


































 medicine
medicineSimilar presentations:










Моногенные болезни
1.
Бойнова И.В.МОНОГЕННЫЕ БОЛЕЗНИ
2.
Моногенные болезни (МБ) – гетерогенная по клиническим проявлениям группанаследственных заболеваний, в основе которых лежат мутации одного гена потери, удвоения, транслокации и т.д.
Результат: нарушение структуры/синтеза кодируемого геном белка с
изменением его количества (до отсутствия).
Мутации могут передаваться из поколения в поколение,
м. возникать в половых клетках родителей спонтанно.
В настоящее время известно около 5000 нозологических единиц МБ.
Это 42–65 детей на 1000 новорожденных (4,2-6,5%).
В структуре общей смертности детей до 5 лет на их долю приходится 10-14%.
3.
Существуют моногенные формы АГ, б. Альцгейаера иПаркинсона, эпилепсии, иммунодефицитов, онкозаболеваний и др.
Моногенный тип заболевания отличается от
спорадического более тяжелым течением и ранним
дебютом.
Большинство МБ болезней распознаются в перинатальном
или раннем детском возрасте, т.к. 25% развиваются в
эмбриональном периоде, и еще 50% проявляются к 3
годам. К концу пубертантного периода диагностируются
уже 90% всех моногенных болезней.
4.
В неврологии описано >350 МБ,дерматологии и офтальмологии – 250.
К.п. это тяжелые неизлечимые болезни.
Только 5% генов связано с моногенными заболеваниями
В зависимости от локализации мутантного гена и характера
доминирования выделяют:
- аутосомно-доминантные,
- аутосомно-рецессивные ,
- сцепленные с полом заболевания (доминантные/рецессивные).
М и Ж = 1:1 при аутосомном типе наследования и с разной частотой в
случае сцепленного с полом/ограниченного полом наследования.
5.
Большинство МБ подчиняется законам Менделя о рецессивности идоминантности гена и пребывании его в гомо- или гетерозиготном состоянии
(менделирующие заб.).
Наследование некоторых МБ не подчиняется з. Менделя - нетрадиционный тип
наследования (митохондриальные заб., б-ни экспансии и др.).
! Отсутствие повторных случаев болезни у членов одной семьи не исключает
наследственности заб-я, т.к. наследуются не заб-я, а аллельные состояния
генов, поэтому в семье м.б. только 1 б-й с насл. заб.
6.
Заболевания с аутосомоно-рецессивным типом наследования(Фенилкетонурия, Галактоземия, АГС, Муковисцидоз,
Гепатолентикулярная дегенерация)
проявляются только при гомозиготном носительстве мутантных
аллелей – один от матери, другой – от отца. При этом происходит
частичная или полная инактивация функции мутантного гена.
К. п. родители сами здоровы, но являются гетерозиготными
носителями мутации. При анализе родословной прослеживается
«горизонтальный» характер наследственной передачи заболевания.
Согласно закону Менделя вероятность рождения больного ребенка
равна 25%. М:Ж=1:1, В одной семье м. б. несколько больных сибсов.
Это не зависит от возраста родителей или очередности беременности.
Заболевания тяжелые, б-е к. п. потомства не оставляют.
В браке гетерозиготных родителей 2/3 здоровых детей гетерозиготны.
В браке гетерозиготного носителя рецессивной мутации с супругом,
без мутантного аллеля, все дети здоровые, но 50%
из них будут гетерозиготными носителями мутации
7.
Заболевания с аутосомно-доминантном типом наследовании(туберозный склероз, коллагенопатии, в т. ч. Б. Марфана, ЭлерсаДанло, несовершенный остеогенеза, хондродисплазии, тугоухость,
офтальмопатии, нарушения дентино- и амелогенез и др.)
Таких заб. Больше, чем АР
Для проявления заболевания достаточно гетерозиготного
носительства мутации. М:Ж=1:1.
Доминантные мутации не приводят к инактивации функции
кодируемого белка, происходит либо снижение количества белка
(гаплонедостаточность), либо появлением у мутантного белка нового
агрессивного свойства.
В браке гетерозиготного носителя доминантной мутации со здоровым
супругом вероятность рождения больного ребенка - 50%. АДз
передаются «по вертикали» только среди родственников одного из
родителей больного.
Если оба родителя ребенка АДЗ здоровыми, то мутация произошла в
половых клетках одного из родителей спонтанно и риск рождения ещё
одного больного ребенка такой же, как в других семьях
8.
Сцепленный с Х-хромосомой тип наследованияМутантный ген расположен в Х-хромосоме.
Если мутация доминантная, то болеют и М, и Ж.
В в 100% случаев от больного отца заболевание унаследует дочь.
В 50% случаев от больной матери заболевание передастся детям
(одинаково и сыну и дочери).
Пример - витамин Д-резистентный рахит (гипофосфатемия).
Чаще Х-сц. мутации рецессивны.
Здоровая мать, гетерозиготная по мутантному аллелю передает его
сыновьям, которые будут больны.
У этих женщин есть больные дяди или братья.
Больные мужчины передают заболевание только через поколение и
только внукам (но не внучкам) через свою здоровую, но
гетерозиготную дочь.
Пример – гемофилия А и В,
миодистрофия Дюшенна-Беккера
9.
Классификация МБ1. По ведущей системной патологии –
по органному и системному типу.
- Болезни НС: системные дегенерации, нервно-мышечные заб-я, факоматозы.
- Болезни ССС: семейная гиперхолестеринемия, наследств. амилоидоз и др.
- Болезни органов дыхания: спонтанный пневмоторакс, первич. легочная
гипертензия, др
- Болезни ЖКТ: синдромы мальабсорбции, целиакия, и др.
- Болезни соединит. ткани и скелета: с. Марфана, мукополисахаридозы и др
- Болезни кожи и ее придатков: ихтиоз, буллезный эпидермолиз и др.
- Болезни почек и МВП: наследственный нефрит, несахарный диабет и др.
- Болезни эндокринных органов: гипотиреоз, гипофизарный нанизм, АГС и др.
- Болезни органов зрения, слуха, половой сферы и др
.
2. По этиологии:
- болезни с установленным первичным молекулярным (биохимическим)
дефектом (10% МБ),
- с неустановленным первичным молекулярным дефектом (90% всех МБ).
10.
3. По типу наследования:В зависимости от локализации патол. гена в аутосоме/половой хромосоме и
его доминантности/рецессивности.
1.
Аутосомно-доминантные б-ни: нейрофиброматоз, с.Марфана,
х.Гентингтона и др.
2.
Аутосомно-рецессивные б-ни: альбинизм, галактоземия, муковисцидоз,
фенилкетонурия, адреногенитальный синдром и др.
3.
Х-сцепленные доминантные б-ни: витамин D-резистентный рахит и др.
4.
Х-сцепленные рецессивные б-ни: гемофилия А, В, миодистрофия
Дюшена и др.
11.
По нарушению вида обмена веществ: - наиболее распространенная.Выделено > 700 форм
1. НБО аминокислот (гомоцистинурия, фенилкетонурия и др.).
2. НБО углеводов (галактоземия, мукополисахаридозы и др.).
3. НБО липидов (гиперлипидемия, гиперхолестеринемия и др.).
4. НБО пуринов и пиримидинов (подагра и др.).
5. НБ биосинтеза кортикостероидов (адреногенитальный синдром и др.).
6. НБ порфиринового и билирубинового обмена (с. Жильбера, порфирии и др.).
7. НБО металлов (болезнь Вильсона-Коновалова и др.).
8. НБ эритрона (гемолитические анемии и др.).
9. НБ лимфоцитов и лейкоцитов (септический грануломатоз и др.).
10. НБ транспорта систем почек (вит. D-резистентный рахит, тубулопатии, и др)
12.
Принципы МБ1. Принцип генетической гетерогенности разные мутации в одном и том же
гене приводят к разной клинике, различным типам наследования, локализации
пат. процесса, биохимическим изменения.
Причина - мутации разных локусов.
2. Принцип клинического полиморфизма - различия в клинике, динамике и
лечении МБ.
Этому способствуют следующие факторы:
а) генетические факторы (на основной патол. ген действуют другие генымодификаторы и факторы эндогенной среды);
б) факторы окружающей среды - действие экологии, бытовых условий, профессии,
питания, обычаев, медикаментов и т.д.
3. Хр. прогредиентное течение, обусловленное пост. действием пат. гена.
Заб-е м. начаться с единичных симптомов и постоянно состояние б-х м. ухудшаться
13.
