




































 medicine
medicine biology
biologySimilar presentations:










Хромосомные болезни. Практическое занятие №3
1.
Мацкиева О.В., Самохина В.И., Скрипкина Г.И.Учебное пособие
для студентов
стоматологического факультета
«Медицинская генетика в стоматологии»
(V семестр)
Омск - 2016
2.
УДК 616.31:575(075)ББК 52.54я73+56.6я73
Учебное пособие к практическим занятиям предназначено для
повышения качества обучения студентов стоматологического
факультета на кафедре детской стоматологии ОмГМУ.
Пособие хорошо иллюстрировано, составлено согласно новому
учебному плану; Является дополнением к имеющимся учебнометодическим материалам.
В пособие вошли материалы, отражающие основные наследственные
синдромы, одним из проявлений которых являются черепно-лицевые
и зубочелюстные аномалии.
Рецензенты:
Антонова А.А. - д.м.н., профессор, зав. каф. стоматологии детского
возраста ДГМУ
Турица А.А. - к.м.н., доцент кафедры пропедевтики детских
болезней и поликлинической педиатрии ОмГМУ
Учебное пособие обсуждено и утверждено на заседании центрального
координационного методического совета Омского государственного
медицинского университета. Протокол №6 от 24 мая 2016г.
3.
•Практическое занятие № 3•Хромосомные болезни
4.
План занятия5.
• Цель занятия:•формирование общих
представлений о хромосомных
болезнях, изучить основы
хромосомных заболеваний
человека
6.
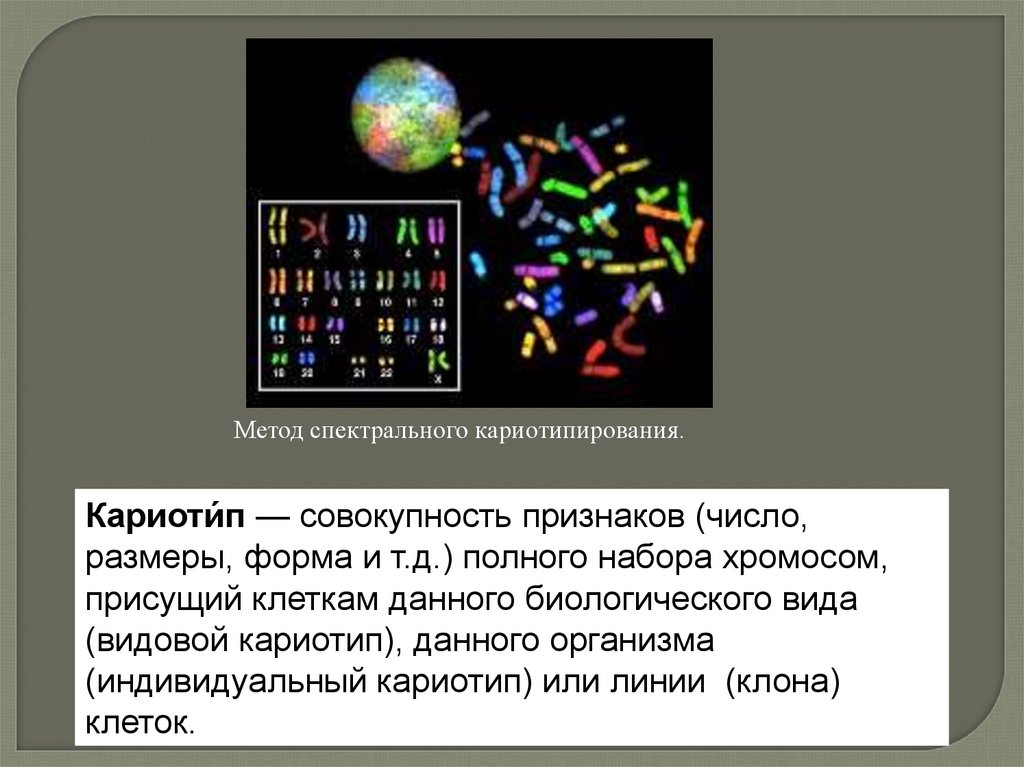
Метод спектрального кариотипирования.Кариоти́п — совокупность признаков (число,
размеры, форма и т.д.) полного набора хромосом,
присущий клеткам данного биологического вида
(видовой кариотип), данного организма
(индивидуальный кариотип) или линии (клона)
клеток.
7.
• К хромосомным относят болезни, обусловленныегеномными мутациями или структурными
изменениями отдельных хромосом. В настоящее
время у человека известно более 700 подобных
заболеваний
8.
Классификация хромосомныхболезней
1. Числовые нарушения по отдельным
хромосомам (анеуплодия)
2. Нарушение кратности гаплоидного
набора хромосом (полиплоидия)
3. Структурные перестройки хромосом
(делеции,
дупликации,
инверсии,
транслокации и др.)
9.
Признаки, характерные дляхромосомных синдромов
- умственная отсталость
- пре - и постнатальная задержка развития
- патология внутренних органов
- лицевые дизморфии
10.
Группы диагностических признаковI группа: физическое недоразвитие, ряд
дизморфий мозгового и лицевого черепа,
косолапость, клинодактилия мизинцев
кистей, пороки развития некоторых
внутренних органов (сердца, легких, почек)
11.
II группа: признаки, встречающиеся в основномпри определенных хромосомных болезнях. Их
сочетание позволяет в большинстве случаев
диагностировать хромосомную аномалию.
(При трисомии 13 хромосомы наиболее часто
встречающиеся признаки - глубокая задержка
умственного
и
физического
развития,
гипертелоризм,
низко
расположенные
деформированные уши, расщелины губы и неба)
12.
III группа: Признаки, характерные толькодля одной хромосомной аномалии
(«кошачий крик» при синдроме 5р-,
алопеция при синдроме 18р-)
13.
Общие особенности хромосомныхболезней
1.Чем больше хромосомного материала утрачено или
приобретено, тем сильнее отклонения в развитии и тем
раньше в онтогенезе они проявляются.
2.Нехватка генетического материала сказывается на
организме тяжелее, чем избыток.
3.Тяжесть клинической картины зависит не только от размера
хромосомы, вовлеченной в патологический процесс, но и её
качественного состава.
4.Прямая корреляция между числом лишних Х-хромосом и
степенью умственной отсталости.
14.
Аутосомные синдромы- Синдром Дауна
- Синдром Эдвардса
- Синдром Патау
15.
Синдром Дауна(трисомия 21 хромосомы)
-частота встречаемости 1:700 – 1:800
-среди умственно отсталых детей больные с
синдромом Дауна составляют 10-12%
-соотношение полов 1:1
-вероятность рождения увеличивается с
возрастом матери (25лет-1/1376; 30 лет-1/960; 35 лет1/424; 40 лет- 1/126; 45 лет- 1/31)
16.
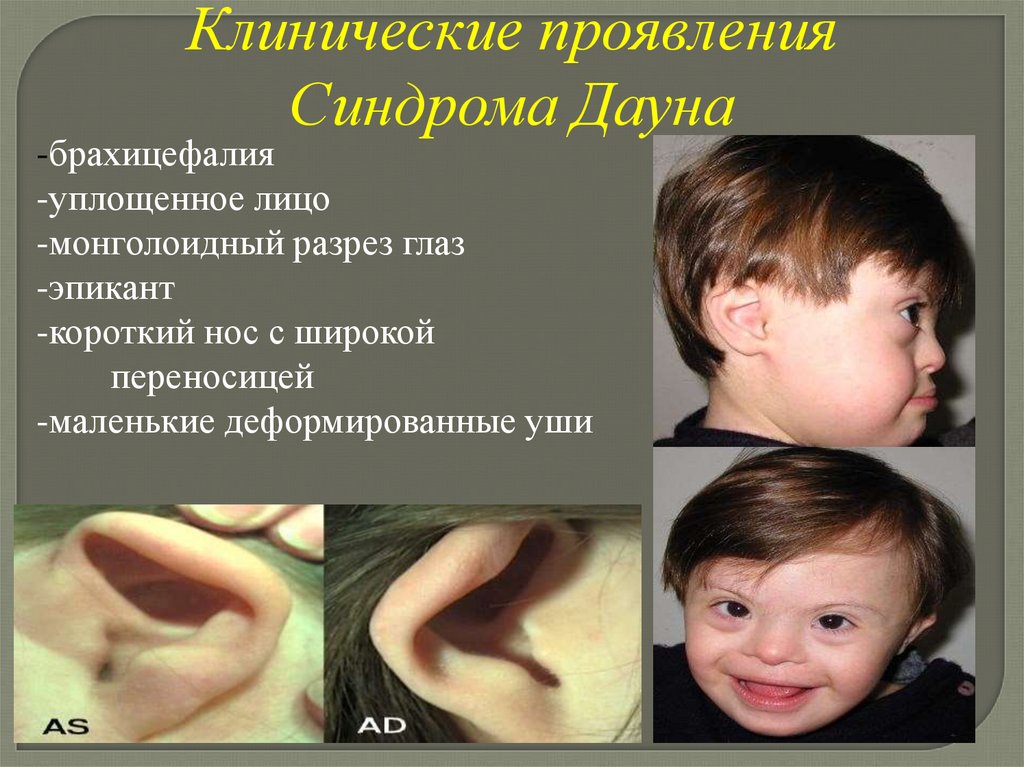
Клинические проявленияСиндрома Дауна
-брахицефалия
-уплощенное лицо
-монголоидный разрез глаз
-эпикант
-короткий нос с широкой
переносицей
-маленькие деформированные уши
17.
