





")






")

")









")


")



")


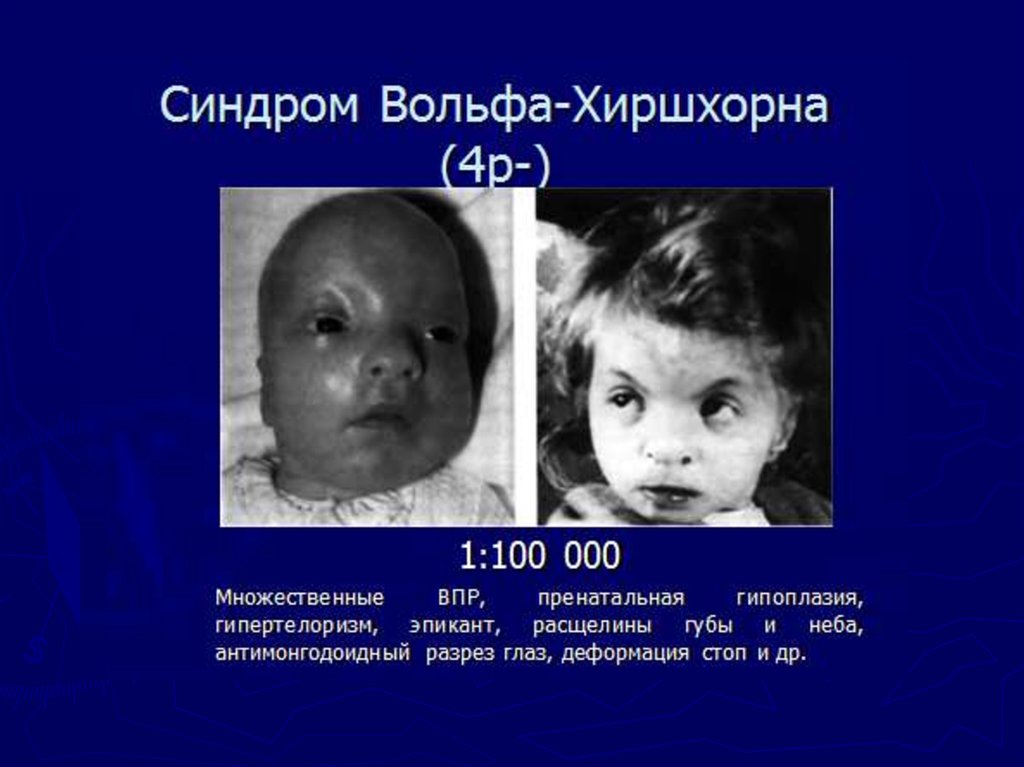










 medicine
medicineSimilar presentations:










Хромосомные болезни
1. ХРОМОСОМНЫЕ БОЛЕЗНИ
2.
► Хромосомныеболезни - это большая группа
врожденных наследственных болезней, в основе
которых лежат хромосомные или геномные
мутации.
► Частота хромосомной патологии составляет 0,61,0% среди новорожденных.
► Самая высокая частота хромосомной патологии
(до 70%) зафиксирована в материале ранних
спонтанных абортусов.
3.
. Все хромосомныеболезни могут быть
разделены на 3
большие группы:
► 1) связанные с
нарушением
плоидности
► 2) обусловленные
нарушением числа
хромосом;
► 3) связанные с
изменением
структуры хромосом
4.