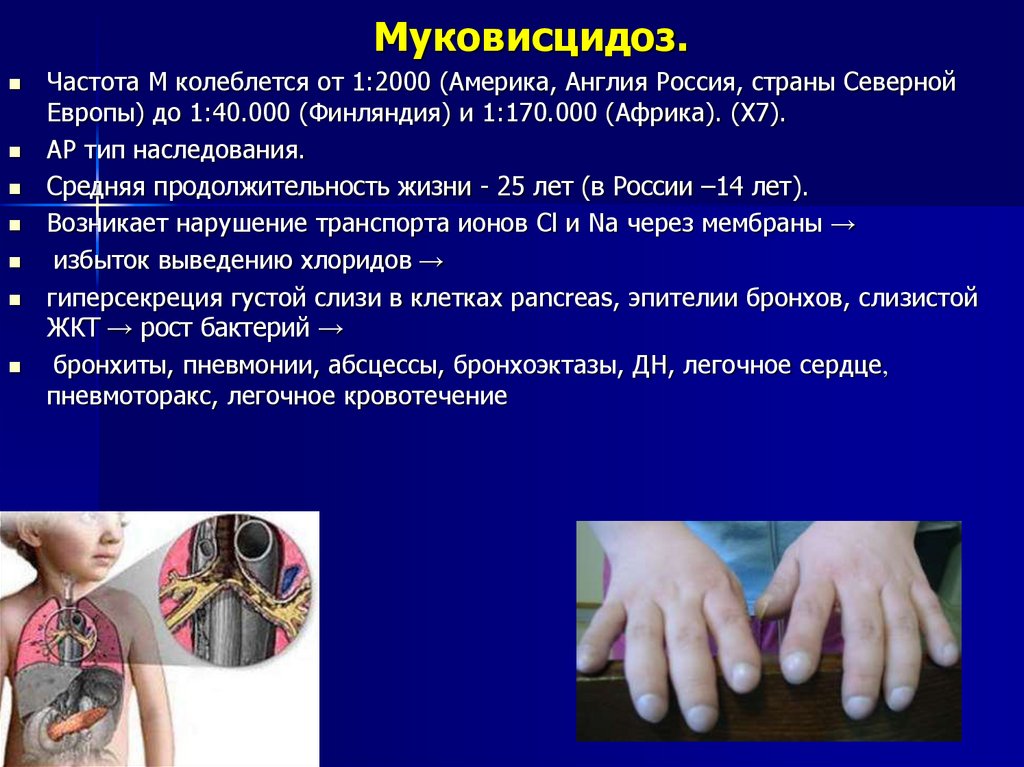
Муковисцидоз.Частота М колеблется от 1:2000 (Америка, Англия Россия, страны Северной
Европы) до 1:40.000 (Финляндия) и 1:170.000 (Африка). (Х7).
АР тип наследования.
Средняя продолжительность жизни - 25 лет (в России –14 лет).
Возникает нарушение транспорта ионов Cl и Na через мембраны →
избыток выведению хлоридов →
гиперсекреция густой слизи в клетках pancreas, эпителии бронхов, слизистой
ЖКТ → рост бактерий →
бронхиты, пневмонии, абсцессы, бронхоэктазы, ДН, легочное сердце,
пневмоторакс, легочное кровотечение
14.
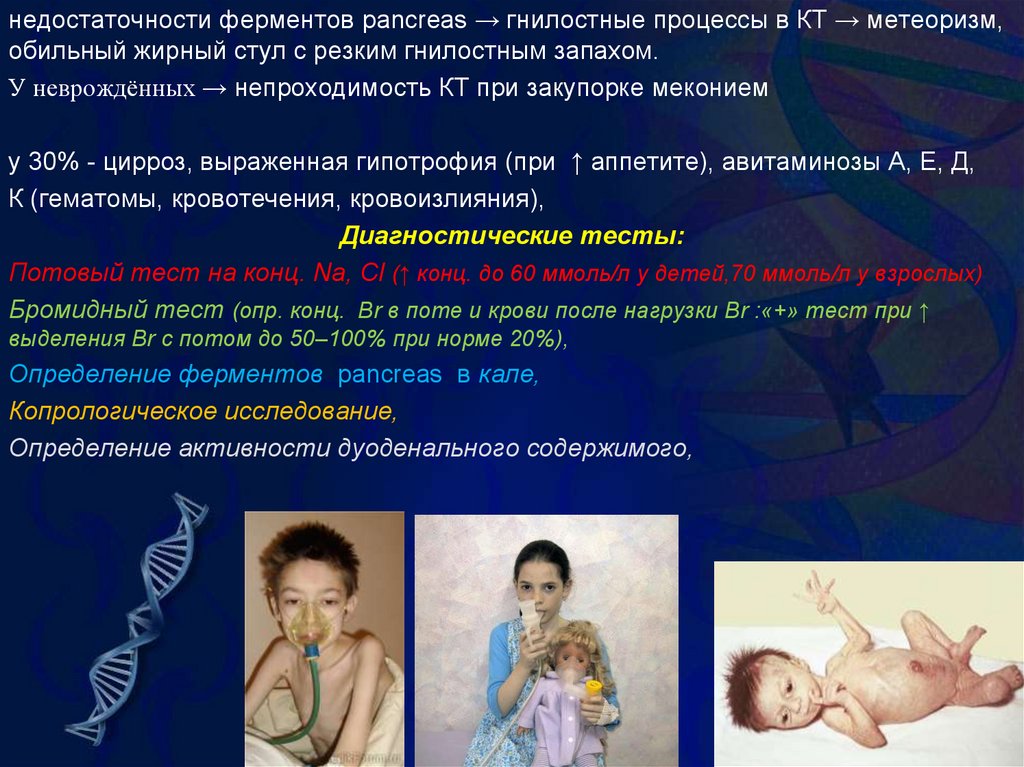
недостаточности ферментов pancreas → гнилостные процессы в КТ → метеоризм,обильный жирный стул с резким гнилостным запахом.
У неврождённых → непроходимость КТ при закупорке меконием
у 30% - цирроз, выраженная гипотрофия (при ↑ аппетите), авитаминозы А, Е, Д,
К (гематомы, кровотечения, кровоизлияния),
Диагностические тесты:
Потовый тест на конц. Na, Cl (↑ конц. до 60 ммоль/л у детей,70 ммоль/л у взрослых)
Бромидный тест (опр. конц. Br в поте и крови после нагрузки Br :«+» тест при ↑
выделения Br с потом до 50–100% при норме 20%),
Определение ферментов pancreas в кале,
Копрологическое исследование,
Определение активности дуоденального содержимого,
15.
Рентгенопленочный тест (на отсутствие трипсина в кале).Скрининг новорожденных (определение иммунореактивного трипсина крови и
альбумина в меконии и определение электролитов в поте).
Возможна пренатальная диагностика по анализу активности в амниотической
жидкости ряда кишечных ферментов.
Лечение
симптоматическое: а/б, бронходилятаторы, гормоны, витамины, ферменты,
диета, ЛФК, вибрационный массаж, постуральный дренаж.
Генотерапия с применением векторных систем (аденовирусы и липосомы) испытания.
С 1980г. в Англии, Франции, США проводится трасплантации легких и сердца.
Победитель фр. «Фабрики звезд» Грегори Лемаршаль,
Британская поп-певица Элис Мартино
16.
-Канадская спортсменка Лиза Бентли победитель 11 соревнований по триатлону.Продолжает заниматься спортом.
-Правнучка легендарного режиссера Альфреда Хичкока Мелисса Стоун. в 16 лет –
пересадка легкого, что продлило жизнь на 8 лет.
-В 2001 г. закончила Школу Киноискусств в 2001 году.
-Работала на студии Universal в Голливуде до самой смерти в 24 года.
-Создала благотвор. фонд, внесла большой вклад в дело открытия гена муковисцидоза.
-Племянница Селин Дион умерла в 16 лет.
- Есть мнение, что Ф. Шопен умер от М. Польские ученые добиваются разрешения на
исследование тканей его сердца, кот. хранится в варшавском костеле. Умер в 39 лет.
17.
Врожденный гипотиреозсопровождается задержкой псих. развития, поэтому надо начинать лечить уже
в первые 3 мес жизни.
- первичный (паталогия щитовидной железы), нарушается биосинтез
тиреоидных гормонов: тироксина (Т4) и трииодтирозина (ТЗ),
- вторичный (патология гипофиза).
Причина - опухоль (хромофобная аденома, краниофарингиома)/сосудистые
нарушения → наруш. синтеза ТТГ.
- третичный (повреждение гипоталамуса) → нарушение продукции
тиреотропного рилизинг – гормона (ТТРГ).
18.
При некомпенсированном Г нарушено формирование скелета:запоздалое окостенение и закрытие эпифизарных щелей, задержка прорезывания зубов.