Синдром Дауна18.
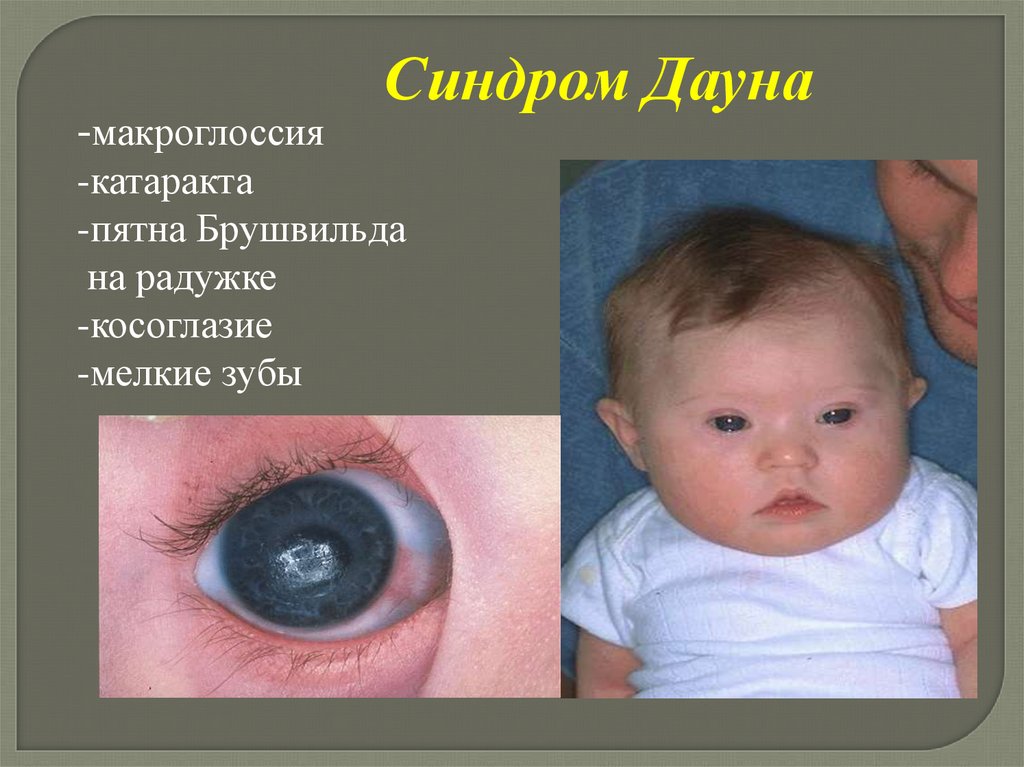
-макроглоссияСиндром Дауна
-катаракта
-пятна Брушвильда
на радужке
-косоглазие
-мелкие зубы
19.
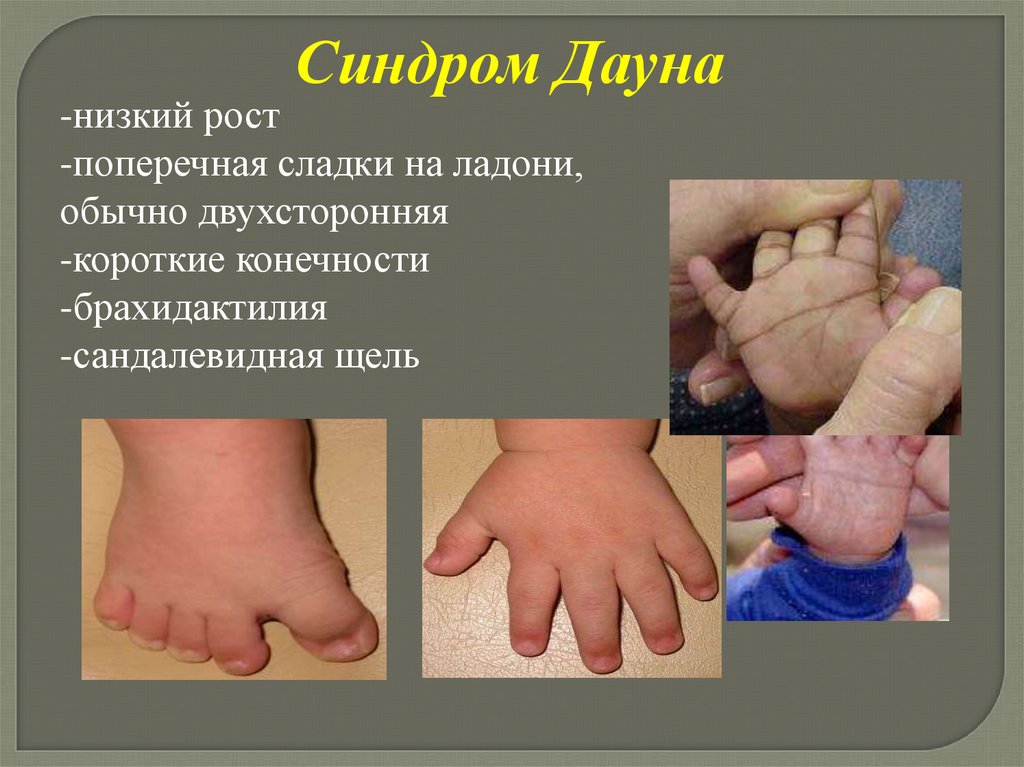
Синдром Дауна-низкий рост
-поперечная сладки на ладони,
обычно двухсторонняя
-короткие конечности
-брахидактилия
-сандалевидная щель
20.
Для Синдрома Даунахарактерно:
-пороки сердечно-сосудистой системы (ДМЖП, тетрада
Фалло, незаращение артериального протока)
-пороки желудочно-кишечного тракта (дуоденальная
обструкция)
-пороки желудочно-кишечного тракта (дуоденальная
обструкция)
-пороки развития почек и мочевыводящих путей
-слабость иммунной системы
21.
22.
Синдром Эдвардса(трисомия 18 хромосомы)
-частота 1:7 000 – 10 000 живорожденных
-среди девочек встречается в 3 раза чаще
Цитогенетические варианты:
- Простая регулярная трисомия (80%)
- Мозаичная форма (10%)
- Другие (10%)
23.
Синдром Эдвардса24.
