















 medicine
medicineSimilar presentations:










Орфанные заболевания. Мукополисахаридозы
1.
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕУЧРЕЖДЕНИЕ ВЫСШЕГО ОБРАЗОВАНИЯ
«РОСТОВСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ»
МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Кафедра детских болезней №2
Орфанные заболевания.
Мукополисахаридозы.
Заведующий кафедрой –
проф. Лебеденко А.А.
Доцент – к.м.н. Мальцев С.В.
2020 г.
2.
ОПРЕДЕЛЕНИЕМукополисахаридозы (МПС) - группа наследственных
болезней обмена веществ, связанных с нарушением
метаболизма гликозаминогликанов (ГАГ), приводящее к
мультиорганному поражению.
Обусловлены данные заболевания мутациями генов,
контролирующих процесс внутрилизосомного гидролиза
макромолекул.
В зависимости от первичного
генетического дефекта, приводящего к
снижению активности лизосомных
ферментов, выделяют несколько типов
мукополисахаридозов.
3.
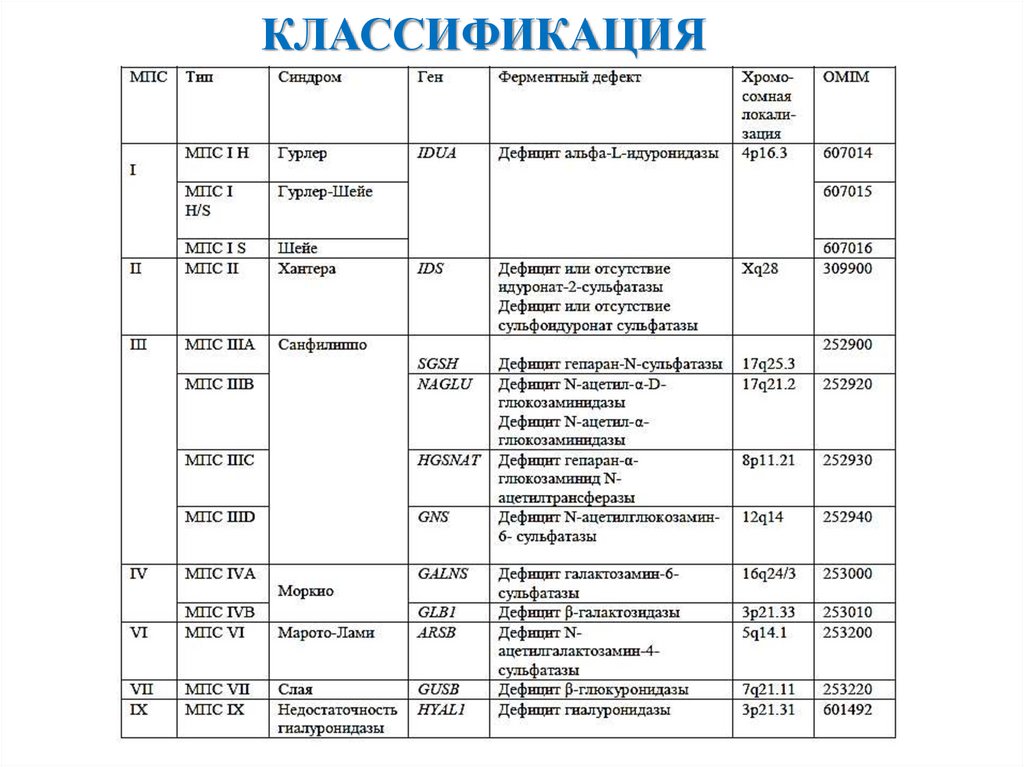
КЛАССИФИКАЦИЯ4.
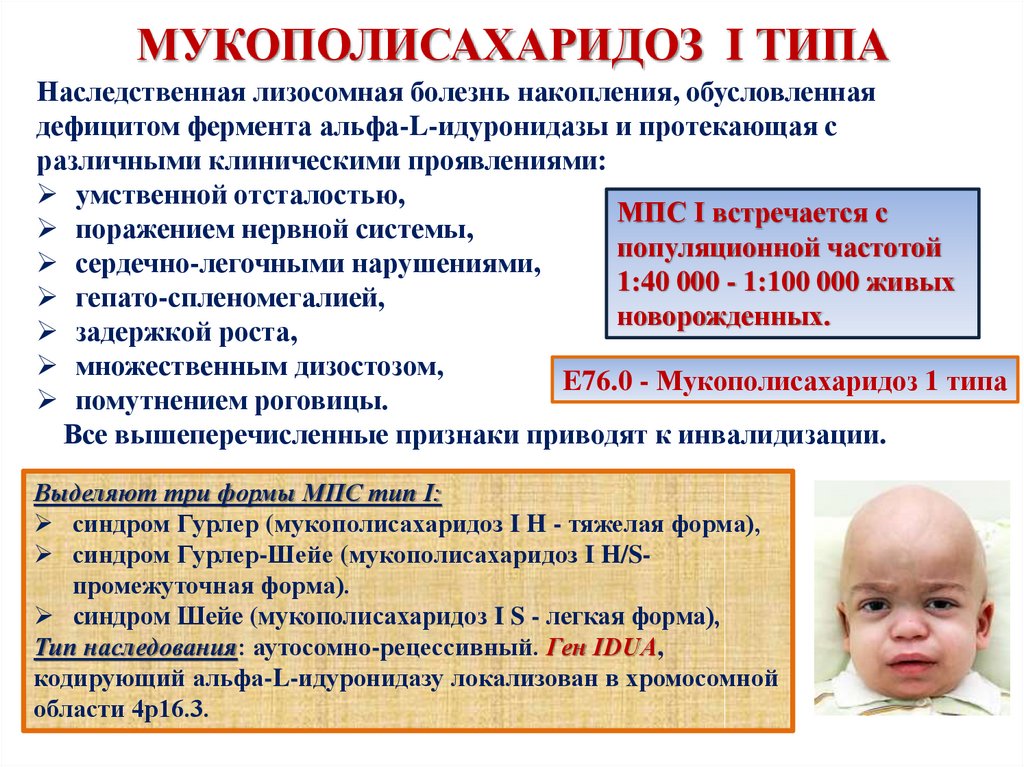
МУКОПОЛИСАХАРИДОЗ I ТИПАНаследственная лизосомная болезнь накопления, обусловленная
дефицитом фермента альфа-L-идуронидазы и протекающая с
различными клиническими проявлениями:
умственной отсталостью,
МПС I встречается с
поражением нервной системы,
популяционной частотой
сердечно-легочными нарушениями,
1:40 000 - 1:100 000 живых
гепато-спленомегалией,
новорожденных.
задержкой роста,
множественным дизостозом,
E76.0 - Мукополисахаридоз 1 типа
помутнением роговицы.
Все вышеперечисленные признаки приводят к инвалидизации.
Выделяют три формы МПС тип I:
синдром Гурлер (мукополисахаридоз I H - тяжелая форма),
синдром Гурлер-Шейе (мукополисахаридоз I H/Sпромежуточная форма).
синдром Шейе (мукополисахаридоз I S - легкая форма),
Тип наследования: аутосомно-рецессивный. Ген IDUA,
кодирующий альфа-L-идуронидазу локализован в хромосомной
области 4p16.3.
5.
АЛГОРИТМ ДИАГНОСТИКИМПС I ТИПА
6.