Внешне: брахицефалия, нанизм, прогнатизм (выступ ниж. челюсть), широкое лицо,
плоский нос, глубокая запавшая переносица.
Пораж. ЦНС: вялость, сонливость, апатичность, замедление псих. реакции на внешние
раздражители, ↓ концентрации внимания, памяти, идиотия
М.б жалобы на парестезии, невралгии, нарушения обоняния и вкуса, ↓ аппетита,
запоры (из-за замедления перистальтики),
Р-сы снижены.
Объективно: брадикардия и глухость тонов, ↓ сАД, анемия, ↓ ЧД, сухость кожи,
ломкость и выпадение волос, ↑ щитовидной железы.
Задержка полового развития, ранний атеросклероз.
19.
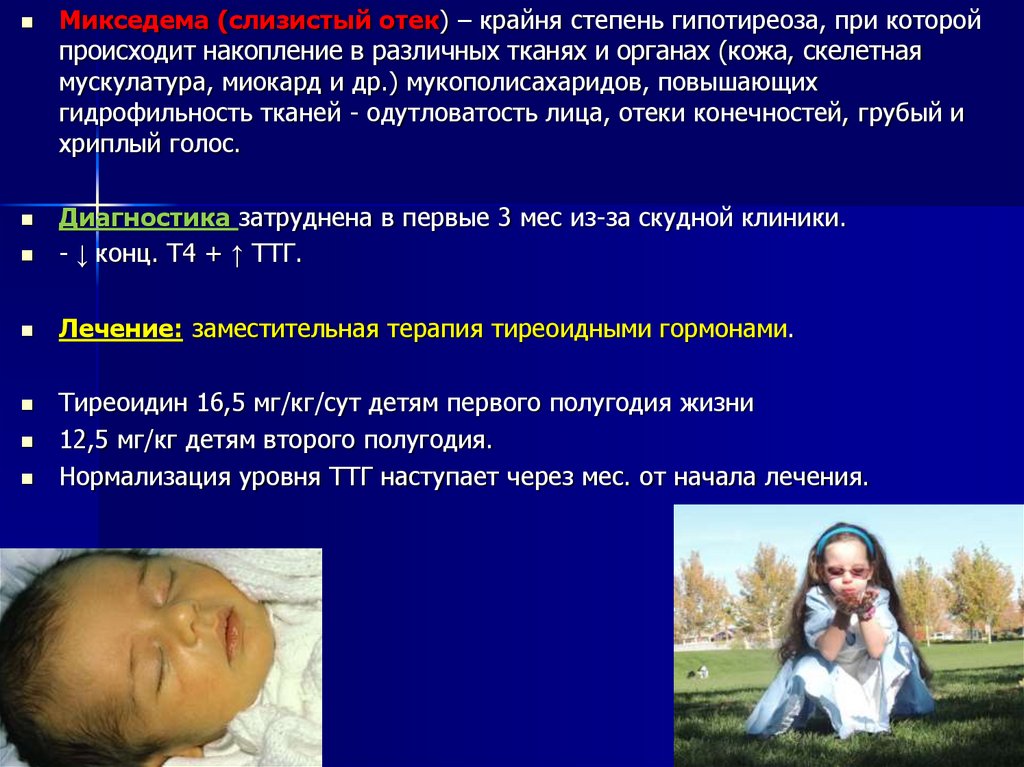
Микседема (слизистый отек) – крайня степень гипотиреоза, при которойпроисходит накопление в различных тканях и органах (кожа, скелетная
мускулатура, миокард и др.) мукополисахаридов, повышающих
гидрофильность тканей - одутловатость лица, отеки конечностей, грубый и
хриплый голос.
Диагностика затруднена в первые 3 мес из-за скудной клиники.
- ↓ конц. Т4 + ↑ ТТГ.
Лечение: заместительная терапия тиреоидными гормонами.
Тиреоидин 16,5 мг/кг/сут детям первого полугодия жизни
12,5 мг/кг детям второго полугодия.
Нормализация уровня ТТГ наступает через мес. от начала лечения.
20.
ФенилкетонурияАР тип насл.
Высокая частота болезни в европейских странах (1:5-10 тыс);
тяжелый исход болезни (необратимая УО) при поздней диагностике (до 6 мес);
высокая частота гетерозиготного носительства гена – 1:50.
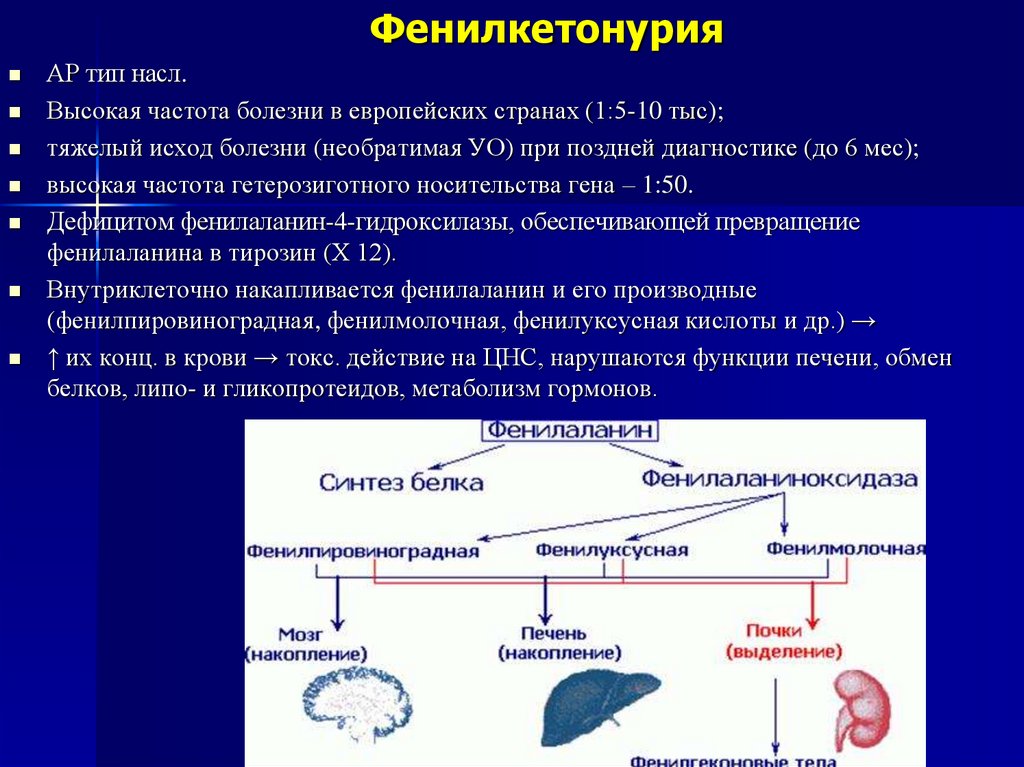
Дефицитом фенилаланин-4-гидроксилазы, обеспечивающей превращение
фенилаланина в тирозин (Х 12).
Внутриклеточно накапливается фенилаланин и его производные
(фенилпировиноградная, фенилмолочная, фенилуксусная кислоты и др.) →
↑ их конц. в крови → токс. действие на ЦНС, нарушаются функции печени, обмен
белков, липо- и гликопротеидов, метаболизм гормонов.
21.
