.1.
АРИЛСУЛЬФАТАЗА-В.
Накопление в организме
дерматансульфата.
Клиника: низкий рост,
множественный дизостоз,
контрактуры.
Дыхательные расстройства,
клапанные пороки сердца.
Потеря слуха, помутнение роговицы.
Интеллект сохранен. Но
прогрессирует гидроцефалия –
слепота.
Д-ка: дерматансульфат в моче,
активность ферментов в лейкоцитах.
Лечение: ортопедические операции,
ферментзамещение Galsulfase NAGLASYME (Biomarin
Pharmaceutical Inc)
62.
ЧВ= 1:1 250 000. АР т/н.Ген GUSB, 7q11.21.
БЕТА-ГЛЮКУРОНИДАЗА
Клинка: грубые черты лица, дисплазия ОДС (килевидная грудная клетка,
тораколюмбальный кифоз), гепатоспленомегалия, пороки сердца,
кардиомиопатия.
Д-ка: низкая активность фермента в лейкоцитах, МГД.
Лечение: ферментзамещение – Vestronidase alfa (Mepsevii) –в США.
63.
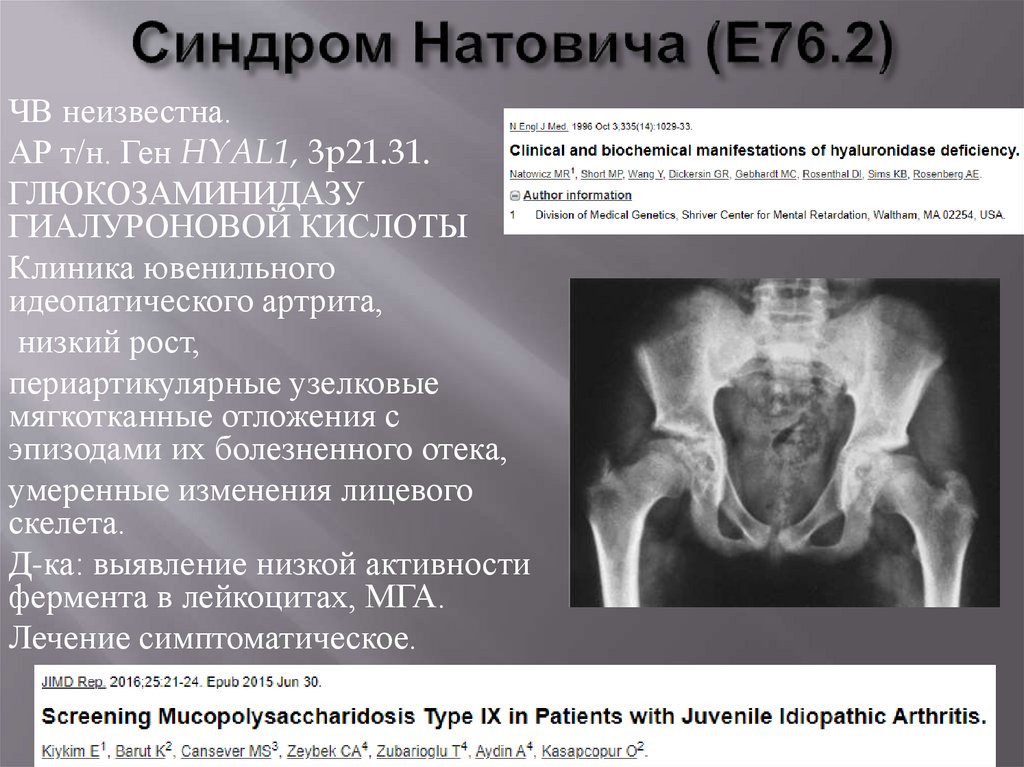
ЧВ неизвестна.АР т/н. Ген HYAL1, 3p21.31.
ГЛЮКОЗАМИНИДАЗУ
ГИАЛУРОНОВОЙ КИСЛОТЫ
Клиника ювенильного
идеопатического артрита,
низкий рост,
периартикулярные узелковые
мягкотканные отложения с
эпизодами их болезненного отека,
умеренные изменения лицевого
скелета.
Д-ка: выявление низкой активности
фермента в лейкоцитах, МГА.
Лечение симптоматическое.
64.
При сфинголипидозах – внутриклеточное накопление сфинголипидов вголовном и костном мозге, легких, печени и селезенке.
Сфинголипиды – компоненты клеточных мембран.
В зависимости от их строения различают:
Фосфолипидозы – болезнь Ниманна-Пика, б. Фабри.
Ганглиозидозы – б. Тея-Сакса, б. Сандхоффа.
Цереброзидозы – б. Гоше, Краббе.
65.
ЧВ типа А и В = 1:40 000, типа С = 1:100 000.БНП типы А и В - ген SMPD1, 11р15 (сфингомиелиназа),
тип С – гены NPC1 (95%), 18q11.2 (внутриклеточный транспортер
холестерина 1), NPC2, 14q24.3 (ВТ холестерина 2).
Клиника: тип А – неонатальный - гипотонус мышц,
лимфаденопатия, гепатоспленомегалия, УО, глухота, слепота.
Тип В – поздний инфантильный - гепатоспленомегалия и задержка
роста, УО не наблюдается.
Тип С – манифестация от 1 до 50 лет. Гепатоспленомагалия,
холестатическая желтуха. Судорожный синдром, парез взора,
дизартрия, атаксия, УО, тетрапарез.
Д-ка: активность фермента, МГА.
Лечение: субстратредуцирующая терапия – Miglustat (Zaveska) –
ингибитор глюкозилцерамидсинтазы (синтез сфинголипидов).
66.
ЧВ=1:40 000. Ген GLA, Xq22.1(альфа-галактозидаза А).
Накопление
глоботриаозилцерамида.
Клиника: грубые черты лица.
Жгучая нейропатическая боль в
кистях и стопах.
Нефросклероз.
Помутнение роговицы.
Снижение слуха.
Аритмия, гипертрофия миокарда.
Ангиокератомы.
Д-ка: активность фермента в
лейкоцитах, МГА.
Лечение: замещение фермента
REPLAGAL (agalsidase alfa)
или FABRAZYME
(agalsidase beta).
67.
АР тип наследования.Ген HEXA, 15q23, альфа субъединица
гексаминидазы.
ЧВ=1:320000, еври-ашкеназы 1:3500.
Накопление в ЦНС ганглиозидов.
Детская форма – смерть до 4 лет (через
полгода после рождения прогрессируют
слепота, глухота, способность глотать,
паралич вследствие атрофии мышц).
