






















































































 medicine
medicine biology
biologySimilar presentations:



")






Моногенные заболевания. Практическое занятие №3
1.
Мацкиева О.В., Самохина В.И., Скрипкина Г.И.Учебное пособие
для студентов
стоматологического факультета
«Медицинская генетика в стоматологии»
(V семестр)
Омск - 2016
2.
УДК 616.31:575(075)ББК 52.54я73+56.6я73
Учебное пособие к практическим занятиям предназначено для
повышения качества обучения студентов стоматологического
факультета на кафедре детской стоматологии ОмГМУ.
Пособие хорошо иллюстрировано, составлено согласно новому
учебному плану; Является дополнением к имеющимся учебнометодическим материалам.
В пособие вошли материалы, отражающие основные наследственные
синдромы, одним из проявлений которых являются черепно-лицевые
и зубочелюстные аномалии.
Рецензенты:
Антонова А.А. - д.м.н., профессор, зав. каф. стоматологии детского
возраста ДГМУ
Турица А.А. - к.м.н., доцент кафедры пропедевтики детских
болезней и поликлинической педиатрии ОмГМУ
Учебное пособие обсуждено и утверждено на заседании центрального
координационного методического совета Омского государственного
медицинского университета. Протокол № 6 от 24 мая 2016г.
3.
•Практическое занятие № 3:•Моногенные
заболевания
4.
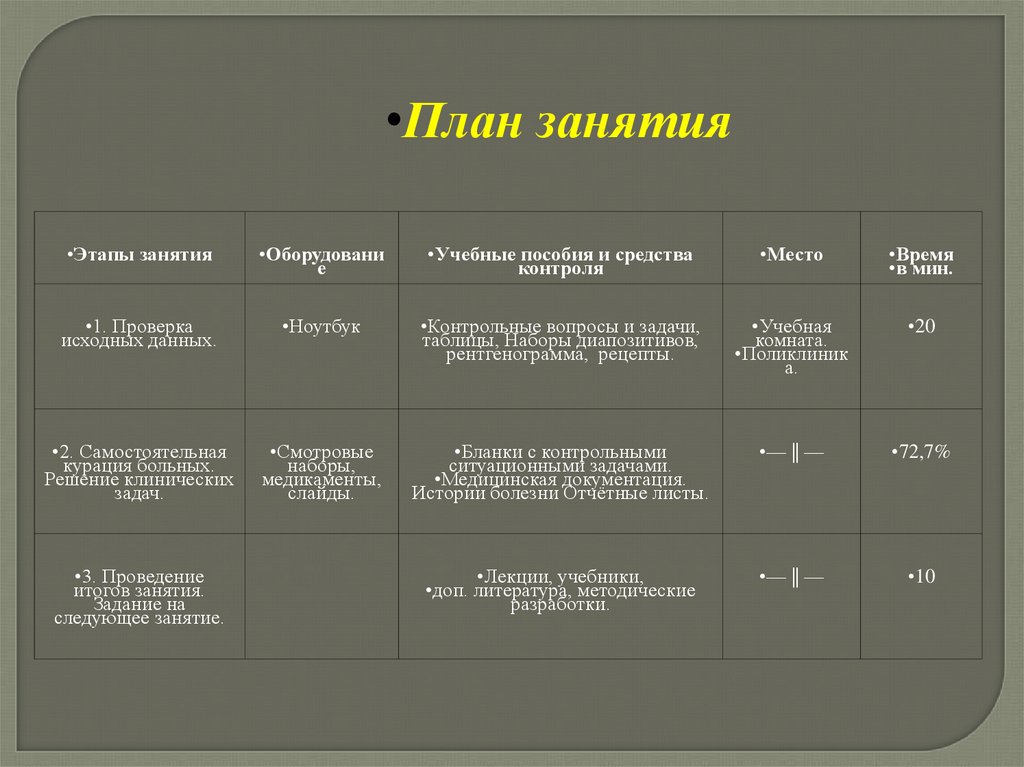
•План занятия•Этапы занятия
•Оборудовани
е
•Учебные пособия и средства
контроля
•Место
•Время
•в мин.
•1. Проверка
исходных данных.
•Ноутбук
•Контрольные вопросы и задачи,
таблицы, Наборы диапозитивов,
рентгенограмма, рецепты.
•Учебная
комната.
•Поликлиник
а.
•20
•2. Самостоятельная
курация больных.
Решение клинических
задач.
•Смотровые
наборы,
медикаменты,
слайды.
•Бланки с контрольными
ситуационными задачами.
•Медицинская документация.
Истории болезни Отчётные листы.
•— || —
•72,7%
•Лекции, учебники,
•доп. литература, методические
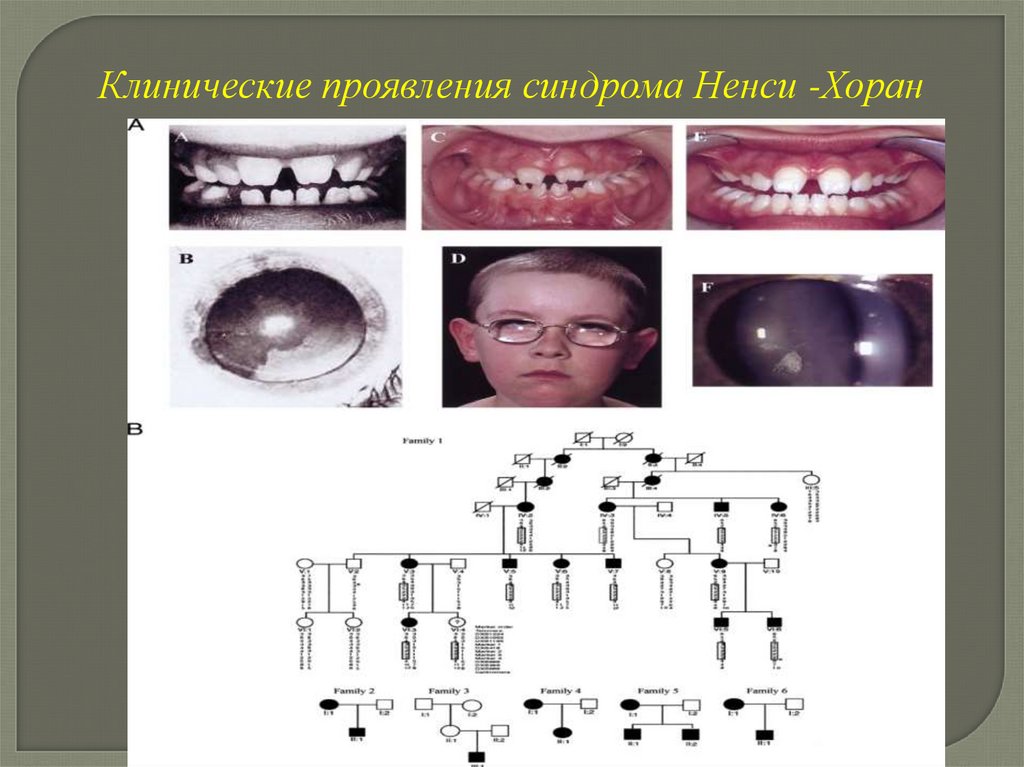
разработки.
•— || —
•10
•3. Проведение
итогов занятия.
Задание на
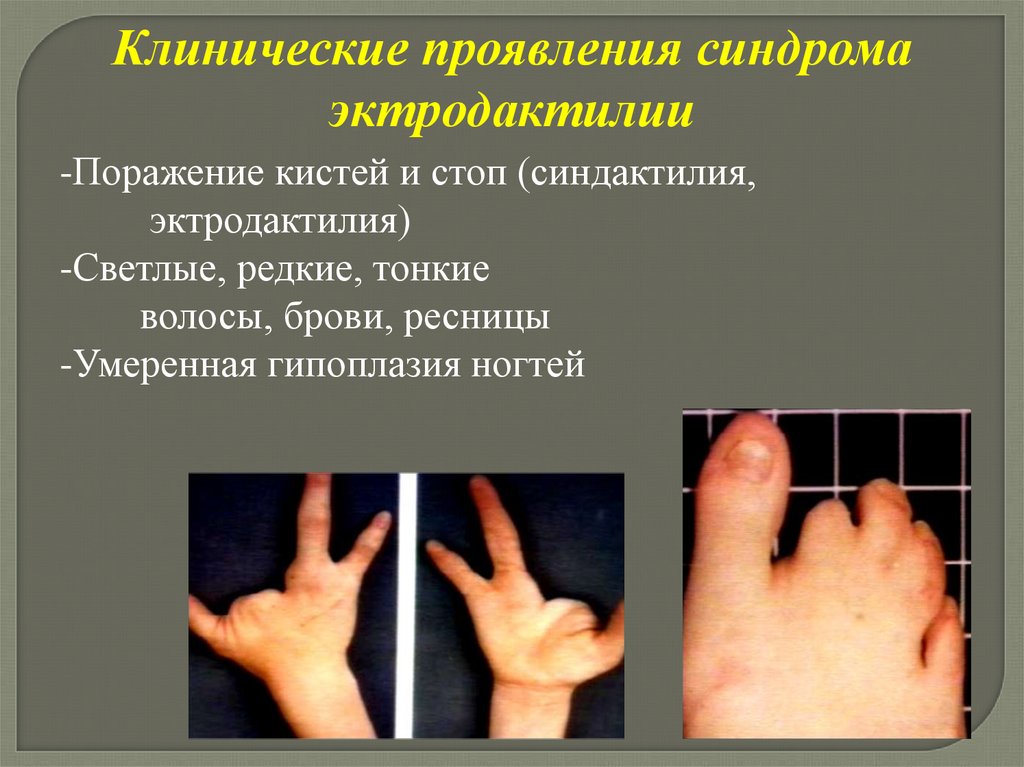
следующее занятие.
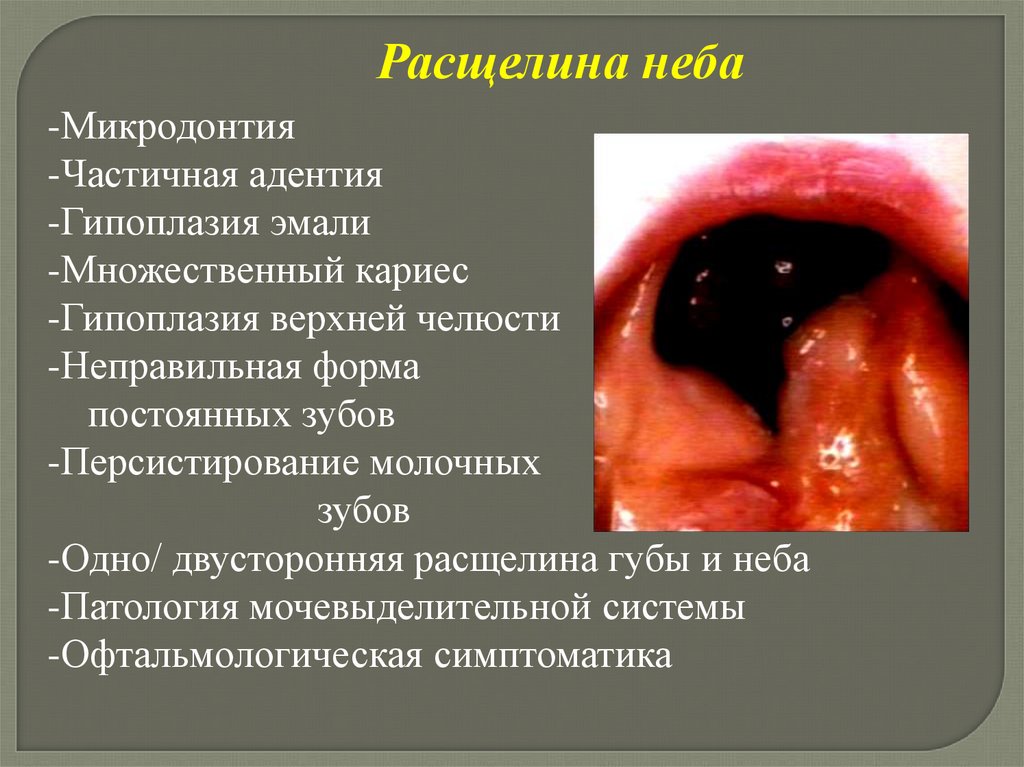
5.
• Цель занятия:•изучить основы моногенных заболеваний
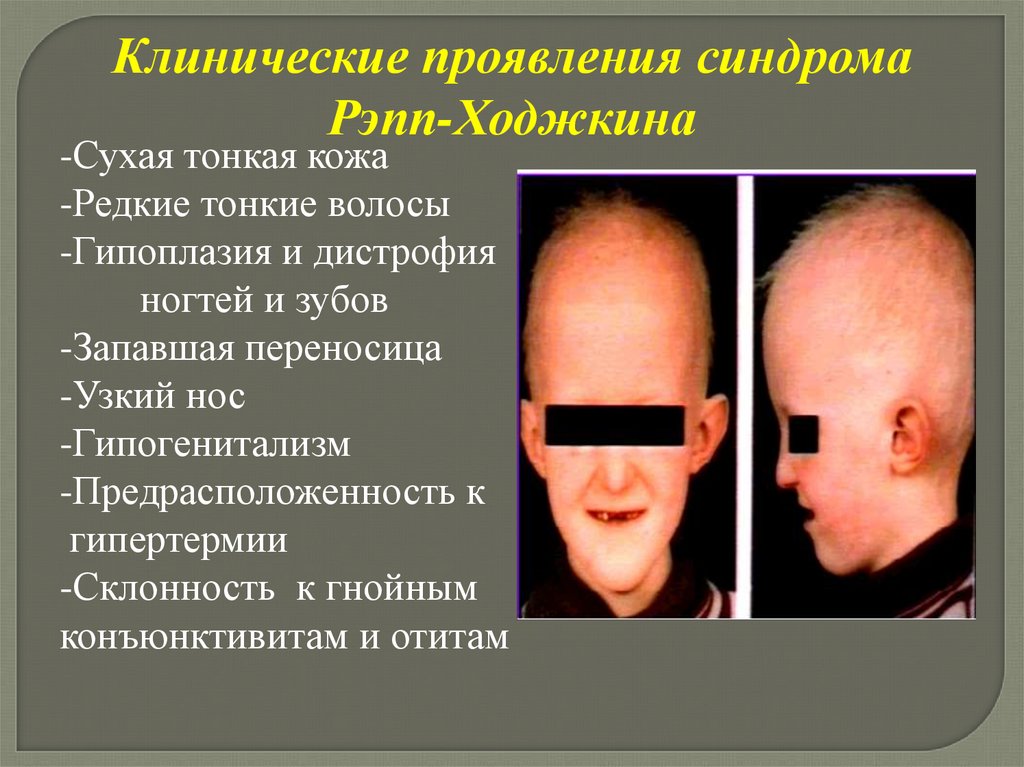
человека, научиться выделять моногенные
формы зубочелюстных аномалий и
синдромов
• с расщелинами губы и нёба
6.
7.
•Моногенные болезни (МБ) -этобольшая группа заболеваний,
возникающих в результате
повреждения ДНК на уровне гена
(генные мутации)
8.
•Частота моногенных болезней зависитот:
• - расы;
• - национальности;
• - территории проживания и уровня изоляции
(изоляты, полуизоляты, панмиксия);
• - типа брака: панмиксный – свободное вступление в
брак; близкородственный; положительный и
отрицательный подбор супружеских пар (по фенотипу)
9.
• Характерные признаки моногенныхболезней
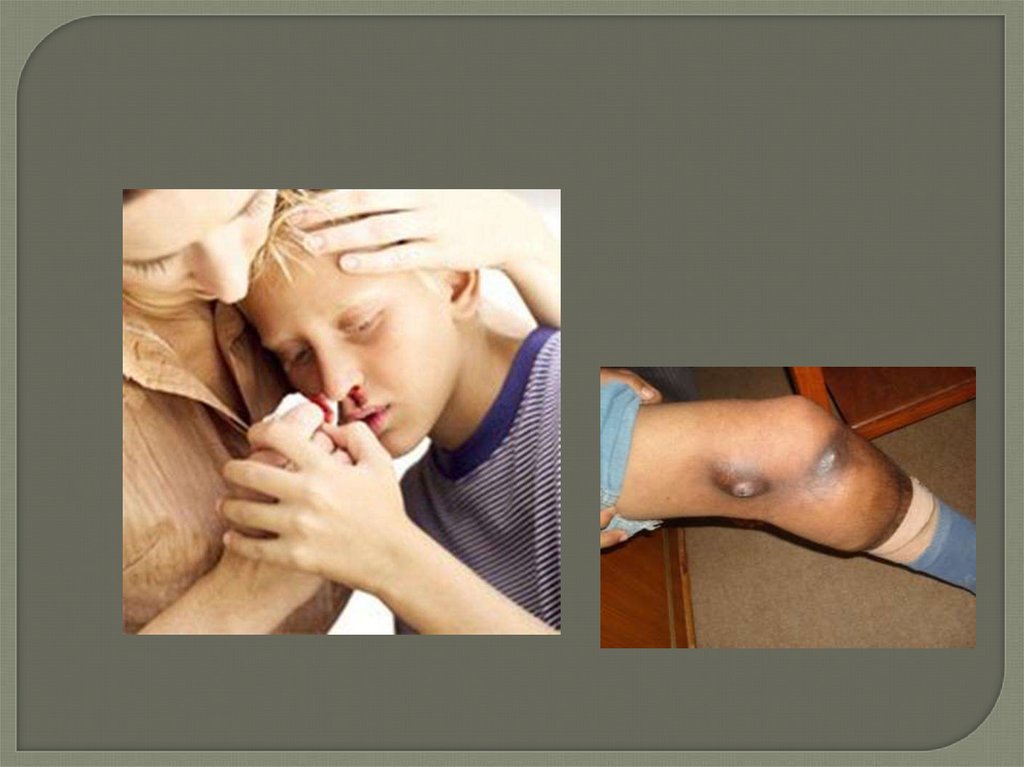
• 1. Наследуются в полном соответствии с законами
Менделя
(аутосомно-доминантное,
аутосомнорецессивное, Х-сцепленное доминантное и рецессивное,
голандрическое)
• 2. Генетическая гетерогенность (внутригенная –
аллельная, т.е. разные мутации внутри гена приводят к
развитию одного и того же заболевания и межгенная –
локусная, т.е мутации в разных генах приводят к
развитию одного и того же заболевания)
• 3. Клинический полиморфизм- наличие или
отсутствие определённого симптома заболевания или
степени его выраженности (полнота и тяжесть
симптоматики)
10.