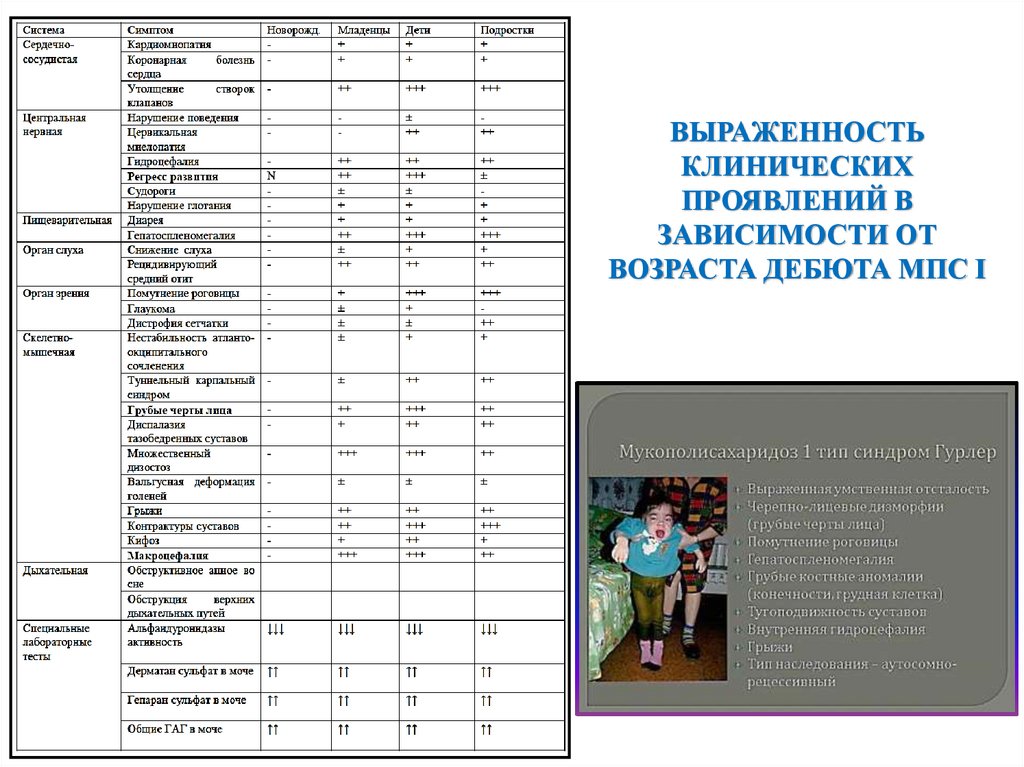
ВЫРАЖЕННОСТЬКЛИНИЧЕСКИХ
ПРОЯВЛЕНИЙ В
ЗАВИСИМОСТИ ОТ
ВОЗРАСТА ДЕБЮТА МПС I
7.
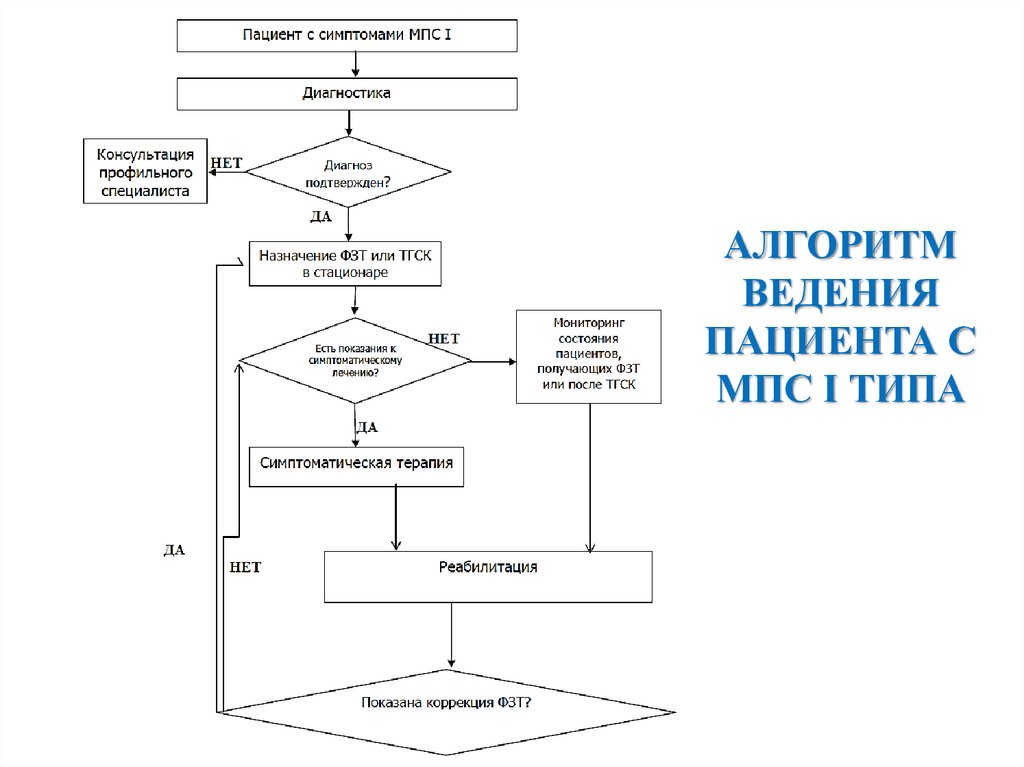
АЛГОРИТМВЕДЕНИЯ
ПАЦИЕНТА С
МПС I ТИПА
8.
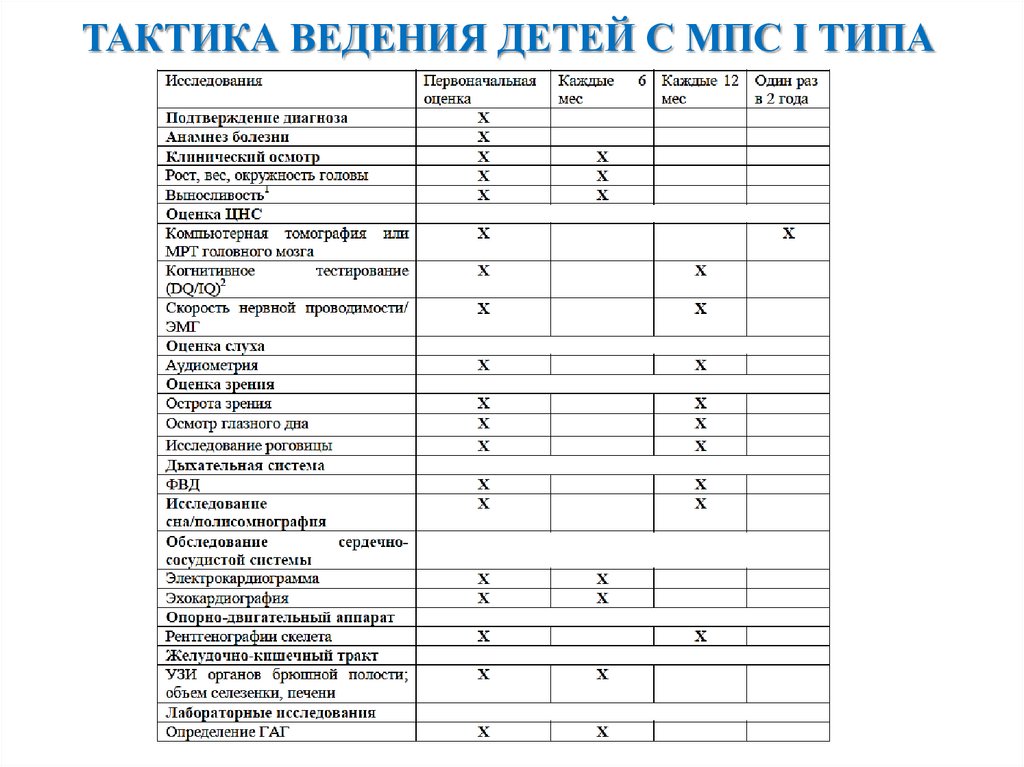
ТАКТИКА ВЕДЕНИЯ ДЕТЕЙ С МПС I ТИПА9.