Подростковая форма – смерть до 16 лет
(дисфагия, атаксия – шаткость походки,
спастика мышц, дизартрия).
Взрослая форма – возникает от 25 до 30
лет (прогрессируют атаксия, дисфагия,
дизартрия, снижение когнитивных
навыков, спастика).
Д-ка: БХ (активность гексаминидазы),
МГА, глазное дно – «вишневая кость».
Лечение симптоматиическое.
68.
Ген GBA, 1q22. БЕТАГЛЮКОЦЕРЕБРОЗИДАЗА.Накопление глюкоцереброзида – клетки Гоше.
Тип I (ненейронопатический) –1:50 000,
гепатоспленомегалия,
панцитопения,поражение костей (боли,
некрозы, патологические переломы.
Тип II (нейронопатичская инфантильная) –
1:100 000, острое начало у младенцев гипертония, судороги, олигофрения, апноэ.,
быстрое прогрессирование дисфункции ствола
мозга - смерть до 2 лет.
Тип III (нейронопатическая ювенильная) –
1:100 000, подострое течение,
прогрессирующая энцефалопатия (мышечные
подергивания, моклонус, судороги, деменция,
апраксия мышц глаз.
Д-ка – активность бета-глюкоцереброзидазы,
МГА.
Лечение – замещение фермента (CEREZYME,
GLURASIM (Imiglucerase) или VPRIV
(velaglucerase alfa), ELELYSO (Taliglucerase
alfa), субстратредуцирующая - (Miglustat,
Eliglustat).
69.
Мальчик 3 года, БГ 3 типБГ тип 1 до и после
ферментзаместительной
терапии
Девочка 11 мес,
БГ тип 2
70.
ЧВ=1:100 000, ген GALC, 14q31.ГАЛАКТОЦЕРЕБРОЗИДАЗА.
Накопление в ЦНС
галактозилерамидов.
Дебют в 3 – 6 месяцев –
возбудимость, нарушение
кормления, гипертермии.
Далее спастический тетрапарез,
слепота, глухота.
Д-ка – активность
галактоцереброзидазы в лц, МГА.
Лечение симптоматическое.
71.
Пероксисома - обязательная органелла эукариотической клетки,ограниченная мембраной, содержащая от 15 до 50 различных
ферментов, катализирующих окислительно-восстановительные
реакции. Размер от 0,2 до 1,5 мкм. Не содержат ДНК.
Набор функций пероксисом : окисление жирных кислот, фотодыхание,
разрушение токсичных соединений, синтез желчных кислот,
холестерина, а также эфиросодержащих липидов, построение
миелиновой оболочки нервных волокон, метаболизм фитановой
кислоты и т. д. Наряду с митохондриями являются главными
потребителями O2 в клетке.
72.
1. Пероксисомы получили такое название благодаря тому,что обычно в их состав входит один или несколько
ферментов, использующих молекулярный кислород для
отщепления атомов водорода от некоторых органических
субстратов в окислительной реакции с
образованием пероксида водорода:
2. Использует образующийся пероксид
для окисления множества субстратов (фенолов, муравьиной
кислоты, формальдегида,этанола) фермент каталаза:
Этот тип окислительных реакций особенно важен в
клетках печени и почек , пероксисомы которых
обезвреживают множество ядовитых веществ ,
попадающих в кровоток. Почти половина этанола ,
попадающего в организм, окисляется этим способом.
73.
В пероксисомах происходит окисление длинноцепочечныхжирных кислот с образованием ацил-CoA, который переходит
в цитозоль для повторного использования в метаболических
реакциях (является важным звеном в цикле трикарбоновых
кислот). Известно, что жирные кислоты длиной более чем 22
углеродных атома окисляются исключительно в этом
органоиде.
В результате дефекта функционирования пероксисом
происходит накопление этих кислот в клетках организма. У
пациентов с пероксисомными болезнями биохимические
нарушения главным образом заключаются в дефектах α- и βокисления жирных кислот и синтеза
плазмалогенов (структурных фосфолипидов миелиновых
волокон)
74.
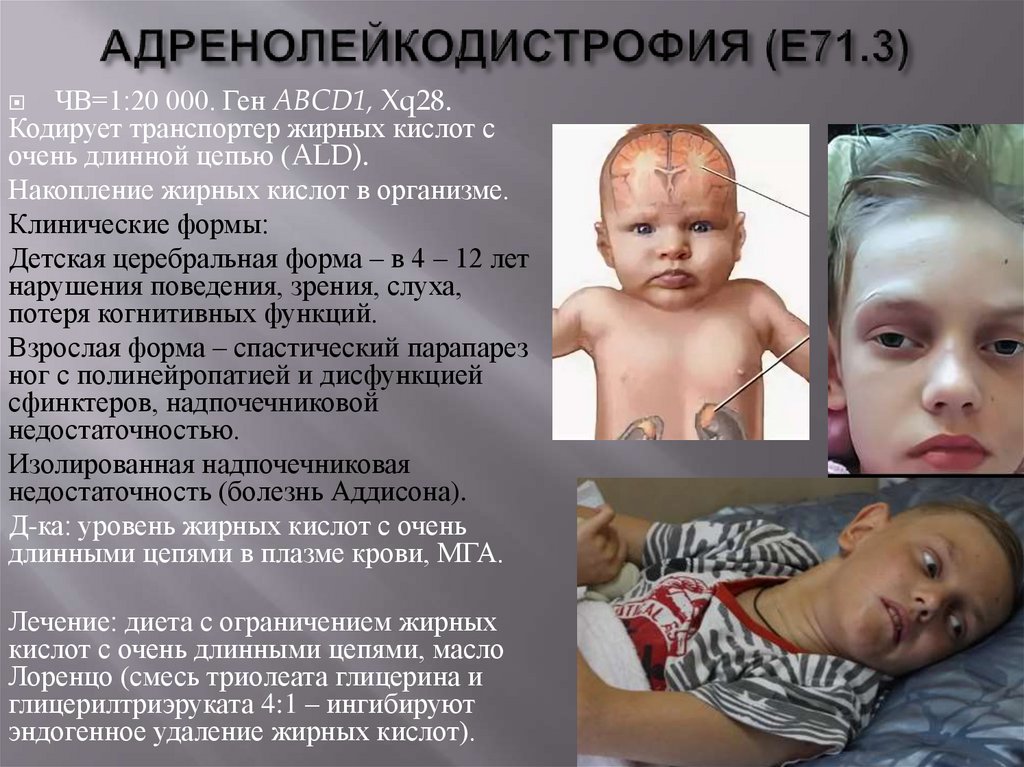
ЧВ=1:20 000. Ген ABCD1, Xq28.Кодирует транспортер жирных кислот с
очень длинной цепью (ALD).