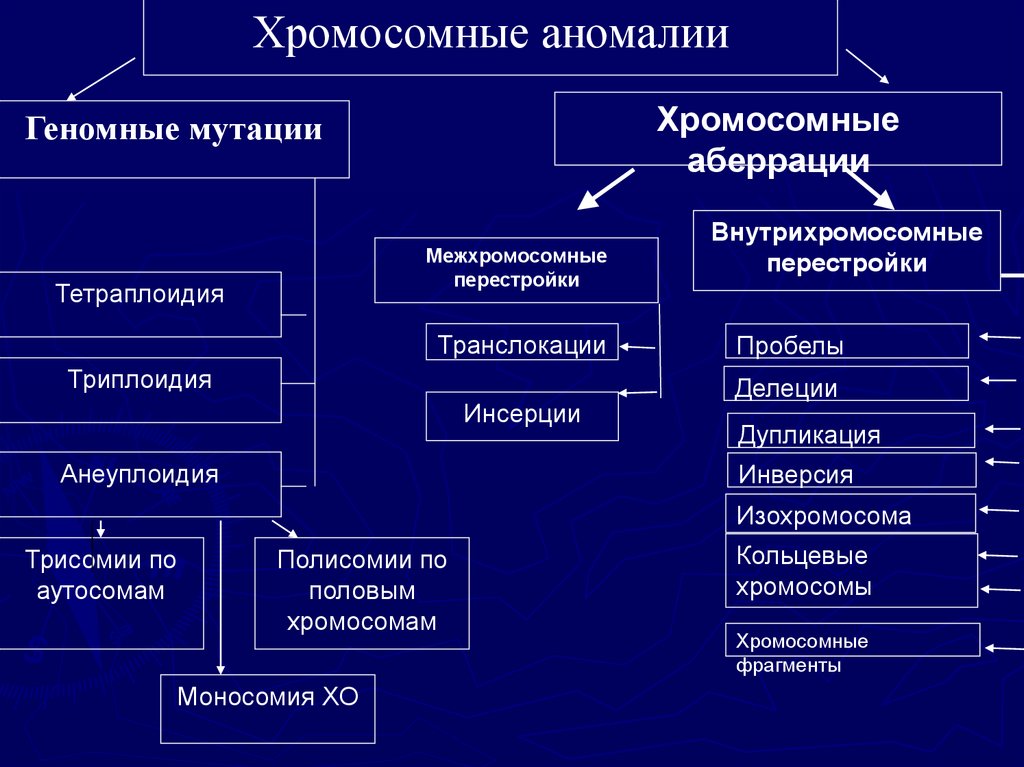
Хромосомные аномалииХромосомные
аберрации
Геномные мутации
Межхромосомные
перестройки
Тетраплоидия
Транслокации
Триплоидия
Инсерции
Анеуплоидия
Внутрихромосомные
перестройки
Пробелы
Делеции
Дупликация
Инверсия
Изохромосома
Трисомии по
аутосомам
Полисомии по
половым
хромосомам
Моносомия ХО
Кольцевые
хромосомы
Хромосомные
фрагменты
5. Влияние соотношения родительских геномов на характер эмбрионального развития человека
кистозная трансформацияворсин хориона;
6. Причины хромосомных мутаций
В основе хромосомных мутаций лежат повреждение
первичной структуры хромосом и последующая их
перестройка в пределах одной (делеции, инверсии) или двух
(транслокации) хромосом, восстанавливающая
непрерывность хромосом.
Анеуплоидии возникают за счет нерасхождения хромосом
или отставания их в анафазе.
Полиплоидии обусловлены либо оплодотворением
яйцеклетки двумя спермиями, либо неразделением
цитоплазмы после редупликации диплоидного
хромосомного набора.
Для нерасхождения хромосом, по-видимому, существует
наследственное предрасположение. Риск повторного
рождения ребенка с хромосомной болезнью в 5-10 раз выше,
чем в общей популяции
7. Эффекты хромосомных аномалий:
1. Летальность (30-40% яйцеклеток гибнет на стадиибластоцисты
из-за
мутаций;
суммарный
вклад
хромосомных аномалий во внутриутробную гибель у
человека – 45%)
2.
Множественные врожденные пороки развития
формируются в раннем эмбриогенезе на основе
нарушенного гисто- и органогенеза. Это создает общность
клинической картины при различных хромосомных
болезнях: задержка физического и психического развития,
черепно-лицевые дисморфии, пороки сердца, мочеполовой
и нервной систем.
3. В соматических клетках – апоптоз или злокачественная
трансформация
8. Синдром Шерешевского-Тернера (45, ХО)
Единственная форма моносомии уживорожденных
Клинические признаки:
► низкий рост,
► Гипогонадизм (первичная аменорея,
бесплодие, стертые вторичные
половые признаки)
► крыловидные кожные складки на шее,
► врожденные пороки сердца и почек,
гипоплазия ногтей, снижение остроты
зрения и слуха, поперечная ладонная
складка, незначительное снижение
умственного развития.