•Классификация моногенных болезней• - По генетическому принципу
- По патогенетическому принципу
• - По клиническому принципу
11.
Генетический принцип
наследования моногенных болезней
- аутосомно-доминантный тип наследования
- аутосомно-рецессивный тип наследования
- Х-сцепленный доминантный тип
наследования
- Х-сцепленный рецессивный тип
наследования
- Y-сцепленный тип наследования
12.
•Патогенетический принципнаследования моногенных болезней
•1.Болезни с нарушением обмена веществ
(НОВ)
(типы
обмена:
углеводный,
аминокислотный,
липидный,
пуриновый
пиримидиновый, витаминов, металлов и др.)
и
•2. Врожденные пороки развития (ВПР)
моногенной природы
•3. Комбинированные состояния (сочетание
ВПР и НОВ)
13.
•Клинический принцип наследованиямоногенных болезней
• Этот принцип основывается на прикреплении
болезни к той или иной группе в зависимости от
системы или органа, наиболее вовлеченных в
патологический процесс
• Различают моногенные болезни:
• сердечно - сосудистой системы, мочеполовой
системы, желудочно-кишечного тракта, легких,
опорно-двигательного аппарата, нервно-мышечной
системы, челюстно-лицевой области, крови,
кожные, глазные, эндокринные, психические и т.д.
14.
Моногенные синдромыс
аутосомно-доминантаным типом
наследования
15.
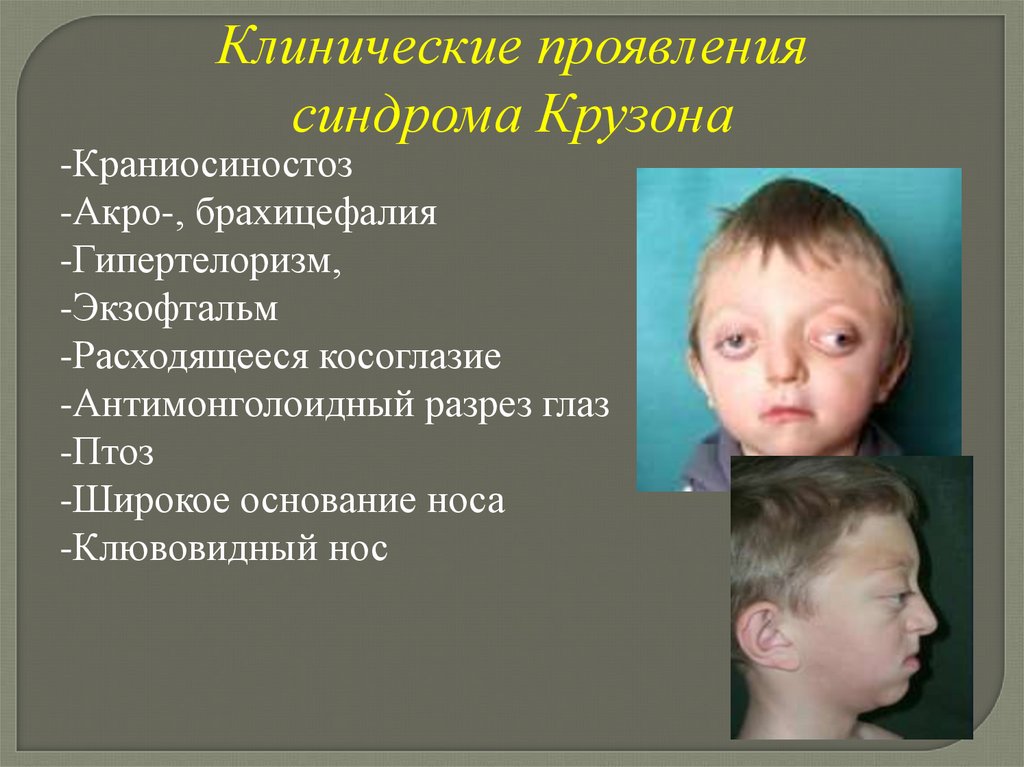
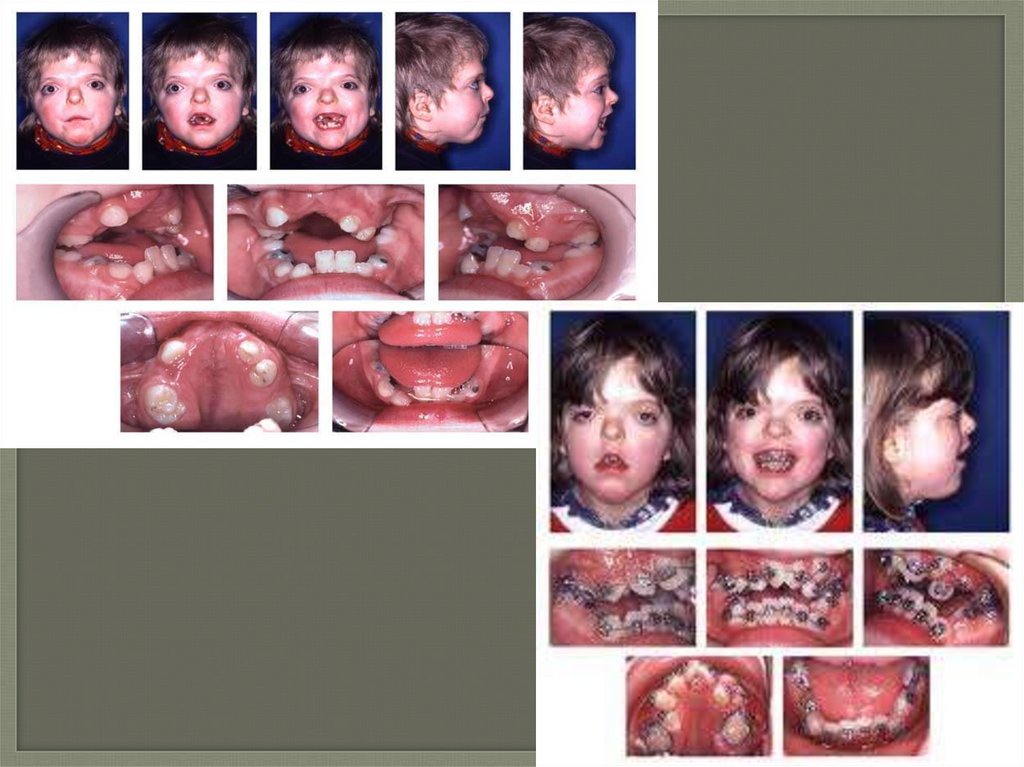
Клинические проявлениясиндрома Крузона
-Краниосиностоз
-Акро-, брахицефалия
-Гипертелоризм,
-Экзофтальм
-Расходящееся косоглазие
-Антимонголоидный разрез глаз
-Птоз
-Широкое основание носа
-Клювовидный нос
16.
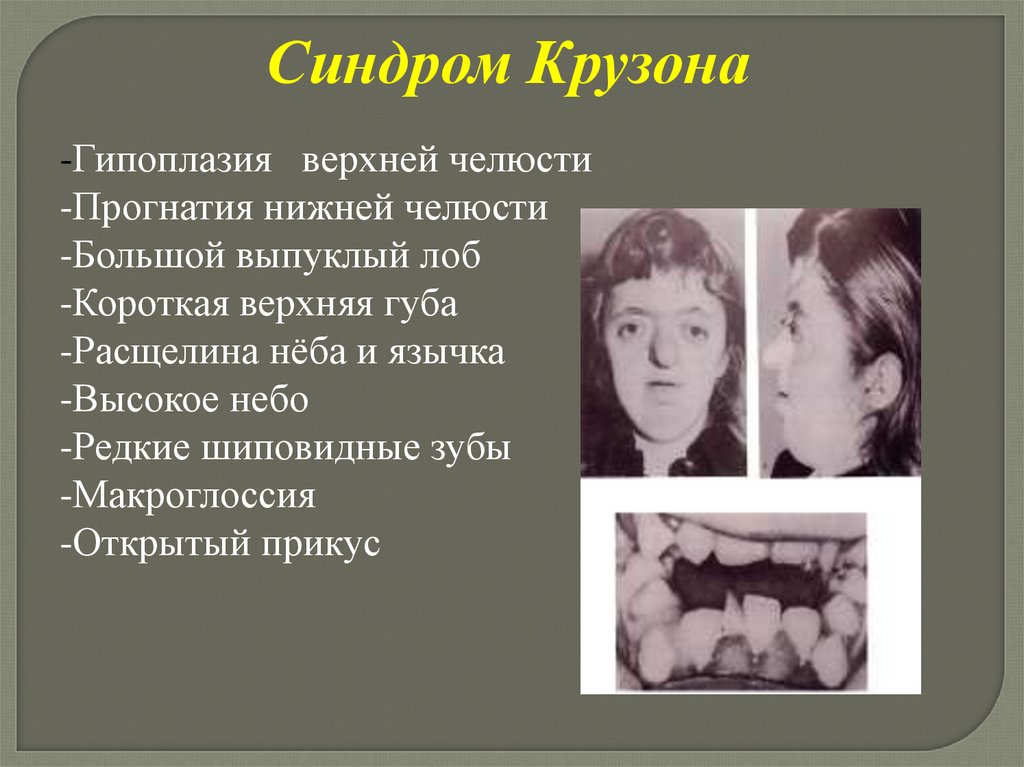
Синдром Крузона-Гипоплазия верхней челюсти
-Прогнатия нижней челюсти
-Большой выпуклый лоб
-Короткая верхняя губа
-Расщелина нёба и язычка
-Высокое небо
-Редкие шиповидные зубы
-Макроглоссия
-Открытый прикус
17.
Синдром Крузона-Атрезия слухового прохода с глухотой
-Нарушение обоняния
-Эпилептические припадки
-Синдактилия, полидактилия
-Двухфаланговые пальцы
-Параличи черепномозговых нервов
-Гипофункция гипофиза
-Умственная отсталость
18.
19.
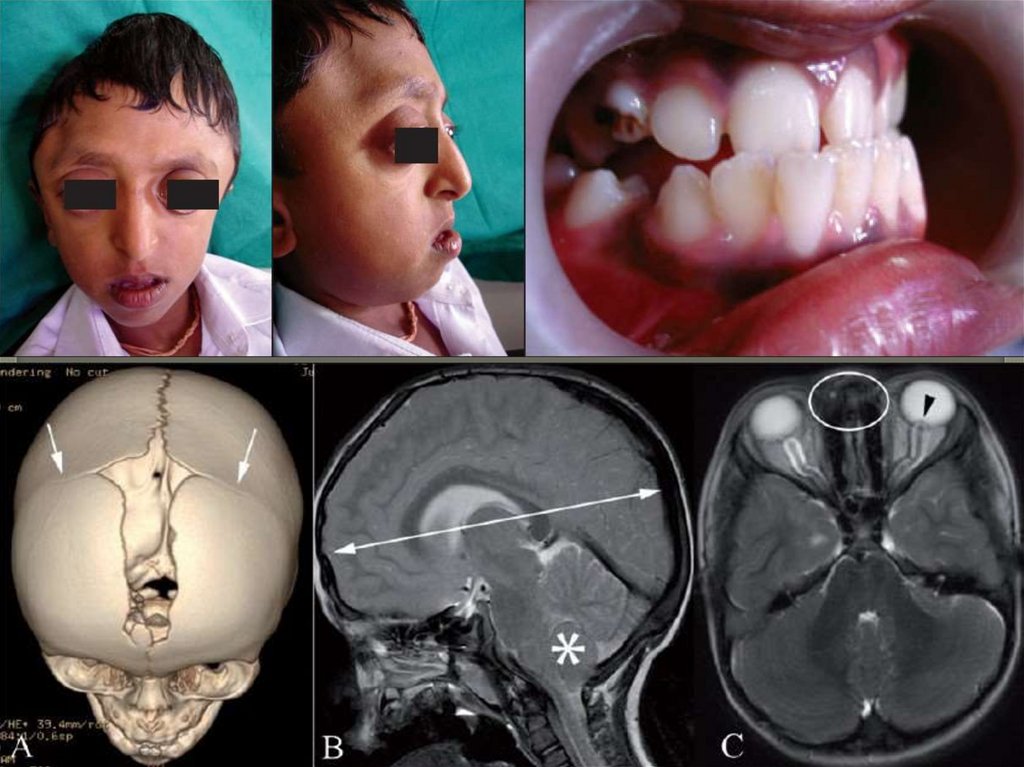
KBG-Синдром(Синдром Германна – Паллистера –
Тидди – Опица)
Тип наследования аутосомно-доминантный
Соотношение мужского и женского пола
21:8
20.
Клинические проявления КВGсиндрома-Низкий рост
-Микроцефалия
-Округлая форма лица
(в детстве)
-Длинный фильтр
-Большие оттопыренные уши
-Гипертелоризм
-Эпикант
-Диспластичные, открытые
вперед ноздри
21.
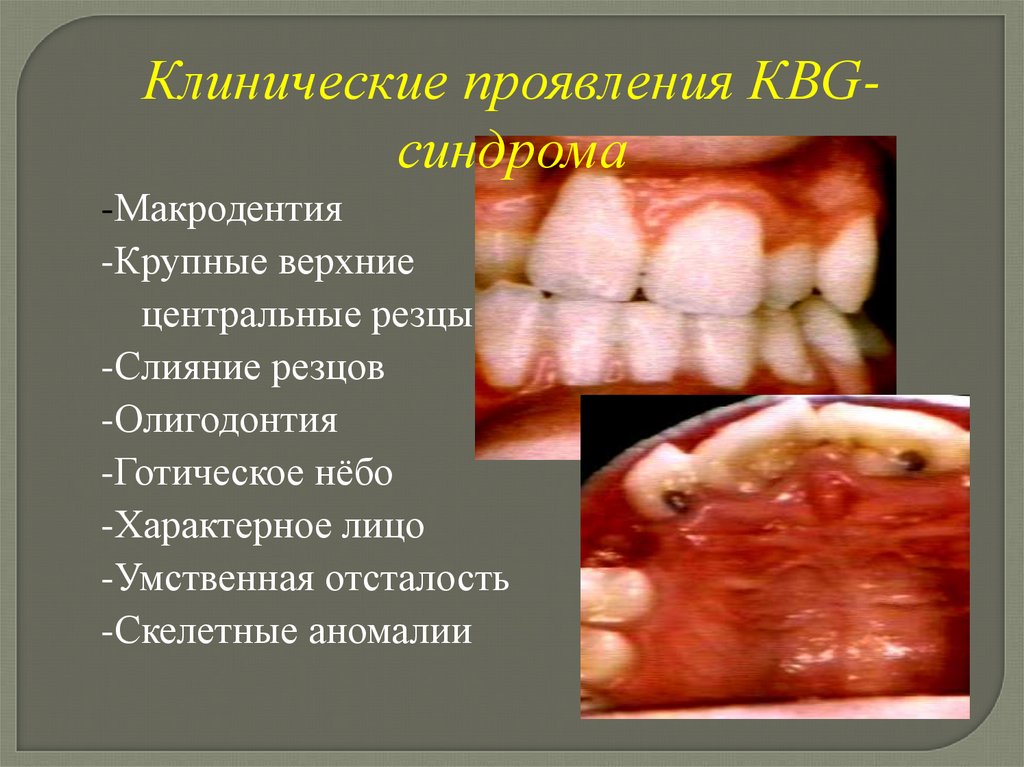
Клинические проявления КВGсиндрома-Макродентия
-Крупные верхние
центральные резцы
-Слияние резцов
-Олигодонтия
-Готическое нёбо
-Характерное лицо
-Умственная отсталость
-Скелетные аномалии
22.
Синдром блефаро-хейлодактилииТип наследования аутосомнодоминантный
Соотношение полов 1:1
23.
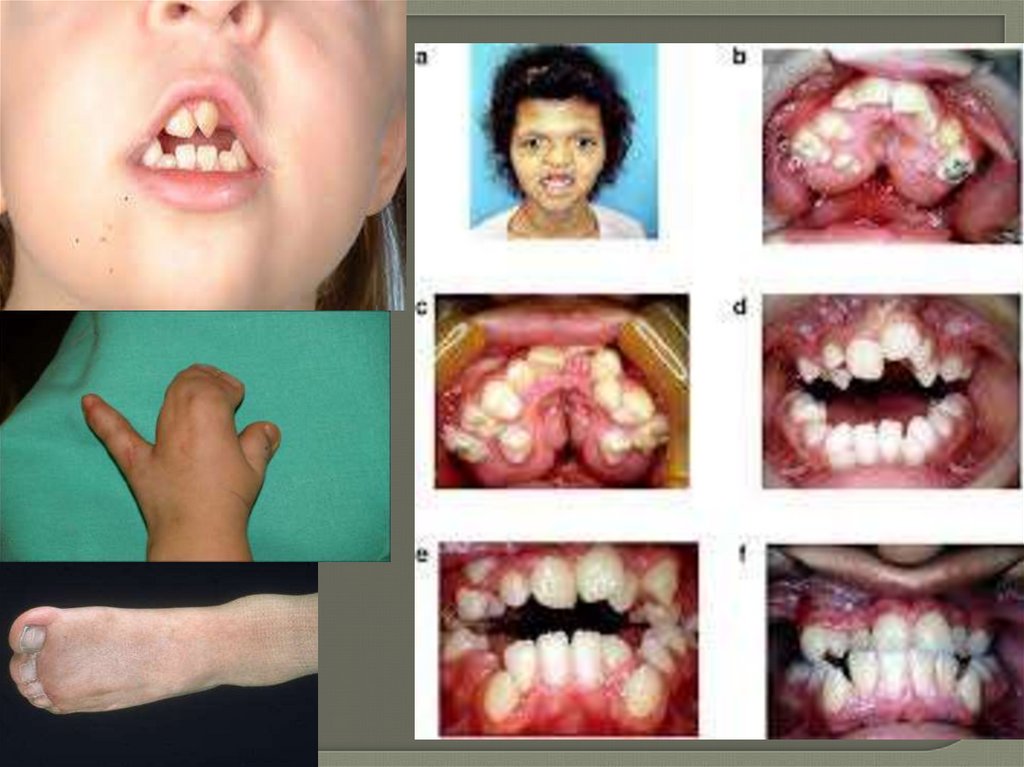
Клинические проявления синдромаблефаро-хейло-дактилии
-Расщелина губы/неба
-Гипертелоризм
-Порок развития слезной
системы
-Микродонтия
-Олигодонтия
-Анодонтития
24.
