
")

.")

.")
.")

.")

.")
")

.")
.")








 во время Муковисцидоза у детей:")









 medicine
medicineSimilar presentations:










Наследственные болезни накопления: Мукополисахаридозы. Муковисцидоз. Причина, патогенез, клиника, диагностика, лечение,прогноз
1. Международный казахско-турецкий университет имени Х.А.Яссауи Шымкентский медицинский институт Тема Наследственные болезни
ИМЕНИ Х.А.ЯССАУИШЫМКЕНТСКИЙ МЕДИЦИНСКИЙ ИНСТИТУТ
ТЕМА НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ:
МУКОПОЛИСАХАРИДОЗЫ. МУКОВИСЦИДОЗ
ПРИЧИНА,ПАТОГЕНЕЗ,КЛИНИКА,ДИАГНОСТИКА,
ЛЕЧЕНИЕ,ПРОГНОЗ.
Подготовила : Якубжанова Захро
Группа :ЖМО506
Проверила : Салходжаева Г.К
2. Мукополисахаридозы (МПС)
МУКОПОЛИСАХАРИДОЗЫ (МПС)Мукополисахаридозы (МПС) группа
метаболических заболеваний
соединительной ткани, связанных с
нарушением обмена
гликозаминогликанов (ГАГ),
проявляющихся дефектами костной,
хрящевой, соединительной тканей.
Обусловлены данные заболевания
мутациями генов, контролирующих
процесс внутри лизосомного гидролиза
макромолекул ГАГ
Все мукополисахаридозы наследуются
аутосомнорецессивно и лишь болезнь
Хантера (МПС второго типа)
наследуется Хсцепленно рецессивно.
3. Различают следующие основные типы мукополисахаридозов:
РАЗЛИЧАЮТ СЛЕДУЮЩИЕ ОСНОВНЫЕ ТИПЫМУКОПОЛИСАХАРИДОЗОВ:
МПС I типа: Н типа – синдром Гурлер,
HS синдром Гурлер –Шейе,
S синдром Шейе;
МПС II типа – синдром Хантера, тяжелая и легкая форма;
МПС III типа – синдром Санфилиппо:
III Атипа – синдром Санфилиппо A;
III В типа – синдром Санфилиппо B;
III С типа – синдром Санфилиппо C;
IIID типа – синдром Санфилиппо D;
МПС IV типа – синдром Моркио:
IV A типа – синдром Моркио A;
IV В типа – синдром Моркио В
МПС VI типа – синдром МаротоЛами (легкая и тяжелая)
МПС VII типа – синдром Слая
МПС IX типа – синдром Натовикс
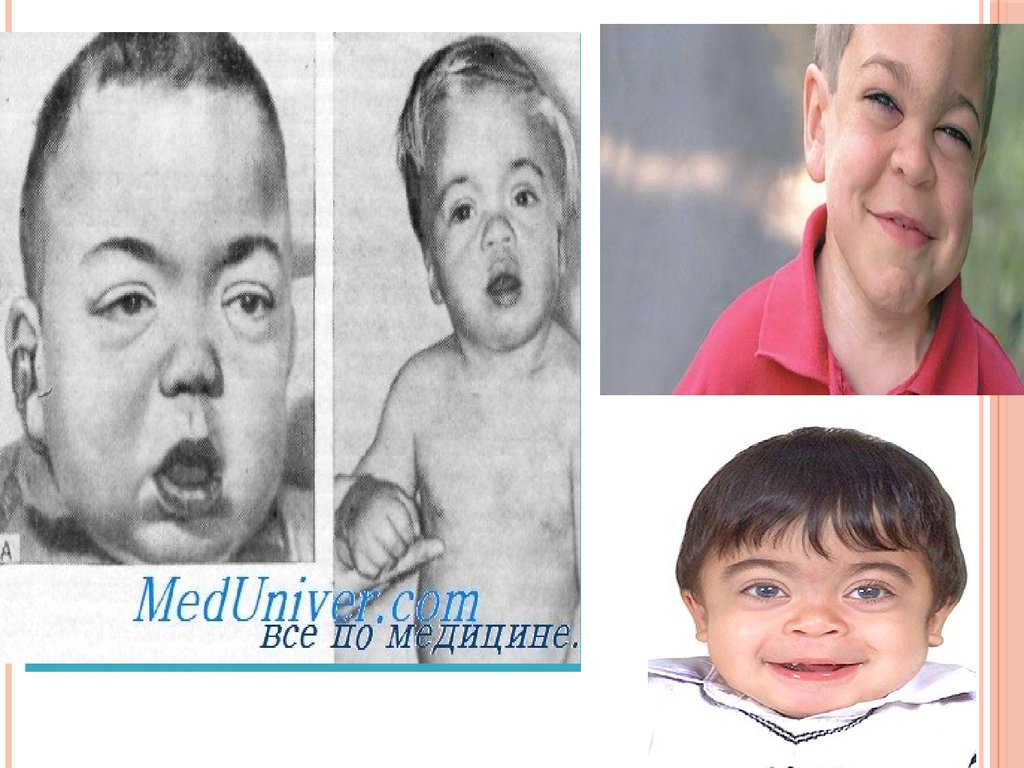
4. Мукополисахаридоз типа I-Н (синдром Гурлер).
МУКОПОЛИСАХАРИДОЗ ТИПА IН(СИНДРОМ ГУРЛЕР).
Впервые описан немецким
педиатром Гурлер (G. Hurler) в 1919
г.
Часто наблюдается у детей, родители
которых находятся в кровном
родстве.
Частота среди новорожденных
1:20000 — 1:25000.
При тяжелой форме синдрома Гурлер дети
умирают в возрасте 6–10 лет от сердечной и
легочной декомпенсации.
При легкой форме доживают в среднем до 30 лет.
5.
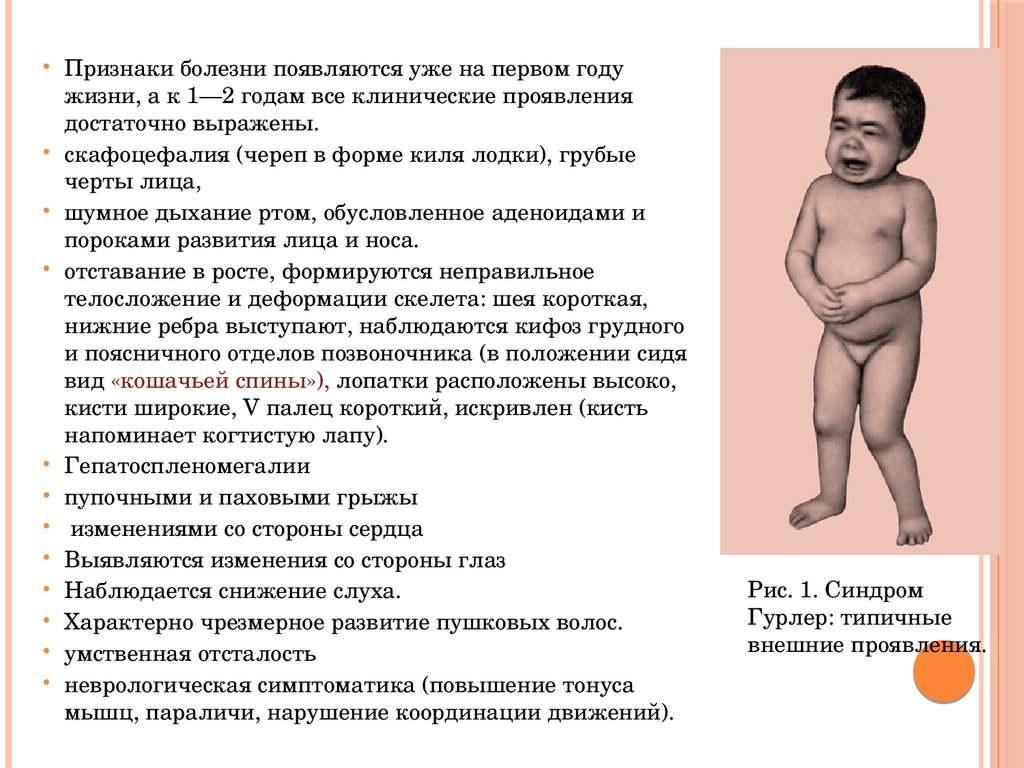
Признаки болезни появляются уже на первом годужизни, а к 1—2 годам все клинические проявления
достаточно выражены.