Накопление жирных кислот в организме.
Клинические формы:
Детская церебральная форма – в 4 – 12 лет
нарушения поведения, зрения, слуха,
потеря когнитивных функций.
Взрослая форма – спастический парапарез
ног с полинейропатией и дисфункцией
сфинктеров, надпочечниковой
недостаточностью.
Изолированная надпочечниковая
недостаточность (болезнь Аддисона).
Д-ка: уровень жирных кислот с очень
длинными цепями в плазме крови, МГА.
Лечение: диета с ограничением жирных
кислот с очень длинными цепями, масло
Лоренцо (смесь триолеата глицерина и
глицерилтриэруката 4:1 – ингибируют
эндогенное удаление жирных кислот).
75.
Refsum disease (G60.1)Ген PHYH, 6q22 – ФИТАНОИЛ-КОА-ГИДРОКСИЛАЗА.
ЧВ=1:25 000.
Накопление фитановой кислоты в в ЦНС, ПНС, органах.
У детей прогрессируют полинейропатия, мозжечковая
атаксия, глухота, потеря обоняния, снижение и
концентрическое сужение полей зрения, пигментный
ретинит, ксеродерма кожи. Интеллект сохранен.
Д-ка: БХ (фитановая кислота в крови), МГА.
Лечение: строгая диета с ограничением молока, зеленых
растений, животных жиров, некоторой рыбы (тунец,
треска, пикша). Плазмаферез для удаления фитановой
кислоты.
76.
СИНДРОМЦЕЛЬВЕГЕРА (Q87.8)
АР т/н. ЧВ=1:50 000.
Гены PEX1, 2, 3, 5, 6, 10, 12.
60% - c.2528G>A,
c.2097_2098insT в гене
PEX1.
Клиника: гипотонус мышц,
судороги, желтуха и
холестаз, укорочение
проксимальных отделов
конечностей.
Глухота, ретинопатия,
катаракта.
77.
Скафоцефалия (преждевременноезарастание швов черепа).
Гипоплазия носа.
Выпуклый куполообразный лоб.
Низкопосаженные уши.
Ретрогнатия (сдвиг нижней челюсти
кзади)
Гипертелоризм. Плоские края орбит.
Атрофия зрительного нерва.
Эпикант. Низкий рост.
Длинные широкие пальцы.
Кальцификация эпифизов.
Д-ка: БХ крови (повышение
соотношения С26/С22 и С24/С22
жирных кислот в крови).
Лечение симптоматическое.
78.
1.2.
3.
4.
5.
6.
7.
8.
Устранение поступления субстрата с пищей.
Коррекция выведения продукта (болезнь Леша-Нихена –
аллопуринол, болезнь Вильсона-Коновалова – пеницилламин,
гемохроматоз – дефероксамин).
Замещение конечного продукта (врожденный гипотиреоз – тироксин,
адреногенитальный синдром – кортизол).
Замещение функции фермента (б. Фабри – агалсидаза, б. Гоше –
имиглуцераза, с. Гурлера – альдуразим, с. Хантера – элапраза, с.
Моркио – элосульфаза, с. Марото-Лами - галсульфаза).
Субстратредуцирующая терапия (б. Ниманна-Пика – миглустат, б.
Гоше – элиглустат, гомоцистинурия – бетаин, б. Л.Н. - аллопуринол).
Замещение кофактора (гомоцистинурия – витамин В6).
Исключение приема провокаторов дисбаланса обмена веществ
(сульфаниламиды – при острой перемежающей порфирии,).
Замещение недостающего продукта – б. Менкеса медь-гистидин.
79.
Гемофилия А. Ген VIII п.ф.с., Xq28, ЧВ=1:10000.
Гемофилия В. Ген F9, Xq27, ЧВ=1:50 000.
Гемофилия С. Ген F11, 4q35.2, ЧВ=1:100 000.
Клиника: повышенная кровоточивость,
гематомы в различных частях тела.
Посттравматические контрактуры и
анкилозы суставов.
Д-ка: АЧТВ (норма 25-39 сек), ПТВ (норма
11-16 сек), МНО (норма 0,85-1,35), тест
генерации тромбина, тромбоэластография.
Лечение: инъекции недостающего фактора
свертывания из донорской крови или
рекомбинантные.
Разрабатывается методика генотерапии
аденоассоциированными вирусами.
80.
1 тип – гиперхиломикронемия (ген липопротеинлипазы8p22, ген апоС2 – 19q32), ЧВ=1:1000.2а тип – гиперхолестеринемия (ген рецептора ЛПНП9р13, ген апоВ – 2р24), ЧВ=1:500.
2b тип – комбинированная гиперлипидемия – 10%
населения – повышенный уровень триглицеридов,
ацилКоА и апоВ.
3 тип – дис-бета-липопротеинемия (ген апоЕ – 19q13),
1:5000.
4 тип – эндогенная гиперлипидемия – 1% населения,
гены, участвующие в распаде ЛПОНП.
5 тип – гипертриглицеридемия (активация генов синтеза
ЛПОНП + мутации гена липопротеинлипазы-8р22).
81.
82.
83.
84.
В настоящее время программа генотерапевтического леченияADA-недостаточности модифицирована таким образом,
что генетическая конструкция, содержащая нормальный
ген ADA, вводится не в зрелые T-лимфоциты, а в
стволовые клетки костного мозга.
Генная терапия -новая область современной биомедицины,
основанная на введении в организм больного
рекомбинантных генетических конструкций с лечебной
целью.
85.
КЛАССИФИКАЦИЯ ГЕННОЙ ТЕРАПИИ1) по типу клеток-мишеней:
соматическая
фетальная
2) по цели воздействия:
позитивная ( компенсация
негативная (подавление
экспресcии гена)
функций гена)
3) по тактике введения генотерапевтического агента:
1.
ex vivo
2.
in vivo
in utero (введение конструкций в эмбрион)
in situ (локально)
4) по типу векторной системы:
вирусные векторы
невирусные векторы
микроинъекция
“gene gun” (генный пистолет)
5) по применяемым агентам:
нуклеиновые кислоты
белки
иммунотерапия
86.
- достоинства:Вирусные векторы:
• ретровирусы
• аденовирусы
• аденоассоциированный
вирус
• герпесвирусы
• лентивирусы
• и др.