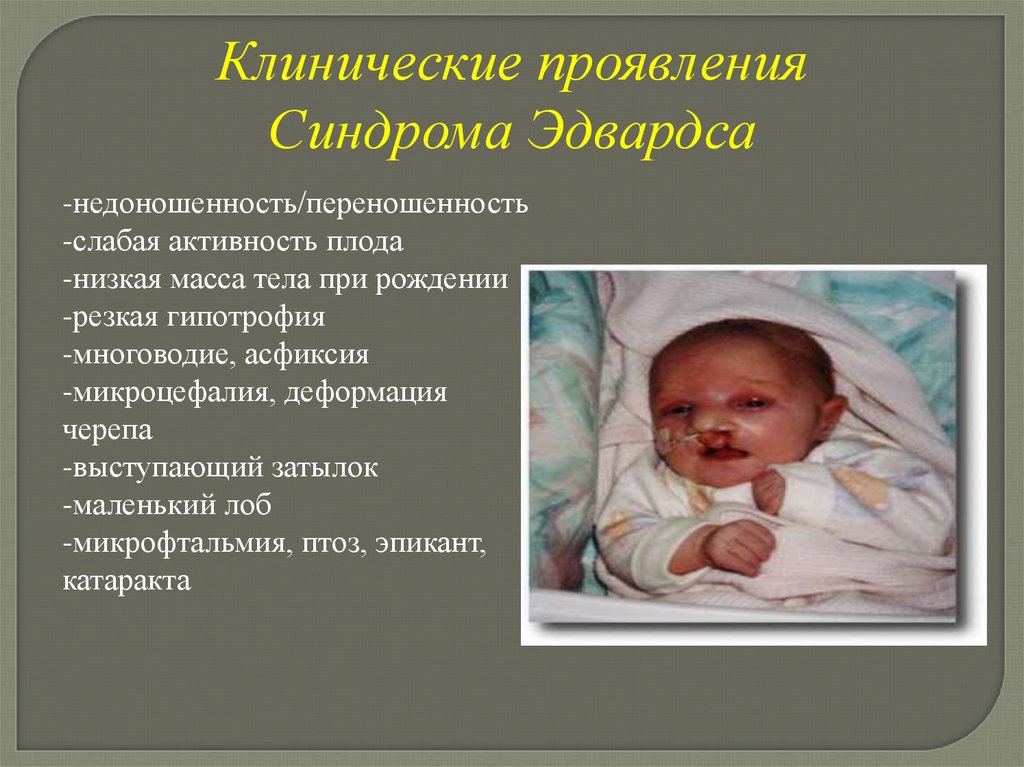
Клинические проявленияСиндрома Эдвардса
-недоношенность/переношенность
-слабая активность плода
-низкая масса тела при рождении
-резкая гипотрофия
-многоводие, асфиксия
-микроцефалия, деформация
черепа
-выступающий затылок
-маленький лоб
-микрофтальмия, птоз, эпикант,
катаракта
25.
Синдром Эдвардса-сгибательная деформация
и перекрест пальцев рук
-стопа – «качалка»
-косолапость
-частичная синдактилия
-гипоплазия ногтей
-микрогнатия
-короткая шея
-седечный горб
-крипторхизм
26.
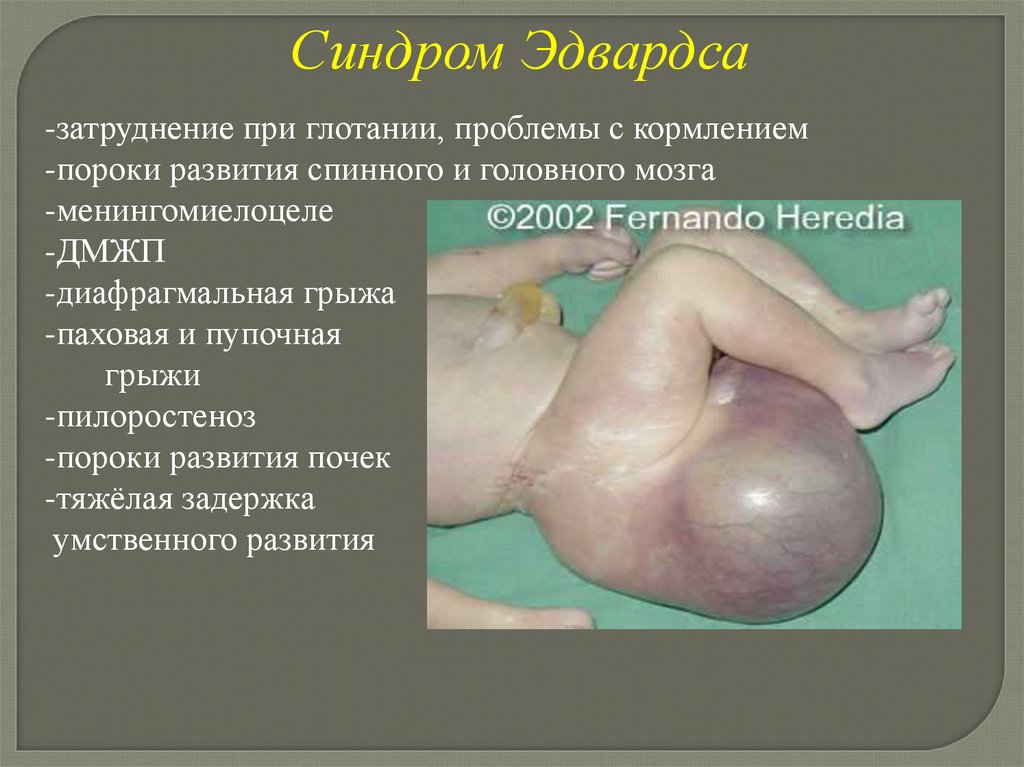
Синдром Эдвардса-затруднение при глотании, проблемы с кормлением
-пороки развития спинного и головного мозга
-менингомиелоцеле
-ДМЖП
-диафрагмальная грыжа
-паховая и пупочная
грыжи
-пилоростеноз
-пороки развития почек
-тяжёлая задержка
умственного развития
27.
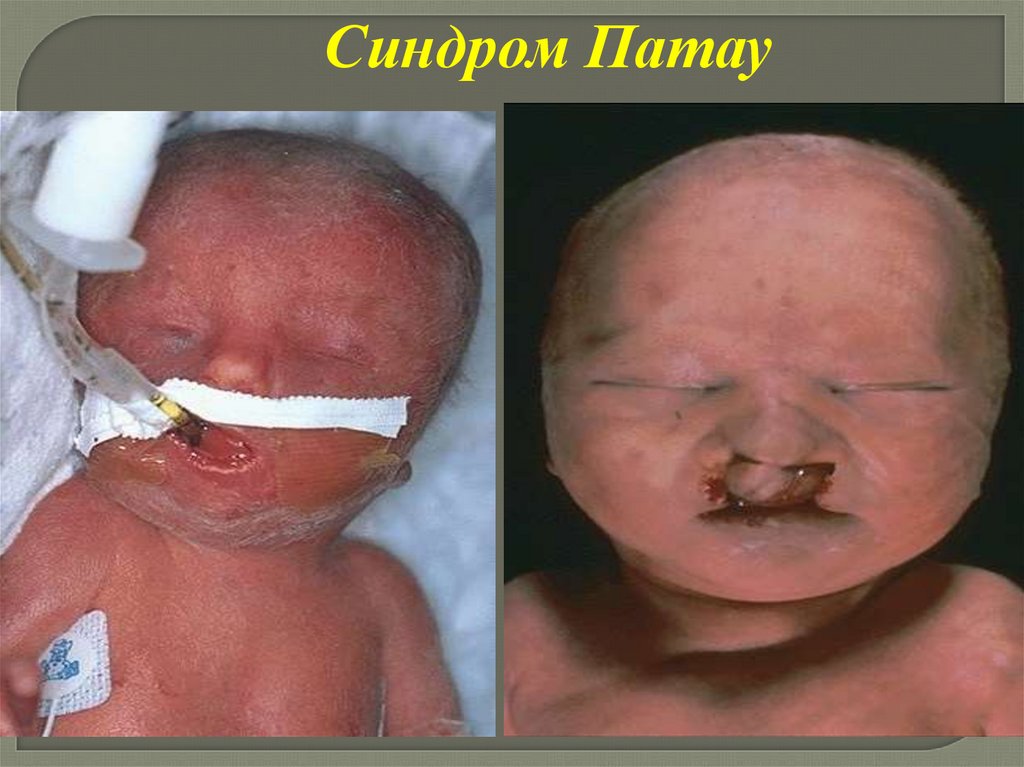
Синдром Патау(трисомия 13 хромосомы)
-частота 1:6000 – 13 000
-соотношение полов 1:1
-риск увеличивается с возрастом матери
Цитогенетические варианты:
-простая регулярная трисомия (80-85%) транслокационные варианты (15-20%)
28.
Синдром Патау29.
Клинические проявлениясиндрома Патау
-пренатальная гипоплазия
-многоводие
-микроцефалия,
-тригоноцефалия
-низко расположенные
деформированные уши
-расщелины губы и неба
30.