скафоцефалия (череп в форме киля лодки), грубые
черты лица,
шумное дыхание ртом, обусловленное аденоидами и
пороками развития лица и носа.
отставание в росте, формируются неправильное
телосложение и деформации скелета: шея короткая,
нижние ребра выступают, наблюдаются кифоз грудного
и поясничного отделов позвоночника (в положении сидя
вид «кошачьей спины»), лопатки расположены высоко,
кисти широкие, V палец короткий, искривлен (кисть
напоминает когтистую лапу).
Гепатоспленомегалии
пупочными и паховыми грыжы
изменениями со стороны сердца
Выявляются изменения со стороны глаз
Наблюдается снижение слуха.
Характерно чрезмерное развитие пушковых волос.
умственная отсталость
неврологическая симптоматика (повышение тонуса
мышц, параличи, нарушение координации движений).
Рис. 1. Синдром
Гурлер: типичные
внешние проявления.
6.
7. ДИФ диагноз
ДИФ ДИАГНОЗВ начале развития болезни у детей раннего возраста
синдром Гурлер необходимо дифференцировать с
врожденными деформациями позвоночника (для
последних не характерна поза «кошачьей спины»,
умственная отсталость, грубые черты лица,
специфические биохимические нарушения);
Гипотиреозом (в анамнезе — затянувшаяся желтуха
и запоры, сухость кожи; тонкие, тусклые, ломкие,
сухие волосы, нет характерных костных
деформаций).
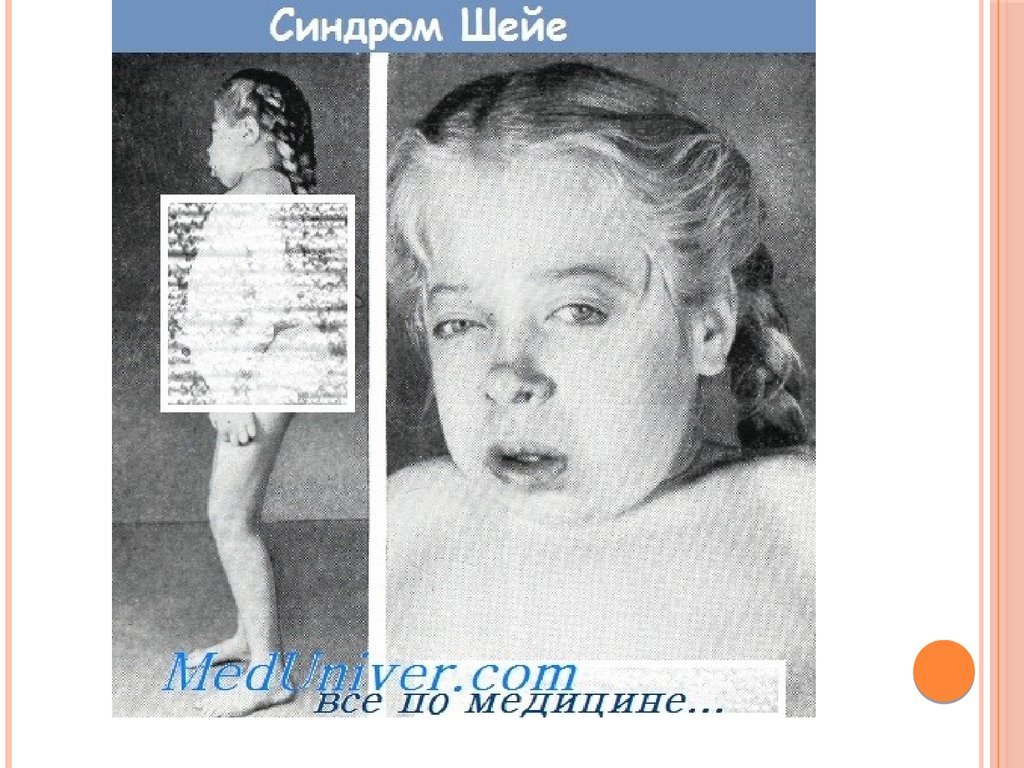
8. Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
МУКОПОЛИСАХАРИДОЗ ТИПА IS (БОЛЕЗНЬШЕЙЕ; ПОЗДНИЙ СИНДРОМ ГУРЛЕР).
Впервые описан
американским
офтальмологом
Шейе (Н.G. Scheie) в
1962 г.
Мукополисахаридоз
ы типов IН и IS
рассматривают как
варианты одной
болезни.
9.
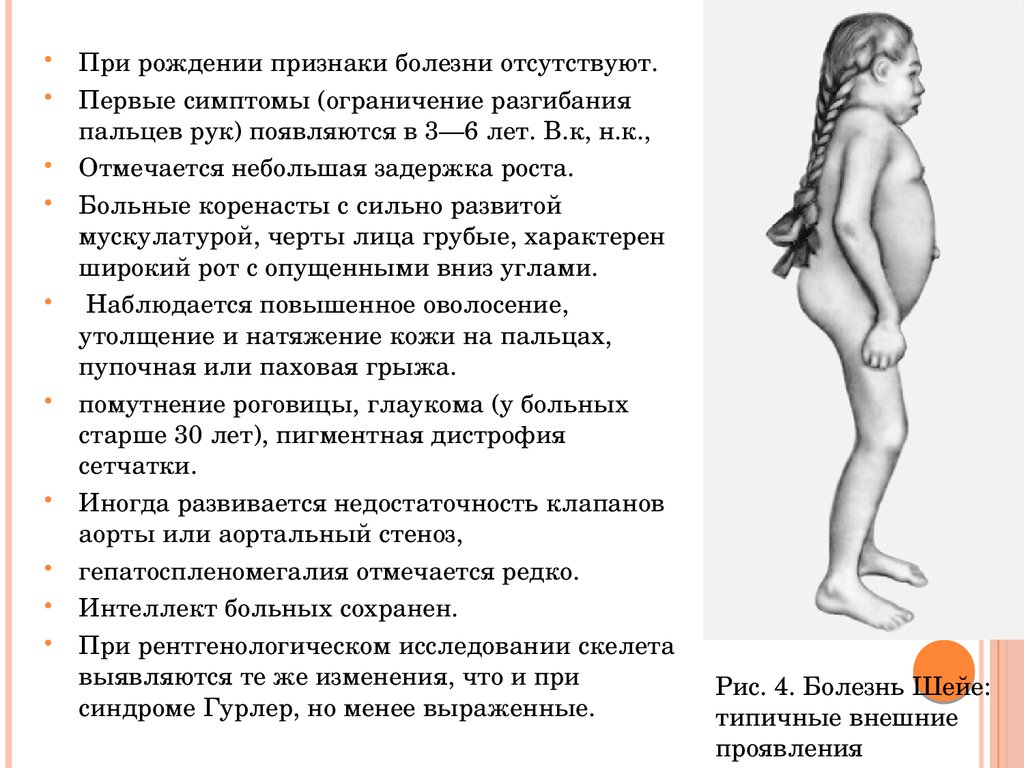
При рождении признаки болезни отсутствуют.Первые симптомы (ограничение разгибания
пальцев рук) появляются в 3—6 лет. В.к, н.к.,
Отмечается небольшая задержка роста.