9. Встречаемость клинических симптомов Ш/Т у больных
► Маленькийрост – 100 %
► Врожденная лемфидема – 65%
► Крыловидные складки – 65%
► Низкий рост волос на шее- 75%
► Уплощенная грудная клетка – 55%
► Короткая шея – 50%
► Изменение ногтей на стопах и кистях –
75%
10.
► Популяционнаячастота
1 : 2000-5000 новорожденных девочек
► Цитогенетика:
Наряду с истинной моносомией 45,ХО
встречаются: делеции короткого или
длинного
плеча
Х-хромосомы,
изохромосомы, кольцевые хромосомы, а
также различные варианты мозаицизма.
11.
12. Лечение – комплексное:
► Реконструктивнаяхирургия (впр
внутренних органов)
► Пластическая хирургия (удаление
крыловидных складок)
► Гормональное лечение (эстрогены,
гормон роста)
► Психотерапия
13.
Синдром Клайнфельтера (47, ХХУ)Описан в 1942 г.
Клинические признаки: высокий
рост, хрупкое телосложение,
гипоплазия яичек, импотенция и
бесплодие, набухание молочных
желез, широкий таз, поперечная
ладонная складка, у взрослых
наблюдается ожирение,
незначительное снижение
умственного развития.
Тип наследования: ХХУ синдром
Популяционная частота – 1 : 1000
мальчиков
14.
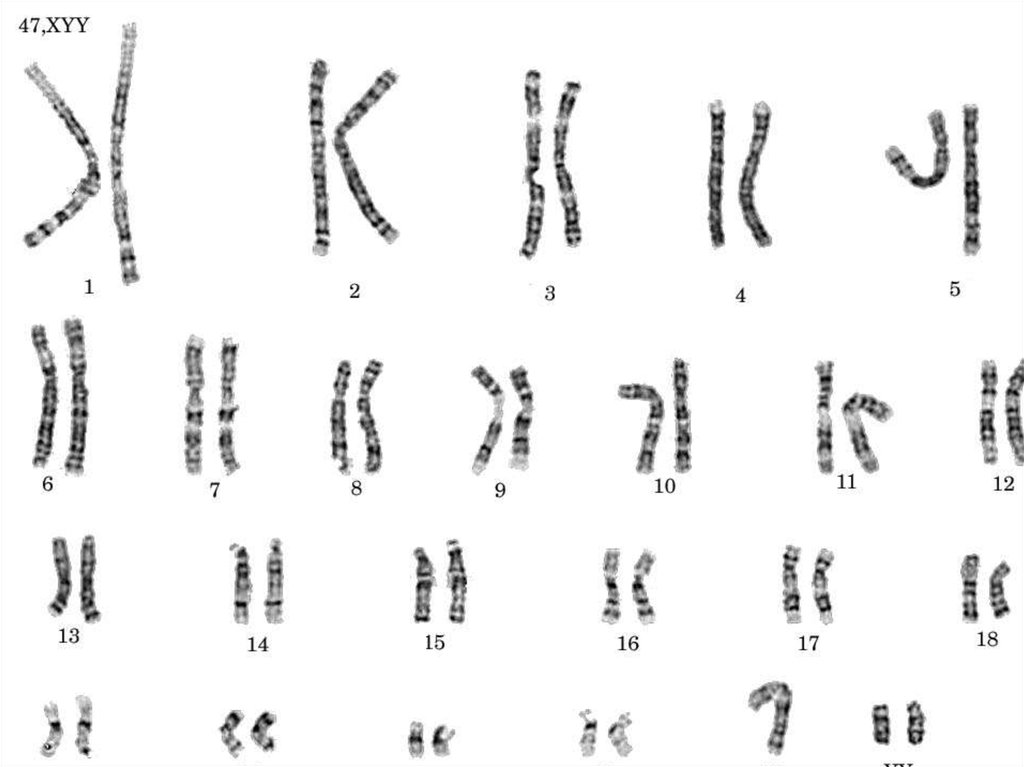
15. Синдром дисомии по У-хромосоме(47, ХУУ)
1:1000 новорожденных мальчиков
Небольшие отличия фенотипа: рост немного выше среднего,
умственно развиты, нет заметных отклонений в половом
развитии, гормональном статусе.
У половины мальчиков – задержка речевого развития,
затруднения при обучении чтению, логопедические проблемы.
Поведенческие особенности: дефицит внимания,
гиперактивность, импульсивность, но без выраженной
агрессии.