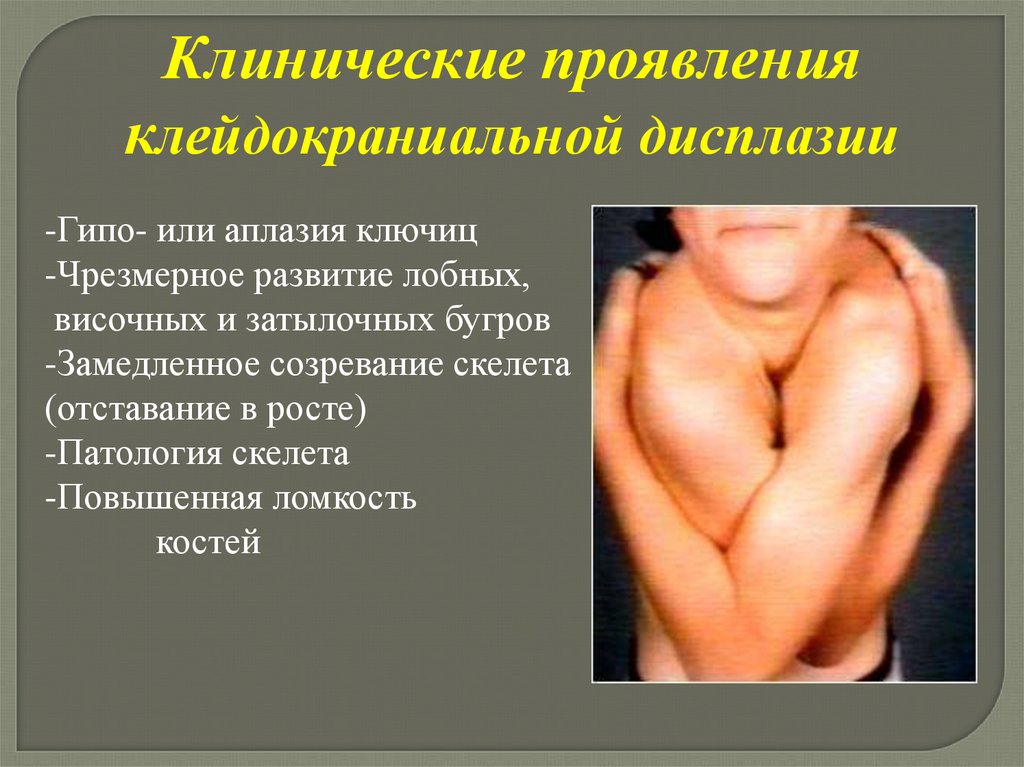
Клейдокраниальная дисплазия(черепно-ключичный дизостоз)
Тип наследования аутосомно-доминантный
с высокой пенетрантностью и различной
экспрессивностью
Ген фактора транскрипции картирован в
6р21
30% - новые мутации
25.
Клинические проявленияклейдокраниальной дисплазии
-Гипо- или аплазия ключиц
-Чрезмерное развитие лобных,
височных и затылочных бугров
-Замедленное созревание скелета
(отставание в росте)
-Патология скелета
-Повышенная ломкость
костей
26.
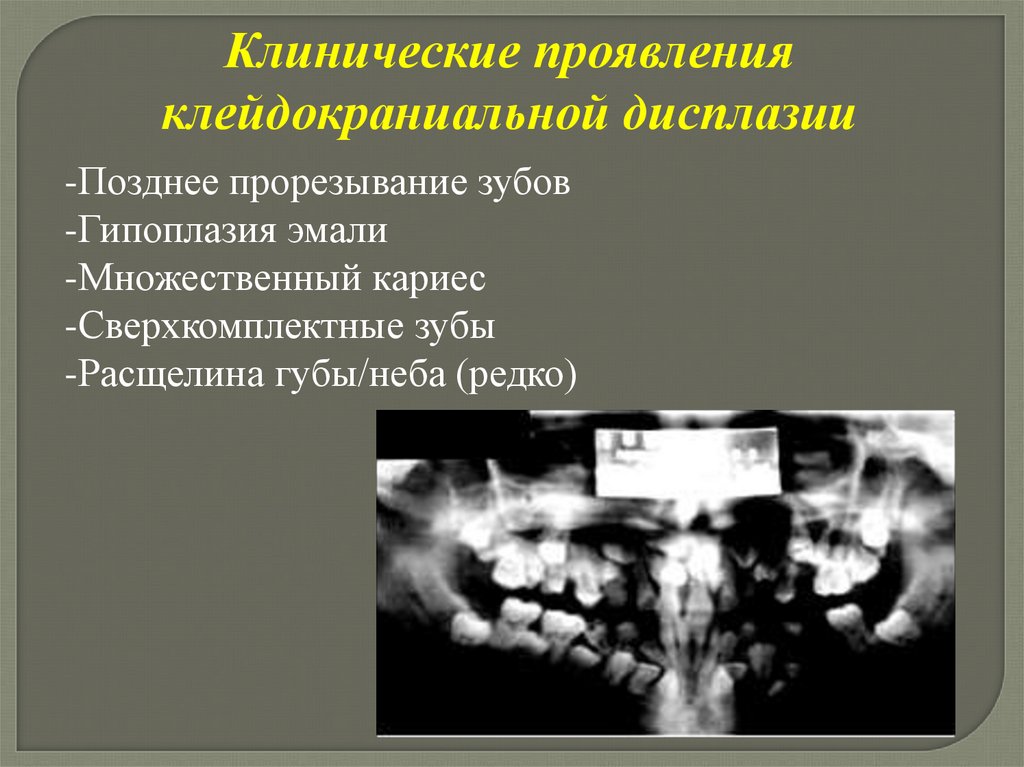
Клинические проявленияклейдокраниальной дисплазии
-Позднее прорезывание зубов
-Гипоплазия эмали
-Множественный кариес
-Сверхкомплектные зубы
-Расщелина губы/неба (редко)
27.
Синдром Ван дер Вуд(Расщелина губы и/или неба и мукозные кисты
на нижней губе)
Частота 1:80 000 - 100 000
Тип наследования аутосомно-доминантный
с пенетрантностью 97% и различной
экспрессивностью
Ген IRF6 картирован в 1q32-q41
28.
Клинические проявлениясиндрома Ван дер Вуда
-Две симметрично расположенные кисты (ямки) на
слизистой нижней губы (88%)
-Свищи или парамедианные синусы
-Выраженная
деформация
нижней
губы,
сопровождающаяся ее удлинением и утолщением
-Расщелина губы
и/или неба (21%)
29.
Врождённая сквозная расщелина верхнейгубы и нёба и врождённый свищ нижней
губы
30.
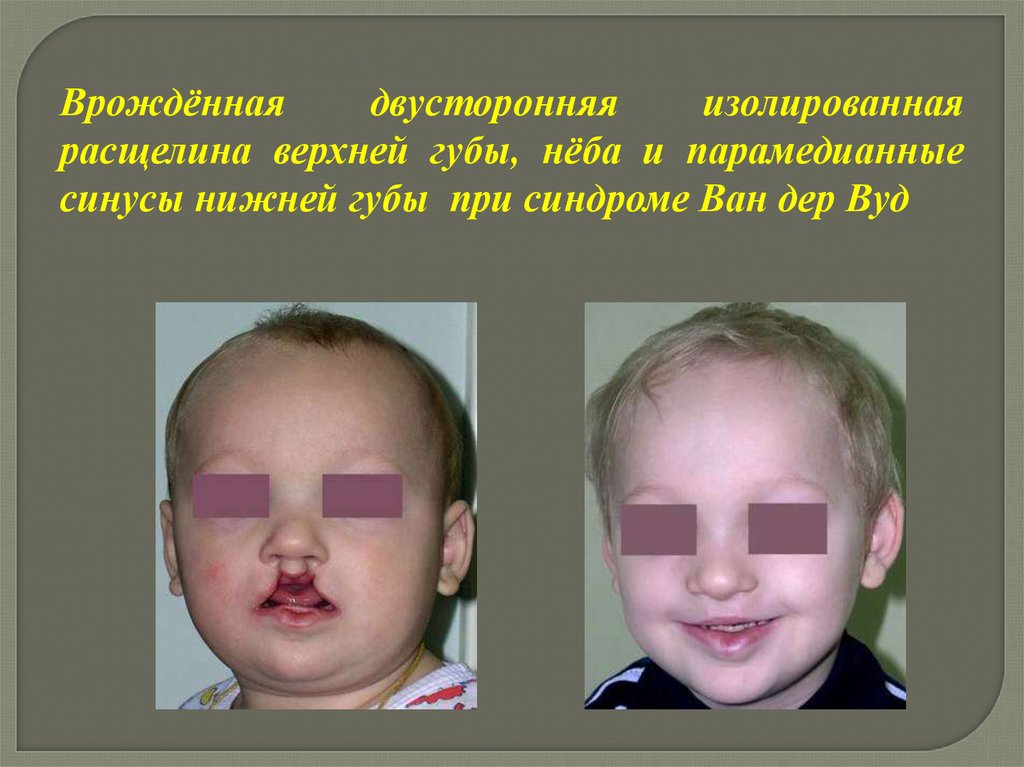
Врождённаядвусторонняя
изолированная
расщелина верхней губы, нёба и парамедианные
синусы нижней губы при синдроме Ван дер Вуд
31.
Синдром Гольденхара(Гемифациальная микросомия)
Частота 1:5600
Тип наследования возможно аутосомно –
доминантный
Ген картирован в локусе 14q32
32.
Клинические проявлениясиндрома Гольденхара
Поражение обычно одностороннее,
аномальное расположение, деформация,
аплазия или гипоплазия ушных раковин
33.
Клинические проявлениясиндрома Гольденхара
-Преаурикулярные папилломы
-Эпибульбарные дермоиды
-Ассиметрия лица
-Гипоплазия верхней и
нижней челюстей
-Макростомия
-Открытый прикус
-Расщелина неба (иногда)
34.
Клинические проявлениясиндрома Гольденхара
-Колобомы верхнего века, радужки
-Дефекты глазодвигательных мышц
-Антимонголоидный разрез глаз
-Микрофтальмия
-Косоглазие
-Аномалии скелета
-Патология ССС
(ДМЖП, ОАП, тетрада Фалло,
коарктация аорты)
-Умственная отсталость 25%
35.
Синдром АпераЧастота 1 : 160 000
Тип наследования аутосомно-доминантный
Ген рецептора 2 фактора роста фибробластов
(FGFR2) в 10q26 (замена серина на
триптофан S252W и пролина на аргинин
P253R
36.
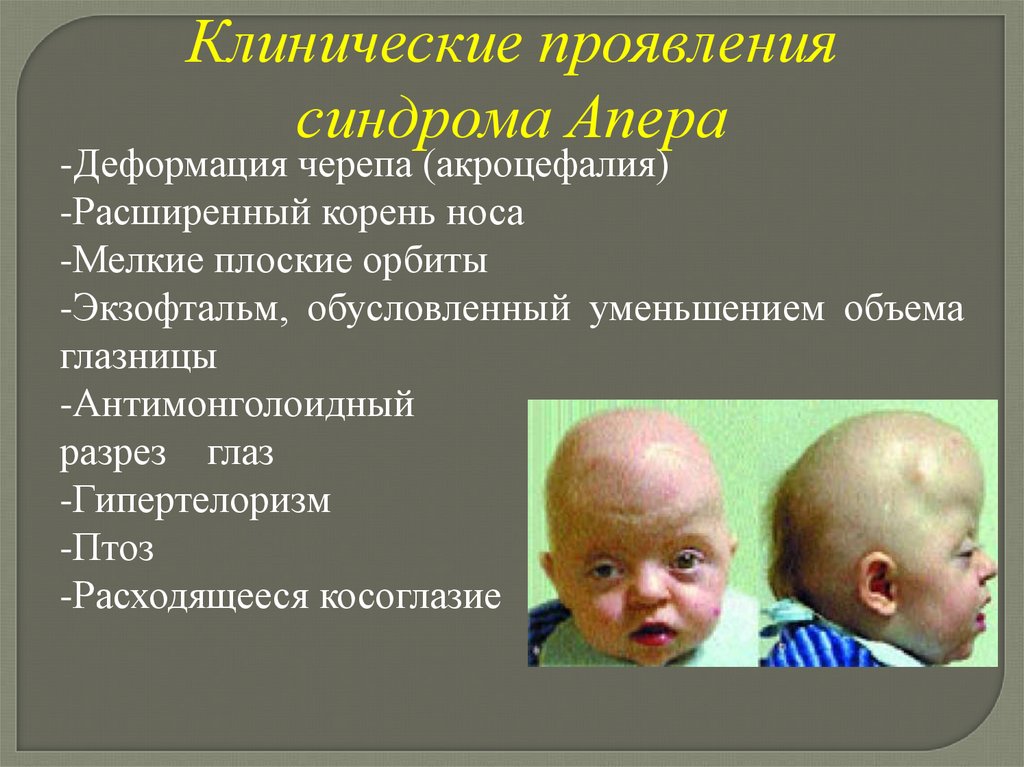
Клинические проявлениясиндрома Апера
-Деформация черепа (акроцефалия)
-Расширенный корень носа
-Мелкие плоские орбиты
-Экзофтальм, обусловленный уменьшением объема
глазницы
-Антимонголоидный
разрез глаз
-Гипертелоризм
-Птоз
-Расходящееся косоглазие
37.
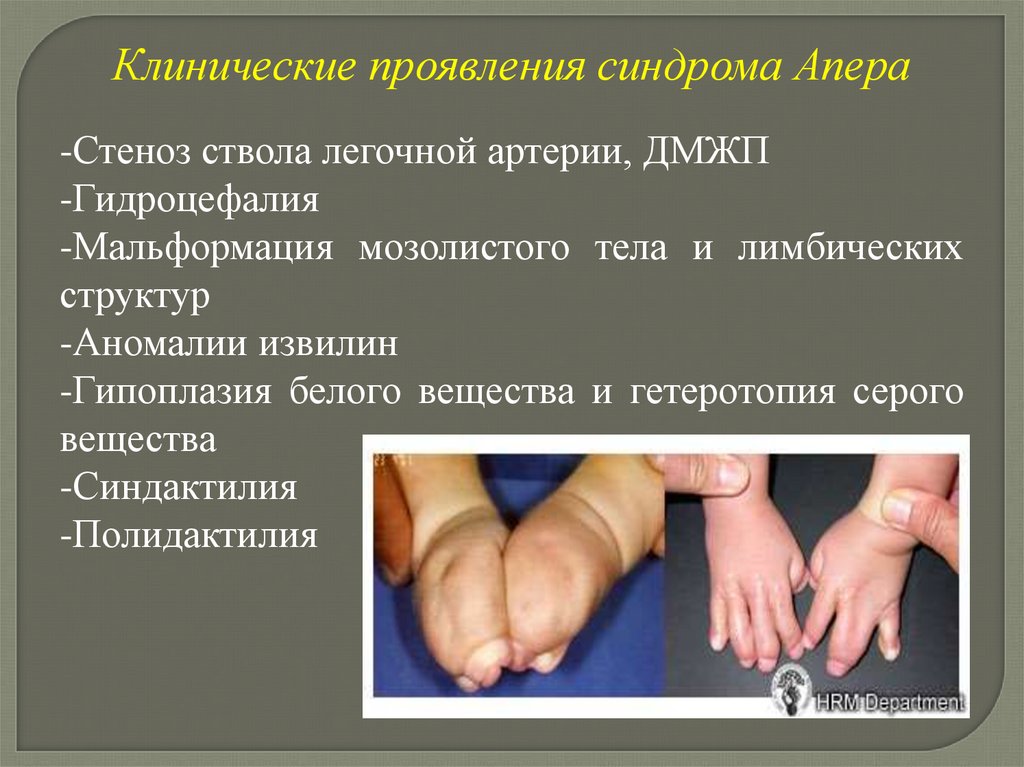
Клинические проявления синдрома Апера-Стеноз ствола легочной артерии, ДМЖП
-Гидроцефалия
-Мальформация мозолистого тела и лимбических
структур
-Аномалии извилин
-Гипоплазия белого вещества и гетеротопия серого
вещества
-Синдактилия
-Полидактилия
38.
Клинические проявления синдрома Апера-Пороки развития позвонков
-Низкий рост
-Пороки сердца
-Дисплазия почек и поджелудочной железы
-Заращение заднего прохода
-Адипозо - генитальная дисплазия
-Пороки развития наружного уха
-Умственная отсталость
39.
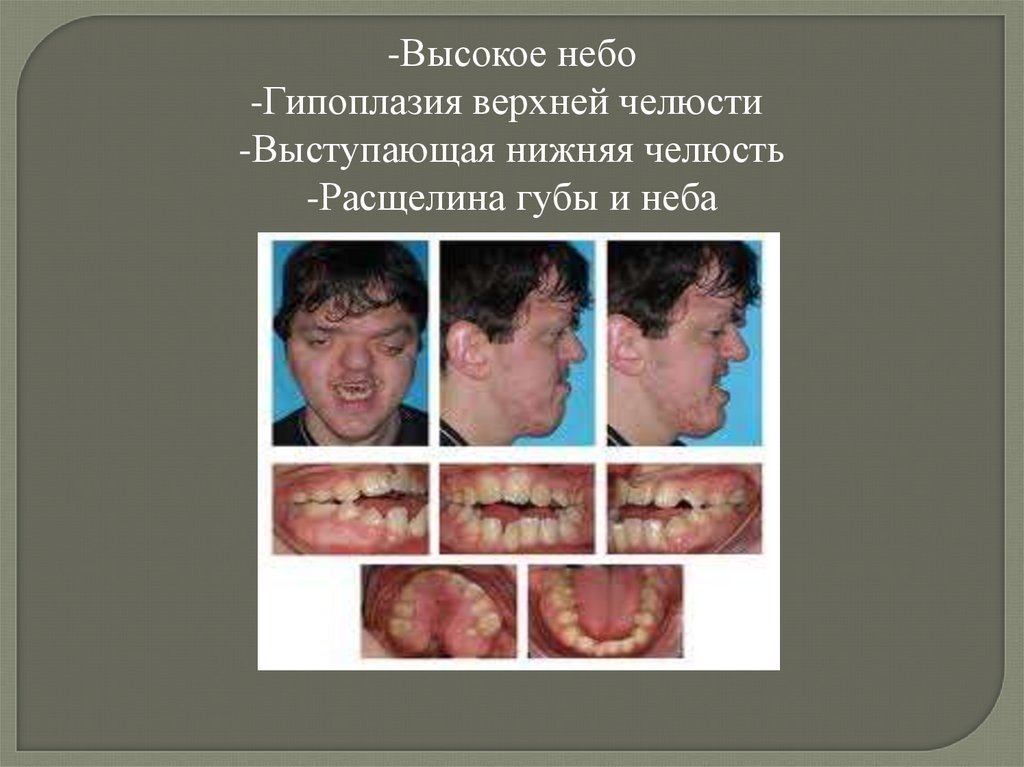
-Высокое небо-Гипоплазия верхней челюсти
-Выступающая нижняя челюсть
-Расщелина губы и неба
40.