Больные коренасты с сильно развитой
мускулатурой, черты лица грубые, характерен
широкий рот с опущенными вниз углами.
Наблюдается повышенное оволосение,
утолщение и натяжение кожи на пальцах,
пупочная или паховая грыжа.
помутнение роговицы, глаукома (у больных
старше 30 лет), пигментная дистрофия
сетчатки.
Иногда развивается недостаточность клапанов
аорты или аортальный стеноз,
гепатоспленомегалия отмечается редко.
Интеллект больных сохранен.
При рентгенологическом исследовании скелета
выявляются те же изменения, что и при
синдроме Гурлер, но менее выраженные.
Рис. 4. Болезнь Шейе:
типичные внешние
проявления
10.
11. Мукополисахаридоз типа II (синдром Гунтера).
МУКОПОЛИСАХАРИДОЗ ТИПА II (СИНДРОМГУНТЕРА).
Клинические симптомы появляются позднее, чем при синдроме
Гурлер (у детей старше 2 лет) и менее выражены.
Чаще болеют мальчики.
Характерны грубые черты лица, скафоцефалия (рис. 3. а), шумное
дыхание, низкий грубый голос, частые острые респираторные
вирусные инфекции.
Кифоз обычно не развивается;
в 3—4 года появляются нарушения координации движений — походка
становится неуклюжей, дети при ходьбе часто падают (рис. 3. б),
изменяется поведение характерна эмоциональная лабильность,
агрессивность.
Отмечаются также прогрессирующая тугоухость, узелковые
поражения кожи спины, остеоартриты, незначительная
гепатоспленомегалия.
В более старшем возрасте появляется легкое помутнение роговицы.
Снижение интеллекта выражено в меньшей степени, чем при
синдроме Гурлер.
12.
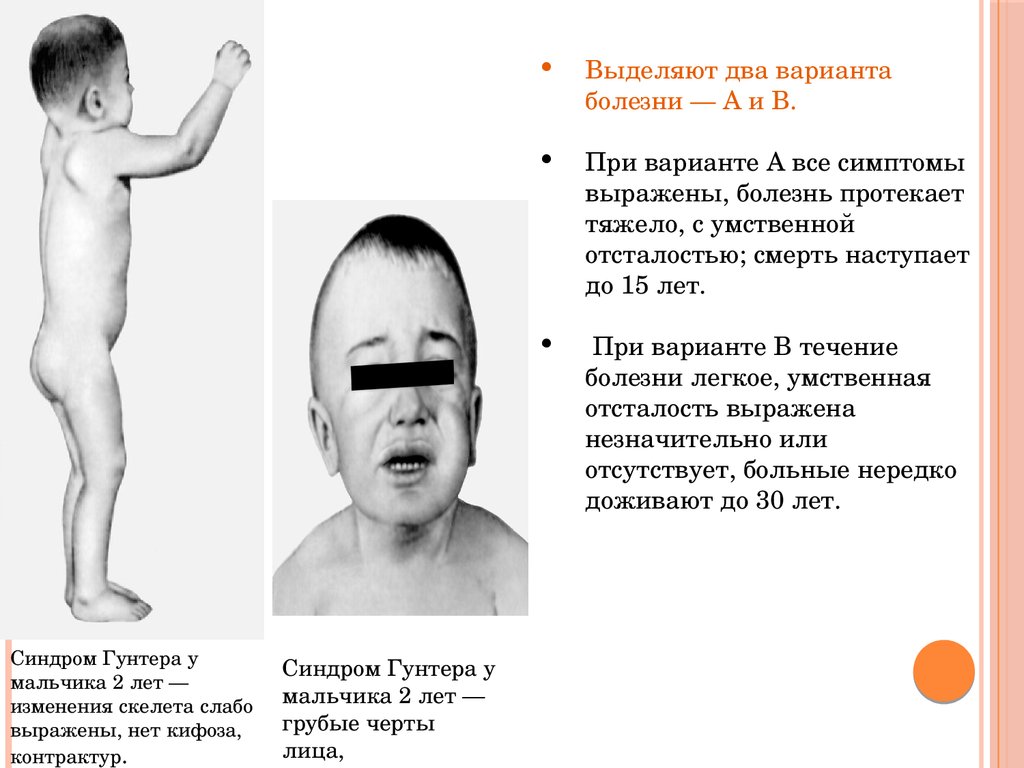
Синдром Гунтера умальчика 2 лет —
изменения скелета слабо
выражены, нет кифоза,
контрактур.
Синдром Гунтера у
мальчика 2 лет —
грубые черты
лица,
Выделяют два варианта
болезни — А и В.
При варианте А все симптомы
выражены, болезнь протекает
тяжело, с умственной
отсталостью; смерть наступает
до 15 лет.
При варианте В течение
болезни легкое, умственная
отсталость выражена
незначительно или
отсутствует, больные нередко
доживают до 30 лет.
13.
14. Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
МУКОПОЛИСАХАРИДОЗ ТИПА III (СИНДРОМСАНФИЛИППО, БОЛЕЗНЬ САНФИЛИППО).
Описан американским педиатром Санфилиппо (S.J. Sanfilippo)
в 1963 г. Частота 1 на 100 000—200 000 новорожденных.
Выраженная ретардация интеллекта,
макроцефалия,
агрессивное поведение.
Относительно слабые скелетные изменения.
Гепатоспленомегалия появляется в школьном возрасте.
Густые, жесткие волосы, сросшиеся брови, нарушение осанки –
гиперлордоз.
Тугоухость кондуктивная или смешанная. Клинически типы
не могут быть дифференцированы. Чаще встречаются типы А
и В. Тип D – самый редкий. Тяжелое течение характерно для
типа А, тип С самый мягкий. Продолжительность жизни до 30
лет
Выраженная ретардация интеллекта
15.
16. Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
МУКОПОЛИСАХАРИДОЗ ТИПА IV(СНИДРОМ МОРКИО, БОЛЕЗНЬ МОРКИО).
Заболевание в 1929 г. независимо
друг от друга впервые описали
уругвайский педиатр Моркио (L.
Morquio) и английский радиолог
Брейлсфорд (J.F. Braiisford).
Частота до 1: 40 000.
Дети рождаются без признаков
болезни.
Первые симптомы появляются в
возрасте 1—3 года, и к 7—8 годам
клиническая картина уже
полностью выражена.
Умирают в возрасте 20–35 лет
17.