• трансфекция большого
количества клеток
• тропизм
• неспособность
реплицироваться в клеткехозяине
• устойчивость к деградации
лизосомами
- недостатки:
иммуногенность
(аденовирусы, герпесвирусы)
потенциальная
туморогенность
(ретровирусы)
87.
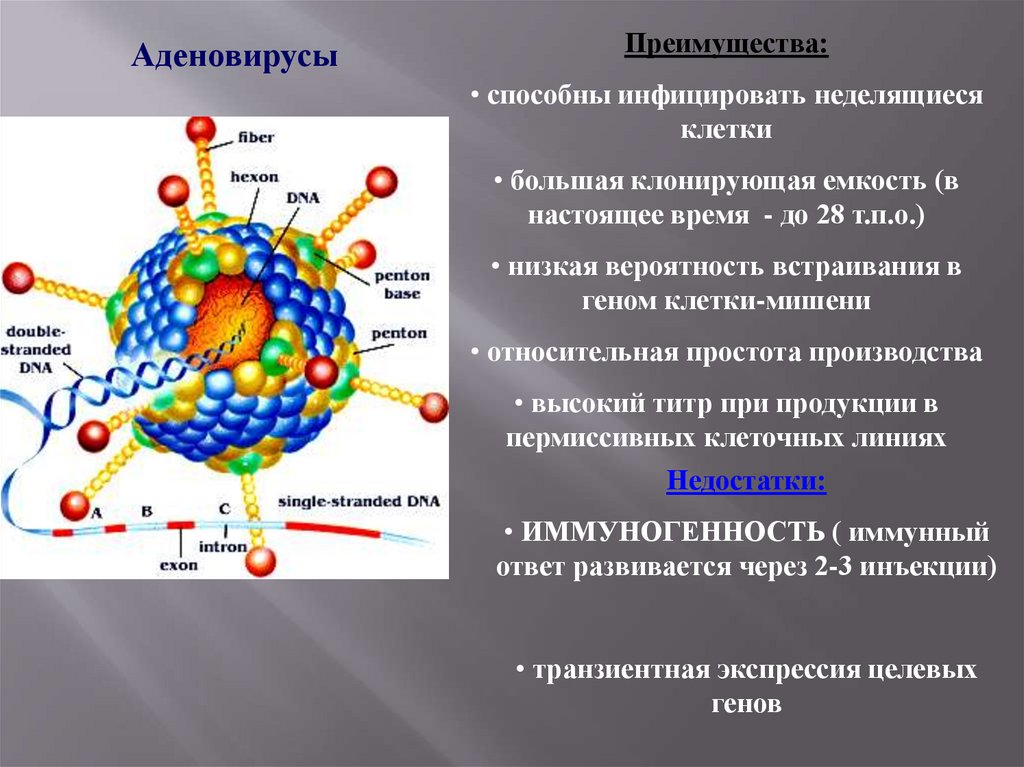
АденовирусыПреимущества:
• способны инфицировать неделящиеся
клетки
• большая клонирующая емкость (в
настоящее время - до 28 т.п.о.)
• низкая вероятность встраивания в
геном клетки-мишени
• относительная простота производства
• высокий титр при продукции в
пермиссивных клеточных линиях
Недостатки:
• ИММУНОГЕННОСТЬ ( иммунный
ответ развивается через 2-3 инъекции)
• транзиентная экспрессия целевых
генов
88.
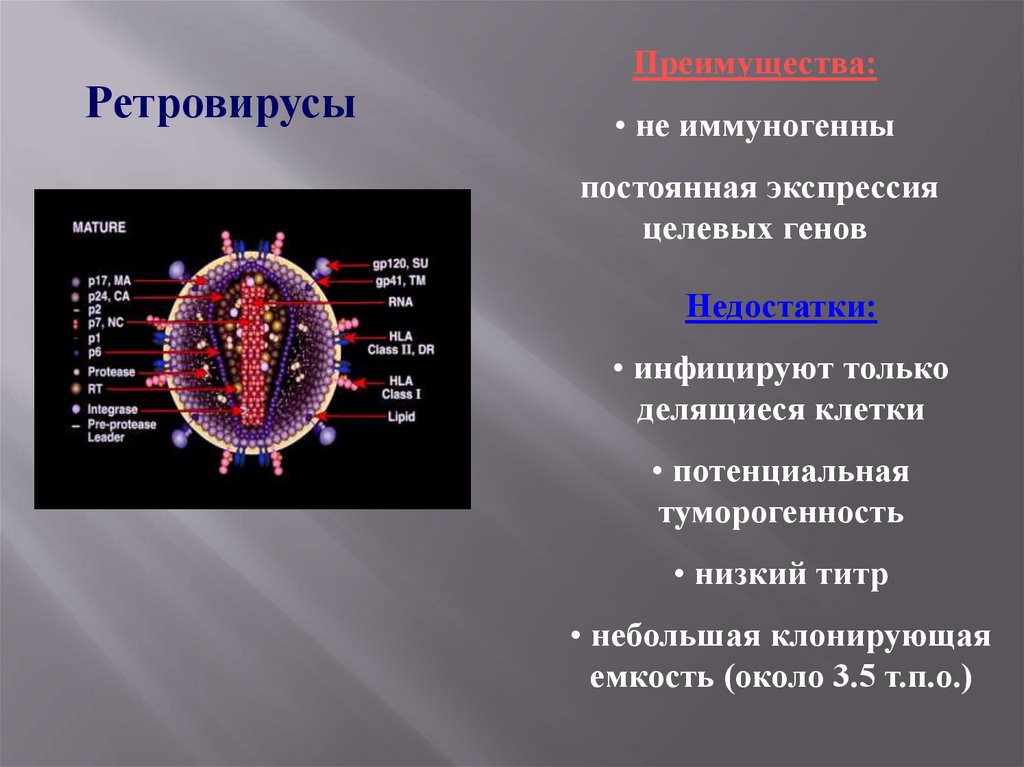
РетровирусыПреимущества:
• не иммуногенны
постоянная экспрессия
целевых генов
Недостатки:
• инфицируют только
делящиеся клетки
• потенциальная
туморогенность
• низкий титр
• небольшая клонирующая
емкость (около 3.5 т.п.о.)
89.
