





















 medicine
medicineSimilar presentations:










Наследственные болезни обмена
1. Наследственные болезни обмена
2. Наследственные болезни обмена
– группа моногенных наследственныхзаболеваний, обусловленных мутациями
генов, кодирующих ферменты, транспортные
или сигнальные белки, что приводит к
нарушению метаболизма клетки.
• Классификация
- тип поврежденного метаболического пути
- локализация в клетке
3. МАР
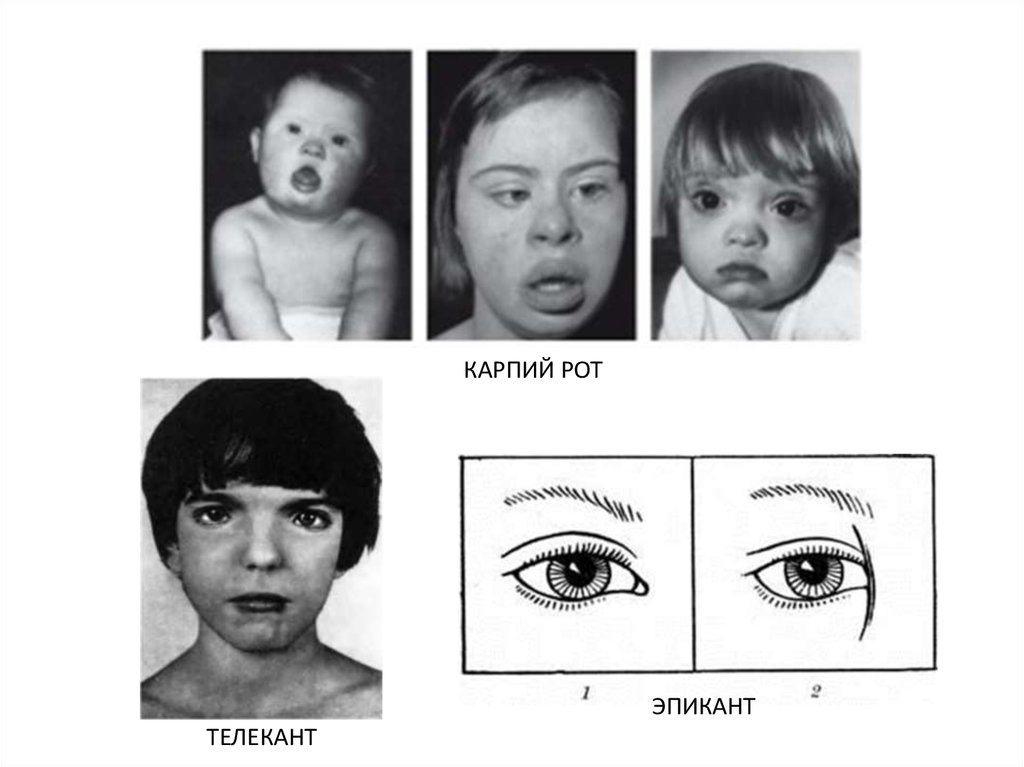
АНТИМОНГОЛОИДНЫЙРАЗРЕЗ ГЛАЗ
4.
КАРПИЙ РОТЭПИКАНТ
ТЕЛЕКАНТ
5. Характерные симптомы
• 1. Наличие светлого промежутка• 2. Симптомокомплекс «вялого ребенка»
• 3. Мышечная гипотония, сменяющаяся
дистонией
• 4. Неонатальные судороги
• 5. Трудности вскармливания
• 6. Нарушения дыхания
• 7. Увеличение печени и селезенки
• 8. Анорексия, рвота, плохая прибавка в весе
6. Фенилкетонурия
• Накопление Фенилаланина и его токсическоевоздействие
• Мышиный запах мочи
• Дети светловолосые, голубоглазые
• Светочувствительность (дерматозы)
7. Лейциноз
• Снижение активности энзимной системы,обеспечивающей декарбоксилирование трех
аминокислот
• Моча с запахом кленового сиропа
8. Вильсона-Коновалова
• Нарушение структуры медьтранспортирующейАТФазы печени
• Токсическое влияние меди на ГМ, почки, печень,
роговицу.
• Патогенетическое лечение – D-пеницилламин +
витамин В6
9. Гемохроматоз
• Гепатомегалиямеланодермия
сахарный
диабет
миокардиопатия
гипогонадизм у
взрослых
артропатии
синдром
мальабсорбции.