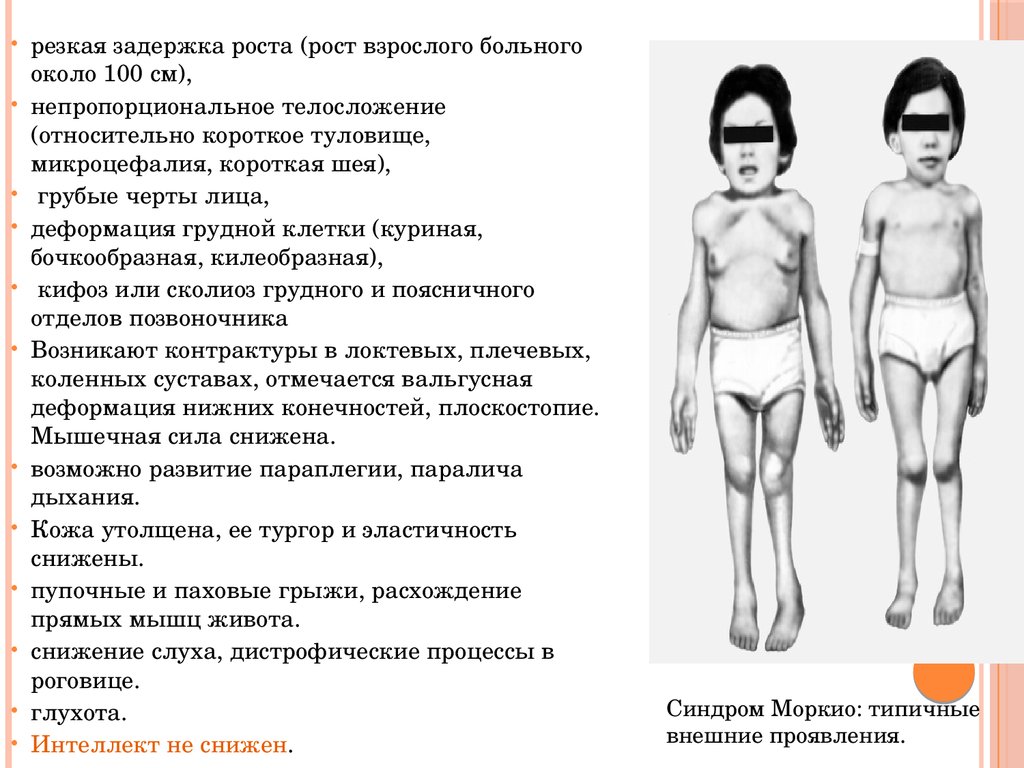
резкая задержка роста (рост взрослого больногооколо 100 см),
непропорциональное телосложение
(относительно короткое туловище,
микроцефалия, короткая шея),
грубые черты лица,
деформация грудной клетки (куриная,
бочкообразная, килеобразная),
кифоз или сколиоз грудного и поясничного
отделов позвоночника
Возникают контрактуры в локтевых, плечевых,
коленных суставах, отмечается вальгусная
деформация нижних конечностей, плоскостопие.
Мышечная сила снижена.
возможно развитие параплегии, паралича
дыхания.
Кожа утолщена, ее тургор и эластичность
снижены.
пупочные и паховые грыжи, расхождение
прямых мышц живота.
снижение слуха, дистрофические процессы в
роговице.
глухота.
Интеллект не снижен.
Синдром Моркио: типичные
внешние проявления.
18. Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
МУКОПОЛИСАХАРИДОЗ ТИПАVI (СИНДРОМ МАРОТО —
ЛАМИ, БОЛЕЗНЬ МАРОТО —
ЛАМИ)
в 1960 г. впервые описан
французскими врачами Марото (Р.
Maroteaux) и Лами (М. Е.J. Lamy).
Первые симптомы появляются у
детей старше 2 лет.
Характерно отставание в росте,
грубые черты лица (как при
синдроме Гурлер, но менее
выражены), малые размеры
верхней челюсти, короткая шея,
бочкообразная грудная клетка,
укороченные ключицы (рис. 7, а).
Синдром Марото — Лами: (типичные
внешние проявления у девочки 9 лет)
— грубые черты лица, бочкообразная
грудная клетка.
19.
Отмечаются сгибательные контрактуры суставовверхних конечностей (больные не могут поднять
руки вверх);
с возрастом появляются контрактуры в суставах
нижних конечностей, нарушается походка (рис. 7,
б).
Часто присоединяются острые респираторные
вирусные инфекции.
Нередко выявляются грыжи,
гепатоспленомегалия.
Могут наблюдаться гидроцефалия, спастические
параличи. Интеллект не страдает.
поражение сердечнососудистой системы
(клапанный аппарат)
прогрессирующая гидроцефалия,
потеря зрения,
неврологическая клиника, связанная со
сдавлением спинномозгового канала.
Изменение кисти в виде «когтистой лапы»,
короткие и утолщенные пальцы
Интеллект сохранен
Рис. 7б). Синдром Марото —
Лами: (типичные внешние
проявления у девочки 9 лет)
— контрактуры верхних и
нижних конечностей.
20. Мукополисахаридоз типа VII (синдром Слая).
МУКОПОЛИСАХАРИДОЗ ТИПА VII (СИНДРОМ СЛАЯ).Описан Слаем (W.S. Sly) в 1973 г.
Клинические проявления схожи с синдромом
Санфилиппо.
Диагноз устанавливают только при детальном
биохимическом исследовании.
Висцеральный фенотип: низкий рост,
гепатоспленомегалия,
пупочные и паховые грыжи,
обструкция верхних дыхательных путей,
поражение сердечнососудистой системы.
Снижение интеллекта.
Помутнение роговицы.
21. Мукополисахаридоз типа VIII (синдром Ди Ферранте).
МУКОПОЛИСАХАРИДОЗ ТИПА VIII (СИНДРОМДИ ФЕРРАНТЕ).
Описан Ди Ферранте (N. Di Ferrante) и др. в 1978 г.
По клиническим проявлениям схож с
мукополисахаридозом типа IV (синдром Моркио), но в
отличие от него при мукополисахаридозе типа VIII
выражена задержка психомоторного и
интеллектуального развития.
Низкий рост,
расщепление неба,
частые отиты,
появлением мягких узелков вокруг суставов, с
периодически появляющейся болезненным отеком
суставов.
22.
Жалобы на:отставание в росте;
грубые черты лица
увеличение размеров
головы;
деформация скелета;
скованность в суставах;
частые инфекции
верхних дыхательных
путей,
шумное дыхание; апноэ
во сне;
пупочная, паховая
грыжи
снижение зрения, слуха;
нарушения понимания
речи, сна,
судороги.
Анамнез:
Дети, как правило, рождаются «нормальными».
Характерен большой вес при рождении.
3–5 месяцев появляется ринит, позже –
рецидивирующие инфекции, отставание в росте,
увеличение головы, иногда – паховые и
пупочные грыжи.
6–8 месяцев черты лица становятся грубыми,
развивается поражение позвоночника.
9–12 месяцев появляется комплекс типичных
клинических проявлений, которые постепенно
прогрессируют. Лицо приобретает характерные
гротескные черты – большой нос с запавшей
переносицей, пухлые губы, маленькие зубы с
широкими зубными промежутками, позднее
прорезывание зубов, макроглоссия. Внешний
вид пациента характеризуется отставанием в
росте , диспропорциональным телосложением, с
укорочением туловища. Возможна глухота,
пупочная грыжа, паховая грыжа, уплотнение и
утолщение кожи, грубые волосы, умеренный
гирсутизм. На поздних стадиях возможно
развитие глухоты, слепоты, деменции [
23. Лабораторные исследования:
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ:ОАК: анемия, лейкопения, у 50% больных в лейкоцитах можно обнаружить зернистость
Альдера.