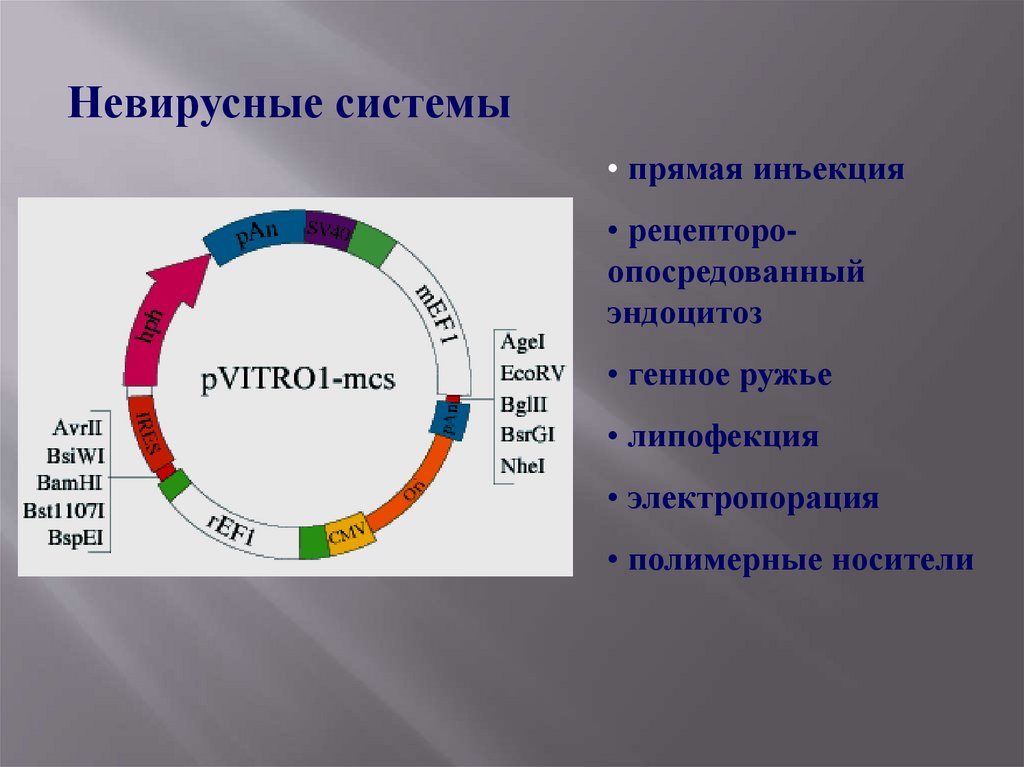
Невирусные системы• прямая инъекция
• рецептороопосредованный
эндоцитоз
• генное ружье
• липофекция
• электропорация
• полимерные носители
90.
Плазмидные векторы- достоинства:
• отсутствие токсичности и
мутагенности
• практически неограниченная емкость
вектора
• дешевизна производства
- недостатки:
• трансфекция ограниченной популяции
клеток
• деградация ДНК лизосомами
• отсутствие тропизма
( в настоящее время есть перспективные разработки)
91.
Генная терапия вмедицине
Клинические испытания
генотерапевтических препаратов.
• I фаза. Оценка токсичности генной
конструкции.
• II фаза. Ограниченные испытания на
небольшом контингенте больных.
• III фаза. Широкомасштабные
мультицентровые клинические испытания.
92.
ПРИМЕРЫ:• муковисцидоз (кистозный фиброз поджелудочной
железы) - перенос гена МТР (муковисцидозный
трансмембранный регулятор) с помощью
аденовирусного вектора и липосом.
•мышечная дистрофия Дюшена - введение нормальных
копий кДНК гена дистрофина с помощью
ретровирусных векторов.
• рестеноз (повторное сужение просвета артерии после
ангиопластики) - введение в сосуд гена сосудистого
эндотелиального фактора роста (VEGF) в виде
плазмидной ДНК.
93.
БолезньИммунодефицит
Дефектный ген
Аденозиндезаминаза
Клеткимишени
Лимфоциты
Семейная
Рецептор
гиперхолестеринемия липопротеинов
низкой плотности
Гепатоциты
Гемофилия В
Фактор IX
Фибробласты
Эмфизема легких
А-1-антитрипсин
Лимфоциты
Фенилкетонурия
Фенилаланин гидроксилаза
Гепатоциты
94.
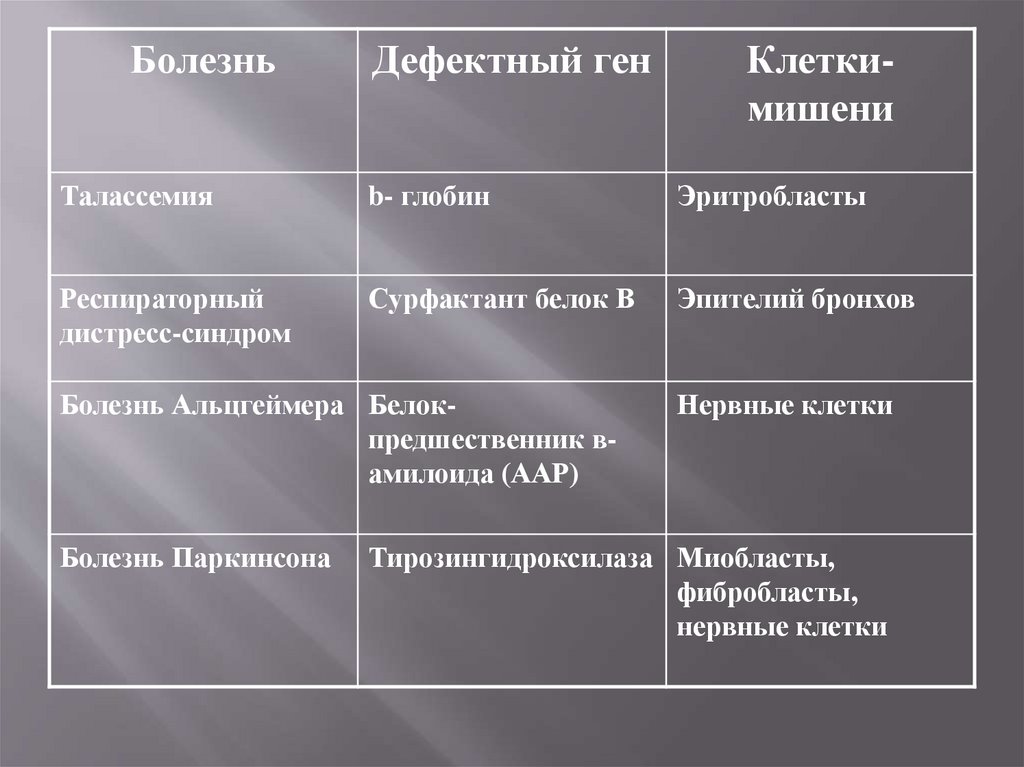
БолезньДефектный ген
Клеткимишени
Талассемия
b- глобин
Эритробласты
Респираторный
дистресс-синдром
Сурфактант белок В
Эпителий бронхов
Болезнь Альцгеймера Белокпредшественник вамилоида (ААР)
Болезнь Паркинсона
Нервные клетки
Тирозингидроксилаза Миобласты,
фибробласты,
нервные клетки
95.