Есть патогенетическое
лечение – хелатная
терапия
10. Х-сцепленная адренолейкодистрофия
• Нарушение транспорта ОДЦЖК впероксисомы, нарушение расщепления
ОДЦЖК
• Токсическое воздействие на миелин и
клетки коры надпочечников
Преимущественно односторонняя
демиелинизация теменнозатылочных областей мозга
11. Лизосомные болезни обмена
• Болезнь Нимана-Пика• Мукополисахаридоз
• Болезнь Гоше
12. Болезнь Нимана-Пика
• Накопление сфингомиелина в лизосомахГМ, печени, РЭС.
• Пенистые клетки
• Симптом «вишневой
косточки»
Тип А
Тип В
Тип С
Проявление сразу после
рождения
Гепатоспленомегалия
Помутнение роговицы
Утрата моторных навыков
и сухожильных рефлексов
Без вовлечения нервной
системы
+ накопление холестерина
Патогенетическое лечение:
Миглустатин
13. Тип С Ювенильная форма
+ Филиппиновый тест14. Мукополисахаридоз
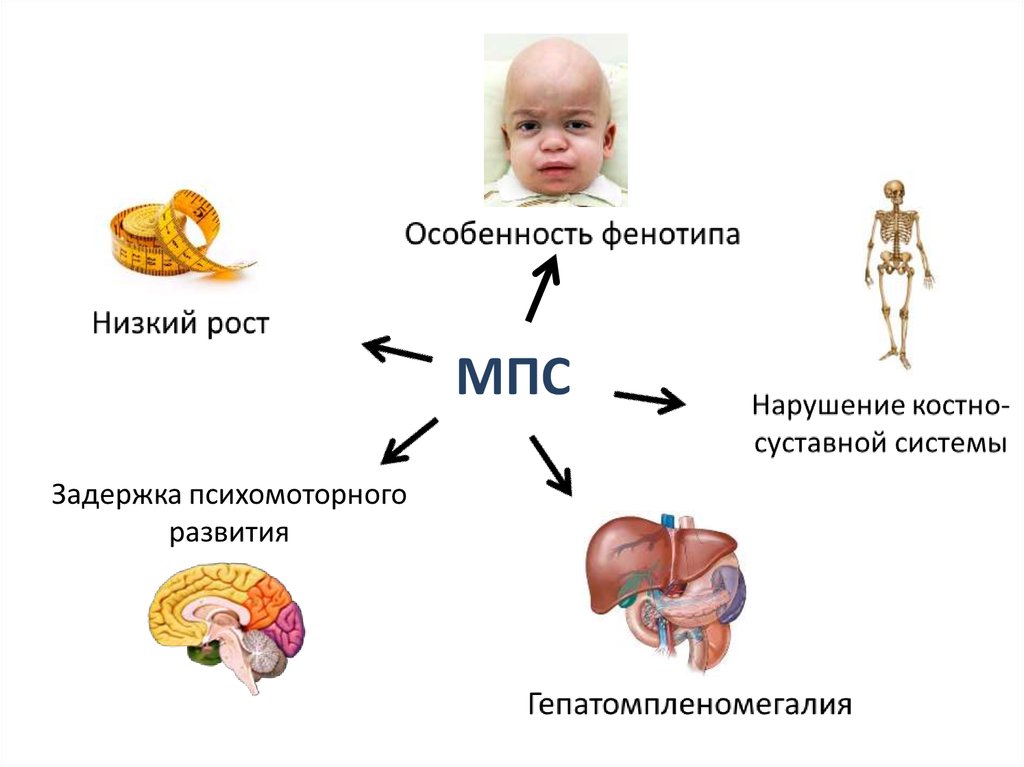
Накопление мукополисахаридов в результате дефектаразличных ферментов
МПС 1
Гурлера
Шейе
Гурлер-Шейе
Дефицит альфа-L-идуронидазы
МПС 6
МПС 2
МПС 4
Хантера
Болезнь
Моркио
Идуронат-2сульфатазы
МПС 3
Санфилиппо
А - N сульфоглюкозаминсульфогидралазы
Б - N-ацетил-Dглюкозаминидазы
С - альфа глюкозамид-Nацетилтрасферазы
D - N-ацетилглюкозамин
-6-сульфатазы
Галактазамин-6сульфатсульфатазы или
b-галактозидазы
Марото-Лами
15.