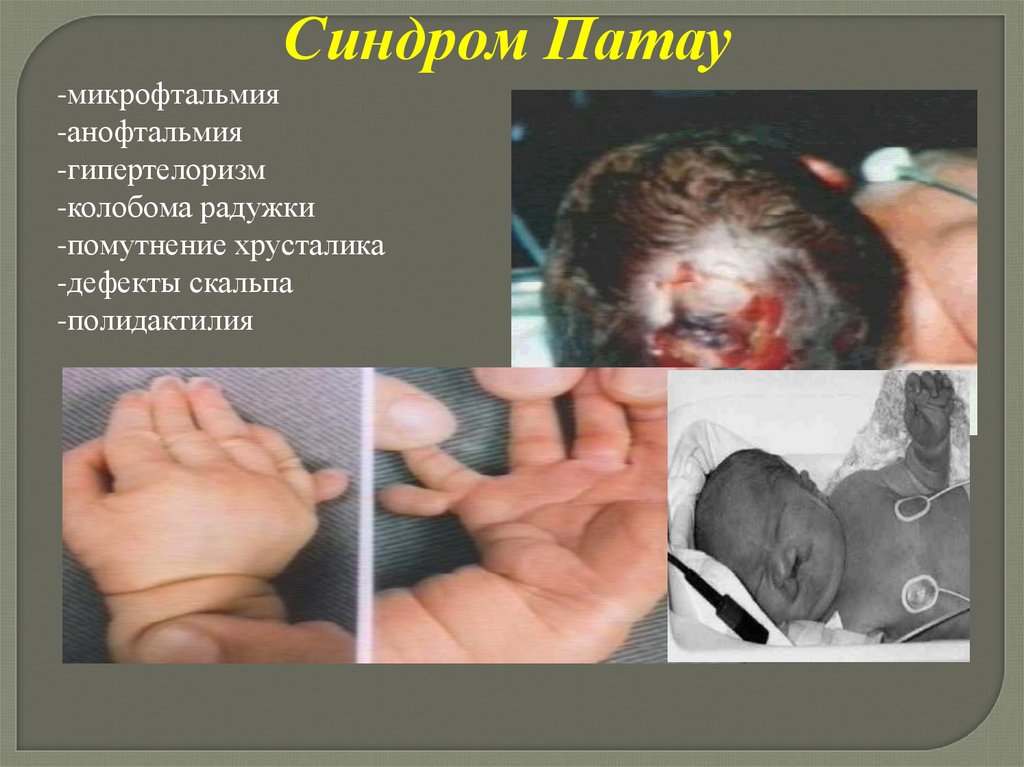
Синдром Патау-микрофтальмия
-анофтальмия
-гипертелоризм
-колобома радужки
-помутнение хрусталика
-дефекты скальпа
-полидактилия
31.
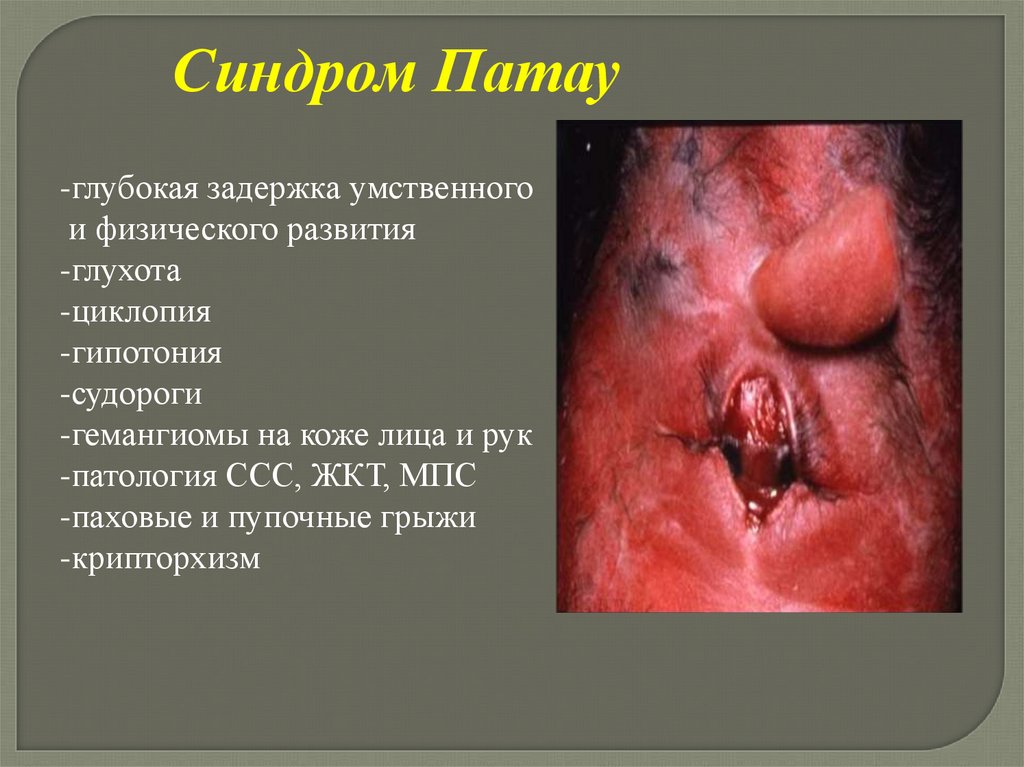
Синдром Патау-глубокая задержка умственного
и физического развития
-глухота
-циклопия
-гипотония
-судороги
-гемангиомы на коже лица и рук
-патология ССС, ЖКТ, МПС
-паховые и пупочные грыжи
-крипторхизм
32.
Синдром Вольфа-ХиршхорнаЦитогенетически обусловлен частичной утратой
короткого плеча 4 хромосомы, критическим районом
является 4р16 (теряется половина короткого плеча)
33.
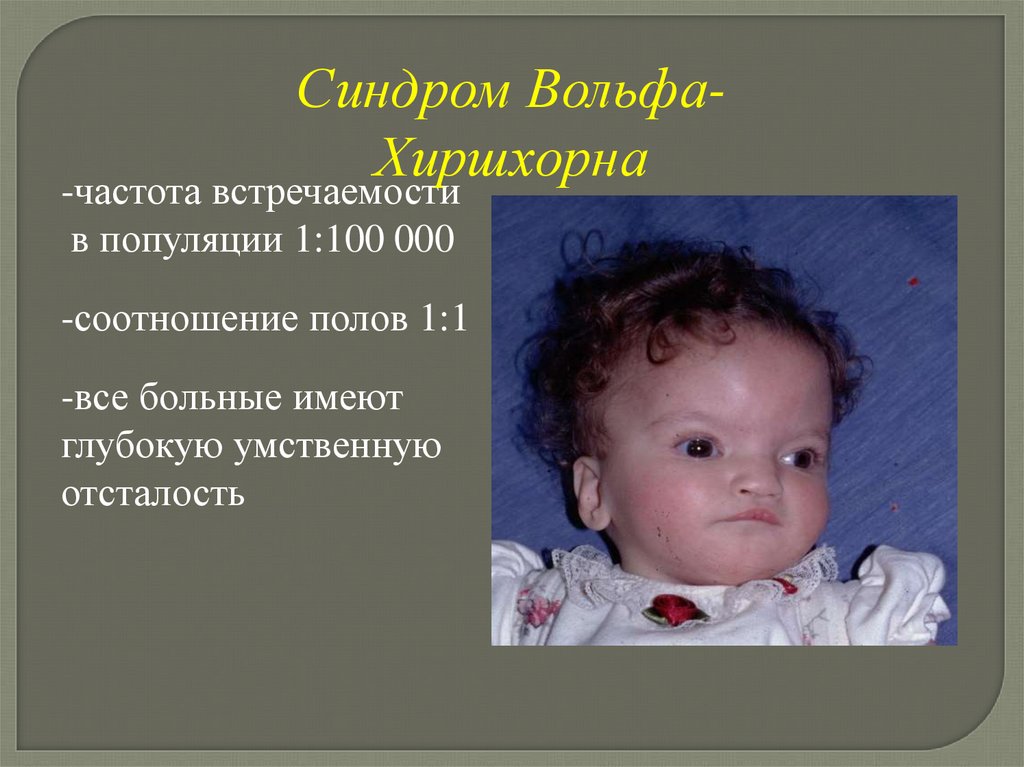
Синдром ВольфаХиршхорна-частота встречаемости
в популяции 1:100 000
-соотношение полов 1:1
-все больные имеют
глубокую умственную
отсталость
34.