Биохимический анализ крови: Гиперхолестеринемия, умеренное повышение уровня
аланинаминотрансферазы (АЛТ) (42 Ед/л), аспартатаминотрансферазы (АСТ) (49 Ед/л);
Определение уровня экскреции ГАГ в моче путем электрофореза: повышение уровня
ГАГ в моче.
Инструментальные исследования:
ЭхоКГ: вторичная дилятационная, гипертрофическая кардиомиопатия. Поражение
клапанного аппарата сердца: клапаны утолщены, отмеачется ограничение их подвижности,
что приводит к недостаточности и/или стенозу. Чаще всего поражается аортальный и
митральный клапаны.
Рентгенография опорнодвигательного аппарата: "рыбьи" позвонки, позвонки имеют
кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность
передневерхних углов, углообразный кифоз, утолщение и укорочение отростков,
кортикальное утончение длинных костей, грудной кифоз, поясничный лордоз, дисплазия
головки бедренной кости и вертлужной впадины.
Рентгенография костей кисти: свидетельствует о недоразвитии ногтевых фаланг,
укорочении и расширении пястных костей, проксимальных и средних фаланг
Рентгенография органов грудной клетки, застойные пневмонии, хронические бронхиты.
Определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся
их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц,
уменьшение и смещение головок плечевых костей.
УЗИ органов брюшной полости: гепатоспленомегалия.
24. Дифференциальная диагностика
ДИФФЕРЕНЦИАЛЬНАЯДИАГНОСТИКА
Синдром Гурлера – 1 тип
Синдром Шейе – 1 тип S
Синдром Хантера – 2 тип
Синдром Санфилиппо – 3 тип
Синдром Моркио 4 тип
Синдром МаротоЛами 6 тип
25.
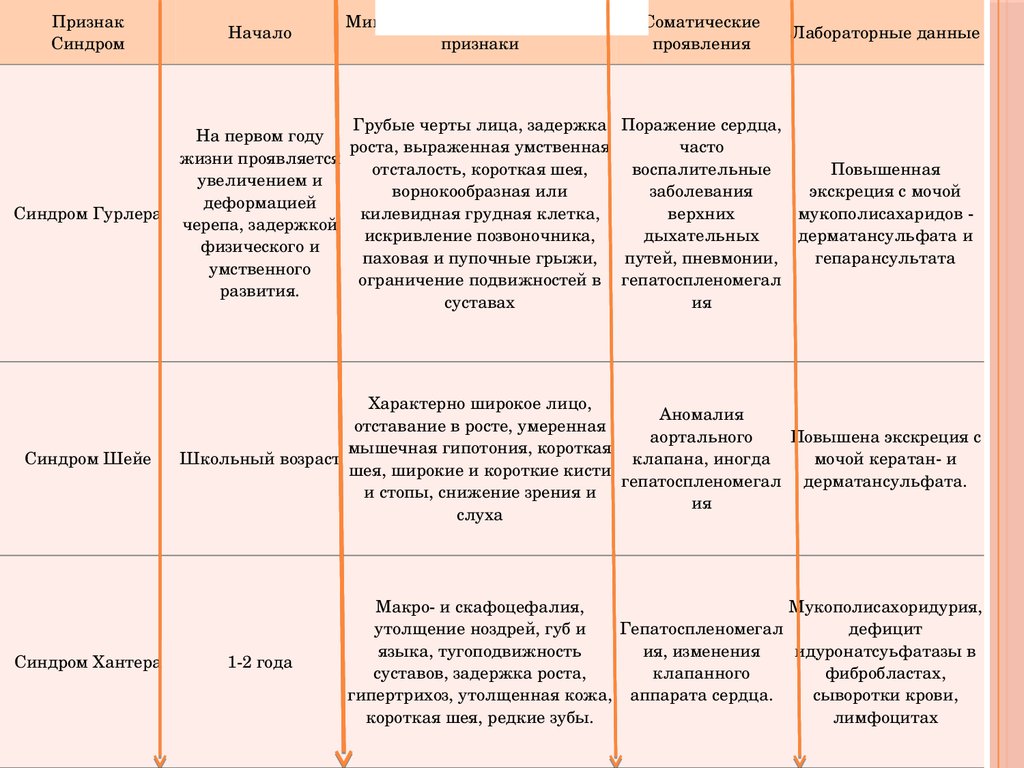
ПризнакСиндром
Синдром Гурлера
Синдром Шейе
Синдром Хантера
Начало
Минимальные диагностические
признаки
Грубые черты лица, задержка
На первом году
роста, выраженная умственная
жизни проявляется
отсталость, короткая шея,
увеличением и
ворнокообразная или
деформацией
килевидная грудная клетка,
черепа, задержкой
искривление позвоночника,
физического и
паховая и пупочные грыжи,
умственного
ограничение подвижностей в
развития.
суставах
Соматические
проявления
Лабораторные данные
Поражение сердца,
часто
воспалительные
Повышенная
заболевания
экскреция с мочой
верхних
мукополисахаридов
дыхательных
дерматансульфата и
путей, пневмонии,
гепарансультата
гепатоспленомегал
ия
Характерно широкое лицо,
Аномалия
отставание в росте, умеренная
аортального
Повышена экскреция с
мышечная гипотония, короткая
Школьный возраст
клапана, иногда
мочой кератан и
шея, широкие и короткие кисти
гепатоспленомегал дерматансульфата.
и стопы, снижение зрения и
ия
слуха
12 года
Макро и скафоцефалия,
Мукополисахоридурия,
утолщение ноздрей, губ и
Гепатоспленомегал
дефицит
языка, тугоподвижность
ия, изменения
идуронатсуьфатазы в
суставов, задержка роста,
клапанного
фибробластах,
гипертрихоз, утолщенная кожа, аппарата сердца.
сыворотки крови,
короткая шея, редкие зубы.
лимфоцитах
26.