16.
17.
18. Синдром Дауна (47 ХУ,+21)
Наиболееизученная
хромосомная болезнь.
Популяционная
частота
синдрома
Дауна
среди
новорождённых равна 1:7001:800, не имеет какой-либо
временной,
этнической
или
географической
разницы
у
родителей
одинакового
возраста.
19.
В литературе описана «пучковость»рождения детей с синдромом Дауна
в определённые промежутки
времени в некоторых странах
(городах, провинциях).
Эти случаи можно объяснить скорее
стохастическими колебаниями
спонтанного уровня нерасхождения
хромосом, чем воздействием
предполагаемых этиологических
факторов (вирусная инфекция,
низкие дозы радиации, хлорофос).
Соотношение мальчиков и
девочек среди новорождённых с
синдромом Дауна составляет 1:1.
20.
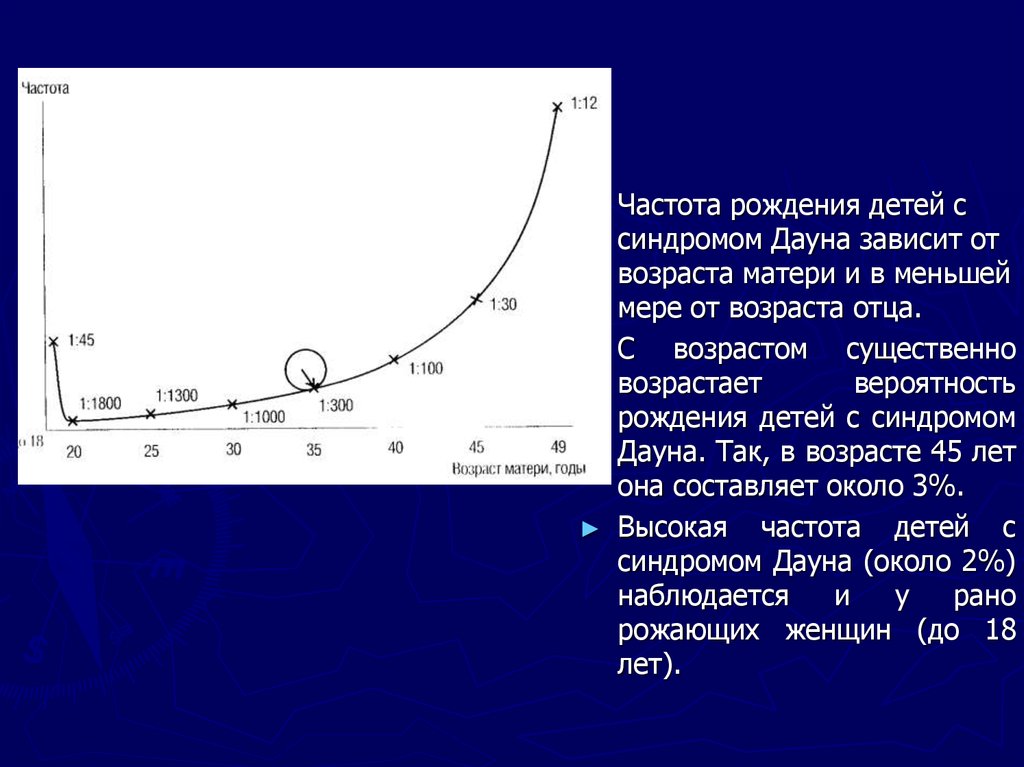
Частота рождения детей с
синдромом Дауна зависит от
возраста матери и в меньшей
мере от возраста отца.
С возрастом существенно
возрастает
вероятность
рождения детей с синдромом
Дауна. Так, в возрасте 45 лет
она составляет около 3%.
Высокая частота детей с
синдромом Дауна (около 2%)
наблюдается
и
у
рано
рожающих женщин (до 18
лет).
21.
Дети с синдромом Дауна рождаются всрок, но с умеренно выраженной
пренатальной гипоплазией (на 8-10%
ниже средних величин). Многие
симптомы синдрома Дауна заметны
при рождении, в последующем они
проявляются более чётко.
► Квалифицированный педиатр ставит
правильный диагноз синдрома Дауна в
родильном доме не менее чем в 90%
случаев.