WBC (white blood cells) = 4 – 9 x 109 /лRBC (red blood cells) = 4 – 5,5 x 1012 /л
HGB (hemoglobin) = 120 – 140 г/л (женщины), мужчины
135 – 160
HCT (hematocrit) = 0,39 – 0,49 (% форменных
элементов по отношению к общему объему крови)
PLT (platelets) = 150 – 400 х 109 /л
ЦП = 0,85 – 1,05
СОЭ (мм/ч) до 60 лет = мужчины до 8; женщины до 12
После 60 лет = мужчины до 15; женщны до 20
96.
По степени тяжести: легкой ( Hb >90 г/л), средней(70 – 90) и тяжелой (менее 70 г/л).
По способности костного мозга к регенерации:
- Норморегенераторная (ретикулоцитов 0,5 – 2%),
- Гиперрегенераторная (>2%) – гемолитические,
постгеморрагические,
- Гипорегенераторная (<0,5%) – Fe- и В12
дефицитные,
- Арегенераторные (0%) - апластические
97.
Гемолитическая анемия возникает при преобладании процессаразрушения эритроцитов над их образованием.
Наследственные гемолитические анемии
Наследственные анемии по локализации генетически обусловленного
дефекта разделяются на три группы:
а) мембранопатии – связанные с нарушением структуры и обновления
белковых и липидных компонентов мембран эритроцитов
(Минковского-Шоффара);
б) ферментопатии – связанные с дефицитом эритроцитарных
ферментов, которые обеспечивают пентозо-фосфатный цикл, гликолиз,
синтез АТФ и порфиринов, обмен нуклеотидов и глютатиона;
в) гемоглобинопатии – связанные с нарушением структуры или синтеза
цепей гемоглобина.
98.
Апластическая анемия Фалькони .Гипоплатическая анемия Ерлиха.
Анемия Даймонда-Блекфена.
Синдром Швахмана-Даймонда.
99.
МЕМБРАНОПАТИИсфероцитоз Минковского-Шоффара
наследственный элиптоцитоз
наследственный пиропойкилоцитоз
наследственный стоматоцитоз
наследственный акантоцитоз
наследственный эхиноцитоз
100.
АД тип наследования. ЧВ= 1 : 5000 населения.Около 25% случаев спорадические.
Мутации гена спектрина мембраны эритроцита
(повышенная проницаемость для ионов Na и
накопление воды).
Спектрин – это белок цитоскелета, выстилающий
внутреннюю поверхность плазматической
мембраны клеток, выполняя важную роль в
поддержании целостности клеточной мембраны
и структуры цитоскелета.
Ген альфа субъединицы спектрина расположен на
1q22-25.
Ген бета субъединицы спектрина расположен на
14q22-23.2.
101.
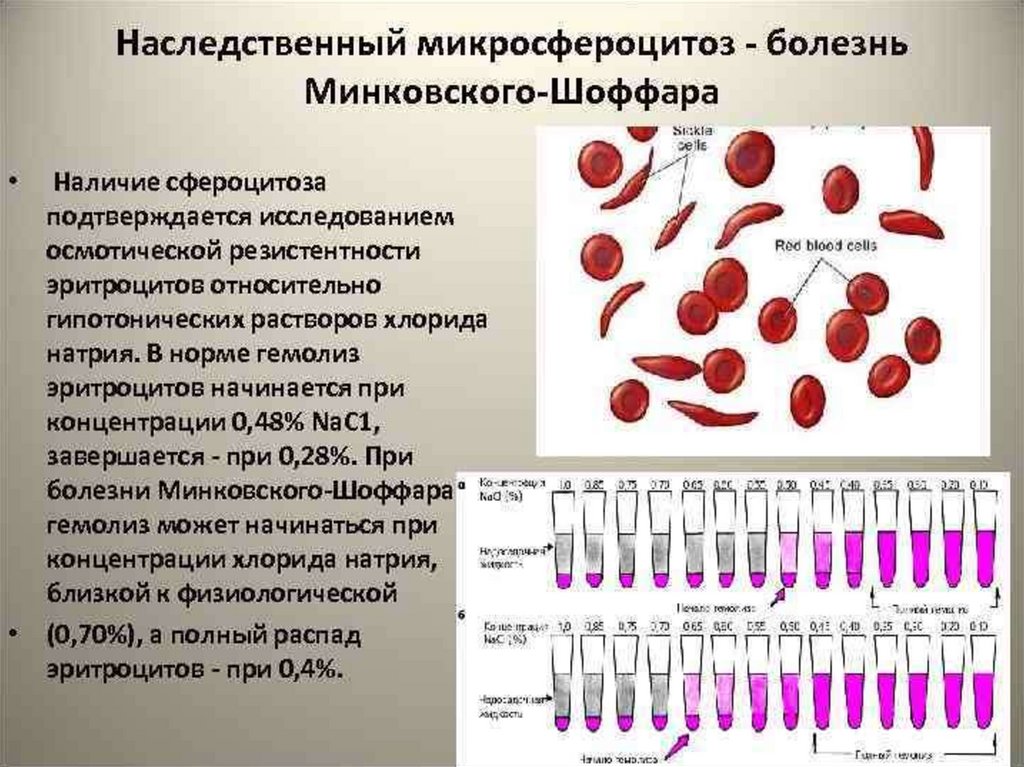
манифестация в раннем детском возрастеДИАГНОСТИКА:
-наследственный анамнез.
-осмотр (башенный череп, готическое небо, изменения
расположения зубов, микрофтальм, деформация
мизинцев).
-объективные данные (спленомегалия, желтуха).
- ОАК (нормохромная анемия, патологический размер
эритроцитов – микросфероциты менее 6,4 мкм (в норме
7,5 мкм), количество ретикулоцитов повышено)
-БХ АК (повышение непрямого (несвязанного)
билирубина)
- УЗИ (ранняя ЖКБ)
102.
103.