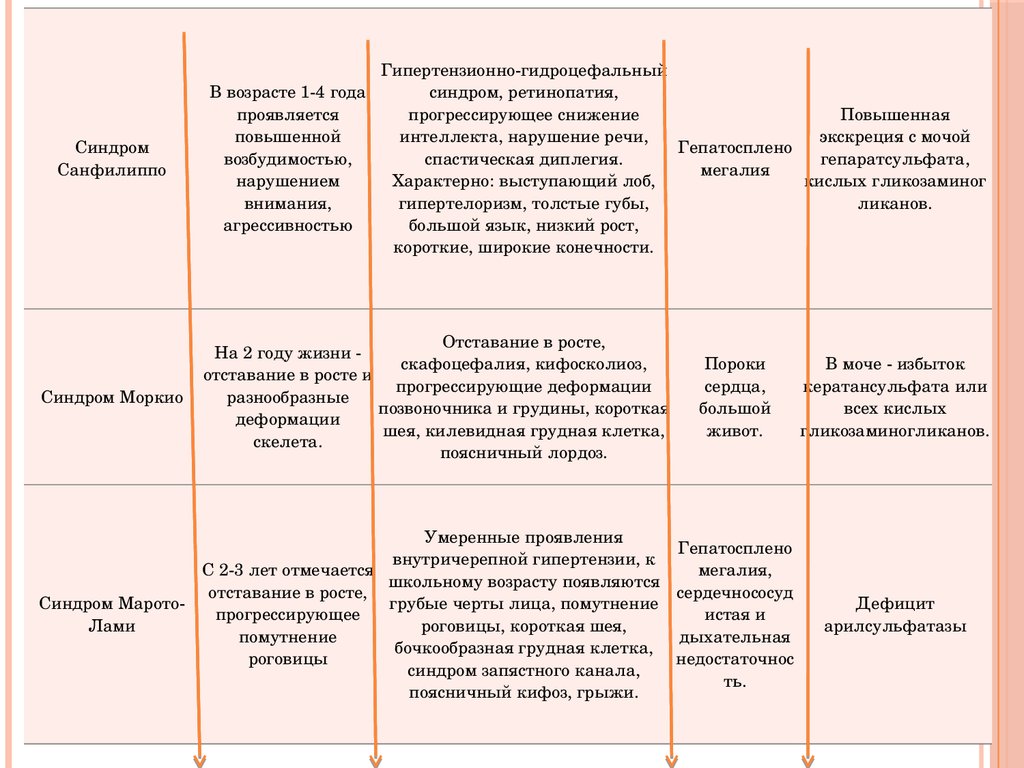
СиндромСанфилиппо
Синдром Моркио
Гипертензионногидроцефальный
В возрасте 14 года
синдром, ретинопатия,
проявляется
прогрессирующее снижение
Повышенная
повышенной
интеллекта, нарушение речи,
экскреция с мочой
Гепатосплено
возбудимостью,
спастическая диплегия.
гепаратсульфата,
мегалия
нарушением
Характерно: выступающий лоб,
кислых гликозаминог
внимания,
гипертелоризм, толстые губы,
ликанов.
агрессивностью
большой язык, низкий рост,
короткие, широкие конечности.
Отставание в росте,
На 2 году жизни
скафоцефалия, кифосколиоз,
отставание в росте и
прогрессирующие деформации
разнообразные
позвоночника и грудины, короткая
деформации
шея, килевидная грудная клетка,
скелета.
поясничный лордоз.
Умеренные проявления
внутричерепной гипертензии, к
С 23 лет отмечается
школьному возрасту появляются
отставание в росте,
Синдром Марото
грубые черты лица, помутнение
прогрессирующее
Лами
роговицы, короткая шея,
помутнение
бочкообразная грудная клетка,
роговицы
синдром запястного канала,
поясничный кифоз, грыжи.
Пороки
сердца,
большой
живот.
В моче избыток
кератансульфата или
всех кислых
гликозаминогликанов.
Гепатосплено
мегалия,
сердечнососуд
истая и
дыхательная
недостаточнос
ть.
Дефицит
арилсульфатазы
27. Лечение :
ЛЕЧЕНИЕ :Режим в зависимости от тяжести и формы заболевания: от общего до
постельного.
Диета стол №15.
Патогенетическая селективная заместительная ферментотерапия для:
МПС Iтипа – Ларонидаза, концентрат д/пригот. рра д/инф 500ЕД/5мл –
100ЕД/кг, 1 раз в неделю пожизненно;
МПС II типа – Идурсульфатаза , концентрат д/пригот. рра д/инф., 6мг/3мл –
0,5мг/кг, 1 раз в неделю пожизненно;
МПС VI типа – Галсульфаза, концентрат д/пригот. рра д/инф., 5мг/5мл – 1
мг/кг 1 раз в неделю пожизненно;
МПС IV А типа – Элосульфаза, концентрат д/пригот. рра д/инф. 5мг/5мл –
2мг/кг 1раз в неделю – пожизненно.
Для купирования отечного синдрома, сообщающейся гидроцефалии,
артериальной гипертензии:
∙ фуросемид 40мг по 1мг/кг 2 раза в день курс 57 дней;
∙ спиронолактон 40мг 13 мг/кг 2 раза в день курс 10 дней;
∙ каптоприл 25 мг 0,1 мг/кг/раз 3 разавдень – курс 10 дней;
∙ винпоцетин, 5 мг – с 3 лет.возраста по2,5 мг 3 раз,курс715
28.
При судорожном синдроме:∙ карбамазепин 0,2гр таблетках 20мг/кг 1 раз в день курс в зависимости от тяжести
течения заболевания
При острых воспалительных процессах с преобладанием экссудативных
явлений (отек слизистой оболочки гортани):
∙ преднизолон 5мг по 0,51 мг/кг 2 раза в день – курс 10 дней.
Для снятия симптомов бронхообструкции:
∙ эуфиллин 10мл – 2.4% 710 мг/кг в сутки в 4 приема, курс 5 дней;
∙ сальбутамол аэрозоль, рр для ингаляций небула содержит2,5 мг по 2,5 мг,
максимально до 5 мг ингаляционно через небулайзер х 4 раза в день курс 57 дней.
Муколитики:
∙ амброксолгидрохлорид 15мг/5мл. До 2 х по 2,5 мл 2 раза в день, от 2 до 5 лет по
2,5 мл 3 раза в день, от 5 12 лет по 5 мл 23 раза в день.
При сердечнососудистой недостаточности в комплексной терапии калия магния
аспарагинат по 250500 мл – 1раза в день в/в кап курс 57 дней;
Общеукрепляющая терапия, при гиповитаминозах:
∙ пиридоксин гидрохлорид, 1 мл 5% 0,02–0,03 г 1–2 раза в день в/м курс 20–25
инъекций;
∙ тиамин хлорид, 5% 1 мл детям старше 8 лет 12,5 мг (0,25 мл 5 % раствора) 1раз в
сутки, курс 1030 инъекций;
∙ фолиевая кислота, 0,001 – с 3х лет по 0,00025 гр (1/4 тб) в сутки, курс 2030 дней.
29.
По показаниям при наличии железодефицитной анемии:∙ актиферрин рр 30мл – по 5 мл 12 раза в день для детей дошкольного возраста, дети школьного
возраста по 5 мо 23 раза в день, курс 812 нед.
В составе комплексной терапии инфекционновоспалительных заболеваний уха, горла, носа с
выраженным болевым синдромом:
∙ диклофенак 25мг 2 мг/кг 2раза в день курс 710 дней.
Рецидивирующие респираторные заболевания уха., горла, носа, верхних дыхательных путей:
∙ цефазолин 0,5гр, 1,0 гр – 2050мг/кг 23 раза в день в/м курс 710 дней;
∙ цефтриаксон амп 1000мг – 5080мг/кг 1 раза в день в/м курс 710 дней.
Для профилактики грибковой инфекции после антибактериальной терапии:
∙ флуконазол 100мг – первый день 6 мг/кг, второй и последующие дни 3 мг/кг, курс 35 дней.
При функциональных и органических поражениях нервной системы, сопровождающихся
повышенной возбудимостью, эмоциональной лабильностью и нарушением сна:
∙ глицин, 0,1 – по 1 таб. 23 раза в день курс 714 дней. Успокаивающее действие – микстура
Павлова раствор 200мл/флакон – по 1 ч/л 3 раза в день курс 710 дней.
Антигипоксическое, антиамнетическое, антиэпилептическое действие:
∙ ноофен, 0,25 – 0,10,2 гр 23 раза в день курс 714 дней.