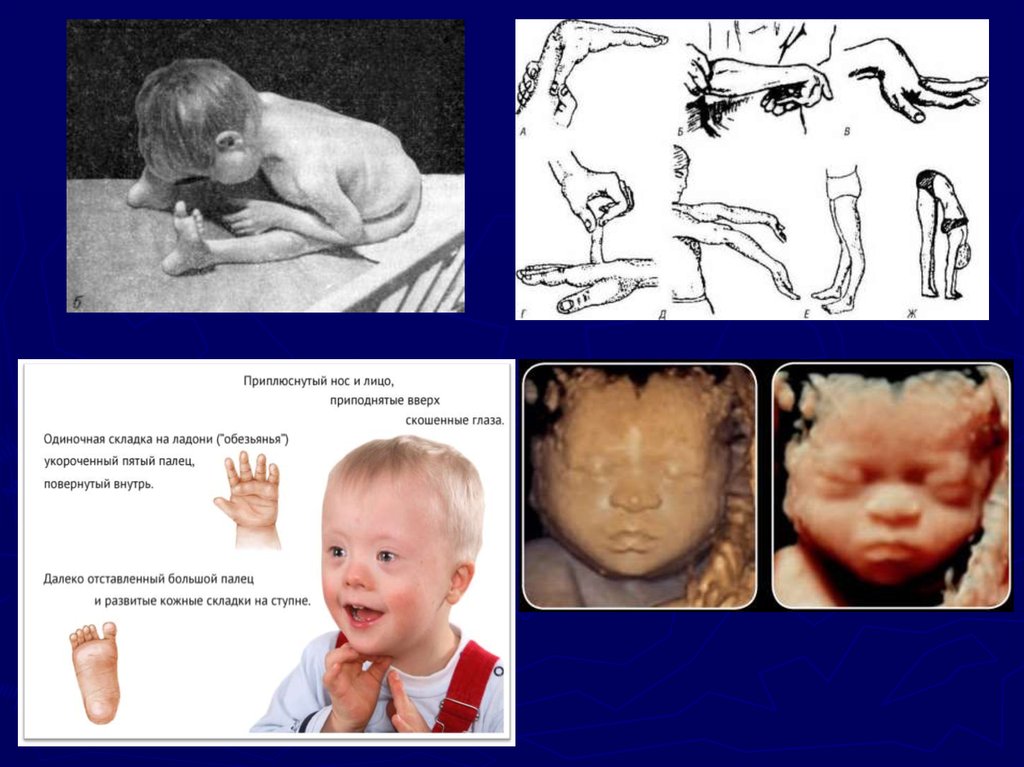
22. Клиническая симптоматика синдрома Дауна
Симптоматика синдрома Дауна
разнообразна: это и врождённые
пороки развития, и нарушения
постнатального развития нервной
системы, и вторичный
иммунодефицит и др.
Из черепно-лицевых аномалий
отмечаются монголоидный разрез
глаз (по этой причине синдром
Дауна долго называли
монголоидизмом), круглое
уплощённое лицо, плоская спинка
носа, эпикант, крупный (обычно
высунутый) язык, брахицефалия,
деформированные ушные
раковины.
23. 10 признаков наиболее важных для установления диагноза
Уплощение профиля лица (90%);► Отсутствие сосательного рефлекса (85%);
► Мышечная гипотония (80%);
► Монголоидный разрез глаз (80%);
► Избыток кожи на шее – тройная шейная складка (80%)
► Разболтанность суставов (80%);
► Диспластичный таз (70%);
► Деформированные ушные раковины (60%);
► Клинодактилия мизинца (60%);
► четырёхпальцевая, или «обезьянья», складка на ладони
(45%)
24.
25.
Большое значение для диагностикиимеет динамика физического и
умственного развития ребёнка. При
синдроме Дауна и то и другое
задерживается.
Рост взрослых больных на 20 см
ниже среднего.
Задержка в умственном развитии
достигает имбецильности, если не
применяются специальные методы
обучения.
Дети с синдромом Дауна ласковые,
внимательные, послушные,
терпеливые при обучении.
Коэффициент умственного развития
(IQ) у разных детей широко
варьирует (от 25 до 75).
26.
Реакция детей с синдромом
Дауна на факторы
окружающей среды часто
патологическая в связи со
слабым клеточным и
гуморальным иммунитетом,
снижением репарации ДНК,
недостаточной выработкой
пищеварительных ферментов,
ограниченными
компенсаторными
возможностями всех систем.