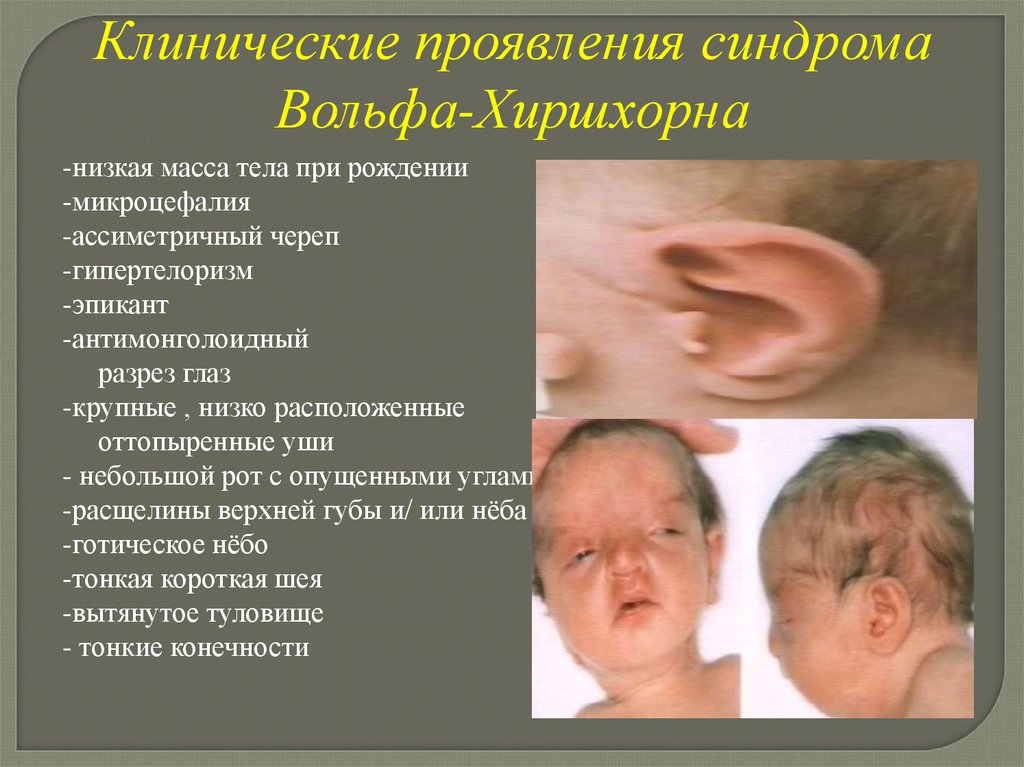
Клинические проявления синдромаВольфа-Хиршхорна
-низкая масса тела при рождении
-микроцефалия
-ассиметричный череп
-гипертелоризм
-эпикант
-антимонголоидный
разрез глаз
-крупные , низко расположенные
оттопыренные уши
- небольшой рот с опущенными углами
-расщелины верхней губы и/ или нёба
-готическое нёбо
-тонкая короткая шея
-вытянутое туловище
- тонкие конечности
35.
Синдром «кошачьего крика»(синдром Лежена)
Цитогенетически обнаруживается укорочение на 1/3
короткого плеча 5-й хромосомы. Участок р15.1-15.2
непосредственно вызывает этот синдром
-частота встречаемости 1:45 000
- возникает спорадически в 85% случаев
- наследуются от фенотипически нормальных родителей –
носителей сбалансированных перестроек в 15 % случаев
36.
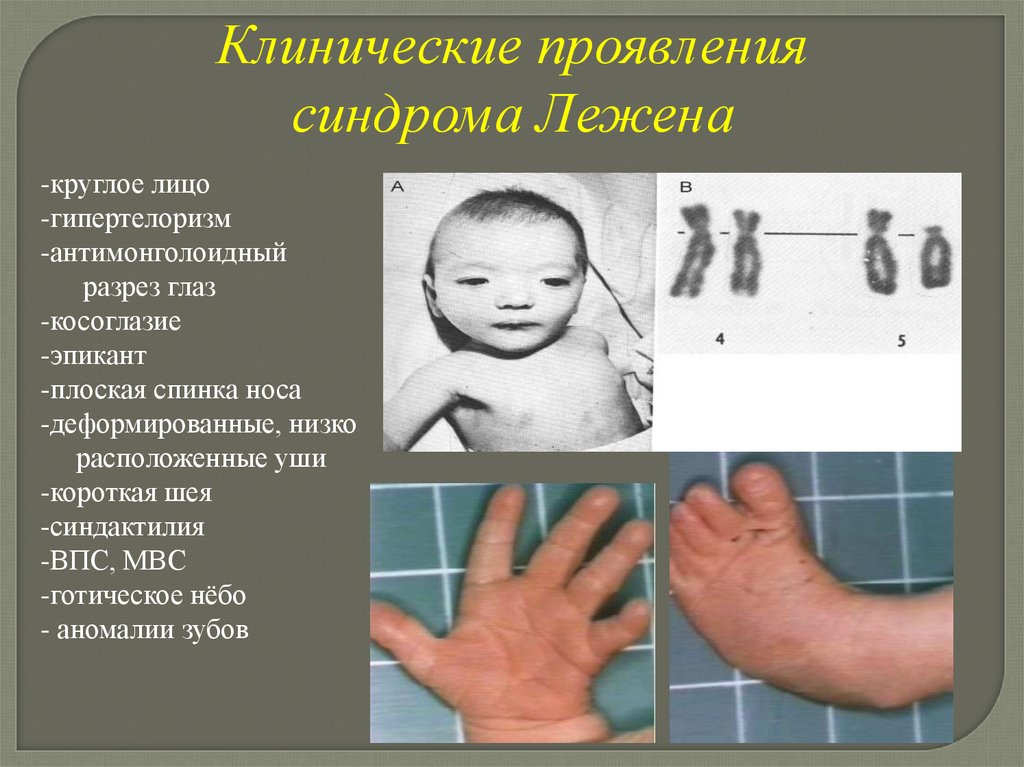
Клинические проявлениясиндрома Лежена
-круглое лицо
-гипертелоризм
-антимонголоидный
разрез глаз
-косоглазие
-эпикант
-плоская спинка носа
-деформированные, низко
расположенные уши
-короткая шея
-синдактилия
-ВПС, МВС
-готическое нёбо
- аномалии зубов
37.
Аномалии половых хромосомПричины: нерасхождение хромосом в мейозе или в
митозе при первых делениях зиготы
Суммарная частота ХА по половым хромосомам от 1,5
до 2,5 на 1 000 новорожденных, большую часть
составляют полисомии ХХХ, ХХУ и ХУУ
38.
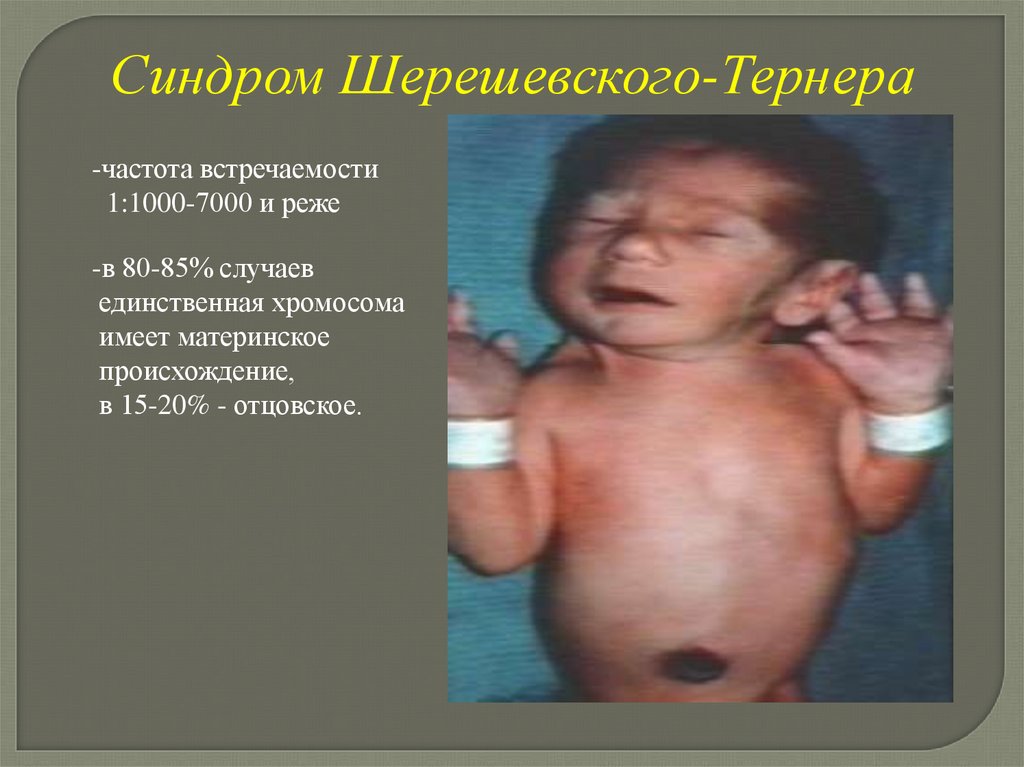
Синдром Шерешевского-Тернера-частота встречаемости
1:1000-7000 и реже
-в 80-85% случаев
единственная хромосома
имеет материнское
происхождение,
в 15-20% - отцовское.