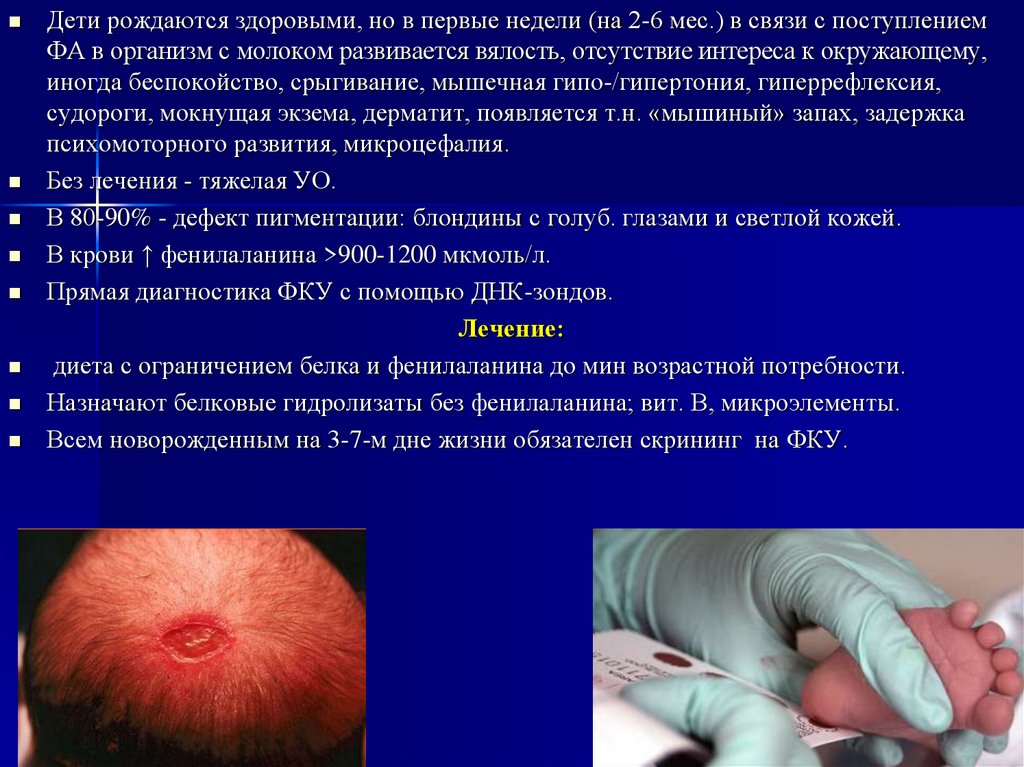
Дети рождаются здоровыми, но в первые недели (на 2-6 мес.) в связи с поступлениемФА в организм с молоком развивается вялость, отсутствие интереса к окружающему,
иногда беспокойство, срыгивание, мышечная гипо-/гипертония, гиперрефлексия,
судороги, мокнущая экзема, дерматит, появляется т.н. «мышиный» запах, задержка
психомоторного развития, микроцефалия.
Без лечения - тяжелая УО.
В 80-90% - дефект пигментации: блондины с голуб. глазами и светлой кожей.
В крови ↑ фенилаланина >900-1200 мкмоль/л.
Прямая диагностика ФКУ с помощью ДНК-зондов.
Лечение:
диета с ограничением белка и фенилаланина до мин возрастной потребности.
Назначают белковые гидролизаты без фенилаланина; вит. В, микроэлементы.
Всем новорожденным на 3-7-м дне жизни обязателен скрининг на ФКУ.
22.
ГалактоземияНарушение обмена простых сахаров.
Первичный дефект: недостаточность фермента галактозо-1-фосфат-уридилтрансферазы
→ накопление в крови, тканях и выделение с мочой галактозо-1-фосфата.
АР тип.
Выявляется при вскармливании младенца материнским/коровьим молоком
содержащими галактозу.
Симптомы: желтуха (↑ прямого билирубина), диспепсия, рвота, гепатоспленомегалия,
цирроз, асцит, обезвоживание, арефлексия, гипотрофия, задержка психомоторного
развития, катаракта.
В моче - галактозурия, протеинурия, аминоацидурия.
Лечение:
искусственное безгалактозное вскармливание.
Прогноз при раннем выявлении благоприятный.
23.
Альбинизм- дефект пигментации ввиду дефекта синтеза тирозиназы, необходимой для
нормального меланинобразования. 1:39.000 – 50.000.
Типы: тиразиназонегативный, тиразиназонезависимый
Сниженная/тотальная депигментация кожи, волос, глаз у всех расовых групп.
Почти неизменна с возрастом (в некоторых случаях возможно накопление меланина,
образование веснушек).
Кожа не загорает, отсутствуют невусы, пигментные пятна.
Выражены нистагм, фотофобия.
Острота зрения резко ↓, с возрастом не улучшается.
↓ резистентность к инфекциям.
Возможны эпилепсия, бесплодие, предрасположенность к раку кожи.
Лечения нет.
Рекомендуются УФ-фильтры.
24.
Адреногенитальный синдром- впервые описан в 1886 году J. Phillips. Частота 1:5.000-15.000 .
Гиперплазия коры надпочечников в результате дефицита 21гидроксилазы (Х 6) →↓кортизола →
избыток секреции АКТГ → ↑ продукции предшественников
кортизола, андрогенов и половых стероидов.
Клинические формы:
вирильная (классическая) 30%.
У девочек с рождения признаки маскулинизации вплоть до
трудности определения пола ребенка
(необходимо исследование кариотипа или полового Х-хроматина),
гиперпигментация в обл. гениталий, мол. желез.
У мальчиков диагностируется с 5 лет и старше при появлении
признаков преждевременного полового развития, что м.б.
причиной нарушений поведения и психики.
У взрослых - бесплодие.
25.
Сольтеряющая. Кроме вышеуказанных симптомов у новорожденных срыгивания,упорная рвота, потеря веса, признаки эксикоза;
коллаптоидные кризы с цианозом, потливостью, потерей сознания.
В крови - гиперкалиемия, метаболический ацидоз, м.б. гипогликемия.
Поздняя (неклассическая). Дебют в подростковом возрасте.
У девочек - ранние менархе, гирсутизм, маскулинное телосложение,
у мальчиков - ускорение костного возраста с ранним закрытием ростковых зон,
преждевременное появление оволосения в лобковой обл.
Латентная (бессимптомная). Клиники нет, кроме бесплодия (не всегда).
В крови: умеренное ↑ предшественников кортизола.
Диагноз АГС м.б. установлен еще во внутриутробном периоде.
Лечение: ЗГТ минерало-, глюкокортикоидами, симптоматическая терапия.
26.
Гепатолентикулярная дегенерация(б. Вильсона-Коновалова)
Описана в 1912г. англ. неврологом Вильсоном. Распространение: 0,6-3:100.000(Х 13).
Дефект одной из медь-транспортирующих АТФаз.
Нарушен обмен Cu, которая не утилизируется тканями, накапливается в токс.
концентрациях, г.о. в печени, ГМ, почках.
Формы:
Брюшная и Ранняя (ригидноаритмогиперкинетическая) – смерть ч/з 2-3года,
Ригиднодрожательная – длительность 6 лет, Дрожательная – 20-30 лет,
Экстрапирамидно-корковая – 6-8 лет, редкая + эпи, парезы.
Клиника: нарушения ф-ции печени, ЭПС (гиперкинезы, ригидность), деменция
Манифестация висцеральных проявлений в 12+/-8 лет,
неврологических - в 23+/-6 лет.
27.
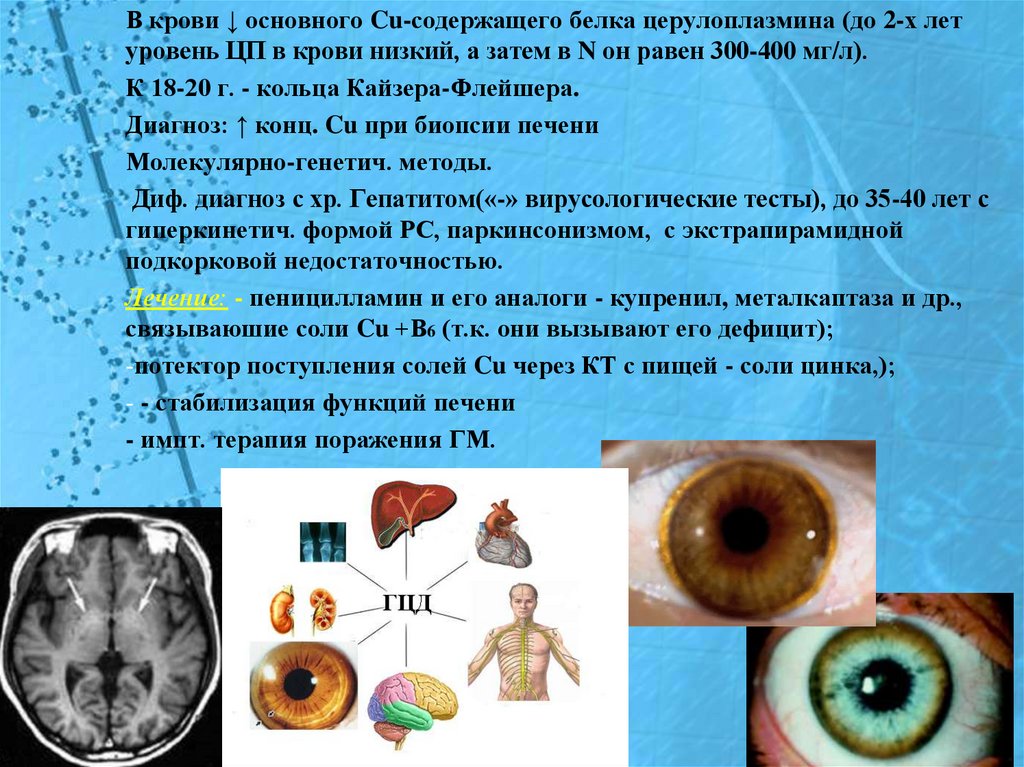
В крови ↓ основного Cu-содержащего белка церулоплазмина (до 2-х летуровень ЦП в крови низкий, а затем в N он равен 300-400 мг/л).