41.
42.
Синдром СотосаТип наследования аутосомнодоминантный
Соотношение полов 1:1
43.
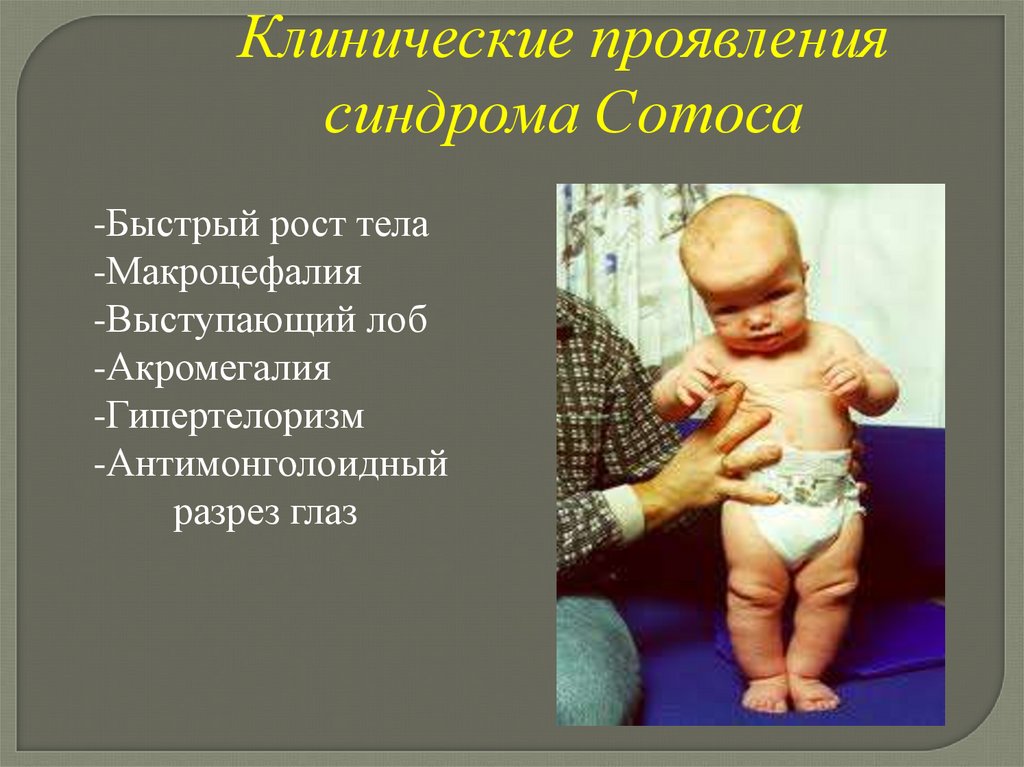
Клинические проявлениясиндрома Сотоса
-Быстрый рост тела
-Макроцефалия
-Выступающий лоб
-Акромегалия
-Гипертелоризм
-Антимонголоидный
разрез глаз
44.
Клинические проявления синдромаСотоса
-Долихоцефалия
- Гиперемия и
одутловатость лица
-Макроглоссия
-Прогения
-Высокое небо
-Сколиоз
-Олигофрения
-Ожирение
-Общее недоразвитие
моторики
45.
Моногенные синдромыс
аутосомно-рецессивным типом
наследования
46.
Синдром КоэнаТип наследования
аутосомно - рецессивный
Ген СОН1 в 8q22-q23
47.
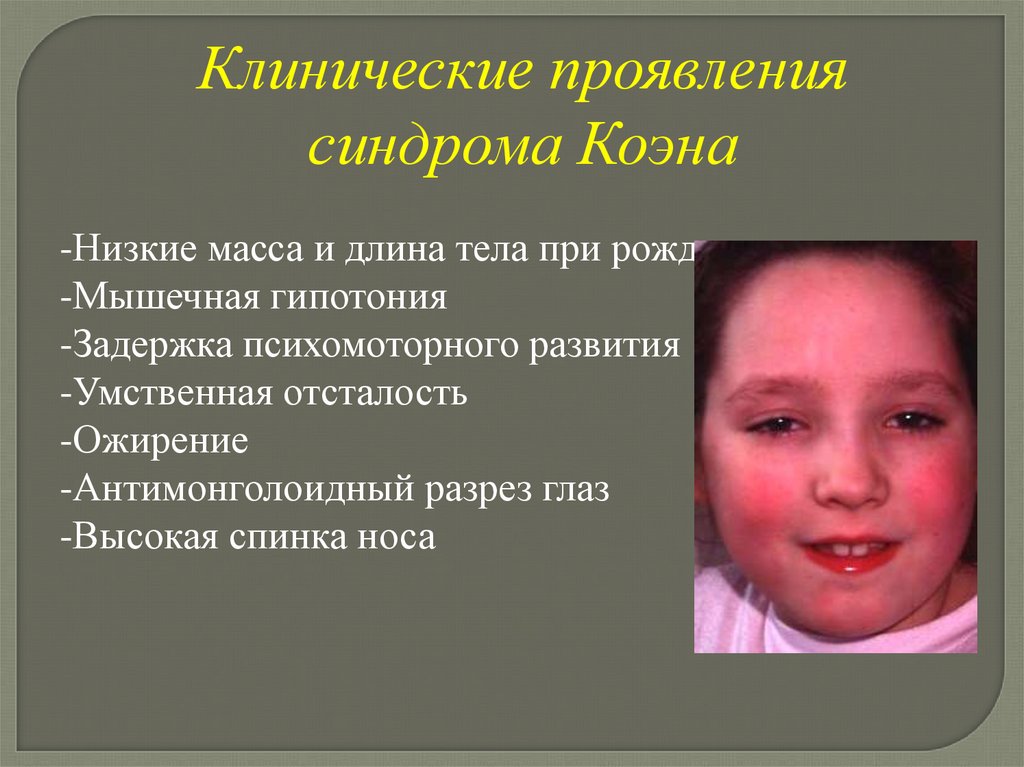
Клинические проявлениясиндрома Коэна
-Низкие масса и длина тела при рождении
-Мышечная гипотония
-Задержка психомоторного развития
-Умственная отсталость
-Ожирение
-Антимонголоидный разрез глаз
-Высокая спинка носа
48.
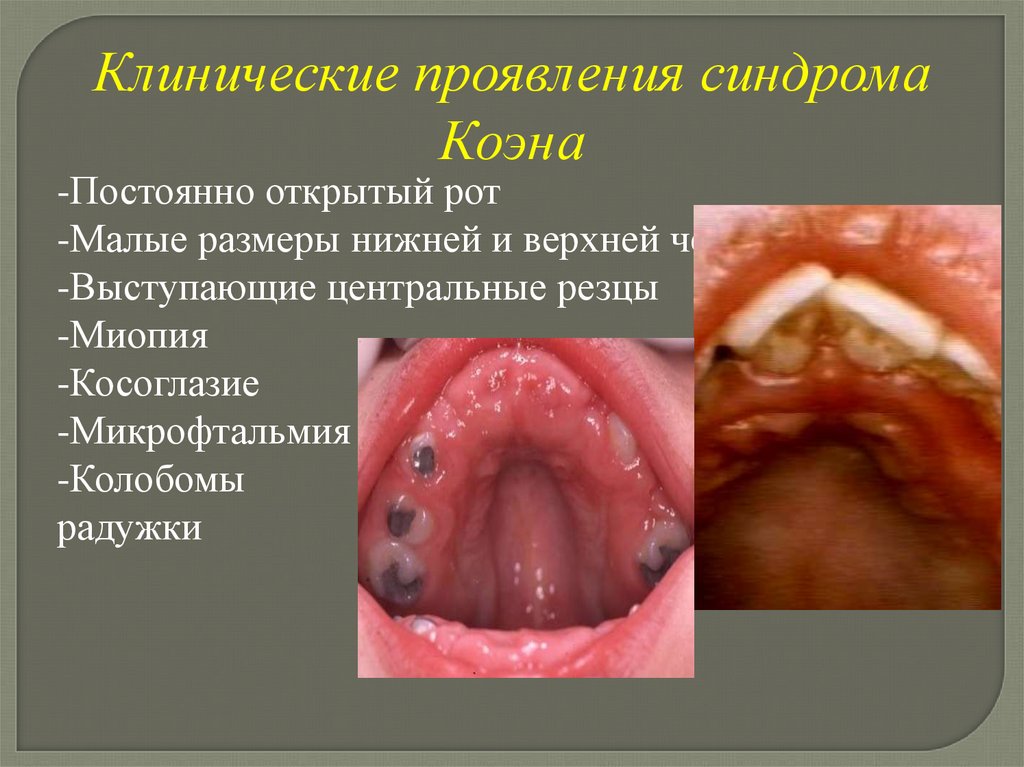
Клинические проявления синдромаКоэна
-Постоянно открытый рот
-Малые размеры нижней и верхней челюстей
-Выступающие центральные резцы
-Миопия
-Косоглазие
-Микрофтальмия
-Колобомы
радужки
49.
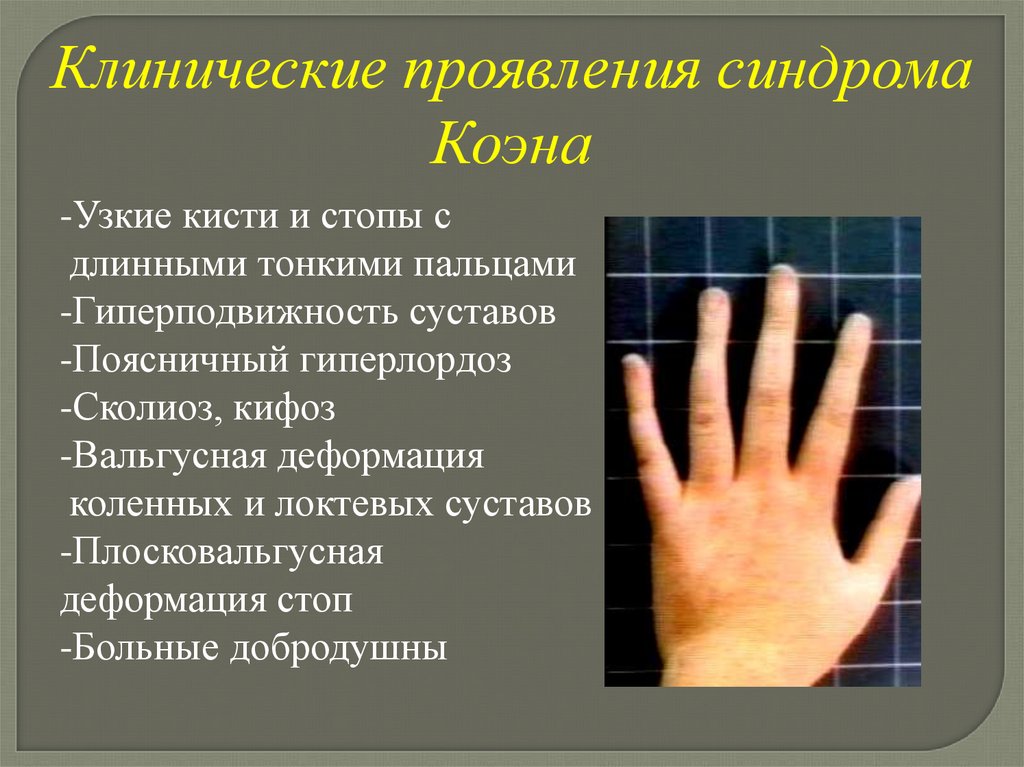
Клинические проявления синдромаКоэна
-Узкие кисти и стопы с
длинными тонкими пальцами
-Гиперподвижность суставов
-Поясничный гиперлордоз
-Сколиоз, кифоз
-Вальгусная деформация
коленных и локтевых суставов
-Плосковальгусная
деформация стоп
-Больные добродушны
50.
Синдром Ротмунда-ТомсонаТип наследования
аутосомно-рецессивный
Ген RTS картирован в xpомосоме 8
51.
Клинические проявлениясиндрома Ротмунда-Томсона
-Пойкилодермия лица и конечностей
-Эритроматоз
-Телеангиоэктазы, участки атрофии кожи
-Двусторонняя катаракта
-Дистрофия волос (ногтей и зубов)
-Гипогонадизм
52.
Клинические проявления СиндромаРотмунда-Томсона
-Нарушения эндохондрального окостенения
-Артериосклероз
-Карликовость
-Множественные аномалии зубов:
микродентия, сверхкомплектные зубы
53.
Моногенные синдромыс Х-сцепленным типом наследования
54.
Синдром АарскогоТип наследования — Х-сцепленный
рецессивный (возможно аутосомнодоминантный)
Ген FGDY 1 локализован в p11.21 Ххромосомы
Соотношение полов — M1: Ж0.
55.
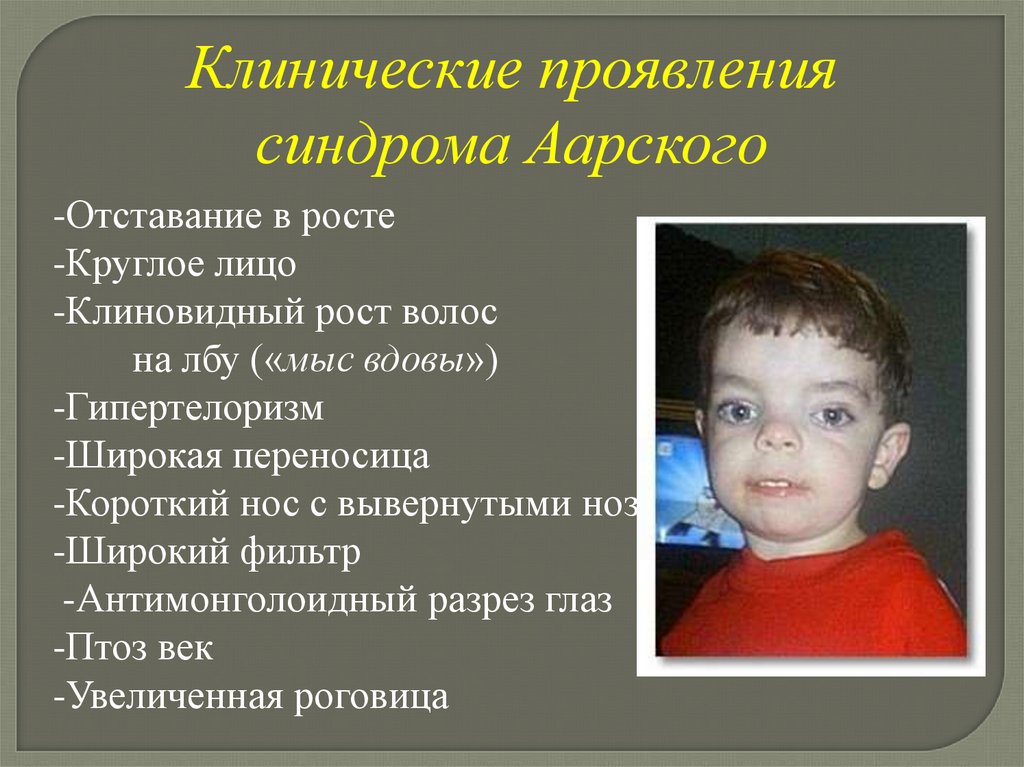
Клинические проявлениясиндрома Аарского
-Отставание в росте
-Круглое лицо
-Клиновидный рост волос
на лбу («мыс вдовы»)
-Гипертелоризм
-Широкая переносица
-Короткий нос с вывернутыми ноздрями
-Широкий фильтр
-Антимонголоидный разрез глаз
-Птоз век
-Увеличенная роговица
56.
Клинические проявления синдромаАарского
-Офтальмоплегия
-Страбизм
-Астигматизм
-Увеличенная роговица
-Разболтанность суставов
-Брахидактилия
-Клинодактилия пятых
пальцев
-Синдактилия
-Поперечная складка ладони
-Широкие стопы
57.