39.
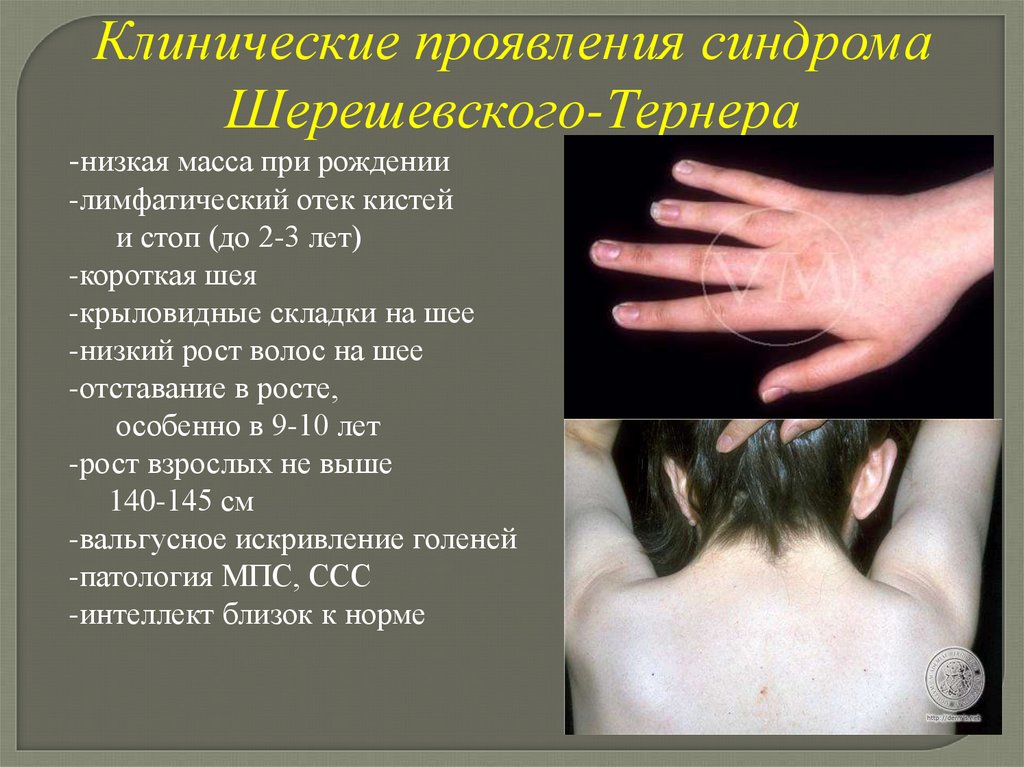
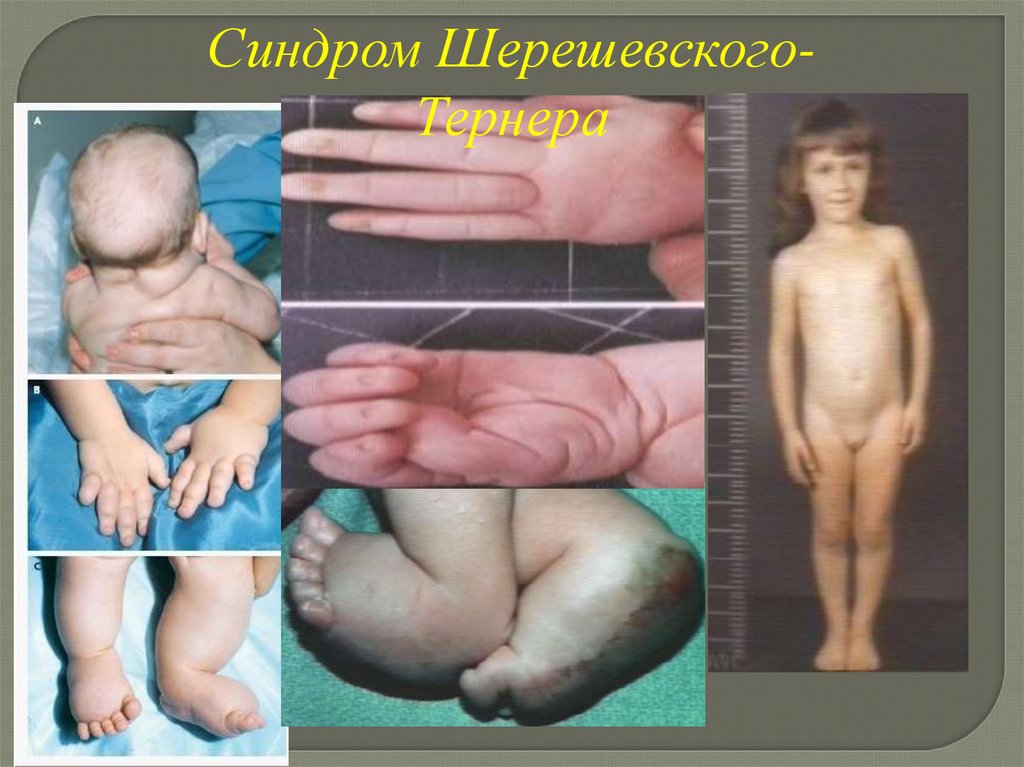
Клинические проявления синдромаШерешевского-Тернера
-низкая масса при рождении
-лимфатический отек кистей
и стоп (до 2-3 лет)
-короткая шея
-крыловидные складки на шее
-низкий рост волос на шее
-отставание в росте,
особенно в 9-10 лет
-рост взрослых не выше
140-145 см
-вальгусное искривление голеней
-патология МПС, ССС
-интеллект близок к норме
40.
Синдром ШерешевскогоТернера41.