При психосоматических и невротических расстройствах, нарушения поведения:
∙ сонапакс, 10 мг – дети 47 лет по 1020 мг/сут. Кратность приема 23 раза/сут; 814 лет по 2030
мг/сут, кратность приема 3 раза/сут; 1518 лет по 3050 мг/сут, кратность приема 3 раза в сут.
∙ хлорпротиксен 15 мг тб 0,52 мг/кг 1раз в день – длительно, только детям старше 6 лет.
30.
Другие виды лечения,оказываемые на
амбулаторном уровне:
Хирургическое вмешательство,
оказываемое в стационарных условиях:
∙
∙ индивидуальные занятия с
логопедом;
∙ занятия с психологом;
∙ кондуктивная педагогика;
∙ ЛФК, массаж,
физиолечение озокеритовые
аппликации, дарсонвализация
волосистой части головы,
электрофорез с эуфиллином на
шейный отдел позвоночника.
∙ ингаляции;
∙ дыхательная гимнастика;
∙ ортопедическая коррекция
(корсет, ортопедическая обувь)
артроскопия;
∙ декомпрессия спинного мозга;
∙ декомпрессия нервных стволов;
∙ хирургическая коррекция сердечно
сосудистой недостаточности (замена клапанов);
∙ хирургическая замена коленного или
тазобедренного сустава;
∙ вентрикулоперитонеальное
шунтирование;
∙ аденоидэктомия;
∙ тонзилэктомия;
∙ трахестомия при выраженной обструкции
верхних дыхательных путей и ночном апноэ;
∙ грыжесечение.
31. Что такое Муковисцидоз у детей
ЧТО ТАКОЕ МУКОВИСЦИДОЗ У ДЕТЕЙНазвание заболевания происходит от
латинских слов mucus «слизь» и viscidus
«вязкий». Муковисцидоз это системное
наследственное заболевание, при котором
поражаются все органы, которые выделяют
слизь: бронхолегочная система,
поджелудочная железа, печень, потовые
железы, слюнные железы, железы
кишечника, половые железы.
Муковисцидоз (cystic fibrosis) — заболевание,
которое происходит в результате мутации гена
МВТР,.
Чаще всего симптоматика муковисцидоза
возникает в таких органах:
Поджелудочная железа;
Печень;
Кишечник;
Дыхательная система, легкие.
32. Патогенез (что происходит?) во время Муковисцидоза у детей:
ПАТОГЕНЕЗ (ЧТО ПРОИСХОДИТ?) ВО ВРЕМЯМУКОВИСЦИДОЗА У ДЕТЕЙ:
Секрет экзокринных желёз становится особенно
вязким, что вызывает большинство патологических
процессов, которые лежат в основе патогенеза
заболевания.
33. Различают следующие формы муковисцидоза:
РАЗЛИЧАЮТ СЛЕДУЮЩИЕ ФОРМЫ МУКОВИСЦИДОЗА:Лёгочная – возникает вследствие изменения состава
слизи и её застоя в лёгких, затруднённого отхода
мокроты. Встречается в 1520% случаев.
Кишечная – обусловливается недостаточностью
секреции органов ЖКТ. Ярко проявляется при
введении прикорма или переводе ребёнка на
искусственное питание. Встречается в 5% случаев.
Смешанная – отличается сочетанием лёгочной и
кишечной симптоматики, составляет 7580% случаев.
Атипичная – характеризуется изолированным
поражением отдельных экзокринных желёз.
34. Легочная
ЛЕГОЧНАЯЖелезы слизистой оболочки дыхательных путей производят большое количество секрета, который изза
повышенной вязкости забивает мелкие бронхи и бронхиолы. Это вызывает застойные явления и
хроническое воспаление, в результате которого бронхиальные железы увеличиваются, а просвет бронхов
сужается, вследствие чего формируются бронхоэктазы (патологические расширения бронхов).
Нарушение проходимости дыхательных путей часто осложняется присоединением
инфекции.Среди патогенных возбудителей наиболее распространены:
Золотистый стафилококк.
Гемофильная (инфлюэнца) и синегнойная палочки.
Инфицирование тканей лёгких и бронхов приводит к гнойному воспалению и усилению непроходимости.
Признаком лёгочной формы муковисцидоза являются деформированные пальцы (симптом барабанных
палочек)
Симптомы лёгочной формы:
общая слабость и вялость;
бледность и синева кожи;
одышка (даже в покое);
кашель, сопровождающийся выделением густой мокроты (сразу после рождения);
пневмония, лихорадка, судороги (у грудничков);
малая прибавка веса при полноценном питании;
деформация грудной клетки, пальцев (в виде барабанных палочек), ногти становятся выпуклыми (у детей
старше 1 года);
синусит, тонзиллит, полипы в носу (у подростков).
Осложнения лёгочной формы:
Лёгочные кровотечения.
Кровохарканье.
Плеврит (воспаление серозной оболочки лёгких).
Пневмоторакс (скопление воздуха в плевральной полости).
Эмпиема плевры (воспаление, сопровождающееся скоплением гноя в плевральной полости).
35.
Признаком лёгочной формы муковисцидозаявляются деформированные пальцы (симптом
барабанных палочек)
36. http://vseroditelyam.ru/mukoviscidoz-u-detej/
HTTP://VSERODITELYAM.RU/MUKOVISCIDOZUDETEJ/
37. Кишечная
КИШЕЧНАЯНарушения со стороны ЖКТ обусловливаются секреторной недостаточностью многих органов пищеварительной системы.
Например, вязкость секрета вызывает закупорку протоков поджелудочной железы ещё на этапе внутриутробного
развития. Скопление ферментов в железе провоцирует её самопереваривание и перерождение в фиброзную ткань уже в
первые недели жизни ребёнка. Преимущественно нарушается усваиваемость белков и жиров. Наличие в кишечнике
непереваренных белков и аминокислот приводит к развитию процессов гниения и разложения, в результате
чего образуются высокотоксичные продукты распада (сероводород, аммиак и т. д.)
Симптомы кишечной формы:
гнилостная диспепсия (комбинированное функциональное расстройство ЖКТ) и метеоризм (скопление газов в
кишечнике), вздутие живота;
частые дефекации с большим объёмом каловых масс, что может превосходить возрастную норму в 28 раз. В отдельных
случаях – недержание кала;
запоры с выпадением прямой кишки (частичным или полным выворотом наружу);
мекониевая непроходимость. Обычно отхождение первого кала (мекония) у новорождённого наблюдается в течение
первых суток после появления на свет, реже – на вторые сутки. При муковисцидозе вязкий меконий закупоривает петли
тонкой кишки, в результате чего отхождение не происходит;
боли в животе разнообразного характера (схваткообразные, резкие, спазматические и т. д.);
снижение тонуса мышц, эластичности и упругости кожи;
сухость в ротовой полости, трудности с пережёвыванием сухой пищи;
полигиповитаминоз (дефицит поступления витаминов разных групп);
увеличение печени (вследствие нарушения функции двенадцатипёрстной кишки).
Осложнения:
Неонатальная (диагностируемая при рождении) желтуха, сопровождающаяся кожным зудом, и фиброз печени.
Цирроз печени и портальная гипертензия (повышение давления в воротной вене, которая идёт от желудка к печени).
Асцит (скопление большого объёма жидкости в брюшной полости), к которому приводит портальная гипертензия.