По этой причине дети с
синдромом Дауна часто
болеют пневмониями, тяжело
переносят детские инфекции.
У них отмечается недостаток
массы тела, выражен
авитаминоз.
27. Хромосомы при синдроме Дауна
► Трисомия21 (95%)
(полная форма или
мозаицизм)
Робертсоновская
транслокация (4%) между
21q и 14 или 22 хромосомами
кариотип 45,XX, rob (14;21)
Очень редко (~1%) бывает
изохромосома - 21q21q
28.
29.
30. Синдром Патау (47,ХУ+13)
Описан в 1961 г.Клинические признаки:
микроцефалия, расщепление
губы и неба, полидактилия, узкая
глазная щель, эпикант, пороки
внутренних органов, гипоплазия
наружных половых органов; 95%
умирают до 1 года.
Тип наследования: трисомия
13
Популяционная частота1 : 7500
31.
32.
33. Синдром Эдвардса (47, ХУ+18)
Частота 1:5000-7000► Соотношение мальчиков и девочек
1:3
Выраженная задержка пренатального
развития при родах в срок.
Множественные пороки развития
лицевой части черепа, сердца, костной
системы, половых органов.
34. Синдром Эдвардса
► Флексорноеположение пальцев
Стопа-качалка (пятка выступает,
свод провисает)
35.
Поражение системы и порок (признак) - Частота %
Мозговой череп и лицо - 100%
микрогения - 96,8%
деформация ушных раковин - 95,6%
долихоцефалия - 89,8%
высокое небо - 78%
расщепленное небо - 15%
микростомия - 71%
Опорно-двигательный аппарат - 98,1%
флексорное положение кистей - 91,4%
стопа-качалка - 76,2%
кожная синдактилия стоп - 49,5%
косолапость - 34,9%
Центральная нервная система - 20,4%
Сердечно-сосудистая система - 90,4%
Органы пищеварения - 54,9%
Мочеполовая система - 33,5%
36.
37. Синдром «кошачьего крика» (моносомия 5р)
Описан в 1963 г.Клинические признаки: необычный
плач, напоминающий кошачье
мяуканье, микроцефалия,
антимонголоидный разрез глаз,
умственная отсталость, лунопообразное
лицо, эпикант, гипертелоризм,
аномалии внутренних органов.
Умирают чаще до 10 летнего возраста.
Тип наследования: моносомия 5 р
Популяционная частота – 1 : 45 000
38.
► Большинствослучаев
синдрома
«кошачьего крика» спорадические, 1015% пациентов – потомки носителей
транслокации.
Точки
разрыва
и
протяженность
утраченного сегмента
хромосомы 5р- различны, но критической
областью является участок 5р15. При
использовании FISH и CGH- матричного
анализа хромосом обнаружено множество
генов, утрачиваемых при del5p15.3.
39.
40.
41.
42.
Синдром Ди Джорджи — это первичный (врожденный, возникшийвнутриутробно или передавшийся от родителей к детям)
иммунодефицит (снижение иммунитета), для которого характерны
аплазия (отсутствие) или гипоплазия (уменьшение размеров)
тимуса (вилочковой железы) и паращитовидных желез,
врожденные пороки сердца, лицевые мальформации (изменения).
Также заболевание может сопровождаться другими аномалиями
(нарушениями) развития (аномалиями скелета, почек, нервной
системы, патологией (болезнью) глаз).
43.
► Полагают,что делеция
22q11.2 играет важную
роль в развитии пороков
сердца
Тетрада Фалло:
1 — сужение устья легочной артерии;
► 2 — дефект в мембранозной части
межжелудочковой перегородки;
► 3 — «аорта-всадник»;
► 4 — гипертрофия правого желудочка.
Клинически тетрада Фалло проявляется ранним
цианозом, задержкой развития, одышкой и
одышечно-цианотическими
приступами,
головокружением и обмороками.
44. Велокардиофациальный синдром
врожденная генетическая аномалия, характеризующаяся
целым рядом различных симптомов: физических и
поведенческих расстройств, а также задержкой развития.
Латинские корни «вело», «кардио» и «фацио» относятся к
трем основным органам, в которых наблюдаются
нарушения развития: нёбо, сердце и лицо. Тяжелого
поражения иммунитета –нет.