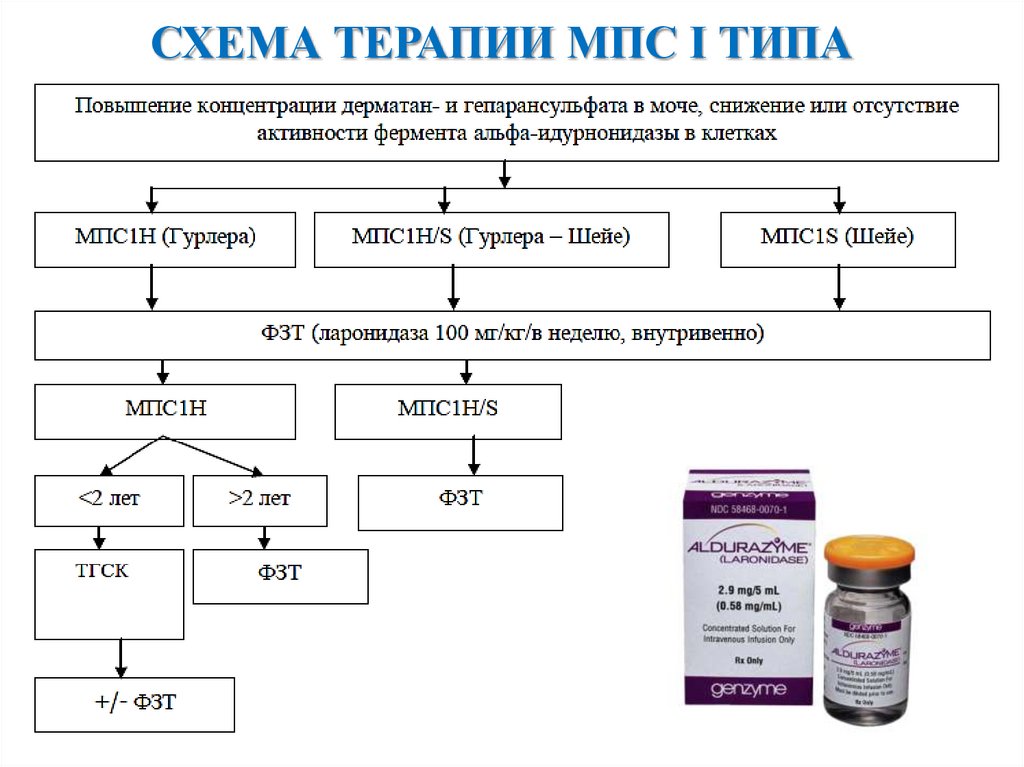
СХЕМА ТЕРАПИИ МПС I ТИПА10.
ПАЛЛИАТИВНАЯ ПОМОЩЬНеобходимо оказание всесторонней помощи (медицинской, психосоциальной и
материальной) детям с неизлечимыми ограничивающими срок жизни
заболеваниями. В состав паллиативных служб входят врачи, медицинские сестры,
психологи и социальные работники. Несмотря на тяжелое состояние и постоянную
потребность в мониторинге, все пациенты преимущественно находятся дома в
кругу своей семьи и друзей. Основной целью работы паллиативных служб
является создание всех необходимых условий для обеспечения нахождения
больных в домашних условиях, а не в стенах лечебного учреждения, что позволяет
не только улучшить качество жизни больных и их семей, но и существенно
снизить государственные затраты на постоянное стационарное лечение таких
пациентов.
ИСХОДЫ И ПРОГНОЗ
Мукополисахаридоз I H - тяжелая форма
В среднем продолжительность жизни пациентов составляет примерно 10 лет.
Дыхательная и сердечная недостаточность, обструктивные процессы верхних
дыхательных путей и инфекции – усугубляют прогноз.
Мукополисахаридоз тип I S - легкая форма
Продолжительность жизни может быть не изменена.
Мукополисахаридоз тип I H/S - промежуточная форма
Скорость прогрессирования заболевания занимает промежуточное положение
между синдромами Гурлер и Шейе.
11.
МУКОПОЛИСАХАРИДОЗ II ТИПА(СИНДРОМ ХАНТЕРА)
Наследственная лизосомная болезнь накопления, с Х-сцепленным рецессивным
типом наследования, которая характеризуется снижением активности лизосомного
фермента идуронат-2-сульфатазы (I2S), вызванным мутацией в гене IDS. Дефицит
фермента приводит к накоплению в лизосомах фракций гепаран- и
дерматансульфата и проявляется прогрессирующими психоневрологическими
нарушениями, поражением паренхиматозных органов, гепатоспленомегалией,
сердечно-лёгочными расстройствами, костными деформациями.
Ген идуронат-2-сульфатазы картирован на
длинном плече Х-хромосомы, в хромосомной
области Xq27.1-q28.
Тип наследования - рецессивный, сцепленный с
Х-хромосомой. Болезнью Хантера страдают, как
правило, только мальчики, однако к
настоящему моменту описано 7 случаев
заболевания у девочек гетерозигот, связанных с
инактивацией второй, нормальной, Ххромосомы или из-за структурных изменений
хромосомы.
МПС II типа
встречается с
популяционной
частотой 1:140 000 1:156 000 живых
новорожденных
мальчиков.
E 76.1 Мукополисахаридоз II
типа
12.
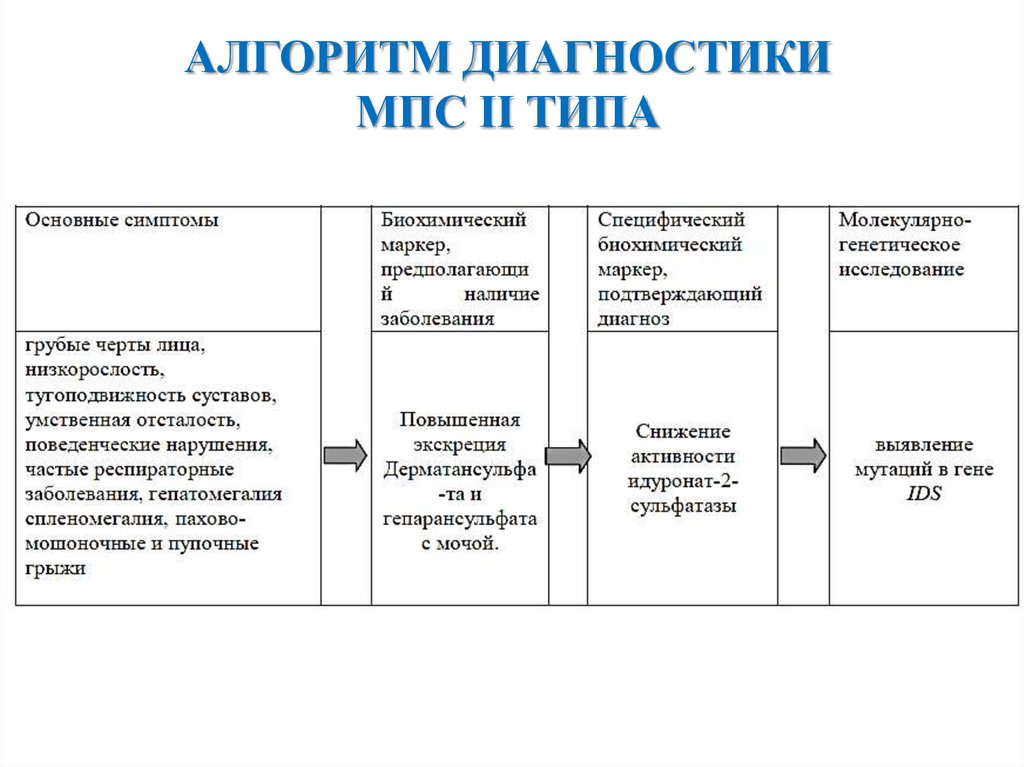
АЛГОРИТМ ДИАГНОСТИКИМПС II ТИПА
13.
КЛИНИЧЕСКИЕСИМПТОМЫ
СИНДРОМА
ХАНТЕРА (МПС
II ТИПА)
14.