К 18-20 г. - кольца Кайзера-Флейшера.
Диагноз: ↑ конц. Cu при биопсии печени
Молекулярно-генетич. методы.
Диф. диагноз с хр. Гепатитом(«-» вирусологические тесты), до 35-40 лет с
гиперкинетич. формой РС, паркинсонизмом, с экстрапирамидной
подкорковой недостаточностью.
Лечение: - пеницилламин и его аналоги - купренил, металкаптаза и др.,
связываюшие соли Cu +В6 (т.к. они вызывают его дефицит);
-потектор поступления солей Cu через КТ с пищей - соли цинка,);
- - стабилизация функций печени
- импт. терапия поражения ГМ.
28.
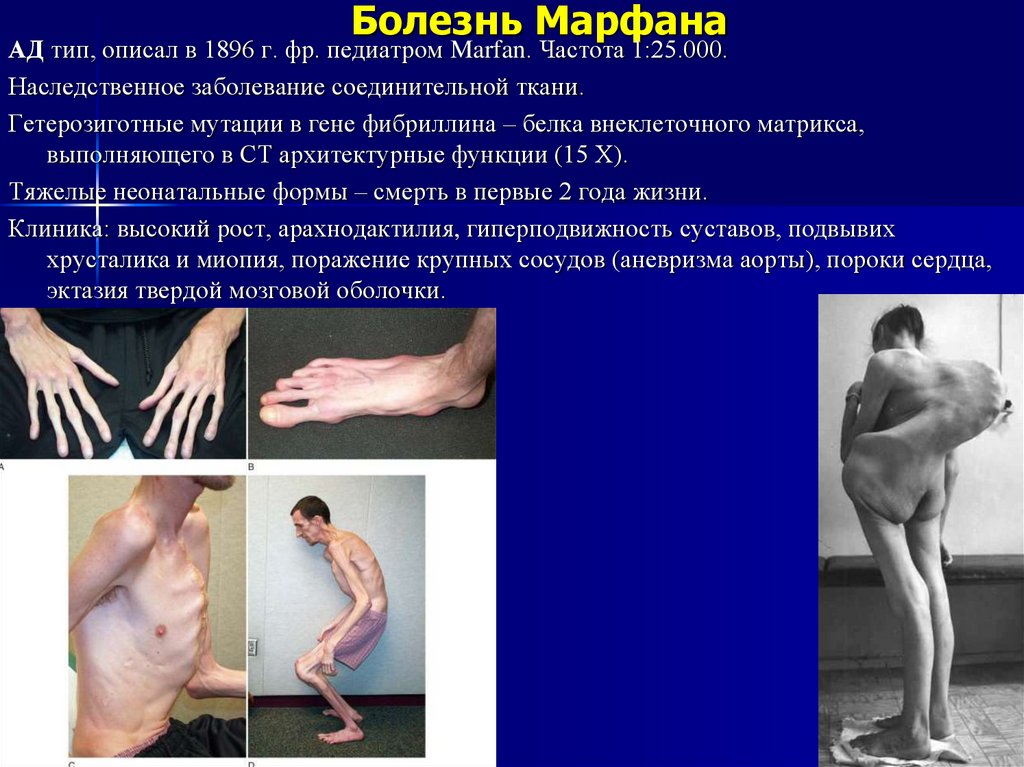
Болезнь МарфанаАД тип, описал в 1896 г. фр. педиатром Marfan. Частота 1:25.000.
Наследственное заболевание соединительной ткани.
Гетерозиготные мутации в гене фибриллина – белка внеклеточного матрикса,
выполняющего в СТ архитектурные функции (15 Х).
Тяжелые неонатальные формы – смерть в первые 2 года жизни.
Клиника: высокий рост, арахнодактилия, гиперподвижность суставов, подвывих
хрусталика и миопия, поражение крупных сосудов (аневризма аорты), пороки сердца,
эктазия твердой мозговой оболочки.
29.
Из-за повышенного содержания адреналина в крови организм постоянно находится«в боевой готовности», в возбужденном состоянии.
Трудоголики.
фараон Древнего Египта Аменхоте́п IV (Эхнато́н) (1375—1336 гг. до н. э.)
Лесоруб Авраам Линкольн: постоянно занимался самообразованием,
Имел выдающиеся способности и потрясающее трудолюбие
Рост – 193 см, огромные стопы, кисти маленькая гр. клетка, длинные гибкие пальцы.
30.
Сын полунищего сапожника Г.Х Андерсен.Необычайное трудолюбие проявилась в школе.
Произведения переписывал по 10 раз, добиваясь точности и легкости стиля.
Описание современников: «Он был высок, худощав и крайне своеобразен по
осанке и движениям. Руки и ноги его были несоразмерно длинны и тонки, кисти
рук широки и плоски, а ступни ног таких огромных размеров, что ему,
вероятно, никогда не приходилось беспокоиться, что кто-нибудь подменит его
калоши. Нос его был так называемой римской формы, но тоже несоразмерно
велик и как-то особенно выдавался вперед».
Страдал фобиями: боялся холеры, пожара, аварий, потери важных документов,
принять не ту дозу лекарства.
31.
Никколо Паганини.Длинные пальцы позволяли ему виртуозно играть на инструментах.
Гете и Бальзак описывали его внешность: «Мертвенно-бледное, как будто вылепленное
из воска лицо, глубоко запавшие глаза, худоба, угловатые движения и, самое главное,
тонкие сверхгибкие пальцы, какой-то невероятной длины, как будто вдвое длиннее,
чем у обычных людей».
Он играл так, что казалось, будто где-то спрятана вторая скрипка, играющая
одновременно с первой.
Мнения: - он в сговоре с дьяволом,
– его искусство является музыкой небес, в которой звучат ангельские голоса.
- в молодости Никколо прибег к помощи хирурга, увеличившего гибкость рук.
32.
Впервые на связь мастерства Паганини с с.М указал амер. врач Майрон Шенфельд встатье в «Журнале Американской медицинской ассоциации»: бледная кожа, глубоко
посаженные глаза, худое тело, неловкие движения, «паучьи» пальцы».
В конце жизни охрип, почти лишился голоса (паралича верхнего гортанного нерва.
Врач Паганини: «Астеническое сложение, сколиоз, «птичье» выражение лица, узкий
череп, выступающий или срезанный подбородок, глаза с синими склерами,
разболтанность суставов, диспропорции в величине туловища и конечностей, кисти и
стопы длинные с тонкими «паукообразными» пальцами.
33.
Шарль де Голль.Деятельный характер проявился в молодости.
Многие сослуживцы в армии до II-й мировой войны прочили его в генералиссимусы.
Его голова всегда возвышалась над строем солдат.
Но сидя за столом, он казался обычным человеком.
34.
К.И. Чуковский.В шаржах часто обыгрывали его длиннорукость, длинноногость, большеносость и
общую нескладность фигуры
«Я всю жизнь работаю. Как вол! Как трактор! Никогда я не наблюдал, чтобы комунибудь другому с таким трудом давалась сама техника писания»
Он многократно переделывал каждую свою строчку.
.
35.
ГемофилияЗаболевание, сцепленное с полом.
нарушение процесса свертываемости из-за ↓/нарушения синтеза факторов свертывания
VIII (А 80-90%) или IX (В-10-15%) или ХI (С– 5%, м.б. у дев.)
Нарушение гемостаза, ↑ времени свертывания.
Первые исторические сведения - в Талмуде, где описана гибель мальчиков после
ритуального обрезания еще во II веке до н.э.
Клиника: дети, страдающие гемофилией, отличаются хрупкостью, бледной тонкой
кожей и слабо развитым подкожной клетчаткой.
Кровотечения неадекватны причине. Кровоизлияния – подкожные, в/м, во внутр.
органы, гемартрозы с ↑t чаще в крупные суставы с послед. деформацией и анкилозом.
У женщин – носителей гена гемофилии («кондукторов»), м.б. склонность к
кровотечениям (обильные менстр., длительные родовые кровотечения).
Диагноз: ↑ времени свёртывания, дефицит антигемофильного глобулина в плазме (в N 0,02-0,03%)
36.
!!! Девочка может страдать Г при гомозиготном носительстве мутацииесли ее отец болен гемофилией, а мать гетерозиготна по мутантному
аллелю.
Лечение патогенетическое: переливние гемопрепаратов, содержащих отсутствующие
факторы - антигемофильную плазму, криопреципитат, концентраты VIII фактора.
В ургентных ситуациях - повторные переливания крови.
Гемофилия в царствующих домах Европы началась с англ. королевы Виктории. Она
является прабабкой царевича Алексея и бабкой Алисы Гессенской.