Энцефалопатия (повреждение и гибель нервных клеток головного мозга) вследствие того, что печень не справляется с
функцией очистки крови и часть токсинов попадает в мозг.
Сахарный диабет (изза нарушения выработки инсулина поджелудочной железой).
Желудочные кровотечения.
Пиелонефрит и мочекаменная болезнь.
Кишечная непроходимость.
38.
39. Диагностические критерии
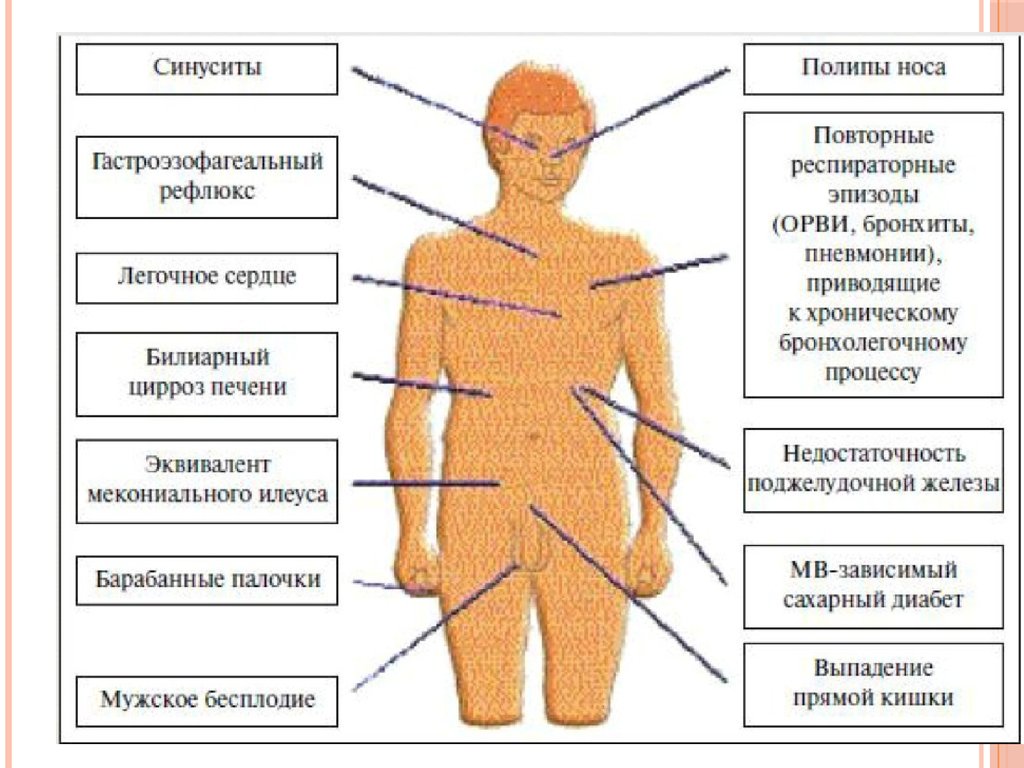
ДИАГНОСТИЧЕСКИЕ КРИТЕРИИЖалобы и анамнез: повторные и рецидивирующие пневмонии
с затяжным течением, хронические изменения со стороны
легочной ткани, особенно двусторонние; бронхиальная астма,
рефрактерная к традиционной терапии; рецидивирующие
бронхиты, бронхиолиты, особенно с высевов Ps.aeruginosa;
мекониальный илеус и его эквиваленты, синдром нарушенного
кишечного всасывания неясного генеза, желтуха обструктивного
типа у новорожденных с затяжным течением, циррозы печени,
сахарный диабет, гастроэзофагальный рефлюкс, холелитиаз,
выпадение прямой кишки, нарушение роста и развития,
задержка полового развития, хронический синусит, полипы носа.
Жалобы на боли в животе, большой живот, слабость,
утомляемость, упорный кашель с отделением мокроты.
Основными симптомами кишечного синдрома являются
метеоризм, обильный, частый (46 раз в сутки), как правило,
замазкообразный или кашицеобразный, серый, блестящий,
жирный, зловонный стул.
40.
Физикальное обследование: при классической картине MBбольные дети имеют характерный внешний вид: «кукольное» лицо;
расширенная, деформированная грудная клетка бочкообразной
формы с выбуханием грудины; большой, вздутый, иногда
«лягушачий живот»; худые конечности с концевыми фалангами в
виде барабанных палочек и часовых стекол». Кожа обычно сухая,
сероватоземлистого цвета, с множественными расчесами.
Отмечается, как правило, навязчивый кашель с гнойной мокротой
в виде комочков, иногда с прожилками крови; цианоз, одышка,
сухие свистящие и влажные разнокалиберные хрипы в легких.
Гепатомегалия. Проявления гиповитаминоза. Снижение массы
тела (БЭН 23 ст.). Нередко у больных выявляется хронический
синусит, а у трети детей старше 8 лет диагностируются полипы
носа. Признаки фиброза печени разной степени выраженности
отмечаются практически у всех больных, но только в 510%
приводят к развитию билиарного цирроза с портальной
гипертензией, требующей хирургического лечения.
41.
Инструментальные исследованияРентгенпленочный тест: полное отсутствие переваривания
желатины или ее разведения в разведениях 1: 20, 1: 40.
Рентгенологическое исследование желудочнокишечного
тракта (ирригоскопия) и эндоскопическое исследование
(ректороманоскопия) дискинезия тонкой кишки, рельеф
слизистой оболочки грубый, «спикулы» или
псевдодивертикулы, большое количество слизи в просвете
кишечника.
Исследование биоптата слизистой оболочки тонкой и толстой
кишок значительное увеличение количества бокаловидных
клеток в слизистой оболочке.
УЗИ органов брюшной полости: диффузные изменения
поджелудочной железы, кистофиброз, изменение размеров.
Рентгенография грудной клетки в двух проекциях: признаки
хронического бронхита или пневмонии.
Спирометрия нарушение функции внешнего дыхания.
42. Лечение
ЛЕЧЕНИЕМуковисцидоз относится к неизлечимым болезням. Пациентов ставят на диспансерное
наблюдение и лечат всю жизнь.
Диетотерапия
Диета приблизительно такая же, как и нормальное здоровое питание. В рационе должно
содержаться большое количество белка. Калорийность должна составлять 120—150% от
нормы, согласно возрасту.
Дополнительное питание
Детям старшего возраста при неадекватном нутритивном статусе вводят дополнительные
высококалорийные продукты:
напитки с высоким содержанием глюкозы
молочные коктейли
а также витамины
При недостаточности массы вводят дополнительное питание. Суточная доза витамина А
для больных муковисцидозом детей любого возраста составляет 500010 000 ЕД, витамина D
– 400800 ЕД. Также необходимы витамины Е и К.
Лечение синдрома дистальной интестинальной обструкции при муковисцидозе
Если состояние ребенка не тяжелое, ему прописывают лактулозу. Для детей до 12 месяцев –
2,5 мл, для детей от 1 до 5 лет – 5 мл, для детей 612 лет – 10 мл, принимать 2 раза в сутки.
Также прописывают Ацетилцистеин, который принимают 3 раза в день, доза от 200 до 600
мг.
Лечение синдрома дистальной интестинальной обструкции включает применение
высокоосмолярных растворов, панкреатических ферментов, регидратационных препаратов.
Если состояние ребенка тяжелое, необходимо лечение в стационаре.