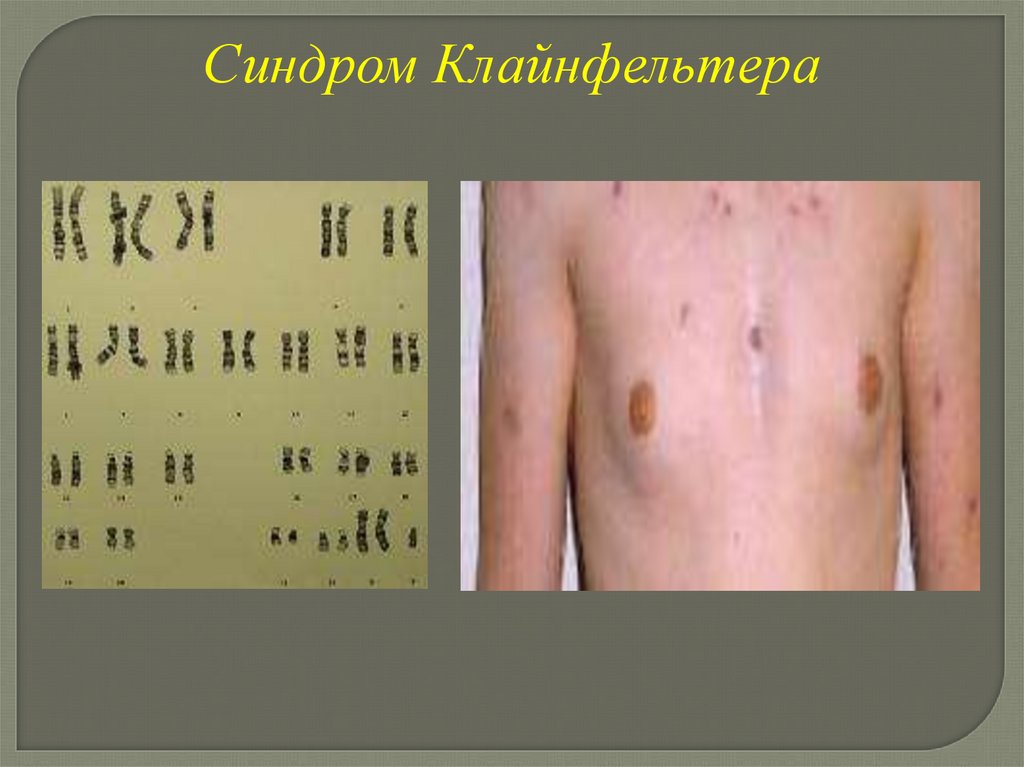
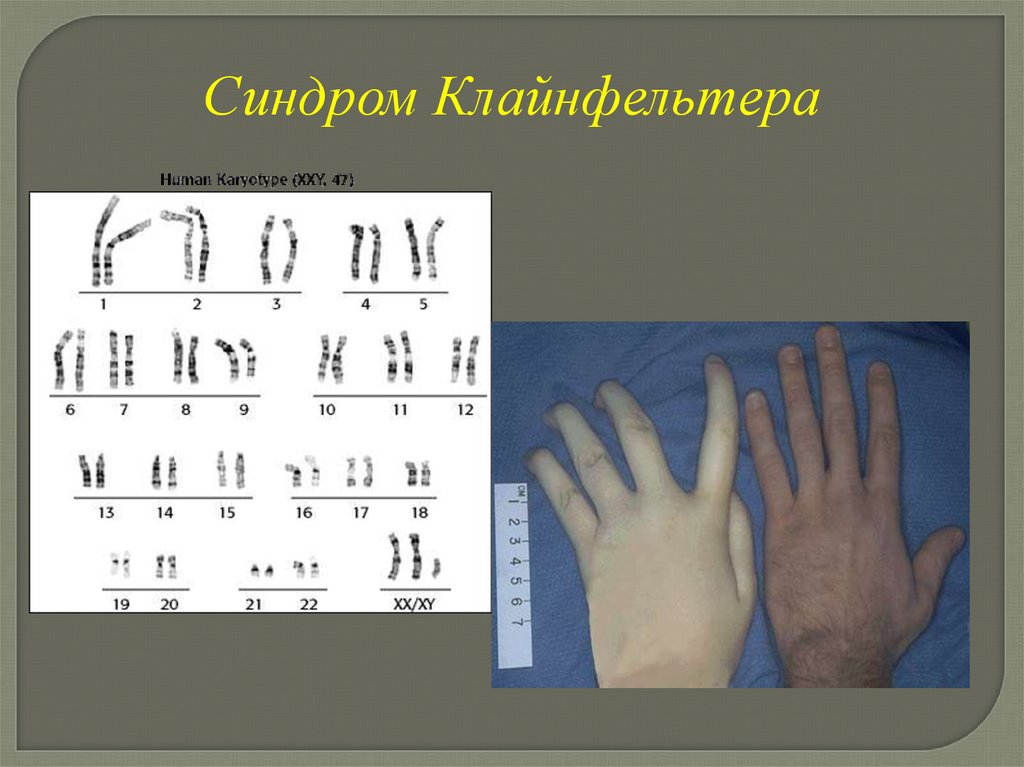
Синдром КлайнфельтераКариотип: 47, ХХУ, 48, ХХХУ, 49, ХХХХУ
Частота в популяции 1,2 на 1 000 новорожденных
мальчиков
42.
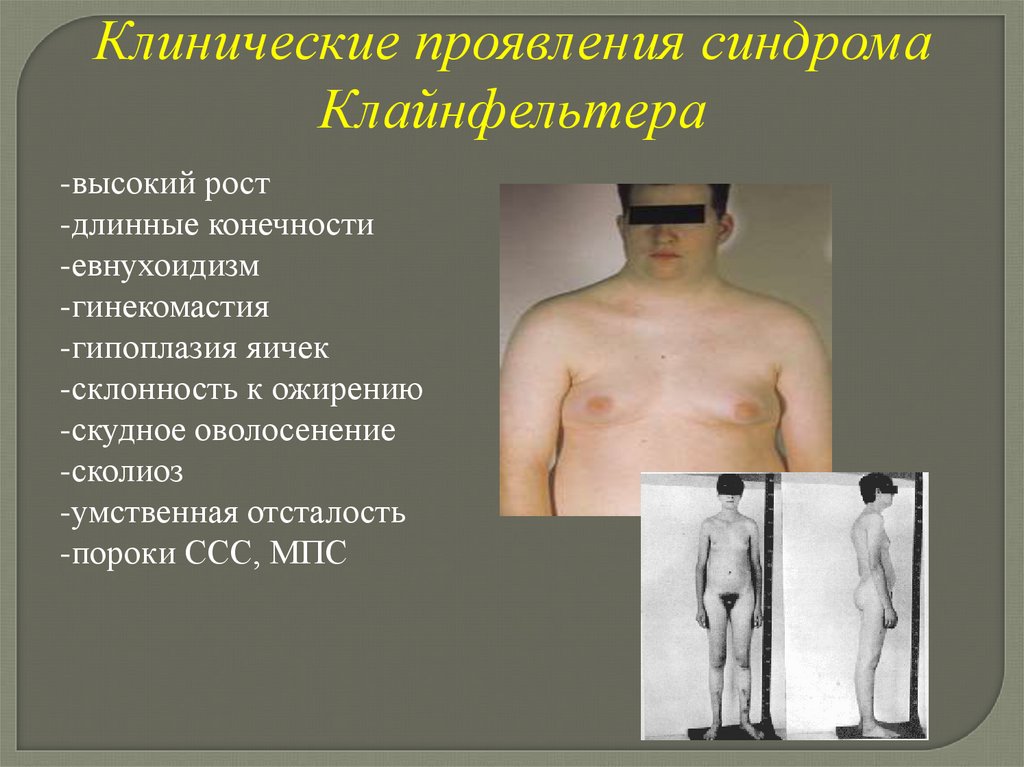
Клинические проявления синдромаКлайнфельтера
-высокий рост
-длинные конечности
-евнухоидизм
-гинекомастия
-гипоплазия яичек
-склонность к ожирению
-скудное оволосенение
-сколиоз
-умственная отсталость
-пороки ССС, МПС
43.
Синдром Клайнфельтера44.
Синдром Клайнфельтера45.
Синдром Дисомии УКариотип 47, ХУУ
Частота 1:1000 новорожденных мальчиков
Клинические проявления
-Рост выше среднего
-умственное развитие
ниже среднего
-агрессивность
-антисоциальное поведение
-бесплодие (50%)
-эндокринный дисбаланс
46.
Болезни с нетрадиционным типомнаследования
Митохондриальные болезни обусловлены генетическими, структурными,
биохимическими дефектами митохондрий, приводящими к
нарушению тканевого дыхания. Передаются в основном по
женской линии к детям обоих полов.
47.
48.
Синдром MELlAS — прогрессирующеенейродегенеративное заболевание, характеризующееся
проявлениями, перечисленными в названии
(«митохондриальная энцефаломиопатия, лактатацидоз,
инсультоподобные эпизоды»), и сопровождается
полиморфной симптоматикой — диабетом, судорогами,
снижением слуха, сердечными заболеваниями, низким
ростом, эндокринопатиями, непереносимостью
физических нагрузок и нейропсихическими
отклонениями.
49.