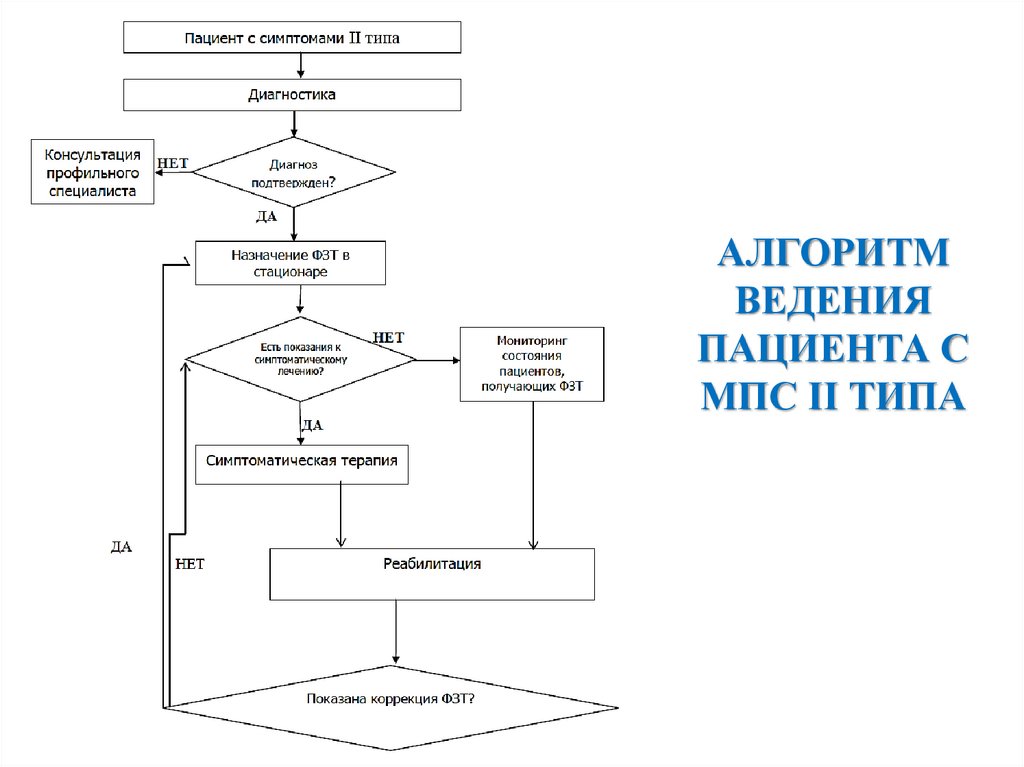
АЛГОРИТМВЕДЕНИЯ
ПАЦИЕНТА С
МПС II ТИПА
15.
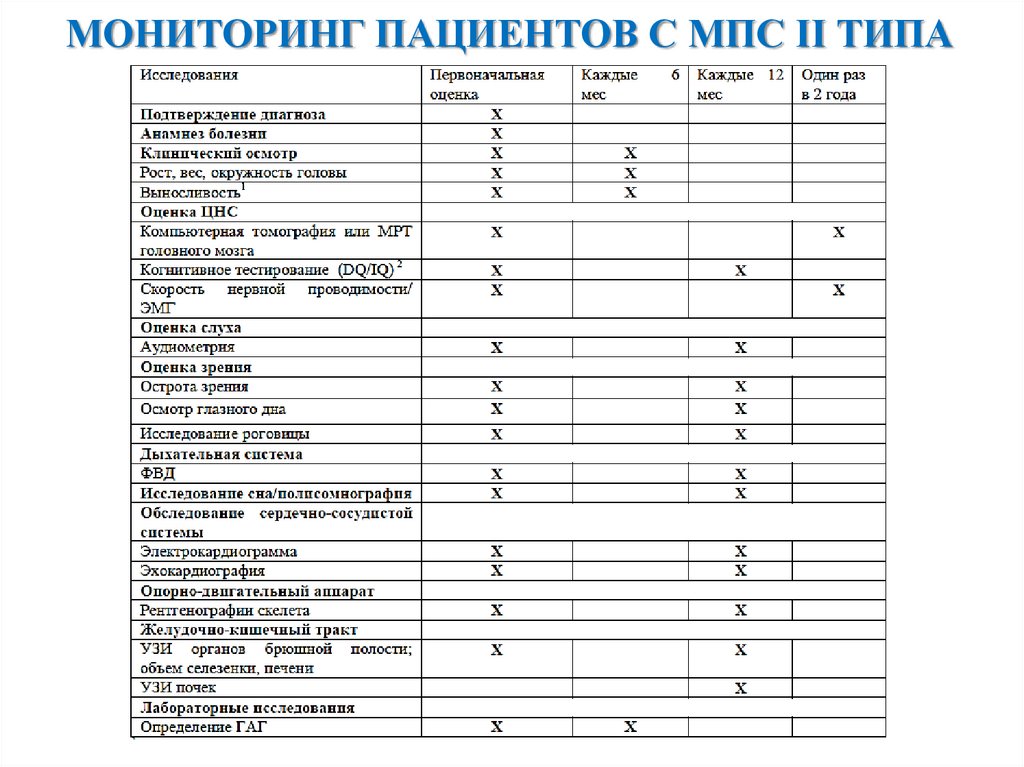
МОНИТОРИНГ ПАЦИЕНТОВ С МПС II ТИПА16.
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС II ТИПАПроведение ферментной заместительной
терапии (ФЗТ) препаратом Идурсульфаза.
ФЗТ проводится постоянно, пожизненно,
непрерывно в дозе 0,5 мг/кг один раз в
неделю в виде внутривенной инфузии на
протяжении 3 ч с максимальной
скоростью введения до 40 мл/ч в
условиях стационара. Весь объем
препарата необходимо развести в 100 мл
раствора натрия хлорида 9 мг/мл (0,9%).
Время инфузии сокращать не следует.
ИСХОДЫ И ПРОГНОЗ
Продолжительность жизни составляет примерно от 10 до 15 лет.
Как правило, причиной смерти являются сердечные и респираторные
осложнения.
У ряда пациентов описаны случаи более продолжительной жизни: до 5060 лет.
17.
МУКОПОЛИСАХАРИДОЗ III ТИПА (СИНДРОМCАНФИЛИППО)
Наследственная лизосомная болезнь накопления, генетически гетерогенная,
обусловленная накоплением гепарансульфата и характеризующаяся
прогрессирующими неврологическими нарушениями, умеренными изменениями
скелета. Все вышеперечисленные признаки приводят к инвалидизации, а при
тяжелом течении болезни - к летальному исходу.
Возможен дефицит разных ферментов, но во
всех случаях в лизосомах накапливается один
тип гликозаминогликанов – гепарансульфат.
Выделяют четыре клинически неразличимых
подтипа, характеризующихся разными
биохимическими дефектами:
Наследуется по аутосомнорецессивному типу, является
генетически гетерогенным
заболеванием. Родители гетерозиготные носители
патологического гена.
Частота 1 на 80 000
новорожденных. Является
третьим по частоте
встречаемости среди всех
известных МПС.
E 76.2 – Мукополисахаридоз
III типа
18.