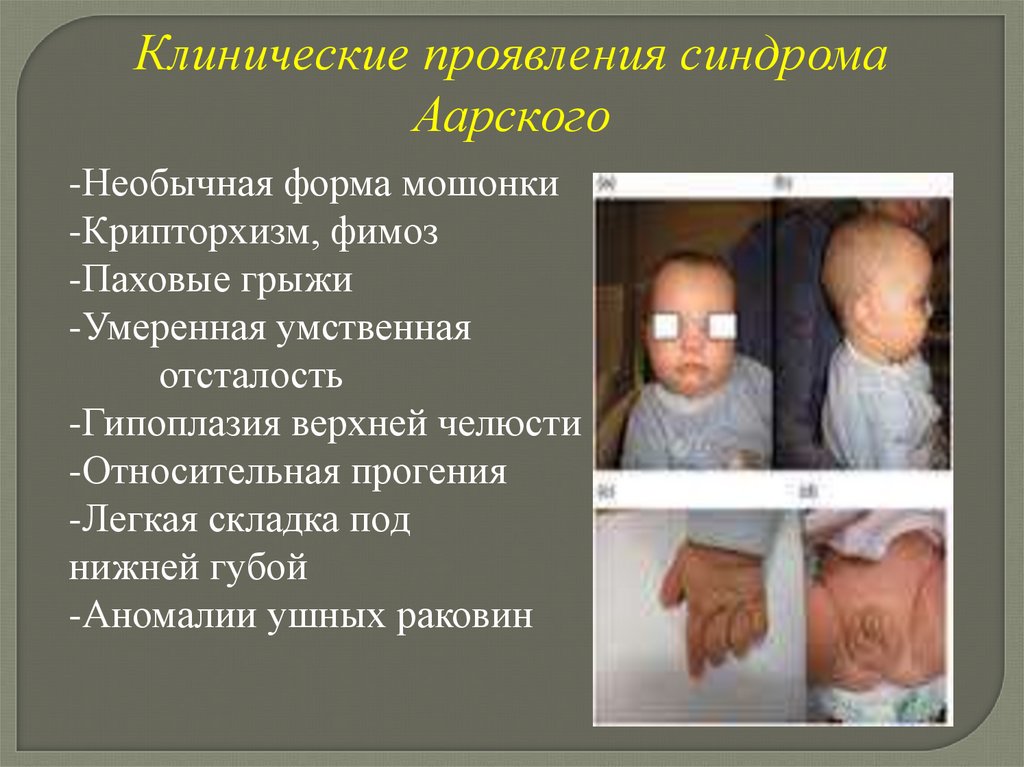
Клинические проявления синдромаАарского
-Необычная форма мошонки
-Крипторхизм, фимоз
-Паховые грыжи
-Умеренная умственная
отсталость
-Гипоплазия верхней челюсти
-Относительная прогения
-Легкая складка под
нижней губой
-Аномалии ушных раковин
58.
Клинические проявления синдромаАарского
59.
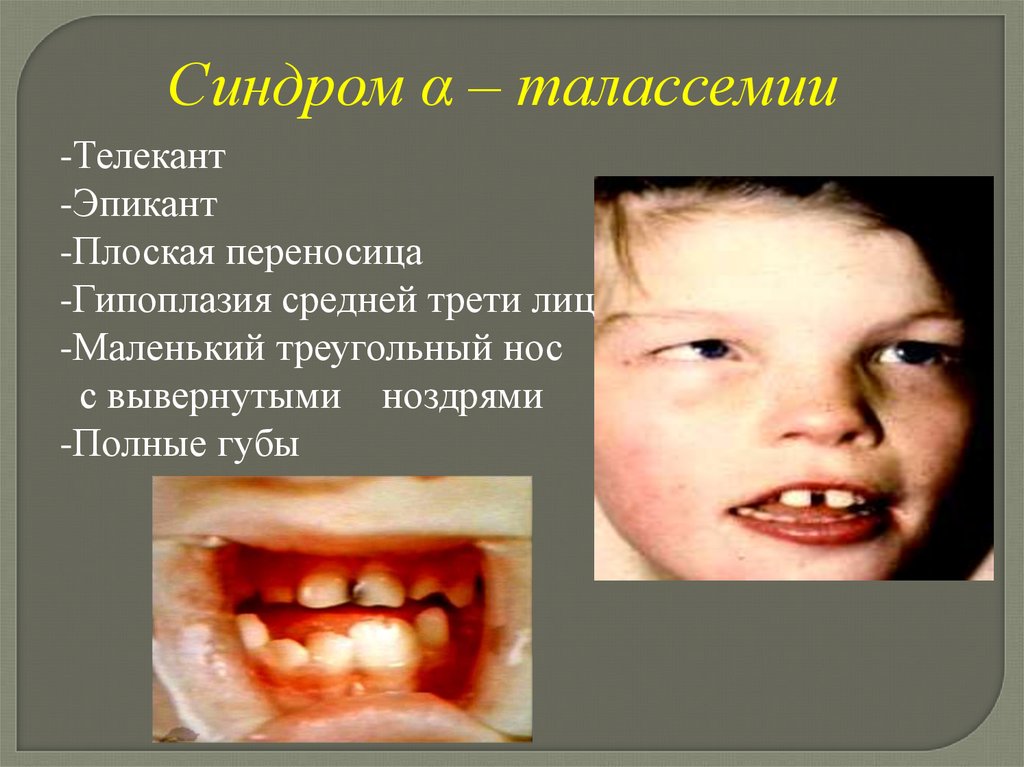
Синдром α – талассемии(Синдром Юберга-Марсиди)
Тип наследования Х-сцепленный
(аутосомно-рецессивный)
Ген геликазы 1ATRX в Xq12-q21
60.
Клинические проявлениясиндрома α – талассемии
-α – талассемия
-Умственная отсталость
-Задержка психического
и речевого развития
-Агенезия почек
-Гидронефроз, гидроуретер
-Повторные инфекции мочевыводящих
путей
61.
Синдром α – талассемии-Телекант
-Эпикант
-Плоская переносица
-Гипоплазия средней трети лица
-Маленький треугольный нос
с вывернутыми ноздрями
-Полные губы
62.
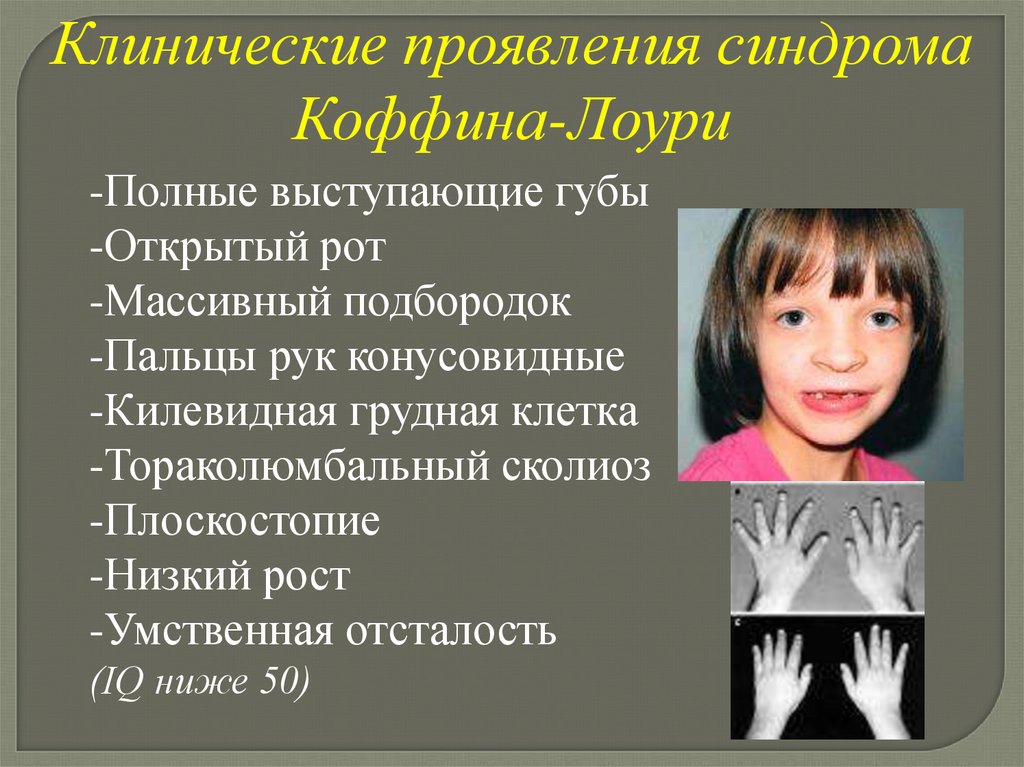
Синдром Коффина-ЛоуриЧастота неизвестна
Соотношение полов — M1:Ж1
Тип наследования — Х-сцепленный
доминантный (с выраженными клиническими
проявлениями у мужчин и более стертой
клинической картиной у женщин)
Ген CLS картирован в p22.2-p22.1
Х-хромосомы
63.
Клинические проявлениясиндрома Коффина-Лоури
-Квадратный лоб
-Выступающие надбровные дуги
-Антимонголоидный разрез глаз
-Гипертелоризм
-Широкая спинка носа
-Открытые вперед ноздри
-Оттопыренные уши
64.
Клинические проявления синдромаКоффина-Лоури
-Полные выступающие губы
-Открытый рот
-Массивный подбородок
-Пальцы рук конусовидные
-Килевидная грудная клетка
-Тораколюмбальный сколиоз
-Плоскостопие
-Низкий рост
-Умственная отсталость
(IQ ниже 50)
65.
Клинические проявления синдромаКоффина-Лоури
-Макродонтия
-Аномалии формы
зубных коронок
66.
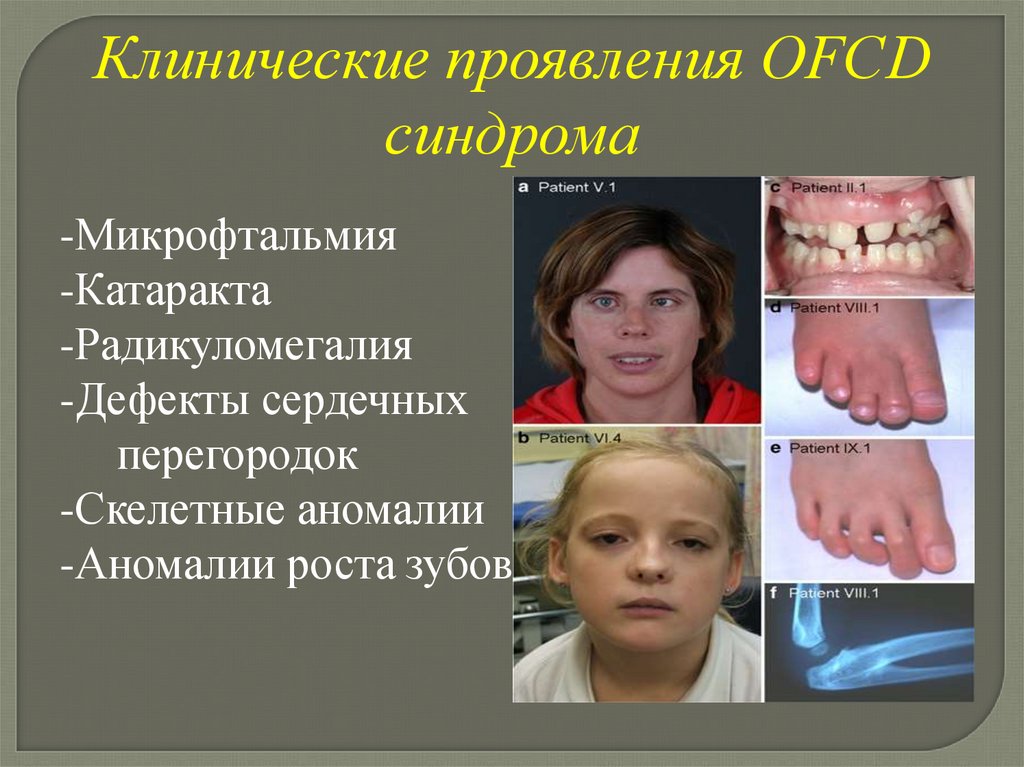
OFCD синдром(Oculofaciocardiodental syndrome глазо-лице-сердечно-зубной синдром)
Тип наследования Х-сцепленный
доминантный
Ген MCOPS2 картирован в Xp11.4
67.
Клинические проявления OFCDсиндрома
-Микрофтальмия
-Катаракта
-Радикуломегалия
-Дефекты сердечных
перегородок
-Скелетные аномалии
-Аномалии роста зубов
68.
Клинические проявления OFCDсиндрома
69.
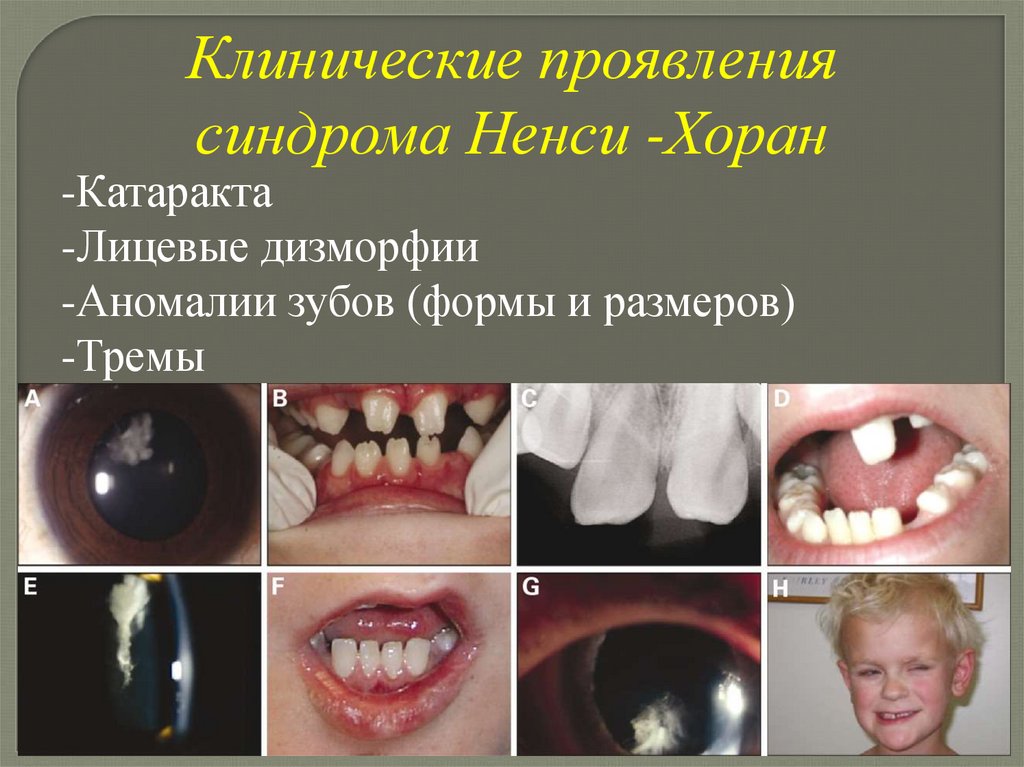
Синдром Нэнси-ХоранТип наследования Х-сцепленный
рецессивный (Х-сцепленный доминантный)
Ген картирован в Xp22.3-р21.1
70.
Клинические проявлениясиндрома Ненси -Хоран
-Катаракта
-Лицевые дизморфии
-Аномалии зубов (формы и размеров)
-Тремы
71.
Клинические проявления синдрома Ненси -Хоран72.
Заболевания с различнымитипами наследования
73.
Эктодермальные дисплазииГетерогенная группа наследственных
заболеваний, обусловленных ненормальным
развитием эктодермы.
Различают ангидротическую и
гидротическую формы, а также ряд
синдромов.
74.
Синдром эктродактилии,эктодермальной дисплазии, расщелины
губы и неба (ЕЕС)
Тип наследования аутосомно-доминантный
с высокой пенетрантностью (93-98%)
Ген ТР 63 картирован в 7q11.2-q21.3
75.
Клинические проявления синдромаэктродактилии
-Поражение кистей и стоп (синдактилия,
эктродактилия)
-Светлые, редкие, тонкие
волосы, брови, ресницы
-Умеренная гипоплазия ногтей
76.
Расщелина неба-Микродонтия
-Частичная адентия
-Гипоплазия эмали
-Множественный кариес
-Гипоплазия верхней челюсти
-Неправильная форма
постоянных зубов
-Персистирование молочных
зубов
-Одно/ двусторонняя расщелина губы и неба
-Патология мочевыделительной системы
-Офтальмологическая симптоматика
77.
Синдром Рэпп-Ходжкина(эктодермальная дисплазия ангидротическая с
расщелиной губы и неба)
Тип наследования аутосомно-доминантный
Ген картирован в 3q27
78.
Клинические проявления синдромаРэпп-Ходжкина
-Сухая тонкая кожа
-Редкие тонкие волосы
-Гипоплазия и дистрофия
ногтей и зубов
-Запавшая переносица
-Узкий нос
-Гипогенитализм
-Предрасположенность к
гипертермии
-Склонность к гнойным
конъюнктивитам и отитам
79.
Клинические проявления синдромаРэпп-Ходжкина
-Гипоплазия верхней челюсти
-Маленький рот
-Расщелины губы и неба
-Расщелины язычка
80.
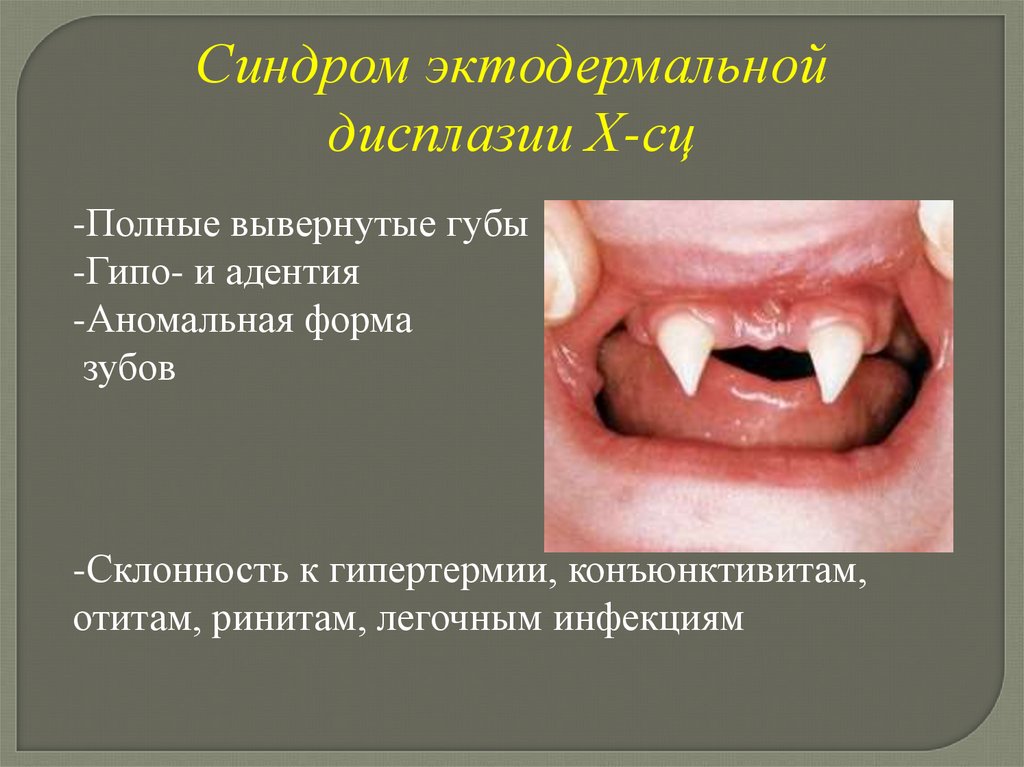
Синдром эктодермальнойдисплазии Х-сц
(синдром Криста-Сименса-Турена)
Тип наследования Х-сцепленный
рецессивный
Ген эктодисплазина-А, картирован в Хq12q13.1
81.