шанс случайного ее возникновения мал. 1:25000
37.
Родословная Виктории изучена до 17-го колена на предмет гемофилии членамибританского Общества евгеники Уильям Буллок и Пол Филдс в 1911 г.
Эти труды находятся в библиотеке Королевского медицинского общества, но никогда
не публиковались: исследователи не смогли найти среди предков королевы Виктории
ни одного б-го Г.
Т.о. Александрина Виктория, будущая королева Англии, дочь Эдуарда Августа, и
принцессы Саксен-Кобургской Виктории - дочерь только кого-то одного из родителей, либо её отец сэр Джон Конрой, либо она дочь пассии Эдуарда, умершей в родах.
Елизавета II- правнучка Эдуарда VII.
Если предположить, что Виктория – внебрачный ребенок, то ни она, ни ее прямые
наследники не вправе занимать британский трон
К тому же по линии короля у всех была порфирия, которая исчезла в роду с
появлением этого ребенка.
1998 г., Англия, изд-во “Bantam Press” книга историка Дж. Рёля,
биохимика М. Уоррена и Д. Ханта “Пурпурная тайна: Гены,
безумие и королевские дома Европы” о роли П.
в генеалогическом лабиринте королевских династий.
38.
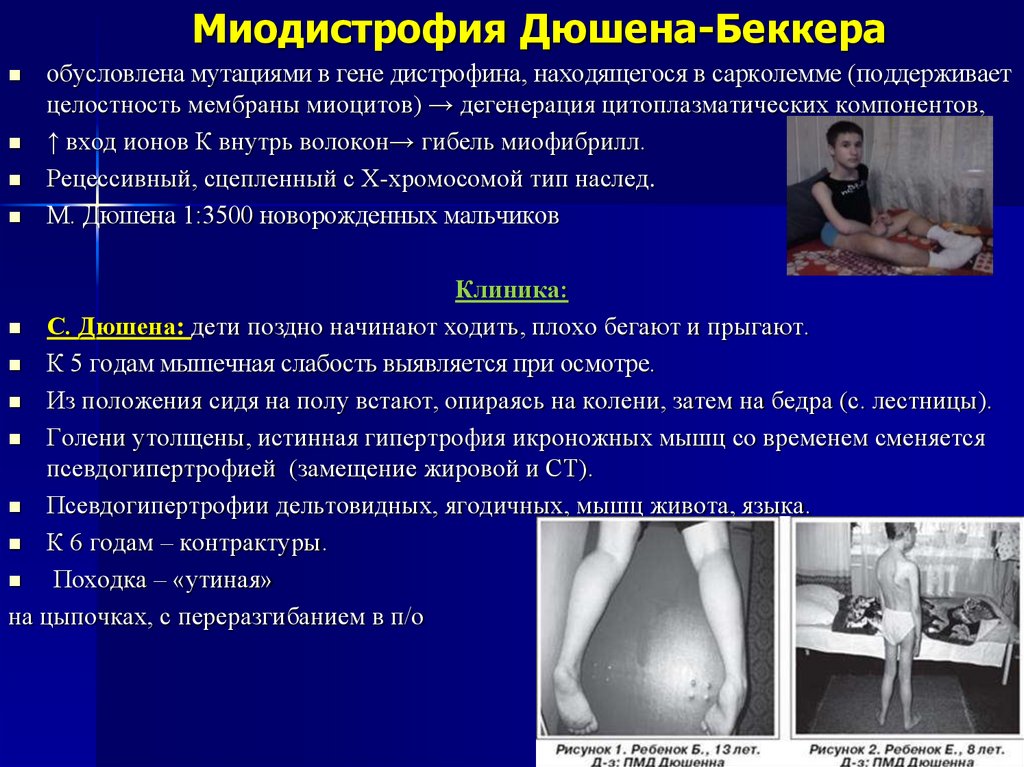
Миодистрофия Дюшена-Беккераобусловлена мутациями в гене дистрофина, находящегося в сарколемме (поддерживает
целостность мембраны миоцитов) → дегенерация цитоплазматических компонентов,
↑ вход ионов К внутрь волокон→ гибель миофибрилл.
Рецессивный, сцепленный с X-хромосомой тип наслед.
М. Дюшена 1:3500 новорожденных мальчиков
Клиника:
С. Дюшена: дети поздно начинают ходить, плохо бегают и прыгают.
К 5 годам мышечная слабость выявляется при осмотре.
Из положения сидя на полу встают, опираясь на колени, затем на бедра (с. лестницы).
Голени утолщены, истинная гипертрофия икроножных мышц со временем сменяется
псевдогипертрофией (замещение жировой и СТ).
Псевдогипертрофии дельтовидных, ягодичных, мышц живота, языка.
К 6 годам – контрактуры.
Походка – «утиная»
на цыпочках, с переразгибанием в п/о
39.
Атрофия восходящая: бедра → тазовый пояс → плечевой пояс → руки.Больше выражена в прокс. отделах и сгибателях шеи.
Развивается поясничный лордоз, крыловидность лопаток, сколиоз, деформация гр.
клетки, что ухудшает функцию легких.
Контрактуры тазобедренных, коленных, локтевых, лучезапястных суставов.
Нарушается моторика ЖКТ, бульбарные расстройства.
С 8-10 лет больным требуются костыли, к 12 годам прикованы к коляске.
К 16-18 годам - тяжелые пневмонии, нередко с летальным исходом.
Др. причины смерти - аспирация пищи, ОСС нед-ть.
Смерть на 2-3-м десятилетии.
М. Беккера - 1 на 20.000 новорожденных мальчиков.
Более мягкий вариант. Начало после 10 лет.
Трудоспособность до 30-35 лет, могут иметь детей.
Интеллект в N.
Описаны единичные случаи у девочек.
Диагностика: повышение в 100-200 раз уровня КФК крови.
Возможна пренатальная диагностика.
Лечение симптоматическое.
40.
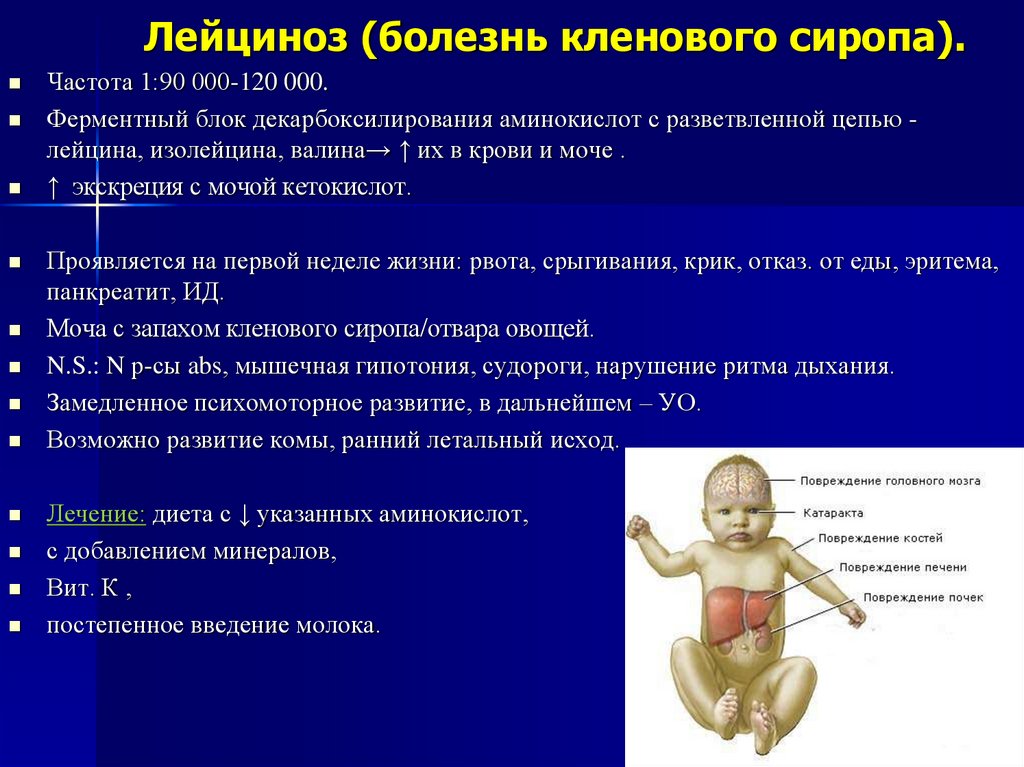
Лейциноз (болезнь кленового сиропа).Частота 1:90 000-120 000.
Ферментный блок декарбоксилирования аминокислот с разветвленной цепью лейцина, изолейцина, валина→ ↑ их в крови и моче .
↑ экскреция с мочой кетокислот.