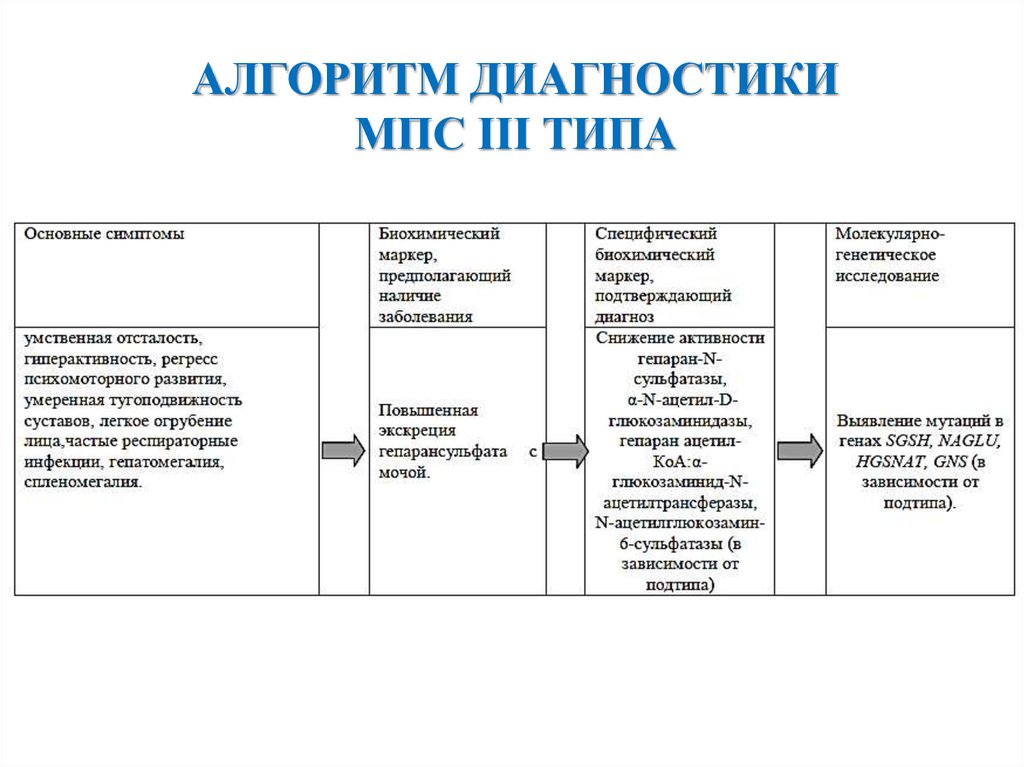
АЛГОРИТМ ДИАГНОСТИКИМПС III ТИПА
19.
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙРАЗЛИЧНЫХ ПОДТИПОВ МПС III В ЗАВИСИМОСТИ ОТ
ВОЗРАСТА ДЕБЮТА ЗАБОЛЕВАНИЯ
20.
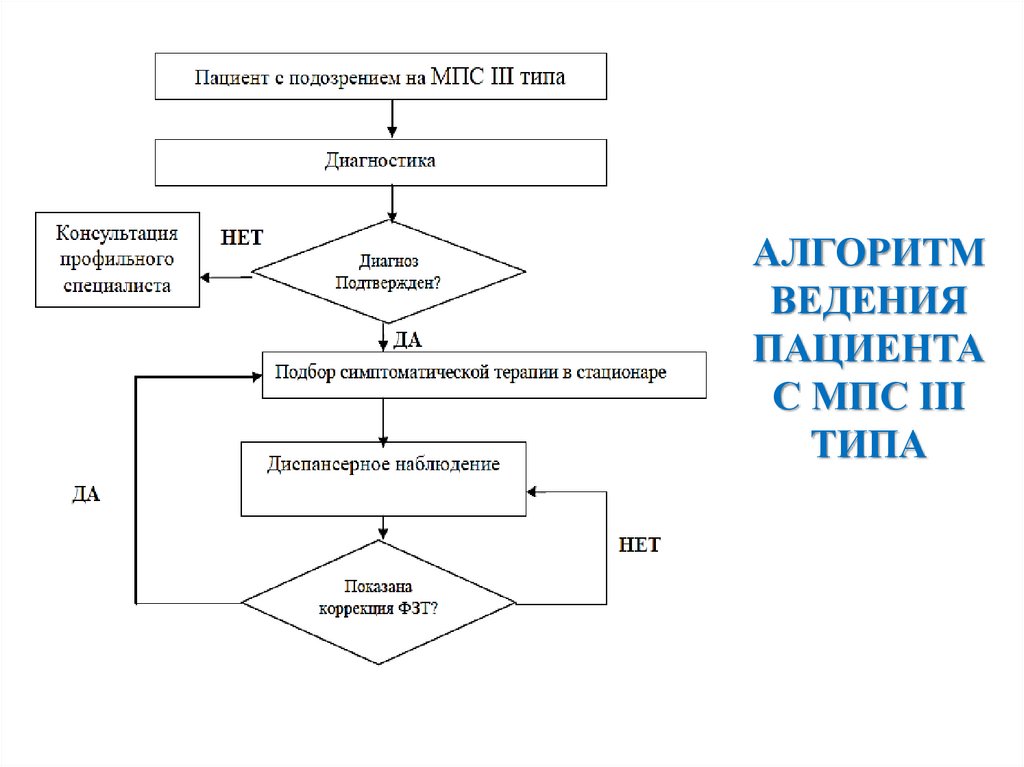
АЛГОРИТМВЕДЕНИЯ
ПАЦИЕНТА
С МПС III
ТИПА
21.
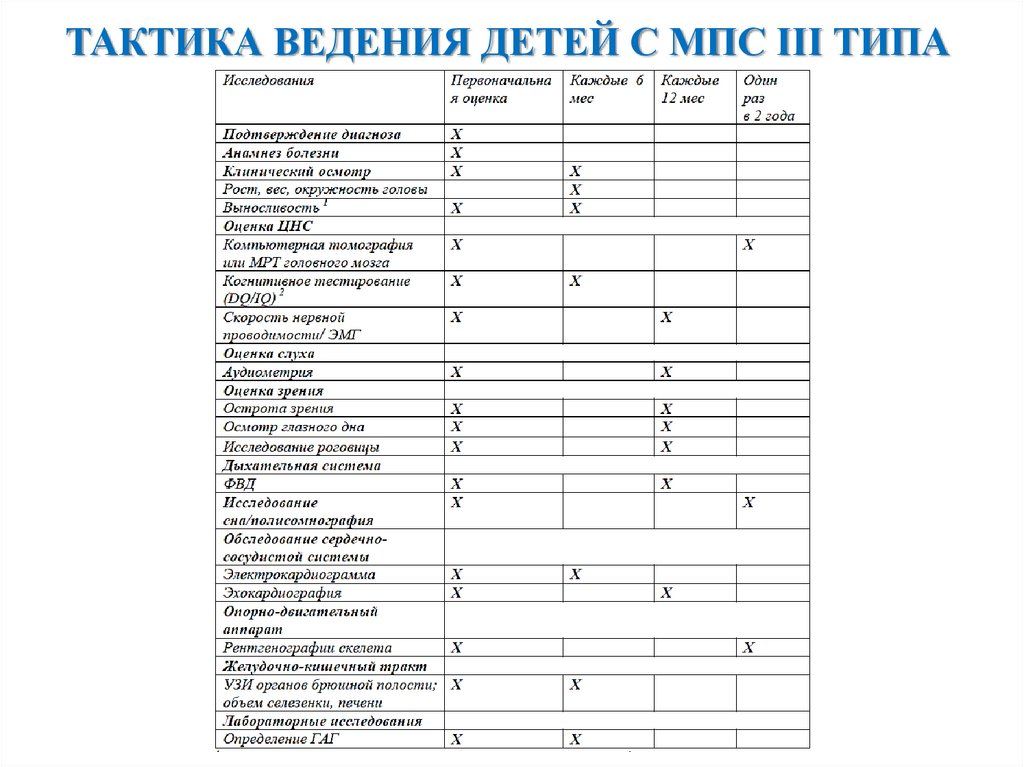
ТАКТИКА ВЕДЕНИЯ ДЕТЕЙ С МПС III ТИПА22.
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС III ТИПАВ настоящее время не существует патогенетического лечения МПС
III типа.
Трансплантация стволовых клеток не имеет достаточных данных
для широкого применения (описано несколько случаев в США).
Трансплантация костного мозга неэффективна.
Пациенты должны получать симптоматическую терапию в
соответствии с выявленными нарушениями и жалобами.