Клинические проявления синдромаэктодермальной дисплазии Х-сц
-Гипоплазия потовых и молочных желез
-Атрофия слезных, бронхиальных желез и
желез ЖКТ и носовой полости
-Тонкие, сухие, светлые, редкие волосы
-Большой лоб
-Запавшая переносица
-Седловидный нос с гипоплазией крыльев
-Запавшие щеки
-Большие деформированные уши
82.
Синдром эктодермальнойдисплазии Х-сц
-Полные вывернутые губы
-Гипо- и адентия
-Аномальная форма
зубов
-Склонность к гипертермии, конъюнктивитам,
отитам, ринитам, легочным инфекциям
83.
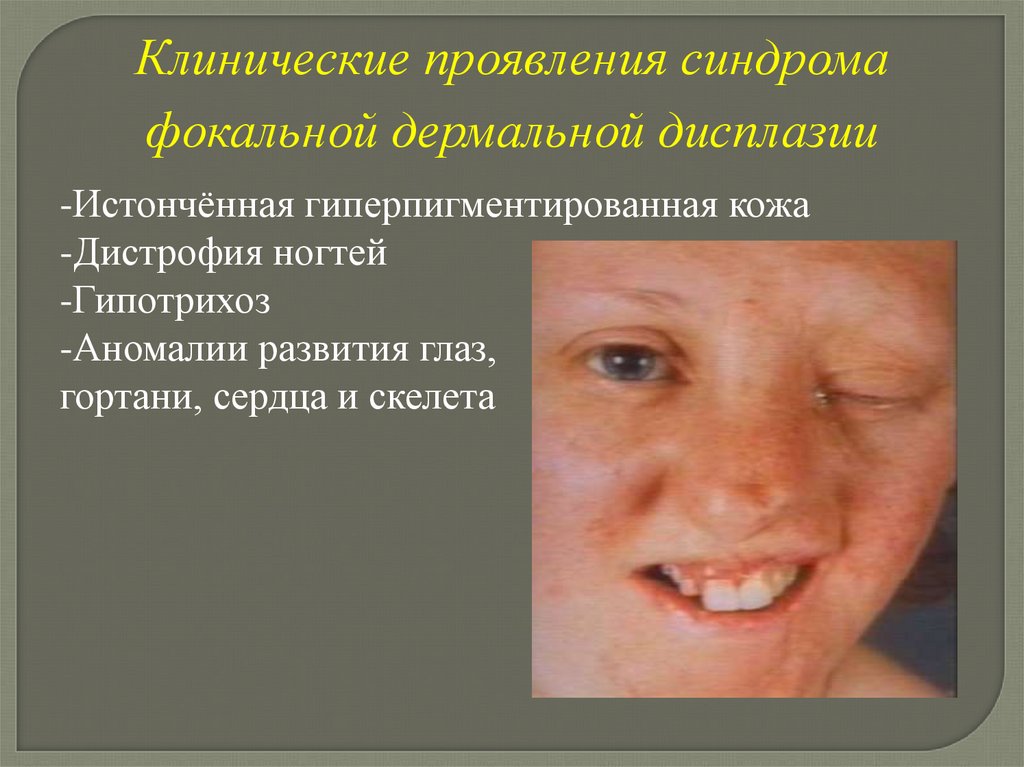
Синдром фокальной дермальнойдисплазии
(Гольтца—Горлина синдром)
Тип наследования
Х-сцепленный доминантный
Гены DHOF, FODH картированы в Хр22
84.
Клинические проявления синдромафокальной дермальной дисплазии
-Истончённая гиперпигментированная кожа
-Дистрофия ногтей
-Гипотрихоз
-Аномалии развития глаз,
гортани, сердца и скелета
85.
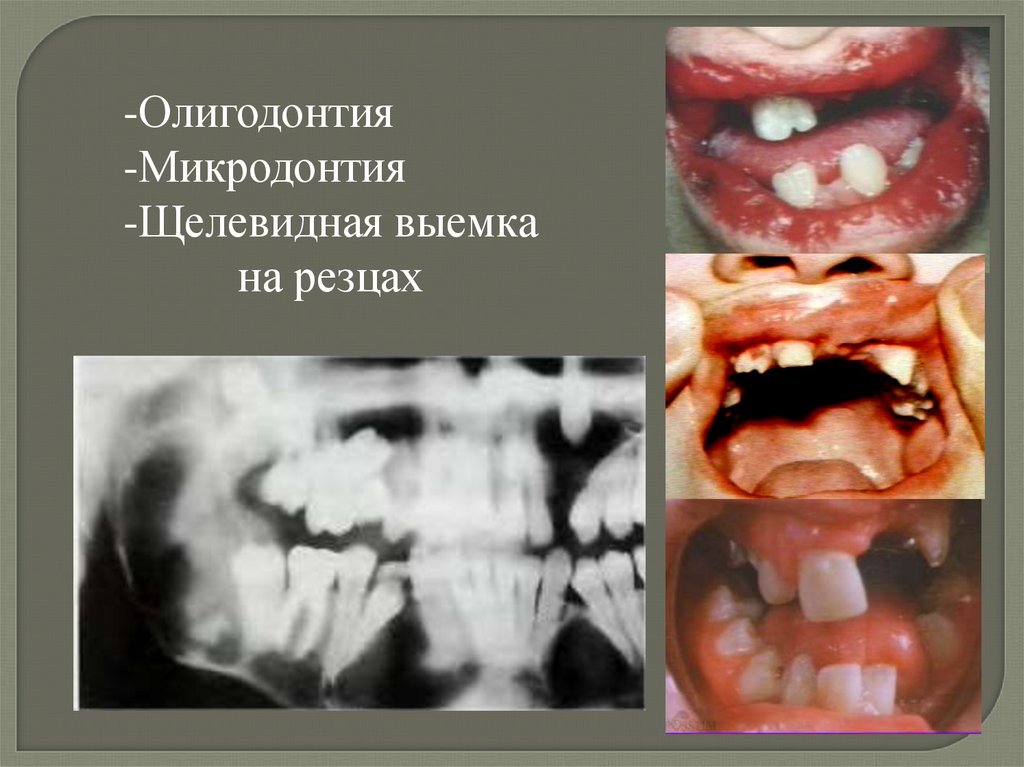
-Олигодонтия-Микродонтия
-Щелевидная выемка
на резцах
86.
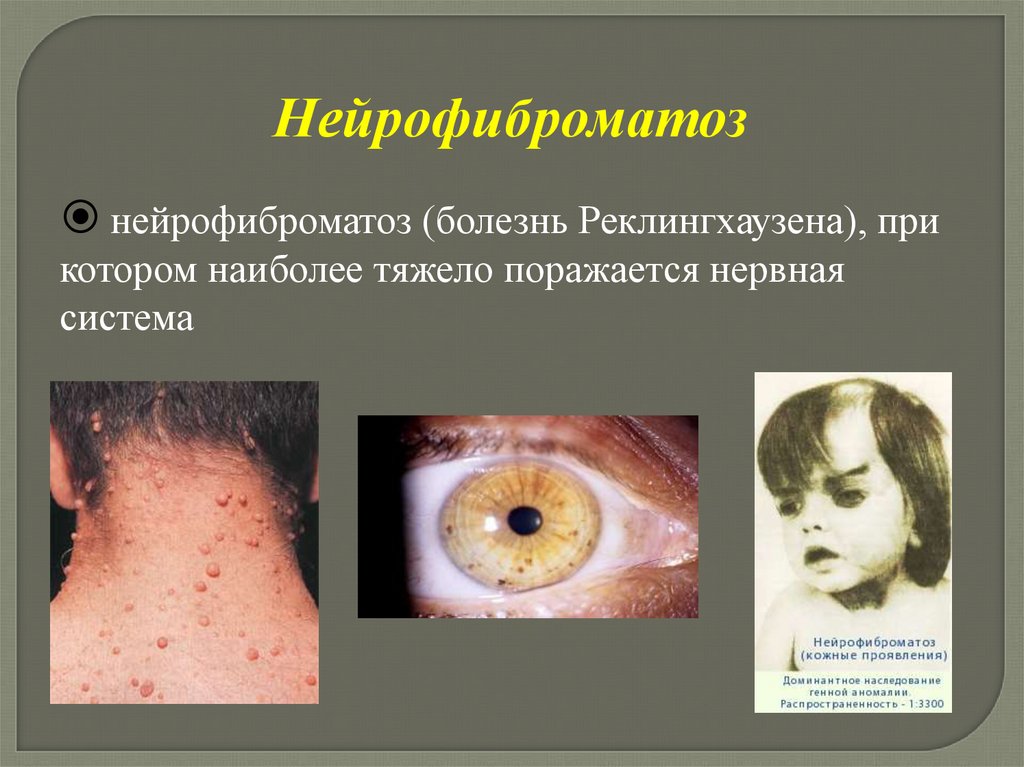
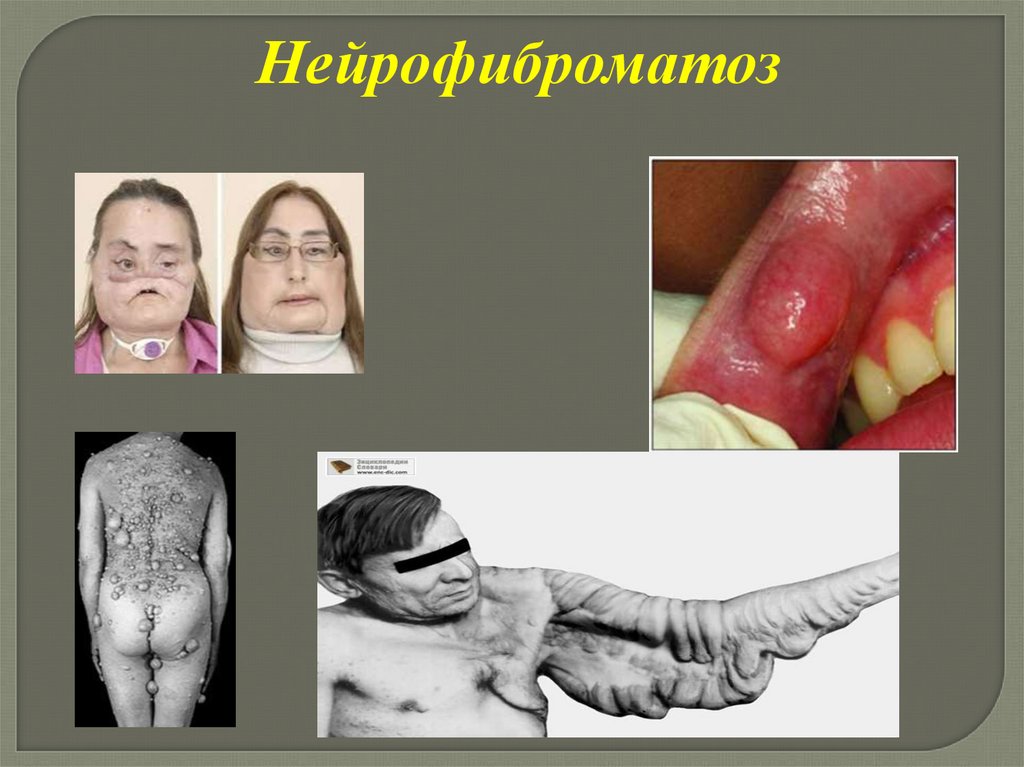
Нейрофиброматознейрофиброматоз (болезнь Реклингхаузена), при
котором наиболее тяжело поражается нервная
система
87.
Синдром Элерса-Данловрожденная гиперрастяжимость соединительной
ткани в связи с нарушением синтеза коллагена,
обусловленным мутациями в разных коллагеновых
генах
88.
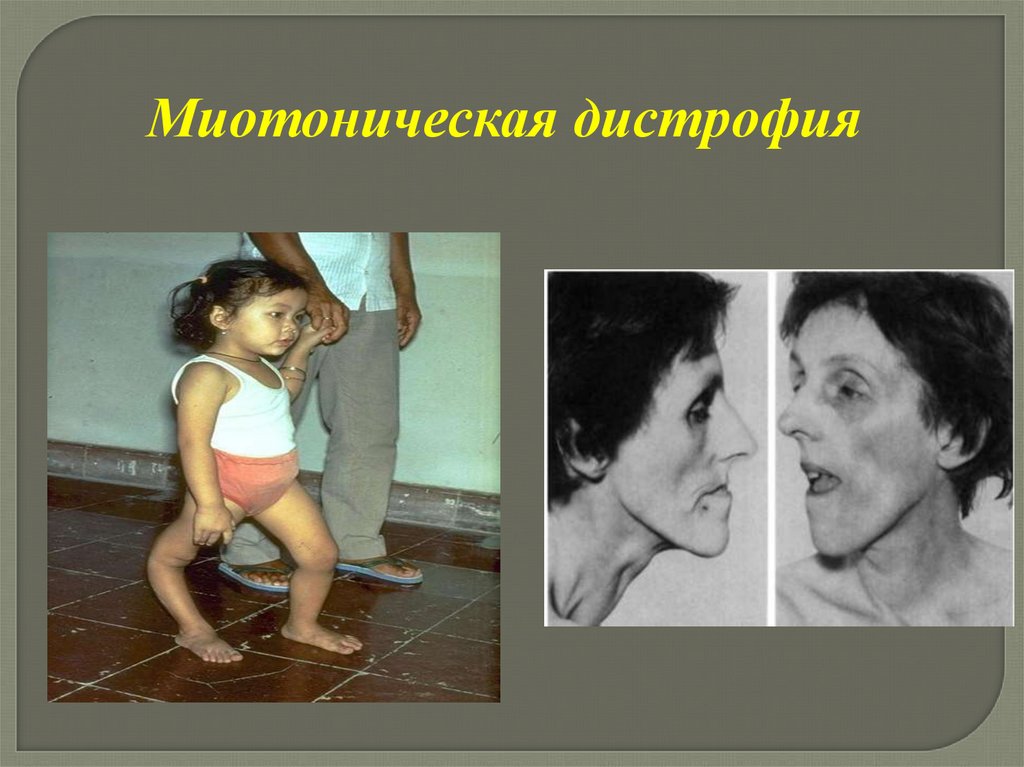
Миотоническая дистрофия89.
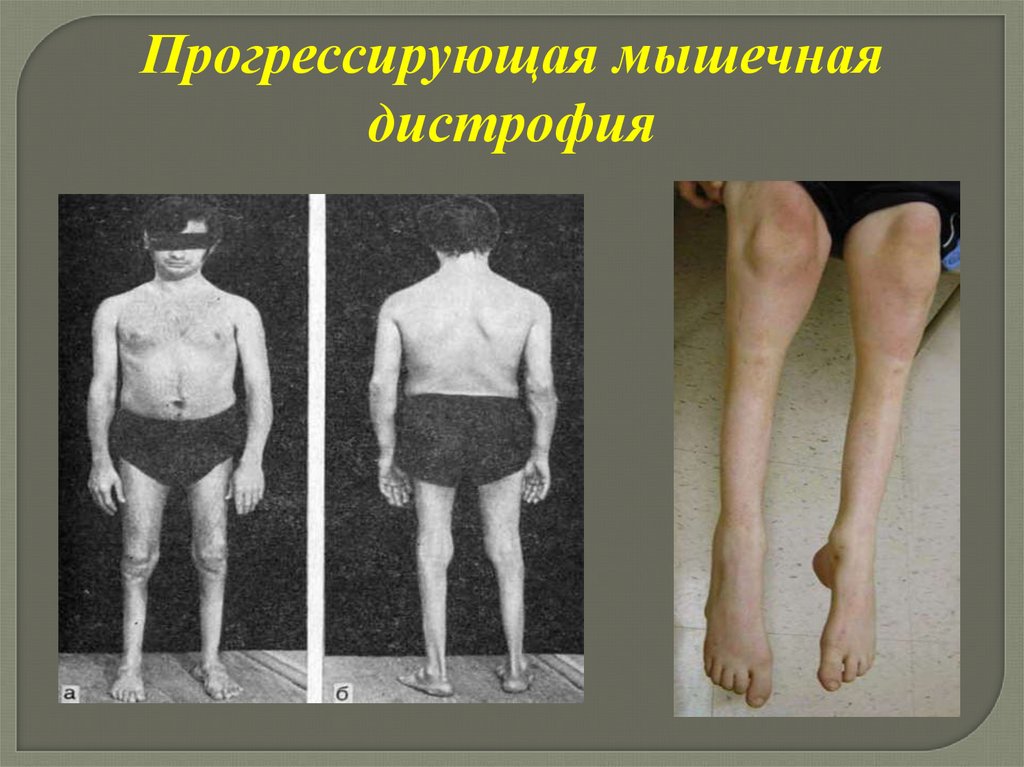
Прогрессирующая мышечнаядистрофия
90.