Лечение:Наследственный сфероцитоз
• Бессимптомные формы
Лечения не требуется
УЗИ контроль состояния желчных путей
• Легкая и среднетяжелая форма
Вне криза – терапии не требуется, УЗИ контроль желчных путей, при
необходимости желчегонная терапия, восполнение дефицита фолатов
Гемолитический криз - трансфузии эр. массы при значительном падении Hb,
инфузионная терапия, фолиевая кислота
Спленэктомия при наличии показаний
• Тяжелая форма
Трансфузии эр. массы
Фолиевая кислота
Желчегонная терапия – по показаниям
Контроль обмена железа – хелаторная терапия при перегрузке
Спленэктомия в плановом порядке
104.
Недостаточность эритроцитарныхферментов:
глюкозо-6-фосфатдегидрогеназа
пируваткиназа
глюкозофосфат изомераза
105.
Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД)катализирует гликолиз в процессе гексозомонофосфатного шунта, метаболизм глютатиона
снижена защита от окислительного стресса
ген - на Х-хромосоме (Xq28) – наследование Х-сцепленное, болеют
мальчики; редко девочки -гомозигы
Девочки-гетерозиготы имеют две популяции эритроцитов: нормальные клетки и
дефицитные, соотношение их вариабельное
~ 400 млн. человек в мире с патологическим геном
частая патология среди народов Закавказья, Средиземноморья, ЮгоВосточной и Юго-Западной Азии, Африки. славяне – 0,5%
106.
Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД)Большинство пациентов с дефицитом Г-6-ФД вне
криза не имеют каких-либо клинических признаков
острая гемолитическая анемия
развивается спустя несколько часов или дней от начала приема
лекарств, контакта с нафталином, развития инфекции, использования
в пищу конских бобов (фавизм)
резкая анемия, ретикулоцитоз (на 4-6 день), тельца Гейнца
желтуха
нет спленомегалии
темная(черная) моча
увеличение непрямого билирубина, свободного гемоглобина плазмы
при тяжелом кризе возможно развитие ОПН
107.
Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД)Диагностика:
Признаки внутрисосудистого гемолиза – увеличение
непрямого билирубина, ЛДГ, свободного
гемоглобина плазмы, снижение гаптоглобина.
Анемия, ретикулоцитоз, тельца Гейнца.
Снижение активности Г-6-ФД в эритроцитах.
Лечение:
устранение провоцирующего фактора
инфузионная терапия, контроль функции почек
трансфузии эр. массы – только в исключительных
случаях
108.
АР тип наследования.В норме гемоглобин человека состоит из 2 альфа-цепей и двух
бета-цепей. То есть гемоглобин А – HbA (α2β2).
Мутации в гене бета-цепи глобина в 6 позиции (глутаминовая
кислота заменена на валин) приводят к образованию
гемоглобина S (гемоглобин склонен к кристаллизации вместо
нормальной четвертичной структуры и растворения в
цитоплазме эритроцита.
Ген HBB (hemoglobin subunit beta) – локализован на 11р12.
Распространенность имеет выраженные различия различных рас
и географического проживания (около 10% афроамериканцев
являются гетерозиготными носителями мутации; в Греции,
Италии и Португалии до 30%). Гомозиготы в США – 1:5000
населения.
109.
Серповидноклеточная анемия. У таких больных вместогемоглобина А синтезируется гемоглобин S. Отличается
он тем, что глютаминовая кислота в нем замещена
валином в шестом положении -цепи.
Эта замена резко снижает растворимость гемоглобина в
условиях гипоксии. Восстановленный гемоглобин S в 100
раз менее растворимый, чем окисленный, и в 50 раз менее
растворимый чем гемоглобин А.
В кислой среде он выпадает в осадок в виде кристаллов и
деформирует эритроциты, придавая им серповидную
форму. Мембрана их теряет прочность, и наступает
внутрисосудистый гемолиз.
Эритроциты больных с СКА содержат гемоглобин S
(HbS), отличающийся по электрофоретической
подвижности и качественному составу от гемоглобина
здорового человека. При СКА в 6 – й позиции В – цепи
гемоглобина глутаминовая кислота заменена на валин.
110.
1.Конституциональные проявления – отставание роста и
развития, полового созревания.
2. Повышенная склонность к тяжёлым инфекциям, особенно
пневмококковым, объясняемую нарушением функции селезёнки
по очищению крови от циркулирующих бактерий вследствие
повторных инфарктов и замещения нормальных тканей органа
фиброзной тканью.
3. Анемические проявления.
Гемолитическая анемия. При гомозиготной форме типична
выраженная анемия, гематокрит 18 – 30 %. В среднем
продолжительность жизни эритроцитов составляет 10 – 15 дней.
Гаптоглобин в плазме либо отсутствует, либо его концентрация
уменьшена, а концентрация свободного гемоглобина умеренно
увеличена. Повышен непрямой билирубин. Имеет место
образование билирубиновых камней ЖП.
Мегалобластные кризы. При ограниченном поступлении с пищей
фолиевой кислоты развивается магалобластический эритропоэз,
а также гиперкоагуляционное состояние из – за гомоцистеинемии
в результате дефицита фолатов.
111.
Апластические кризы. Пациенты с СКА подвержены инфекциям ивоспалительным процессам, подавляющим эритропоэз.
Типичный пример – инфекция парвовирусом В19, вызывающая
гипоплазию и аплазию кроветворения у больного с СКА. Быстро
падает количество ретикулоцитов и гемоглобина на 10 г/л в день.
При отсутствии немедленной терапии развивается застойная СН.
Острый гиперспленизм характерен для новорождённых и детей
раннего возраста. В течение нескольких часов селезёнка
увеличивается и секвестрирует большинство циркулирующих
эритроцитов. Массивная спленомегалия и падение гемоглобина
более 10 г/л могут привести к смерти.
Гипергемолиз. Типичные гемолитические кризы с повышением
ретикулоцитов и падением гемоглобина.
4. Феномен окклюзии сосудов с болевыми кризами.
Рецидивирующие тромбозы определяют в основном
болезненность и смертность пациентов при СКА. Кризы
начинаются внезапно и локализуются в области живота, грудной
клетки. Появляются боли в суставах.
112.
1. Изменения со стороны анализа крови: макроцитоз эритроцитов(MCV>100), ретикулоцитоз (>100,0*10^9/л), лейкоцитоз с
нейтрофилёзом и незначительным левым сдвигом, тромбоцитоз. В
мазке периферической крови: серповидные эритроциты,
полихроматофильный макроцитоз эритроцитов, наличие эритроцитов
с тельцами Жолли (функциональная аспления).