ИСХОДЫ И ПРОГНОЗ
Больные умирают, не достигая
возраста 30 лет, часто вследствие
возникших инфекций нижних
дыхательных путей.
23.
МУКОПОЛИСАХАРИДОЗ IV ТИПА(СИНДРОМ МОРКИО) (СИНОНИМЫ: БОЛЕЗНЬ МОРКИО, СПОНДИЛО-ЭПИФИЗАРНАЯ ДИСПЛАЗИЯ,
ХОНДРООСТЕОДИСТРОФИЯ, ДЕФОРМИРУЮЩАЯ ОСТЕОХОНДРОДИСТРОФИЯ, МОРКИО БРАЙЛСФОРДА СИНДРОМ, МОРКИО - УЛЬРИХА СИНДРОМ, К - МУКОПОЛИСАХАРИДОЗ,
ЭКСЦЕНТРОХОНДРОПЛАЗИЯ, ДУГВЕ - МЕЛХИОРА - КЛАУЗЕНА СИНДРОМ)
Наследственная болезнь накопления, обусловленная дефицитом лизосомных
гидролаз: галактозамин-6-сульфат-сульфатазы (МПС IVА) или β-галактозидазы
(МПС IVВ), обусловлена отложением в соединительной ткани кератансульфата и
характеризуется значительной деформацией скелета и отставанием в росте. Все
вышеперечисленные признаки приводят к инвалидизации, а при тяжелом
течении болезни - к летальному исходу.
МПС IVА, ген GALNS локализован в
хромосомной области 16q24.3.
МПС IVВ, ген GBS локализован в
хромосомной области 3q21.33. Важно
отметить, что мутация гена, кодирующего βгалактозидазу, вызывает также ганглиозидоз
типа I.
Тип наследования: Наследуется по аутосомнорецессивному типу.
Распространенность МПС IVА
1:250 000 новорожденных,
МПС IVВ встречается еще
реже.
E 76.2 – Мукополисахаридоз IV типа
24.
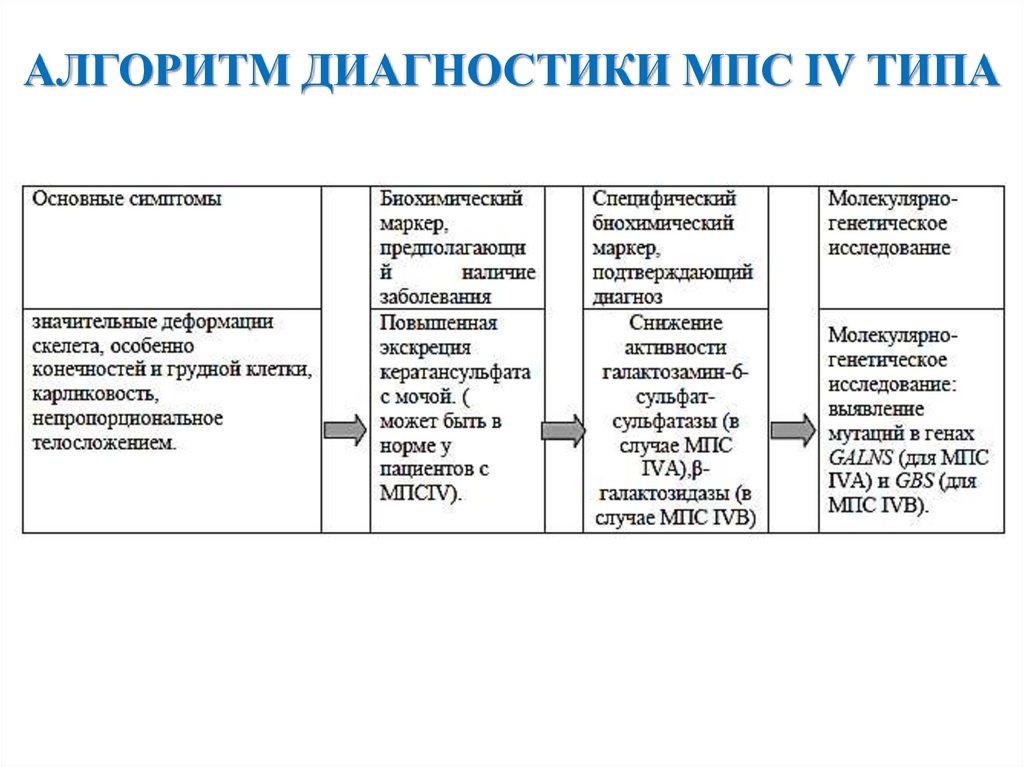
АЛГОРИТМ ДИАГНОСТИКИ МПС IV ТИПА25.
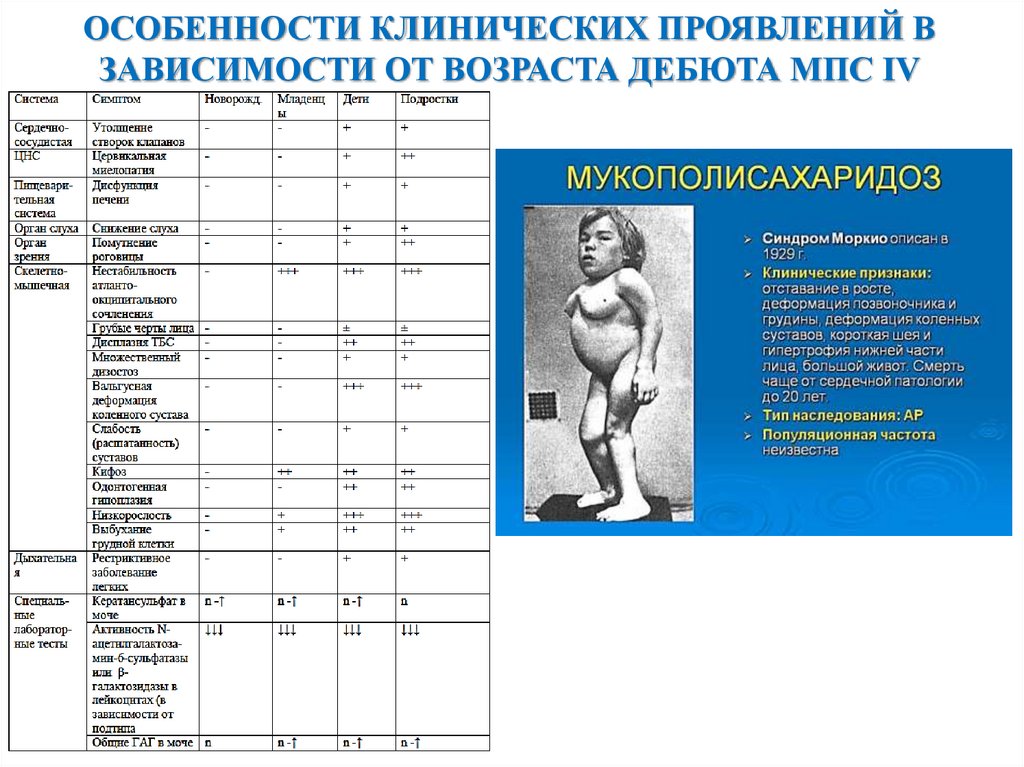
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ ВЗАВИСИМОСТИ ОТ ВОЗРАСТА ДЕБЮТА МПС IV
26.