45.
Нарушение хромосомного баланса неизбежноприводит к нарушению развития организма.
Общие закономерности:
1) трисомии и моносомии по целым хромосомам переносятся
тяжелее, чем частичные;
2) мозаичные формы хромосомных болезней протекают легче, чем
полные (гаметного происхождения);
3) у живорожденных дисбаланс по крупным хромосомам
встречается значительно реже, чем по мелким;
4) недостаток хромосомного материала вызывает более серьезные
нарушения развития, чем его избыток (моносомии по
аутосомам не обнаружено);
5) полные трисомии по аутосомам наблюдаются только по
богатым гетерохроматином хромосомам;
6) аномалии по половым хромосомам ведут к меньшим
нарушениям развития, чем по аутосомам.
46.
Факторы риска рождения детей схромосомными болезнями
Биологические – возраст матери старше 35 лет;
кровное родство родителей, отягощенный по ВПР
анамнез (наличие в семье или у родственников
детей с ВПР)
Экзогенные –
(мутагенами)
контакт
с
генотоксикантами
–
Цитогенеческие – наличие у родителей следующих
варинтованеуплоидия,
робертсоновская
транслокация,
сбалансированные
реципрокные
транслокации, кольцевые хромосомы, инверсии
47. Профилактика
Первичная1.Планирование
Вторичная
Третичная
Прерывание беременности Коррекция проявления
при высокой
патологичных
оптимальный
вероятности
генотипов репродуктивный возраст
заболевания плода или
нормокопирование
21-35 лет;
пренатально
диагностированной
отказ от деторождения в
болезни.
случаях высокого риска
рождения детей с
Это не самое лучшее
врожденной патологией;
решение, но пока
в браках с кровными
единственный
родственниками и между
практический метод
двумя гетерозиготными
при большинстве
носителями
тяжелых и
патологического гена
смертельных
генетических
2. Улучшение среды
дефектах.
обитания человека
деторождения:
48. Терминологический словарь
Акроцефалия - высокий
«башенный» череп.
Алопеция – стойкое или
временное выпадение волос.
Аменорея – отсутствие
менструального цикла.
Аплазия – полное отсутствие
органа или части его.
Атрезия – отсутствие канала или
естеств. отверстий.
Арахнодактилия – необычно
длинные и тонкие пальцы.
Брахидактилия – укорочение
пальцев.
Витилиго – очаговая
депигментация кожи.
Гипертелоризм –широко
расставленные глаза.
Гипертрихоз – избыточный
рост волос.
Гипоплазия – недоразвитие
органа.
Гипогонадизм –
недоразвитие половых желез.
Крипторхизм –отсутствие
одного или обоих яичек.
Макроцефалия – чрезмерно
большая голова.
Микрогения –малые
размеры нижней челюсти.
Микроцефалия – малые
размеры головного мозга.
Полидактилия – увеличение
количества пальцев.
Прогения –чрезмерное
развитие нижней челюсти.
49. Терминологический словарь
Прогерия – преждевременноестарение организма.
Птеригиум – крыловидные складки
кожи.
Птоз – опущение внутренних органов
или века.
Синдактилия – сращение соседних
пальцев.
Страбизм – косоглазие.
Телекант – латеральное смещение
внутренних углов глаз.
Тремор - дрожание конечностей,
головы и даже всего тела.
Энофтальм - глубокопосаженные глаза
Экзофтальм – смещение глазного
яблока вперед,
сопровождающееся
расширением глазной щели.
Эпикант – вертикальная кожная
складка у внутреннего угла
глаза.
Анорексия - уменьшения
аппетита.
Гематома - полость, заполненная
кровью.
Гематурия - кровь в моче.
Нистагм - непроизвольные
ритмичные судорожные
движения глазных яблок.
50.
ЛИТЕРАТУРАБочков
Н.П. Клиническая генетика. –
М.: ГЭОТАР-МЕД, 2006
Наследственные
синдромы и медикогенетическое консультирование:
Справочник /Козлов С.И. и др. – Л.:
Медицина, 1987
Рязанова
Л.А., Алферова И.П. Учителю
о медико-генетическом
консультировании. – Челябинск: Изд-во
ЧГПИ «Факел», 1995
Шевченко
В.А. Генетика человека. – М.:
ВЛАДОС, 2002