2. Электрофорез гемоглобина с количественным определением
гемоглобина A, S, A2, F.
113.
ТерапияИнфузионная терапия, анальгетики
Трансфузионная терапия – только по показаниям (с
осторожностью!!!, возможно увеличение вязкости крови и
усиление гемолиза). Возможно частичное «заменное»
переливание
Хелаторная терапия
Фолиевая кислота, желчегонная терапия
Гидроксимочевина – стимулятор синтеза HbF → снижение
уровня HbS и препятствие его внутриклеточной
полимеризации
Вакцинация – функциональная аспления
Холецистэктомия – по показаниям
Трансплантация костного мозга
114.
ТАЛАССЕМИЯЭто группа аутосомно – рецессивных заболеваний
крови, характеризующихся снижением синтеза
одного из двух полипептидных цепей глобина
(альфа или бета), которые образуют молекулу
взрослого гемоглобина. Это приводит к
уменьшению наполнения эритроцитов
гемоглобином и анемии.
Когда заторможен синтез альфа- или бета-цепей
гемоглобина, развивается талассемия. Для нее
характерные мишенеподобные эритроциты.
У гетерозигот развивается так называемая малая
талассемия, у гетерозигот – большая талассемия
Кулли с высшей степенью гемолиза эритроцитов
115.
Синтез a – цепей глобина кодируется диплоиднымнабором генов aa/aa (4 гена, по два от обоих родителей).
Клиническая картина a – талассемии определяется числом
подвергшихся делеции или мутации генов.
Бессимптомное носительство – отсутствие клинических
симптомов и изменений в гемограмме – имеет место при
делеции одного из 4 – х генов (-а/аа).
Малая талассемия – легкая анемия без признаков гемолиза
или её отсутствие, снижение MCV, MCH (ср. объём
эритроцита, ср. содержание гемоглобина в эритроците) –
возникает в результате делеции двух генов или
неделетирующей мутации второго гена (--/аа, -а/-а, -а/а).
Гемоглобинопатия Н (промежуточная талассемия) –
хроническая гемолитическая анемия – возникает при
делеции трёх генов (--/-а).
Внутриутробная водянка – смерть во внутриутробном
периоде или сразу после рождения – обусловлена делецией
всех четырёх генов (--/--).
116.
Малая талассемия (В+/В) протекает, как правило, без клиническихпроявлений, за исключением более высокой частоты анемии в
период беременностиСинтез В – цепей глобина кодируется
двумя генами В/В ( по одному от обоих родителей). Синтез В
– цепей глобина при данном виде талассемий нарушается
вследствие мутационного изменения функциональной
активности гена. При этом может быть снижение синтеза В –
цепей (В+) или полное отсутствие синтеза (В0).
Большая талассемия (анемия Кули) имеет генотип В0/В0 с
типичной клинической картиной заболевания, проявляющейся с
6 – 8 – й недели жизни.
Промежуточная талассемия (В+/В+ или В0/В) клиническая картина
гемолитичекой анемии менее выражена, потребность в
гемотрансфузиях возникает при интеркуррентных инфекциях.
117.
1) Анемия хронического течения, вызывающая замедление роста, половогоразвития, хроническую сердечную недостаточность и другие связанные с
ней осложнения.
2) Гиперплазия костномозгового кроветворения, прежде всего эритропоэза, в
пределах костного скелета и с возникновением очагов экстрамедуллярного
кроветворения в селезёнке, печени и мягких тканях:
-расширение губчатого вещества костей черепа с рентгенологической картиной,
называемой «стриженный под ёжик», гипертрофия лобной кости с
уплощением переносицы, сужением глазных щелей, верхней челюсти с
выступанием зубов и формированием «лица бурундука»;
- расширение мозгового слоя и истончение кортикальной пластинки позвонков
и длинных трубчатых костей, предрасполагающее к переломам;
- спленомегалия, гепатомегалия;
- очаги миелоидного кроветворения в мягких тканях, паравертебральных
областях с возможным сдавлением спинного мозга.
3) Синдром перегрузки железом из – за повышенной абсорбции железа из ЖКТ
и вследствие трансфузий эритроцитарной массы. Гемосидероз миокарда
приводит к КМП, аритмиям; гемосидероз печени – к развитию
гепатоцирроза, гепатоцеллюлярной карциномы.
4) Хронический гемолиз: сплено-, гепатомегалия, билирубиновые камни в
желчном пузыре, гиперспленизм, повышающий трансфузионную
зависимость.
118.
А. В анализе крови анемия различной степени,микроцитоз эритроцитов (MCV<80), гипохромия
эритроцитов (МСН < 24), наличие мишеневидных
эритроцитов, нормобластов.
Б. Наличие симптомов гемолиза и акселерированного
эритропоэза.
В. Изменения фракций гемоглобина по данным
электрофореза гемоглобина.
Г. Типичные соматические нарушения: аномалии
скелета, сплено-, гепатомегалия, задержка роста,
полигландулярная недостаточность.
Д. Симптомы перегрузки железом.
119.
При тяжёлых формах:1) Регулярные трансфузии эритроцитарной массы (1 – 3
дозы каждые 3 – 5 недель): целевой уровень
гемоглобина составляет 100 – 120 г/л. С целью
предупреждения изоиммунизации, трансфузионных
реакций, инфекционных осложнений и поддержания
высокого комплаенса к лечению целесообразно
использовать системы с лейкоцитарными фильтрами,
устанавливать венозные порты, осуществлять
мероприятия по обеспечению вирусной безопасности
гемотрансфузией.
120.
2) Профилактика и лечение трансфузионнойперегрузки железом при СФ более 1000 нг/мл.
Десферал 500 мг вводят по 50 мг/кг подкожно в
течение 8-12 часов или в/в капельно 5 дней в
неделю. Возможные побочные эффекты:
повреждение сетчатки, катаракта, иерсиниозная
инфекция. Эксиджад 250 (500) мг принимают в дозе
10-30 мг/кг один раз в день внутрь в виде раствора
шипучей таблетки длительно, не менее 1 года.
3) Заместительная гормональная терапия:
использование гормонов роста, половых,
тиреоидных гормонов, ингибиторов остеокластов
(богатая кальцием диета, сапплементация кальцием,
при необходимости в сочетании с витамином Д,
бисфосфонаты).
4) Спленэктомия в случае массивной спленомегалии и
повышения трансфузионной зависимости.
5) Аллогенная трансплантация КМ.