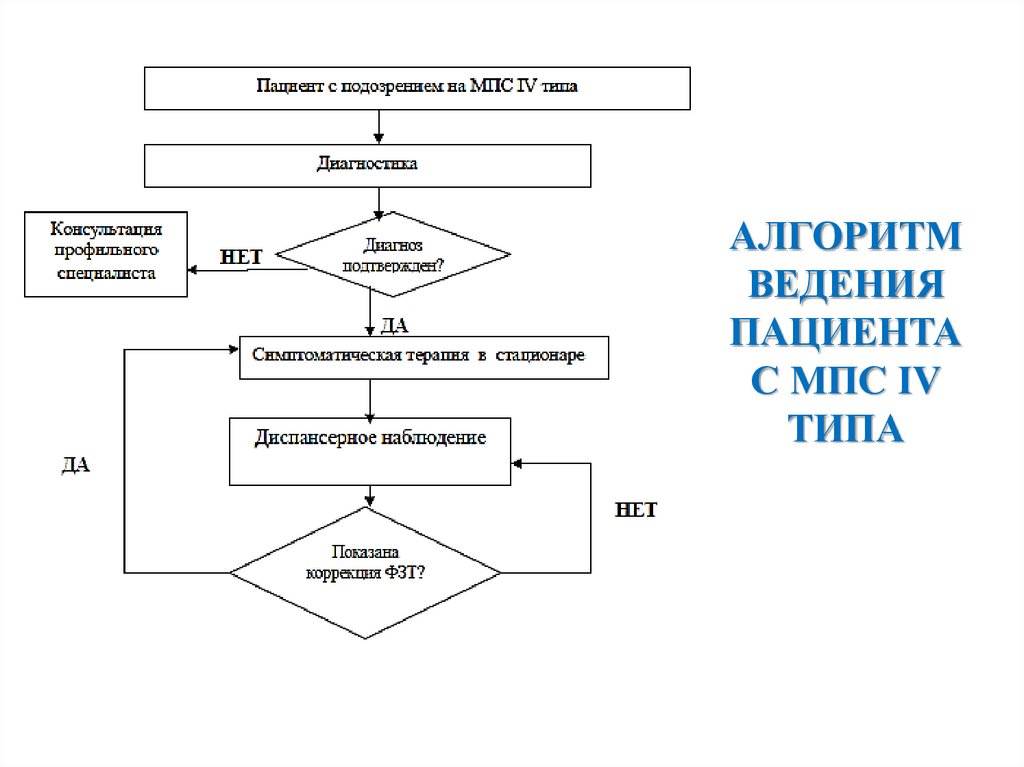
АЛГОРИТМВЕДЕНИЯ
ПАЦИЕНТА
С МПС IV
ТИПА
27.
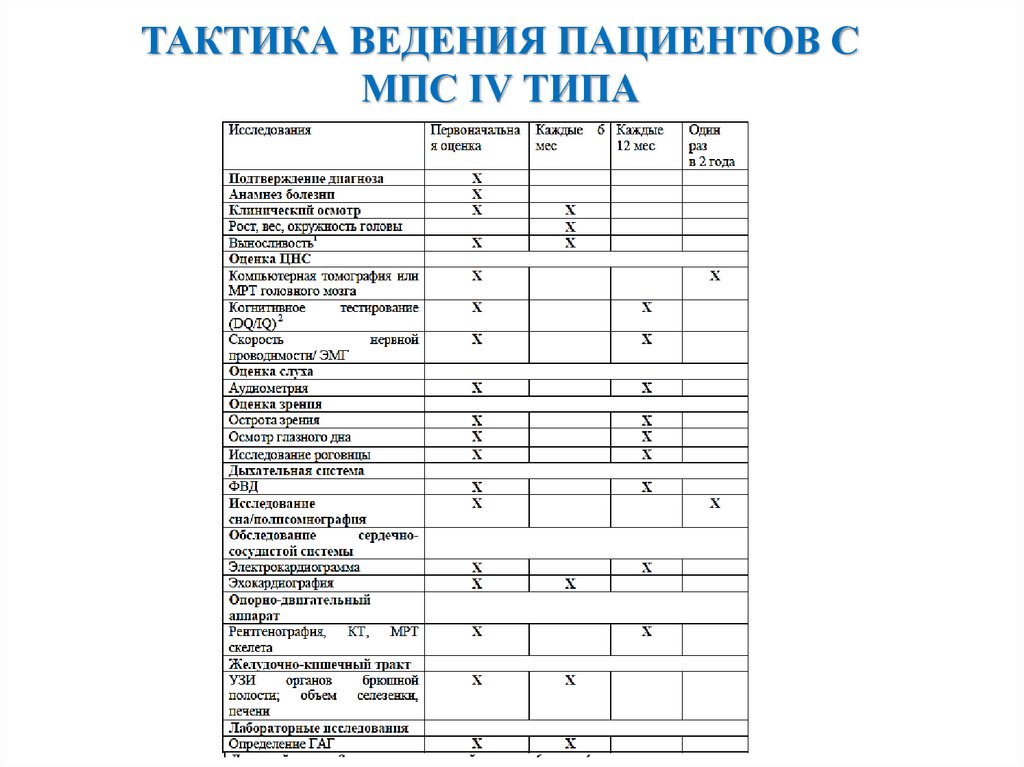
ТАКТИКА ВЕДЕНИЯ ПАЦИЕНТОВ СМПС IV ТИПА
28.
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС IV ТИПАФерментозаместительная терапия разработана, однако, в настоящее
время лекарственный препарат в Российской Федерации не
зарегистрирован. Препарат показал свою эффективность в
международных клинических исследованиях. Его назначение возможно
по жизненным показаниям на основании решения врачебного
консилиума.
Очень важно симптоматическое лечение.
ИСХОДЫ И ПРОГНОЗ
Летальный исход наступает до достижения возраста 20 лет вследствие
сердечно-легочной недостаточности на фоне интеркуррентных
заболеваний. Возможна внезапная смерть в результате смещения
атланто-окципитального сочленения и повреждения ствола мозга.
29.
МУКОПОЛИСАХАРИДОЗ VI ТИПА(СИНДРОМ МАРОТО ЛАМИ)
Наследственная лизосомная болезнь накопления, при которой недостаточность
фермента N-ацетилгалактозамин-4-сульфатазы (арилсульфатазы В) приводит к
нарушению ступенчатой деградации глюкозаминогликана (ГАГ)
дерматансульфата.
Ген ARSB, кодирующий
арилсульфатазу В, локализуется в
хромосомной области 5q14.
Синдром Марото — Лами
наследуется по аутосомнорецессивному типу.
МПС VI типа
встречается с
популяционной
частотой 1:300 000
Мукополисахаридоз VI типа - E 76.2
30.
АЛГОРИТМ ДИАГНОСТИКИМПС VI ТИПА
31.
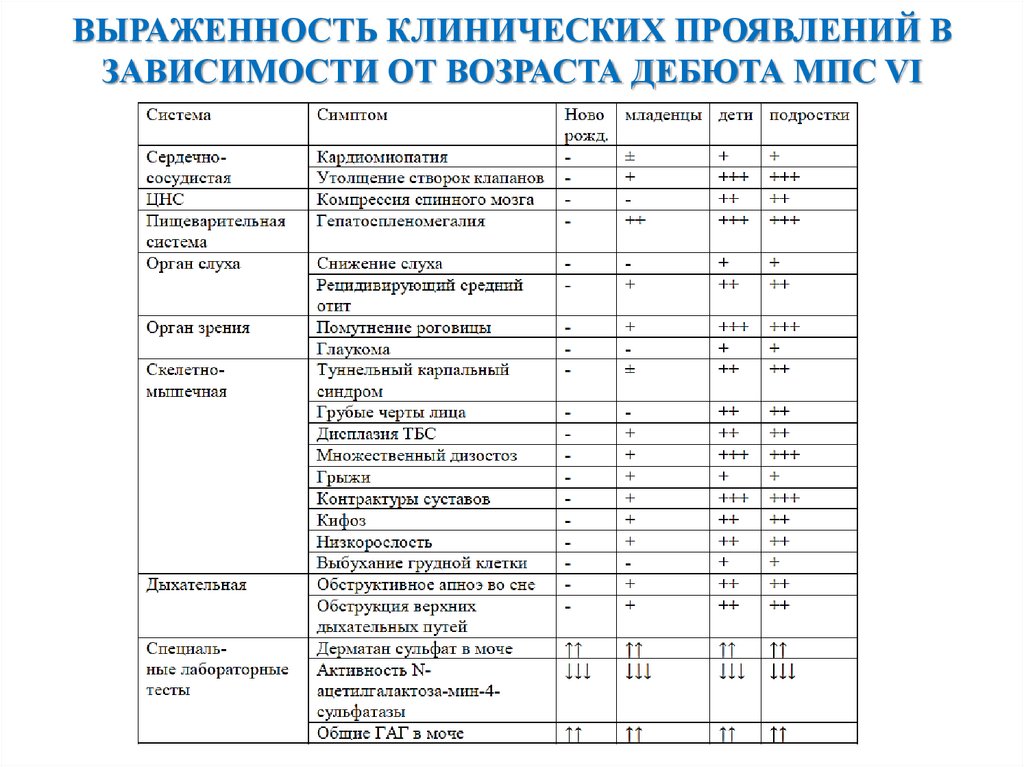
ВЫРАЖЕННОСТЬ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ ВЗАВИСИМОСТИ ОТ ВОЗРАСТА ДЕБЮТА МПС VI
32.