МПС16. МПС 1 типа
СиндромГурлера-Шейе
Синдром
Гурлера
Синдром
Шейе
17. МПС 2 Синдром Хантера
Светлы жесткие волосыУзелково-папулезное поражение кожи
К 8 годам – умственная отсталость
Расторможенность поведения
Прозрачная роговица
18. МПС III Синдром Санфилиппо
К 3 годам жизни развитиепрекращается
Физическое развитие
соответствует возрасту
Расстройства поведения
19. МПС IV Болезнь Моркио
Отсутствие снижения интеллектаДеформация скелета
Глухота
20. МПС VI Синдром Марото-Лами
Отсутствие сниженияинтеллекта
Тугоподвижность
суставов
Рестриктивные
заболевания легких
Дисплазия головки
бедренной кости
21. Болезнь Гоше
• Накоплениеглюкоцереброзида
• Гепатоспленомегалия
1 тип
ненейропатический
Геморрагический
синдром
2 тип
Острый
нейропатический
Гиперрефлексия
Мышечная гипотония
Регресс психомоторного
развития
клонико-тонические
судороги
3 тип
Подострый
нейропатический
Генерализованные
клонико-тонические
судороги
Панцитопения
Для 1 и 3 типа – заместительная терапия Церезин*
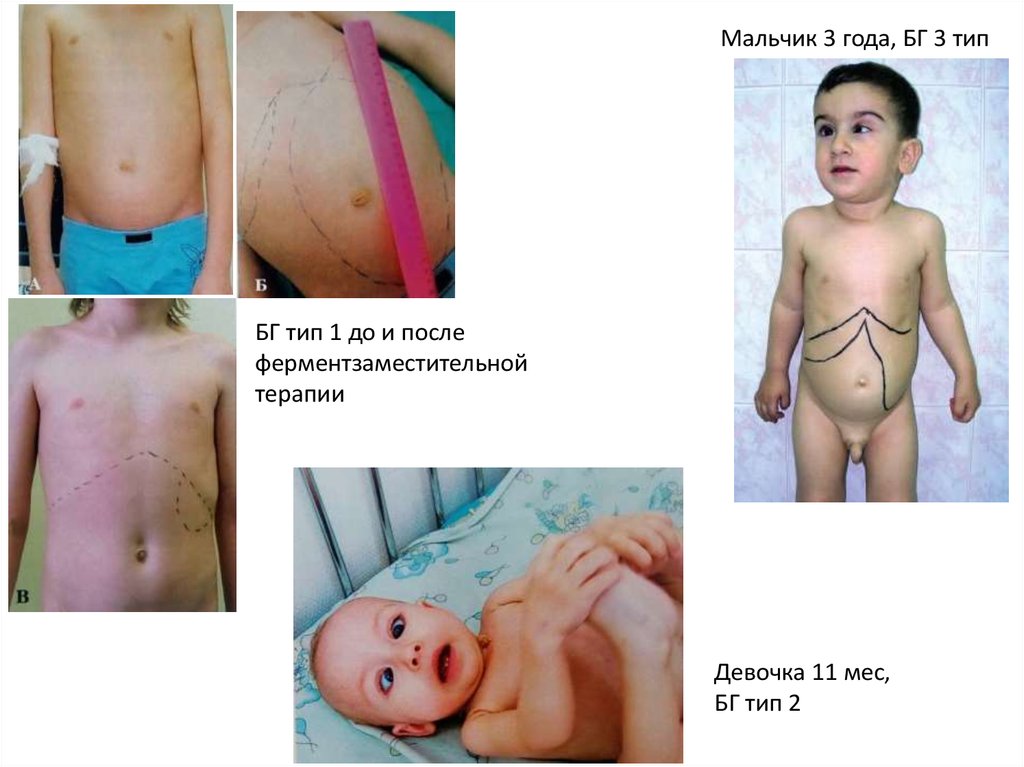
22.
Мальчик 3 года, БГ 3 типБГ тип 1 до и после
ферментзаместительной
терапии
Девочка 11 мес,
БГ тип 2
23. Митохондриальные заболевания
• Феномен гетероплазии – присутствиемутантных и нормальных мтДНК в одной
клектке
• Клиническая картина зависит от %
мутантных мтДНК
24. Синдром Кернса-Сейра
Наружная офтальмоплегия
Миопатия
Атаксия
Нейросенсорная глухота
Феномен «рваных красных волокон» в
мышечных биоптатах
25. Синдром Пирсона
Поражение костного мозга - ПАНЦИТОПЕНИЯСиндром Кернса-Сейра
Синдром Пирсона
Мутантная мтДНК
Мышцы 80%
Кроветворные клетки 5%
Мутантная мтДНК
Мышцы 5%
Кроветворные клетки 80%