ФенилкетонурияСвязанная с недостаточностью печеночного фермента
фенилаланингидроксилазы, локус которой расположен в длинном плече
хромосомы 12. Дети с фенилкетонурией рождаются здоровыми, но в
первые же недели после рождения в связи с поступлением фенилаланина
в организм с молоком матери развиваются клинические проявления
заболевания: повышенная возбудимость, гиперрефлексия, повышенный
тонус мышц, судорожные эпилептиформные припадки; от ребенка
исходит «мышиный» запах. Позже развиваются умственная отсталость,
микроцефалия
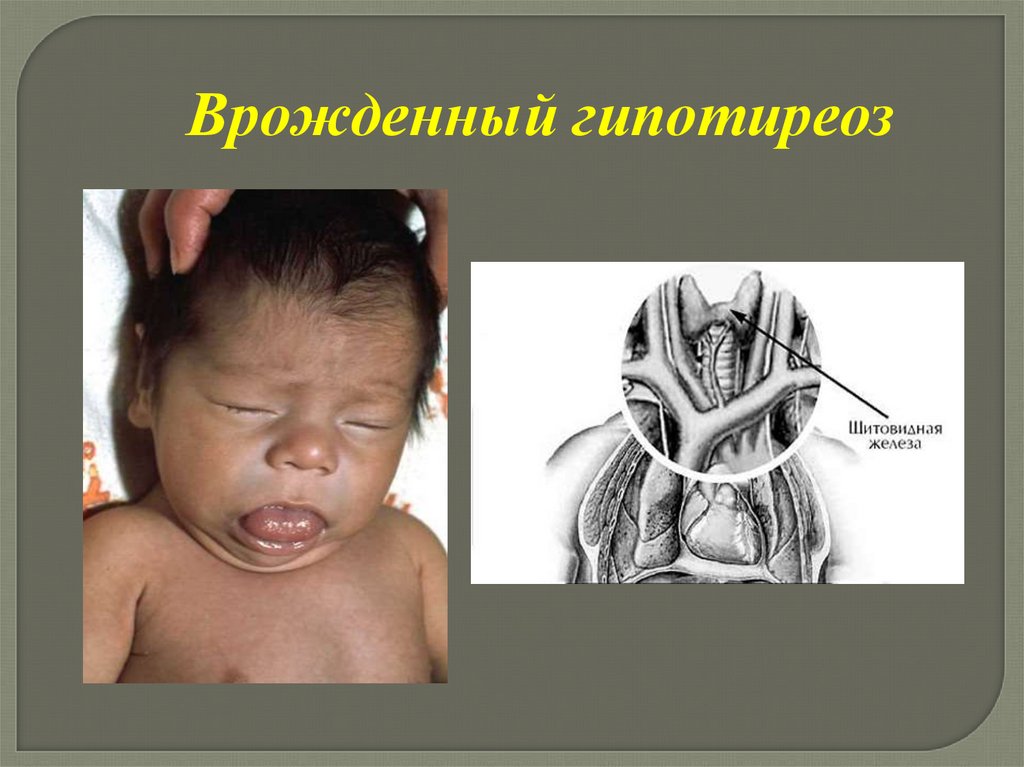
91.
Врожденный гипотиреоз92.
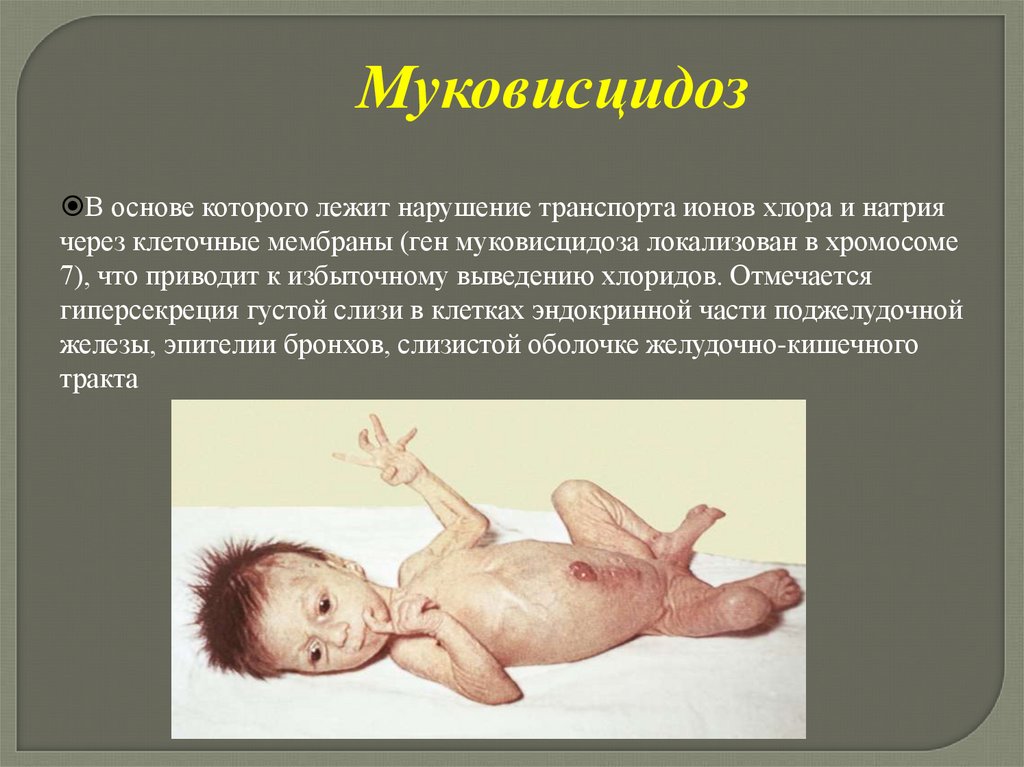
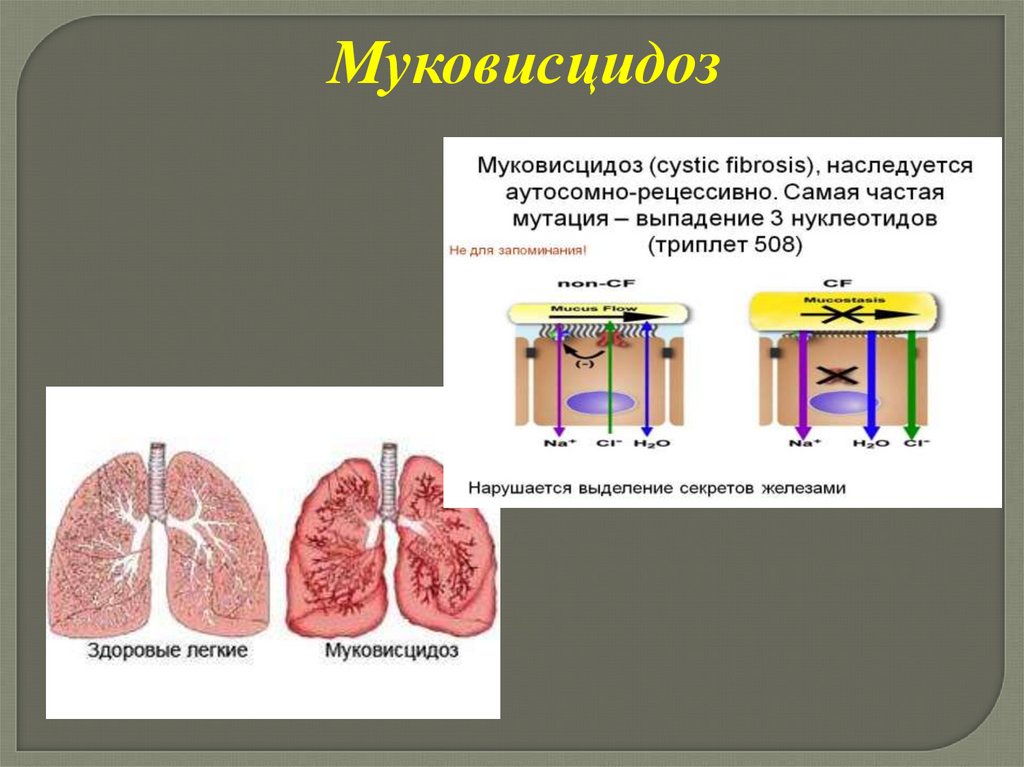
МуковисцидозВ основе которого лежит нарушение транспорта ионов хлора и натрия
через клеточные мембраны (ген муковисцидоза локализован в хромосоме
7), что приводит к избыточному выведению хлоридов. Отмечается
гиперсекреция густой слизи в клетках эндокринной части поджелудочной
железы, эпителии бронхов, слизистой оболочке желудочно-кишечного
тракта
93.
Муковисцидоз94.
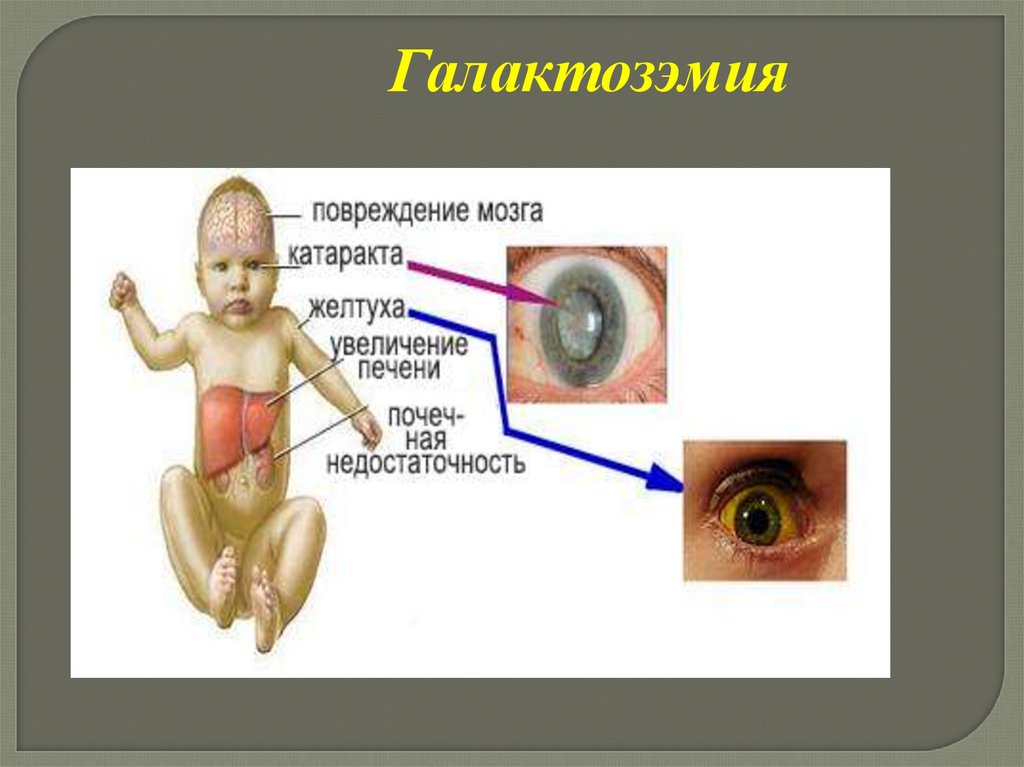
95.
Галактозэмия96.
Андрогенитальный синдромотносится к группе наследственных нарушений
синтеза стероидных гормонов. Наиболее
распространенная форма врожденной гиперплазии
коры надпочечников — дефицит 21-гидроксилазы,
ген локализован в коротком плече хромосомы 6
97.
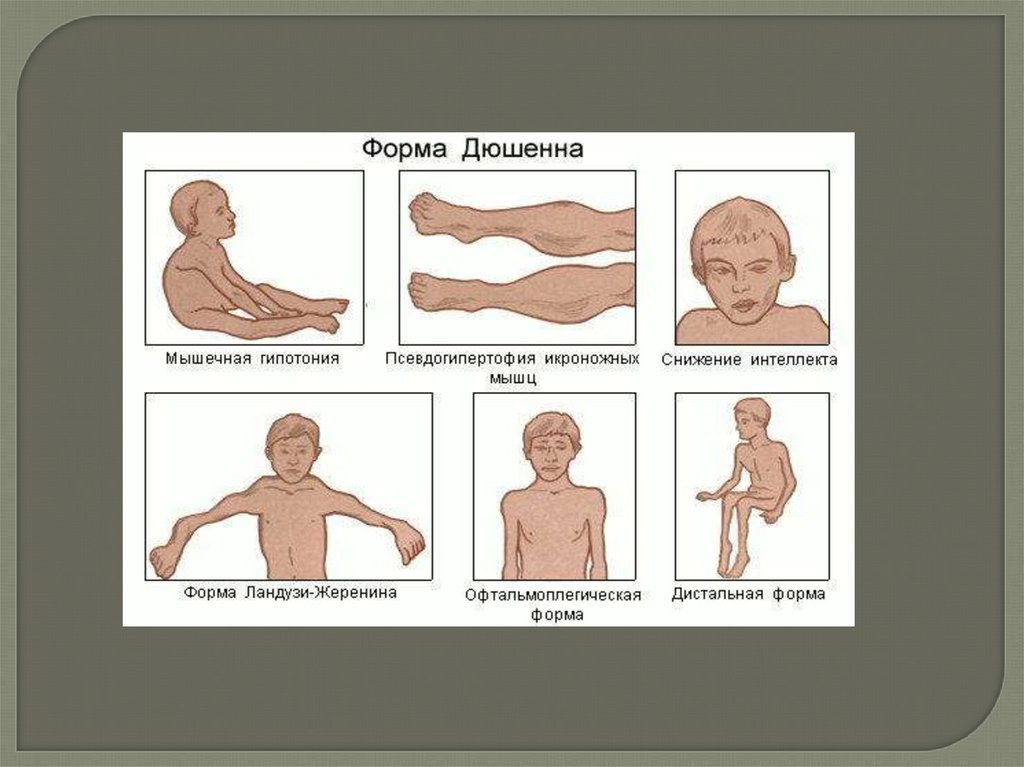
Миопатия ДюшеннаВызванная мутацией в гене, ответственном за синтез белка дистрофина
(ген расположен в локусе X^21). Заболевание проявляется
прогрессирующей мышечной слабостью, дистрофией и некрозом
отдельных мышечных волокон
98.
99.
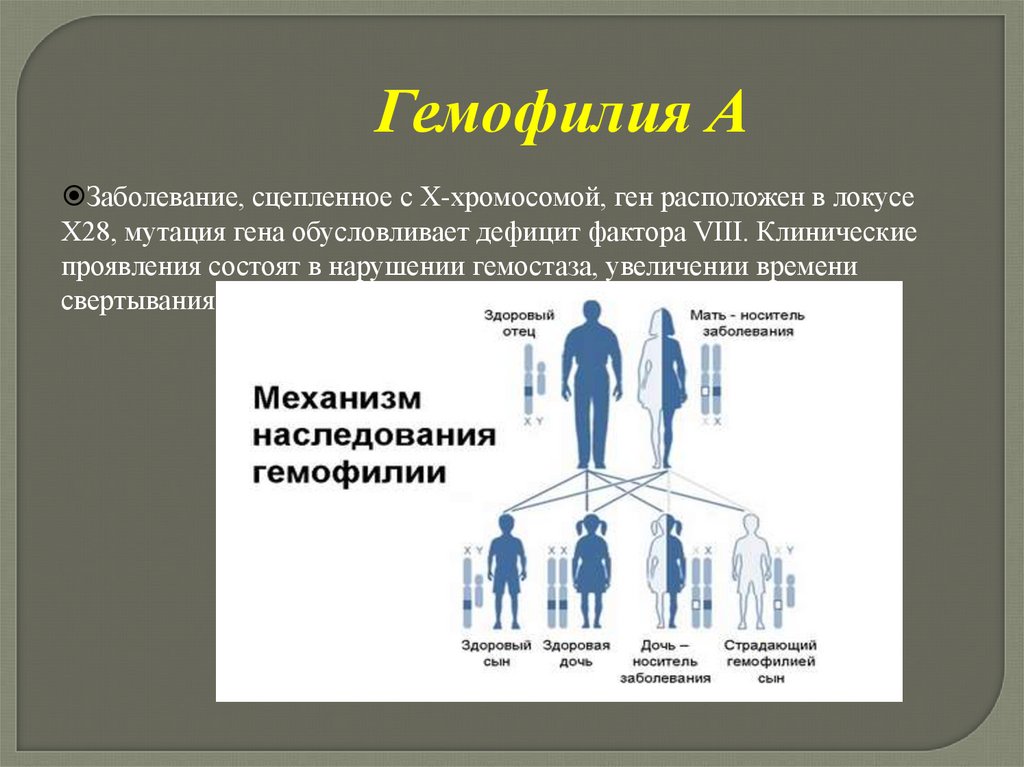
Гемофилия АЗаболевание, сцепленное с Х-хромосомой, ген расположен в локусе
Х28, мутация гена обусловливает дефицит фактора VIII. Клинические
проявления состоят в нарушении гемостаза, увеличении времени
свертывания.
100.
101.
Нейрофиброматоз102.
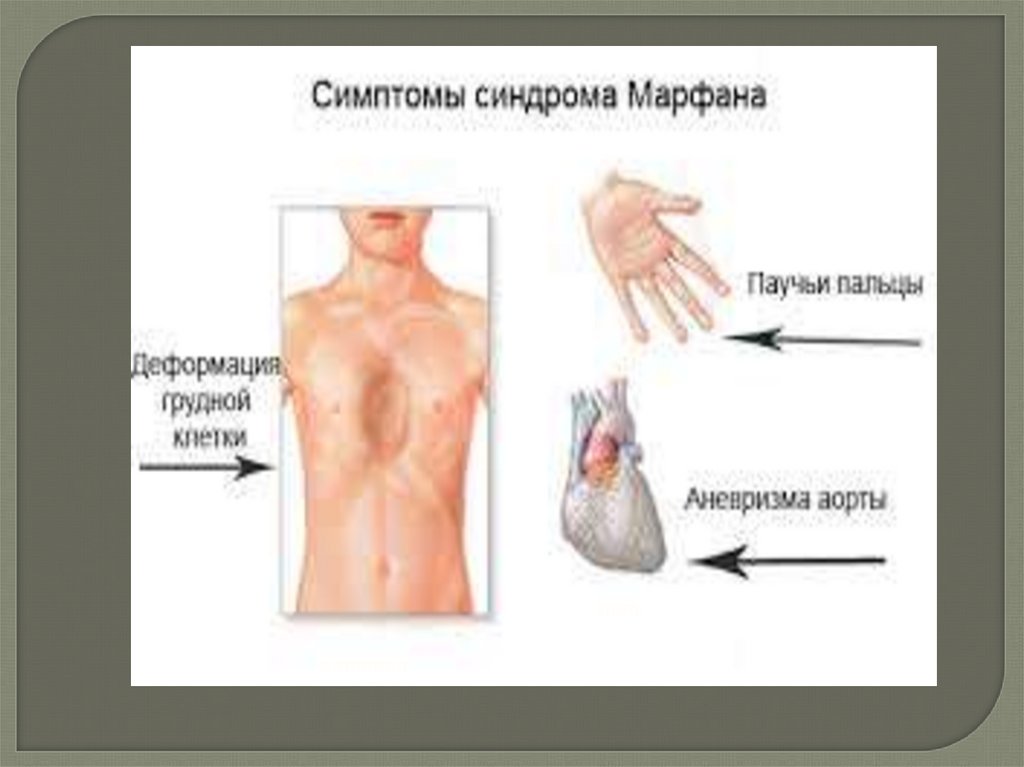
Синдром Марфана– наследственное заболевание, характеризующееся
поражением
соединительной
ткани, что
может
проявляться патологиями разных систем организма,
включая скелет, глаза, кровеносные сосуды, нервную
систему, кожу, легкие и др.