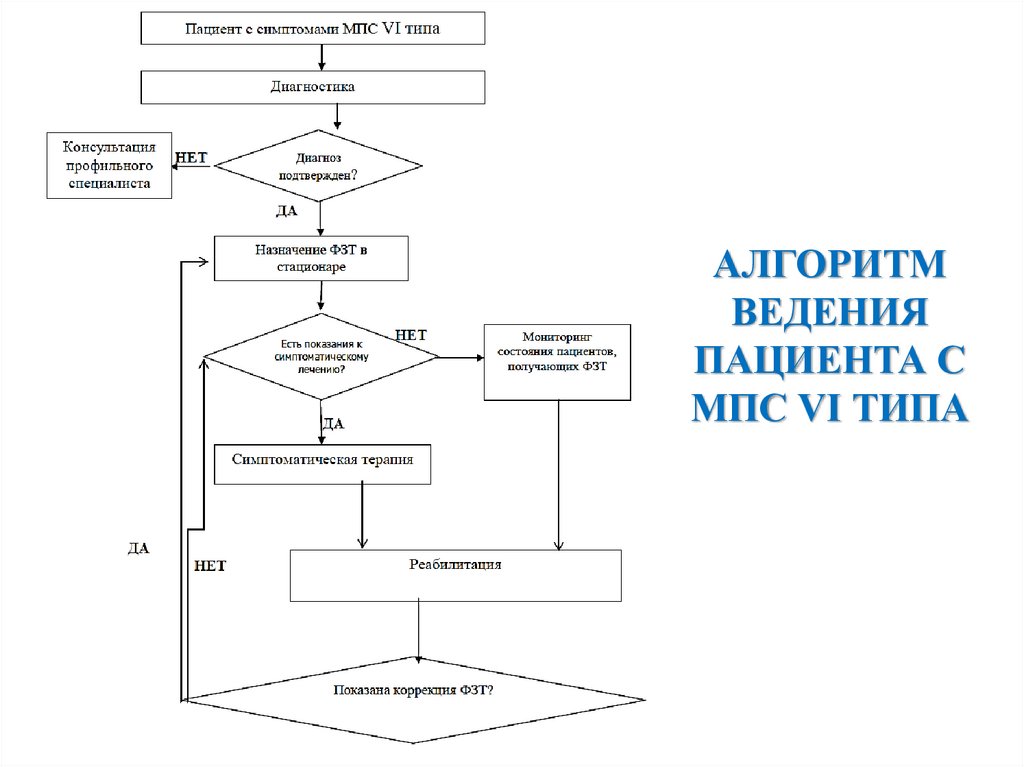
АЛГОРИТМВЕДЕНИЯ
ПАЦИЕНТА С
МПС VI ТИПА
33.
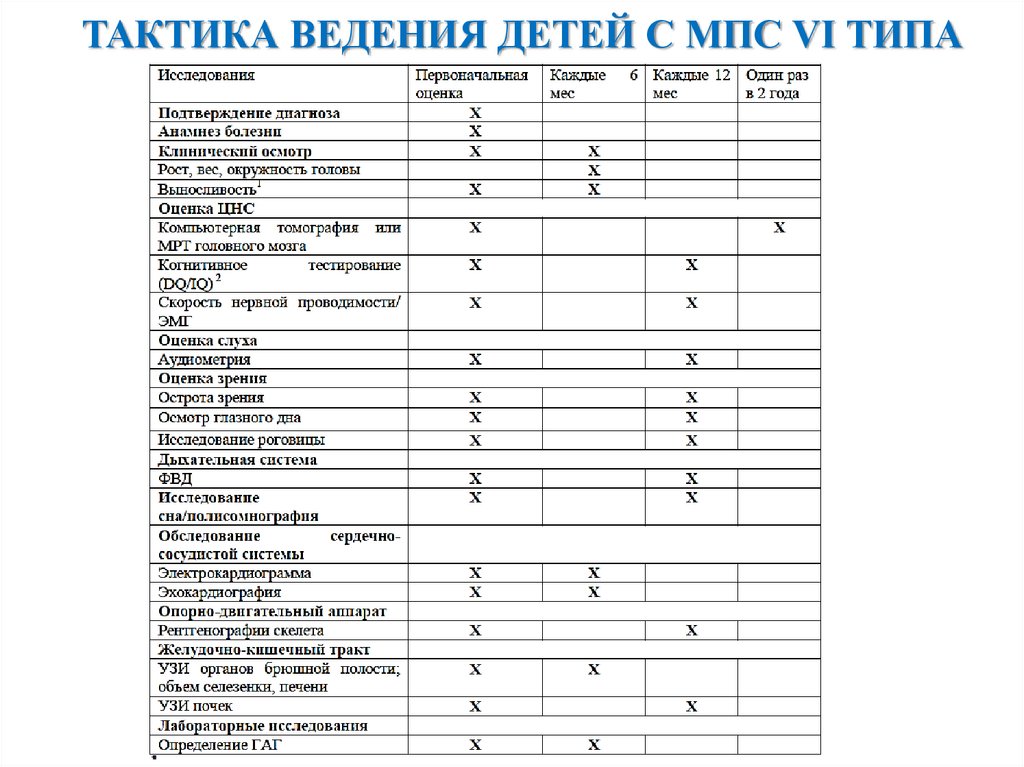
ТАКТИКА ВЕДЕНИЯ ДЕТЕЙ С МПС VI ТИПА34.
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС VI ТИПАПроведение ферментной заместительной терапии (ФЗТ). ФЗТ
проводится препаратом Галсульфаза.
Комментарии: препарат вводится в дозе 1мг/кг один раз в неделю в виде
внутривенной инфузии в течение 4 ч в условиях стационара. Препарат
перед инфузией необходимо развести в растворе натрия хлорида 9 мг/мл
(0,9%). Общий объем инфузии определяется на основании
индивидуального веса пациента. Он составляет 100 мл инфузионного
раствора натрия хлорида 9 мг/мл (0,9%), если вес пациента меньше или
равен 20 кг или 250 мл, если вес пациента больше 20 кг. 2,5% раствора
вводят в течение первого часа, остальной объём (примерно 97,5%) в
течение последующих 3 ч. ФЗТ в виде внутривенных инфузий проводят
еженедельно пожизненно.
35.
ИСХОДЫ И ПРОГНОЗ МПС VI ТИПАПрогрессирование заболевания - скелетные деформации, заболевания
суставов, сердечно-легочные осложнения, слепота и компрессия
спинного мозга приводит к инвалидизации пациента.
В литературе описаны случаи пациентов с выраженными
физическими изменениями, резвившимися уже на первом году жизни;
однако иногда отмечается медленное прогрессирование болезни в
течение многих десятилетий.
Продолжительность жизни может составить примерно 30 лет.
Основная причина смерти – осложнения со стороны сердечнососудистой системы.