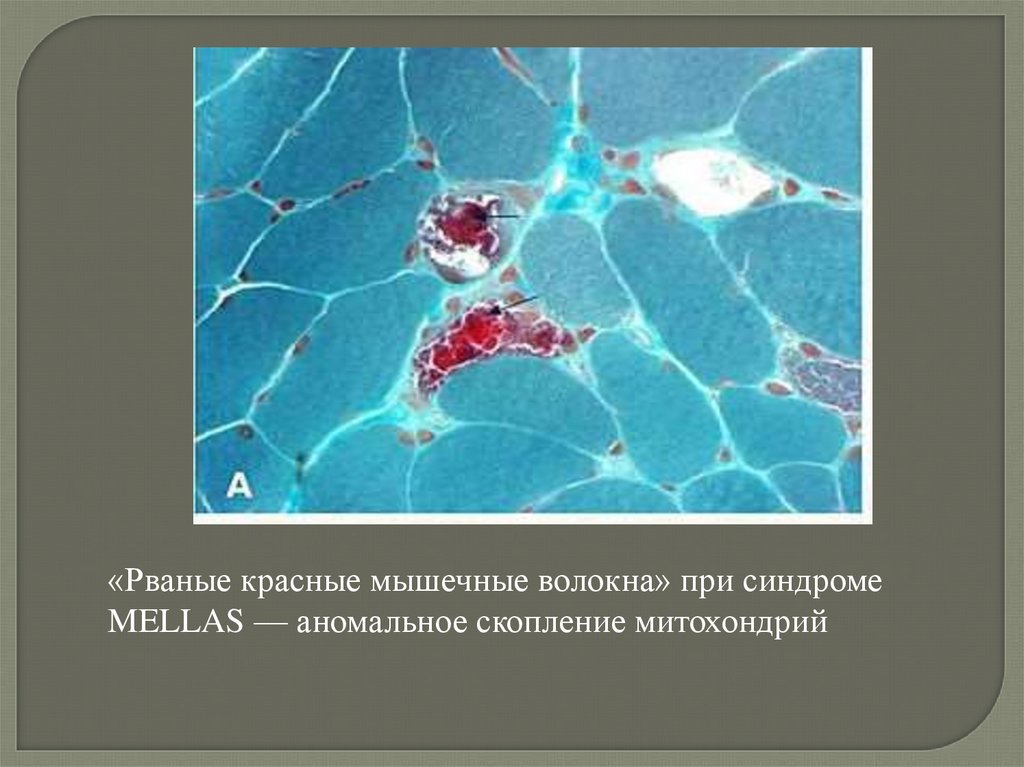
«Рваные красные мышечные волокна» при синдромеMELLAS — аномальное скопление митохондрий
50.
Болезни геномного импринтингаГеномный импринтинг приводит к неодинаковой
экспрессии материнской и отцовской копий гена.
В основе этой группы НБ лежат:
1.Мутации в импритинговом центре
2. Явление однородительской дисомии, т. е. наследование
обеих копий целой хромосомы или ее части (дупликация или
делеция) от одного родителя (при отсутствии
соответствующего генетического материала от другого
родителя). (Синдром Прадера-Вилли, Синдром Ангельмана,
Синдром Рассела-Сильвера)
51.
Синдром Рассела-Сильвера52.
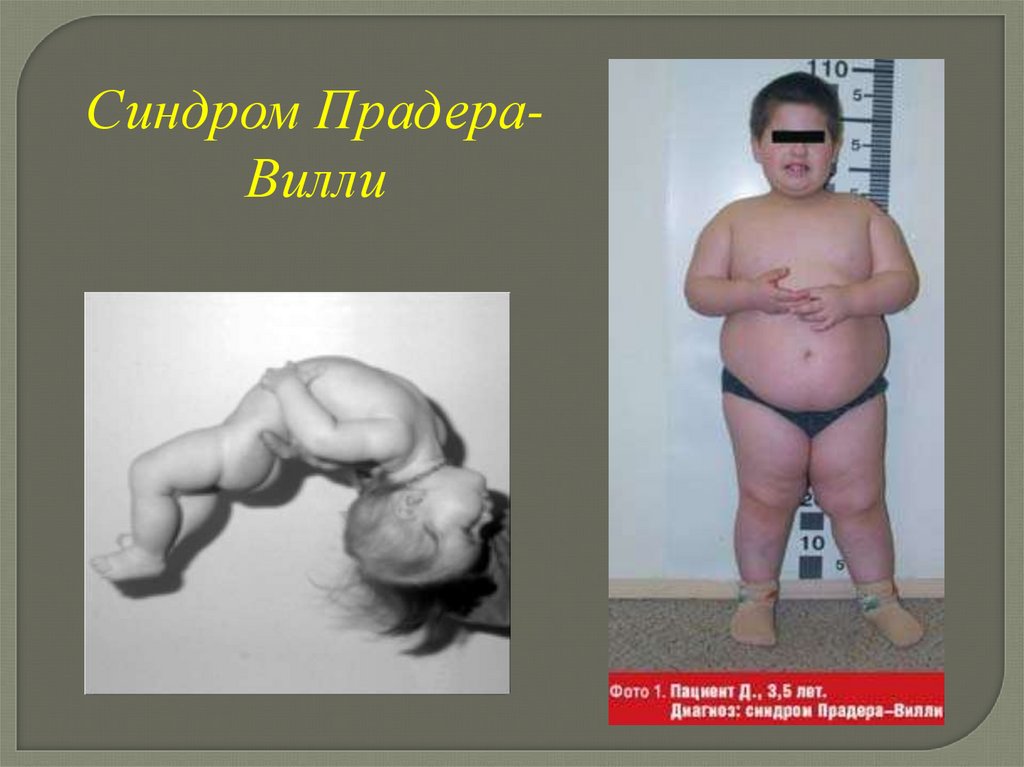
Синдром ПрадераВилли53.
СиндромАнгельмана
54.
Болезни экспансии тринуклеотидных повторовВ основе данных болезней лежат «динамические
мутации» – резкое и стабильное увеличение или
уменьшение числа тандемных повторов (например,
CGG) в транскрибируемой части гена в последующих
поколениях, что проявляется в явлении антиципации,
т.е. утяжеление болезни в каждом последующем
поколении и более раннее начало (синдром ломкой Ххромосомы,
атрофическая
миотония,
хорея
Гентингтона
,
Х-сцепленная
бульбоспинальная
амиотрофия, спиноцеребральные дегенерации)
55.
Синдром Мартин-Белла(ломкой Х-хромосомы)
56.
•Контрольные вопросы:•1.Классификация хромосомных болезней, их
особенности и основные диагностические
признаки.
•2. Клиническая характеристика наиболее
распространенных хромосомных синдромов
•3. Методы диагностики хромосомных синдромов
•4. Принципы лечения хромосомных болезней
57.
Рекомендуемая литература:Основная:
1. Медицинская и клиническая генетика для
стоматологов: учебное пособие для медицинских
вузов /Л.В. Акуленко [и др.]; под ред. О.О.
Янушевича, 2008, М.: ГЭОТАР-Медиа
2. Медицинская и клиническая генетика для
стоматологов: учебное пособие (УМО по мед. и
фармац. образованию вузов России)/Л.В. Акуленко
[и др.]; под ред. О.О. Янушевича, 2015, Москва:
Гэотар-Медиа
58.
ДополнительнаяНаследственные болезни. Национальное руководство: [с прил. на
компакт-диске] / Рос. о-во мед. генетиков, Ассоц. мед. о-в по качеств/ Н.П.
Бочков, Е.К. Гинтер, В.П. Пузырев, 2012, Москва : ГЭОТАР-Медиа
2. Детская неврология: учебник для студентов учреждений высшего
профессионального образования в 2 т./Н.П. Бочков, В.П. Пузырев, С.А.
Смирнихина, 2011, Москва: ГЭОТАР-Медиа
3. Клиническая генетика: учебник [с прил. на компакт-диске] (УМО по
медицинскому и фармацевтическому образованию вузов России) 4-е изд.,
дополненное и переработанное/Н.П. Бочков, В.П. Пузырев, С.А.
Смирнихина, 2011, Москва: ГЭОТАР-Медиа
4. Генетика человека с основами общей генетики: учеб. пособие 2-е изд.,
переработанное и дополненное/ Н.А. Курчанов / 2009, СПб.: СпецЛит.
5. Общая и молекулярная генетика [Электронный ресурс]: учеб.
пособие. Режим доступа: htp://www.knigafund.ru/books/18890/И.Ф.
Жимулев / 2007, Новосибирск: Сиб. университет изд-во.
1.
59.
Базы данных, информационносправочные и поисковые системы1.Электронная библиотека ОмГМА: http://weblib.omsk-
osma.ru/;
2.Электронно-библиотечная система
«КнигаФонд»:http://www.knigafund.ru;
3.Электронная библиотека 1-го МГМУ им. И. М. Сеченова:
http://www.scsml/rssi/ru;
4.Научная электронная библиотека:
http://elibrary.ru/defaultx.asp;
5.Медицинская поисковая система PubMed. Режим доступа:
http://www.ncbi.nlm.nih.gov/pubmed/
6. Центральная научная медицинская библиотека. Режим
доступа: http://www.scsml.rssi.ru
http://www.medterapevt.ru/1019.html
ЭБС «Консультант студента».