Проявляется на первой неделе жизни: рвота, срыгивания, крик, отказ. от еды, эритема,
панкреатит, ИД.
Моча с запахом кленового сиропа/отвара овощей.
N.S.: N р-сы abs, мышечная гипотония, судороги, нарушение ритма дыхания.
Замедленное психомоторное развитие, в дальнейшем – УО.
Возможно развитие комы, ранний летальный исход.
Лечение: диета с ↓ указанных аминокислот,
с добавлением минералов,
Вит. К ,
постепенное введение молока.
41.
Нейрофиброматоз РеклингхаузенаАД тип с высокой пенетрантностью и вариабельной экспрессивностью (Х17).
Частота 1:3.000-4.000. Мужчины болеют чаще.
Диагноз устанавливают в первое десятилетие жизни.
Кожный синдром: не < 5 пятен кофейного цвета до 1,5 см в диаметре и >, мелкие
пигментные пятна типа «веснушек» в подмышечных и паховых областях,
42.
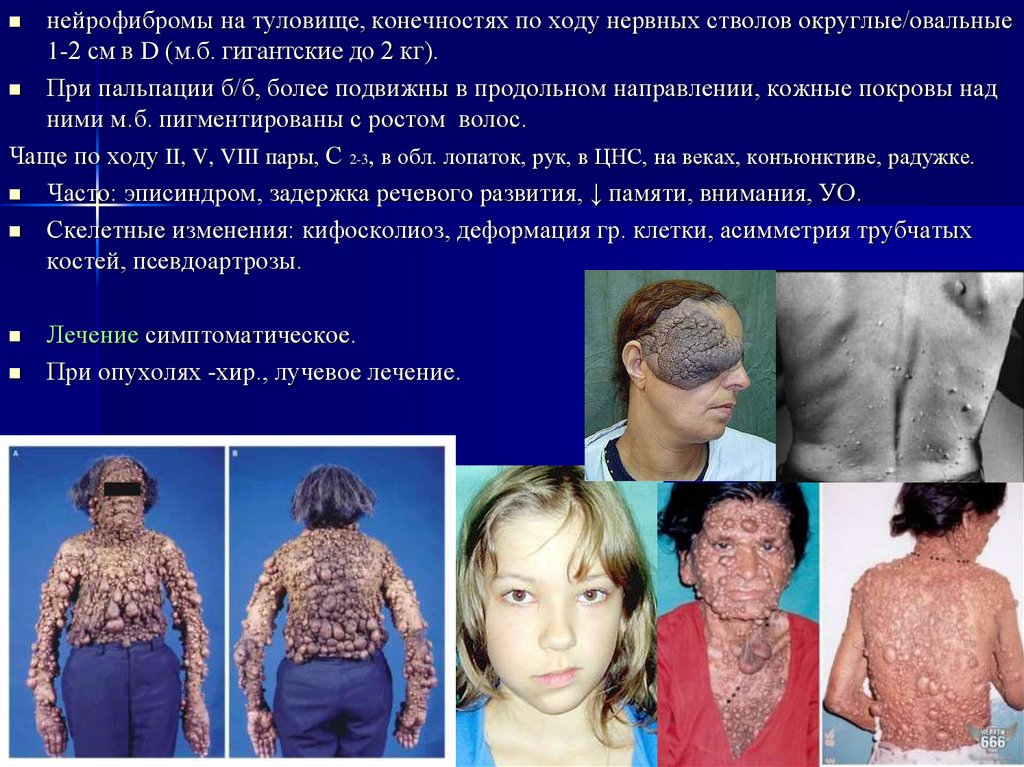
нейрофибромы на туловище, конечностях по ходу нервных стволов округлые/овальные1-2 см в D (м.б. гигантские до 2 кг).
При пальпации б/б, более подвижны в продольном направлении, кожные покровы над
ними м.б. пигментированы с ростом волос.
Чаще по ходу II, V, VIII пары, С 2-3, в обл. лопаток, рук, в ЦНС, на веках, конъюнктиве, радужке.
Часто: эписиндром, задержка речевого развития, ↓ памяти, внимания, УО.
Скелетные изменения: кифосколиоз, деформация гр. клетки, асимметрия трубчатых
костей, псевдоартрозы.
Лечение симптоматическое.
При опухолях -хир., лучевое лечение.
43.
Наиболее ярким доказательством определяющей роли генов в старении являютсямоногенные болезни с признаками ускоренного старения - прогерии.
Одним из подходов к изучению молекулярных основ старения человека является
выяснение причин частичных прогерий.
К.п. они моногенны, а значит, легко поддаются анализу. Недостаток данного подхода:
иногда симптомы П лишь напоминают свойства нормального старения.
Пример: симптомы старения при П более выражены и могут появляться в другой
последовательности, чем в случае с "нормальным" старением:
рост ногтей замедляется при старении, а при П с короткими теломерами
останавливается полностью,
- истончение бровей при старении следует за потерей волос на голове, но, наоборот,
предшествует ему при П.
44.
синдром Вернера (прогерия взрослых) - АР тип.Мутация в гене WRN 8-ой хромосомы → нарушение функции ДНК-геликазы →
нарушается репликация и репарация ДНК, экспрессия генов → ускоренное укорочение
теломер и ↑ чувствительность клеток к апоптозу
Преждевременное старения кожи, сосудистой, костной, репродуктивной системы
До полового созревания пациенты развиваются нормально.
В молодом возрасте: катаракты, склеродермальные и дегенеративные сос. изменения,
диабет, атеросклероз, остеопороз, высокая частота некоторых видов рака, поседение.
Погибают от онко- или СС-патологии.
Продолжительность жизни - 40-50 лет.
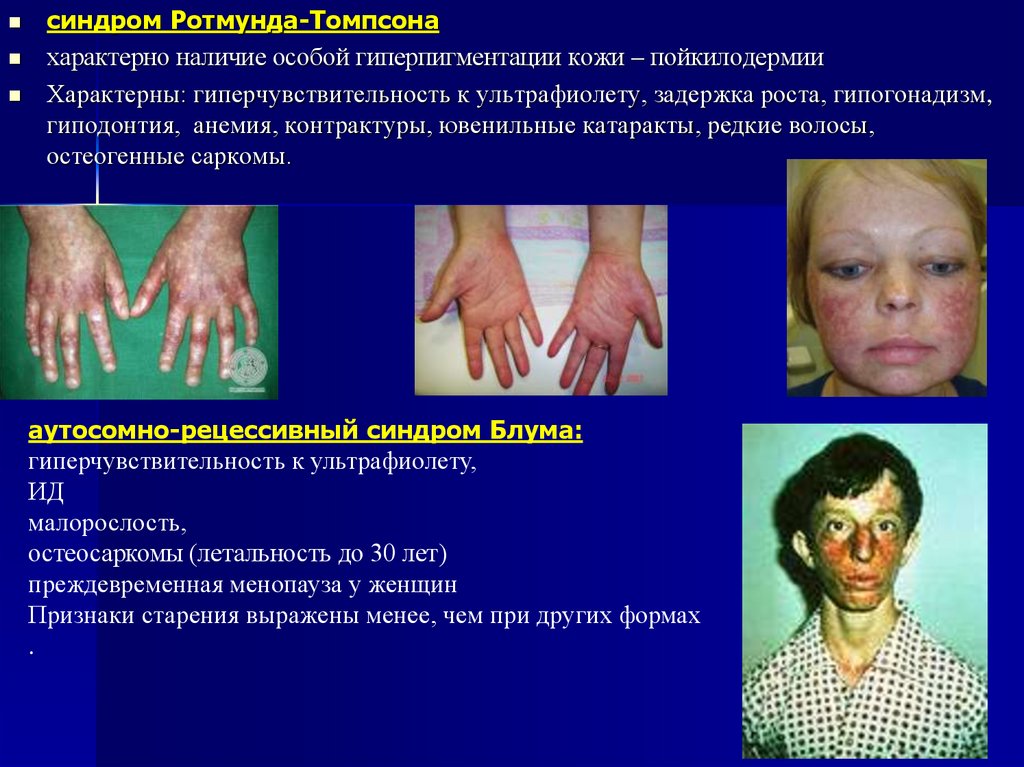
45.
синдром Ротмунда-Томпсонахарактерно наличие особой гиперпигментации кожи – пойкилодермии
Характерны: гиперчувствительность к ультрафиолету, задержка роста, гипогонадизм,
гиподонтия, анемия, контрактуры, ювенильные катаракты, редкие волосы,
остеогенные саркомы.
аутосомно-рецессивный синдром Блума:
гиперчувствительность к ультрафиолету,
ИД
малорослость,
остеосаркомы (летальность до 30 лет)
преждевременная менопауза у женщин
Признаки старения выражены менее, чем при других формах
.