103.
СиндромМарфана
развивается
вследствие дефекта в гене, который
определяет
структуру
фибрина,
играющего
огромную
роль
в
формировании соединительной ткани.
Дефектный
ген
проявляется
у
различных людей в различной
степени. Здоровые родители имеют 1
шанс из 10 000 родить ребенка с
104.
105.
106.
107.
Актер Винсент Скьявелли(Фильм «Пролетая над гнездом кукушки»)
108.
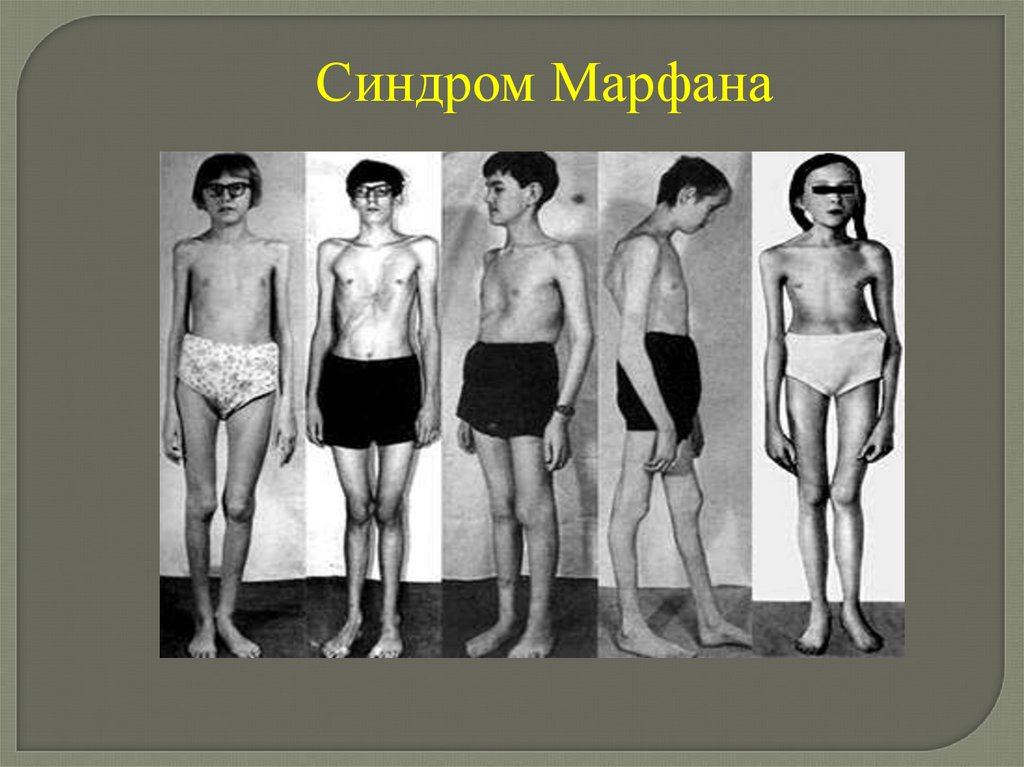
Человек с синдромом Марфана обычноочень высокий и худой. Туловище,
руки, ноги, пальцы рук и ног могут
быть непропорционально длинными.
Другие скелетные аномалии включают
изменение
грудины,
искривление
спины (сколиоз) и плоскостопие
109.
Синдром Марфана110.
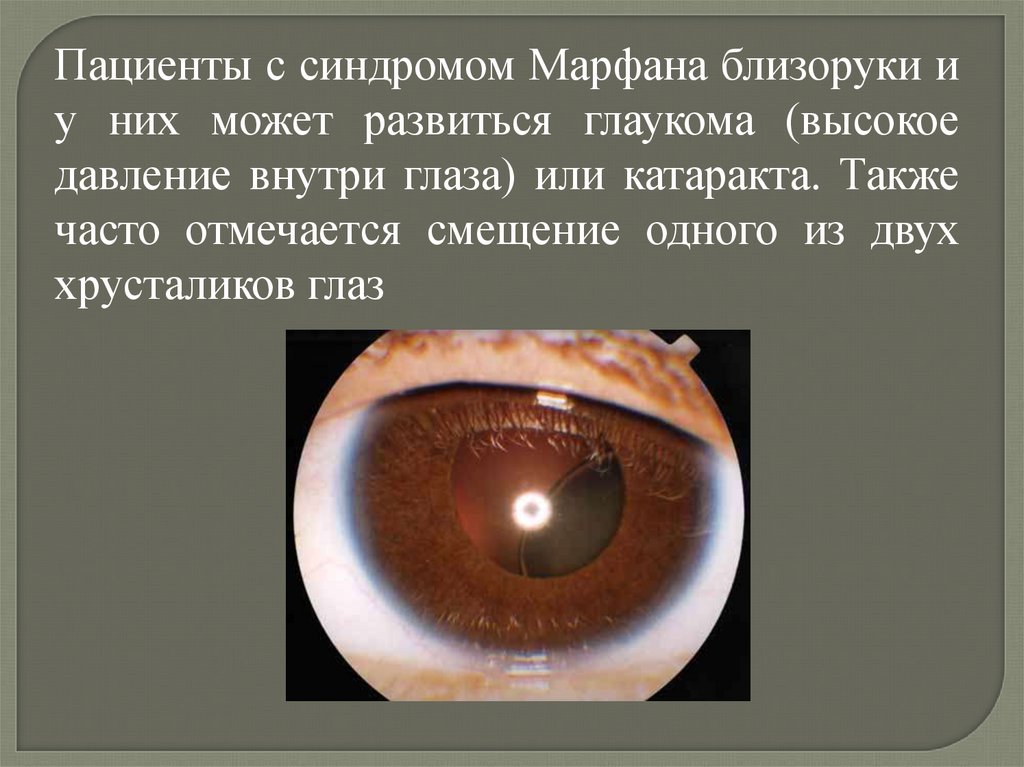
Пациенты с синдромом Марфана близоруки иу них может развиться глаукома (высокое
давление внутри глаза) или катаракта. Также
часто отмечается смещение одного из двух
хрусталиков глаз
111.
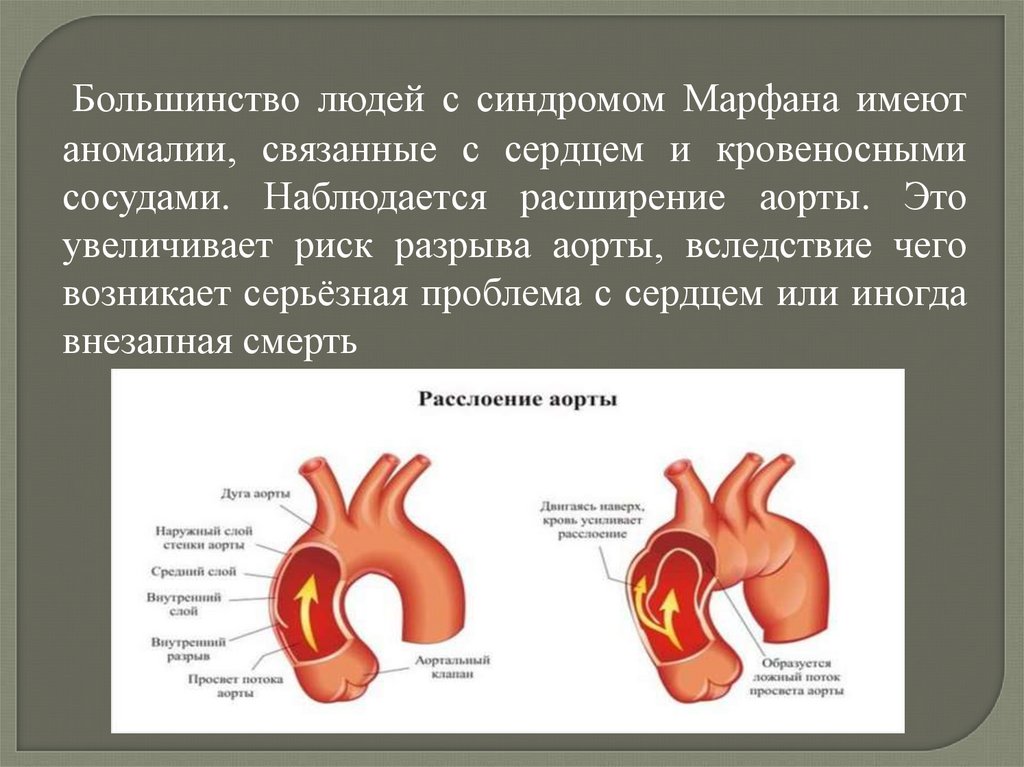
Большинство людей с синдромом Марфана имеютаномалии, связанные с сердцем и кровеносными
сосудами. Наблюдается расширение аорты. Это
увеличивает риск разрыва аорты, вследствие чего
возникает серьёзная проблема с сердцем или иногда
внезапная смерть
112.
Головной и спинной мозг окруженыжидкостью; вокруг имеется оболочка, которая
называется твердой мозговой оболочкой, ее
составляет соединительная ткань. Когда люди
с синдромом Марфана стареют, твердая
мозговая
оболочка
часто
слабеет
и
вытягивается. Это называется дуральная
эктазия.
113.
114.
Для большинства пациентов с синдромом Марфанахарактерно растягивание кожи, даже без изменения
массы тела. Это может появиться в любом возрасте и не
представляет опасности
Резиновая кожа
Гарри Тернера
115.
ДиагностикаНаиболее точным лабораторным признаком СМ
является генетическая идентификация мутаций в
гене FBN1 + показатели почечной экскреции
метаболитов соединительной ткани: оксипролина,
оксилизилгликозаминов, гликозаминогликанов и их
фракционного состава (повышенный распад
коллагена, а его уровень может определять тяжесть
заболевания;
электрокардиография;
рентгенография;
компьютерная
томография;
ангиография;
зхокардиография;
магнитнорезонансная томография (МРТ); генеалогический
анализ.
116.
Консервативное лечениеТак как ведущая причина смерти больных СМ - разрыв
расслаивающей аневризмы аорты, то консервативное
лечение направлено в первую очередь на его
предотвращение. Еще в начале 70-х годов прошлого
столетия было показано, что риск расслоения аорты у
больных с СМ можно снизить путем длительного
применения β-блокаторов (пропранолол, атенолол и
метопролол). При наличии непереносимости или
противопоказаний
к
применению
β-блокаторов
используют антагонисты кальция или ингибиторы
ангиотензин
превращающего
фермента
(АПФ).
Стимуляция преждевременного полового созревания
при помощи гормонотерапии может затормозить
дальнейший рост и уменьшить проявления СМ у очень
117.
Хирургическое лечениеВ настоящее время при СМ в основном
применяется два типа вмешательств на аорте:
комбинированная трансплантация по Bentall,
при которой пересаживают корень аорты и ее
клапан и операции, сохраняющие аортальный
клапан.
5-летняя и 10-летняя выживаемость при
операции по Bentall - 80% и 60%
соответственно, а операции с сохранением
аортального клапана еще более эффективны:
5-летняя выживаемость превышает 90%.
118.
ПрогнозПродолжительность и качество жизни больных СМ
в основном зависит от объема и выраженности
поражения сердечно-сосудистой системы, скелета и
глаз. Продолжительность и качество жизни больных
СМ в основном зависит от объема и выраженности
поражения сердечно-сосудистой системы, скелета и
глаз. Приемлемым для них является низкий или
средний уровень физической активности. Из-за
риска сердечно-сосудистых осложнений, развития
пневмоторакса
и
возможной
дислокации
хрусталиков, им не рекомендуется заниматься
контактными видами спорта и подводным
плаванием. Оперированные пациенты имеют еще
119.
120.
•Методы диагностики моногенныхболезней
•-Фенотипический анализ
•-Лабораторная диагностика:
• (молекулярная ДНК-диагностика,
биохимическая, иммунологическая)
•-Клинико-генеалогический метод
•-Инструментальная диагностика (УЗИ,
рентгенологическое исследование,
радиологическое и др.)
121.
• Основа современной (прежде всего прямой)
ДНК- диагностики - метод PCR или ПЦР
(Polymerase Chain Reaction или Полимеразная
Цепная Реакция), который позволяет in vitro в
течение часа получить млн. копий заданного
фрагмента молекулы ДНК, что облегчает
идентификацию в геноме пациента (пробанда)
сайта ДНК, представляющего диагностический
интерес
122.
УЗИ- диагностика123.
Скрининг неонатальный124.
Лечение•- Симптоматическое
- Патогенетическое
- Этиологическое
125.
•Симптоматическое лечение• направлено
на коррекцию патологических
симптомов и, не воздействуя на причину болезни,
приводит к улучшению состояния больного,
предотвращению осложнений и снижению темпов
прогрессирования заболевания. При этом могут
быть использованы лекарственные препараты,
хирургическое
вмешательство,
физиотерапевтические,
рентгенологические
и
другие методы.
126.
•Патогенетическое лечение• направлено на коррекцию биохимических
и
патофизиологических
процессов,
нарушенных
в
результате
изменения
концентрации
белкового
продукта
мутантного гена. Эти методы лечения
наиболее эффективны при наследственных
болезнях обмена.
127.
•МЕТОДЫ:•1) Восполнение дефицита фермента/белка
• (гемофилия – переливание крови)
•2) Восполнение дефицита кофактора/витамины или коэнзимы/
• (пиридоксинзависимые судороги – В6, атаксия с дефицитом
витамина Е – вит. Е)
•3) Восполнение дефицита конечного продукта
• (адреногенитальный синдром – кортизол, кортинеф, врожденный
гипотиреоз – тироксин)
•4) Ограничение количества субстрата в пище
• (ФКУ – фенилаланинсодержащие продукты, Тирозинемия – ФА,
тирозин, галактоземия – лактоза и галактоза)
•5) Коррекция выведения продукта
•6) Ограничение применения лекарственной терапии
• (дефицит глюкозо-6-фосфатдегидрогеназы – сульфаниламиды)
128.
•7) Замещение поврежденных тканей• (поликистоз почек – трансплантация почки, тяжелые
формы Х-сцепленного комбинированного иммунодефицита трансплантация костного мозга)
•8) Удаление пораженных тканей
• (семейный аденоматозный полипоз – колонэктомия,
сфероцитарная анемия – спленэктомия, нейрофиброматоз –
удаление фибром)
129.
• Этиологическое лечение• Наиболее
перспективным
и
эффективным
способом
лечения
наследственной
патологии
человека является коррекция генетического дефекта
на уровне гена.
• Это
направление
в
лечении
называется
генотерапия или молекулярное протезирование.
• Коррекцию функции мутантных генов
и
восстановление их экспрессии можно осуществить
двумя путями:
• 1) заменой мутантного гена его нормальной
копией
• 2)введение
нормальной
копии
гена
при
сохранении мутантной
130.
Профилактика
моногенных заболеваний
• 1. Медико-генетическое консультирование семей с
расчетом теоретического (менделевского риска) рождения
ребенка с моногенным заболеванием
• 2. Массовый скрининг беременных женщин с помощью
неинвазивных методов пренатальной диагностики (УЗИ и
определение сывороточных – фетальных маркеров ) и по
показаниям – инвазивных методов пренатальной
диагностики
• 3.
Массовый
скрининг
новорожденных
на
фенилкетонурию, муковисцидоз, адрено-генитальный
синдром, врожденный гипотиреоз, галактоземию
• 4.
Селективный
(выборочный)
скрининг
на
гетерозиготное носительство
131.
•Контрольные вопросы•1. Этиология, эпидемиология, классификация
моногенных болезней
•2. Методы диагностики моногенных болезней
•3. Принципы лечения моногенных болезней
132.
Рекомендуемая литература:Основная:
1. Медицинская и клиническая генетика для
стоматологов: учебное пособие для медицинских
вузов /Л.В. Акуленко [и др.]; под ред. О.О.
Янушевича, 2008, М.: ГЭОТАР-Медиа
2. Медицинская и клиническая генетика для
стоматологов: учебное пособие (УМО по мед. и
фармац. образованию вузов России)/Л.В. Акуленко
[и др.]; под ред. О.О. Янушевича, 2015, Москва:
Гэотар-Медиа
133.
ДополнительнаяНаследственные болезни. Национальное руководство: [с прил. на
компакт-диске] / Рос. о-во мед. генетиков, Ассоц. мед. о-в по качеств/ Н.П.
Бочков, Е.К. Гинтер, В.П. Пузырев, 2012, Москва : ГЭОТАР-Медиа
2. Детская неврология: учебник для студентов учреждений высшего
профессионального образования в 2 т./Н.П. Бочков, В.П. Пузырев, С.А.
Смирнихина, 2011, Москва: ГЭОТАР-Медиа
3. Клиническая генетика: учебник [с прил. на компакт-диске] (УМО по
медицинскому и фармацевтическому образованию вузов России) 4-е изд.,
дополненное и переработанное/Н.П. Бочков, В.П. Пузырев, С.А.
Смирнихина, 2011, Москва: ГЭОТАР-Медиа
4. Генетика человека с основами общей генетики: учеб. пособие 2-е изд.,
переработанное и дополненное/ Н.А. Курчанов / 2009, СПб.: СпецЛит.
5. Общая и молекулярная генетика [Электронный ресурс]: учеб.
пособие. Режим доступа: htp://www.knigafund.ru/books/18890/И.Ф.
Жимулев / 2007, Новосибирск: Сиб. университет изд-во.
1.
134.
Базы данных, информационносправочные и поисковые системы1.Электронная библиотека ОмГМА: http://weblib.omsk-
osma.ru/;
2.Электронно-библиотечная система
«КнигаФонд»:http://www.knigafund.ru;
3.Электронная библиотека 1-го МГМУ им. И. М. Сеченова:
http://www.scsml/rssi/ru;
4.Научная электронная библиотека:
http://elibrary.ru/defaultx.asp;
5.Медицинская поисковая система PubMed. Режим доступа:
http://www.ncbi.nlm.nih.gov/pubmed/
6. Центральная научная медицинская библиотека. Режим
доступа: http://www.scsml.rssi.ru
http://www.medterapevt.ru/1019.html
ЭБС «Консультант студента».