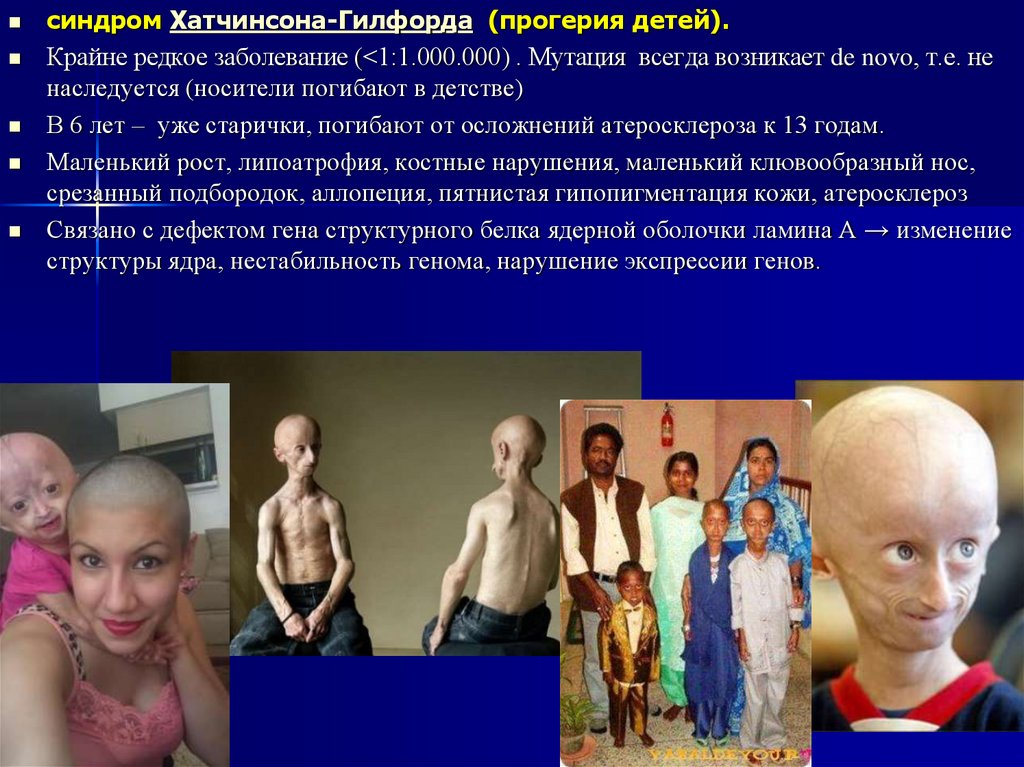
46.
синдром Хатчинсона-Гилфорда (прогерия детей).Крайне редкое заболевание (<1:1.000.000) . Мутация всегда возникает de novo, т.е. не
наследуется (носители погибают в детстве)
В 6 лет – уже старички, погибают от осложнений атеросклероза к 13 годам.
Маленький рост, липоатрофия, костные нарушения, маленький клювообразный нос,
срезанный подбородок, аллопеция, пятнистая гипопигментация кожи, атеросклероз
Связано с дефектом гена структурного белка ядерной оболочки ламина А → изменение
структуры ядра, нестабильность генома, нарушение экспрессии генов.
47.
Я начал стареть, жизнь и так коротка.У многих людей она, как река –
Несется куда-то в манящую даль,
Даруя то радость, то скорбь, то печаль.
Моя же подобна скале с водопадом,
Что падает с неба серебряным градом;
Той капле, которой секунда дана,
Лишь чтобы разбиться о камни у дна.
Но зависти нет к могучей реке,
Что ровно течет по тропе на песке.
Удел их один, – закончив скитанья,
Покой обрести в морях состраданья.
Пусть век мой не долог, судьбы не боюсь,
Ведь, в пар превратясь, вновь к небу вернусь.
29 сентября 2000 г. Бычков Александр
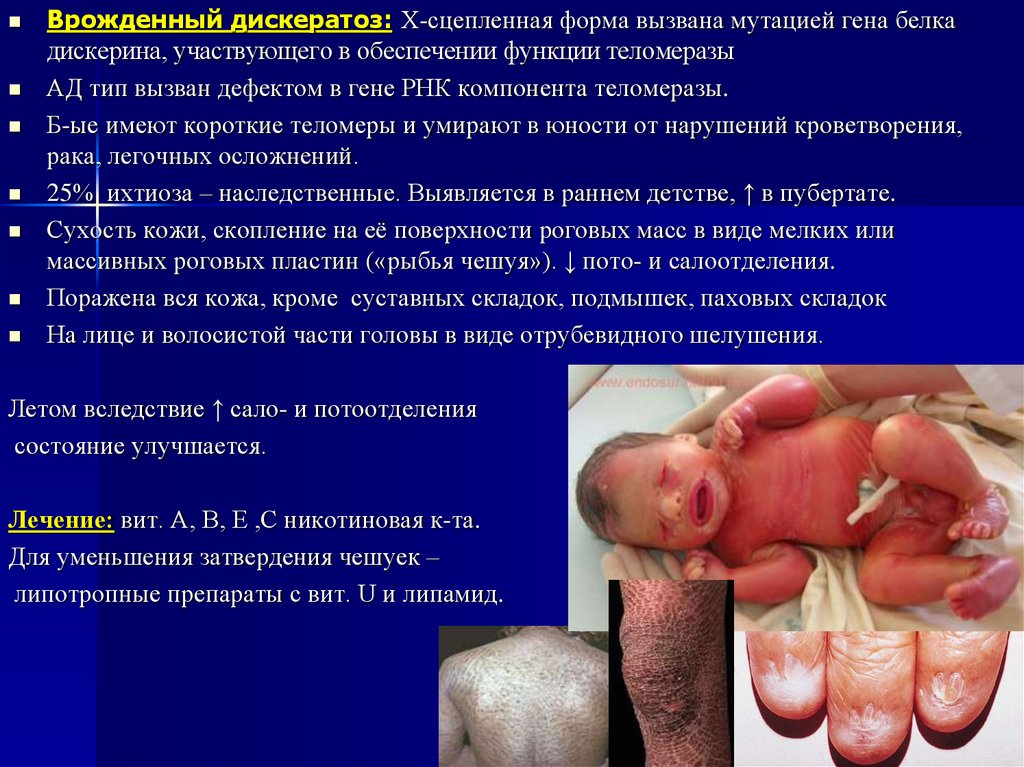
48.
Врожденный дискератоз: Х-сцепленная форма вызвана мутацией гена белкадискерина, участвующего в обеспечении функции теломеразы
АД тип вызван дефектом в гене РНК компонента теломеразы.
Б-ые имеют короткие теломеры и умирают в юности от нарушений кроветворения,
рака, легочных осложнений.
25% ихтиоза – наследственные. Выявляется в раннем детстве, ↑ в пубертате.
Сухость кожи, скопление на её поверхности роговых масс в виде мелких или
массивных роговых пластин («рыбья чешуя»). ↓ пото- и салоотделения.
Поражена вся кожа, кроме суставных складок, подмышек, паховых складок
На лице и волосистой части головы в виде отрубевидного шелушения.
Летом вследствие ↑ сало- и потоотделения
состояние улучшается.
Лечение: вит. А, В, Е ,С никотиновая к-та.
Для уменьшения затвердения чешуек –
липотропные препараты с вит. U и липамид.
49.
Профилактика МЗ: пренатальная д-ка в семьях, где есть больной ребенок.Цель: предотвращение повторного рождения в семье больного, т.к. у гетерозиготных
родителей и их родственников при каждой беременности сохраняется высокий риск
рождения больного ребенка.
Показания для направления на лабораторную диагностику
лиц группы риска по НБО в первые 2 года жизни:
1. Задержка в психомоторном и физ. развитии сразу или после периода N развития
различной продолжительности в сочетании с:
-эпиприпадками, непереносимостью отдельных продуктов (рвота, дегидратация,
диарея), желтухой, мышечной гипо- ,гипертонией, нарушением дыхания, летаргией,
комой, асцитом, необычным цветом/запахом мочи и тела (мышиный запах, запах
потных ног, солода, кленового сиропа), гидроцефалией, глухотой , помутнением
роговицы, катарактой, вывихом хрусталика, искривлением позвоночника,
тугоподвижностью суставов, изменением волос.
гипотрофией (исключить экзогенные причины),- гепато- и/или спленомегалией,
поражением почек (нефролитиаз), экземой.
- лабораторные данные: метаболический ацидоз, алкалоз, сахар, белок, ацетон в моче,
лейко- и/или тромбоцитопения, изменение иммунологич. Показателей
50.
Надеюсь,я Вас не очень утомила!