


































































































































 medicine
medicineSimilar presentations:


")







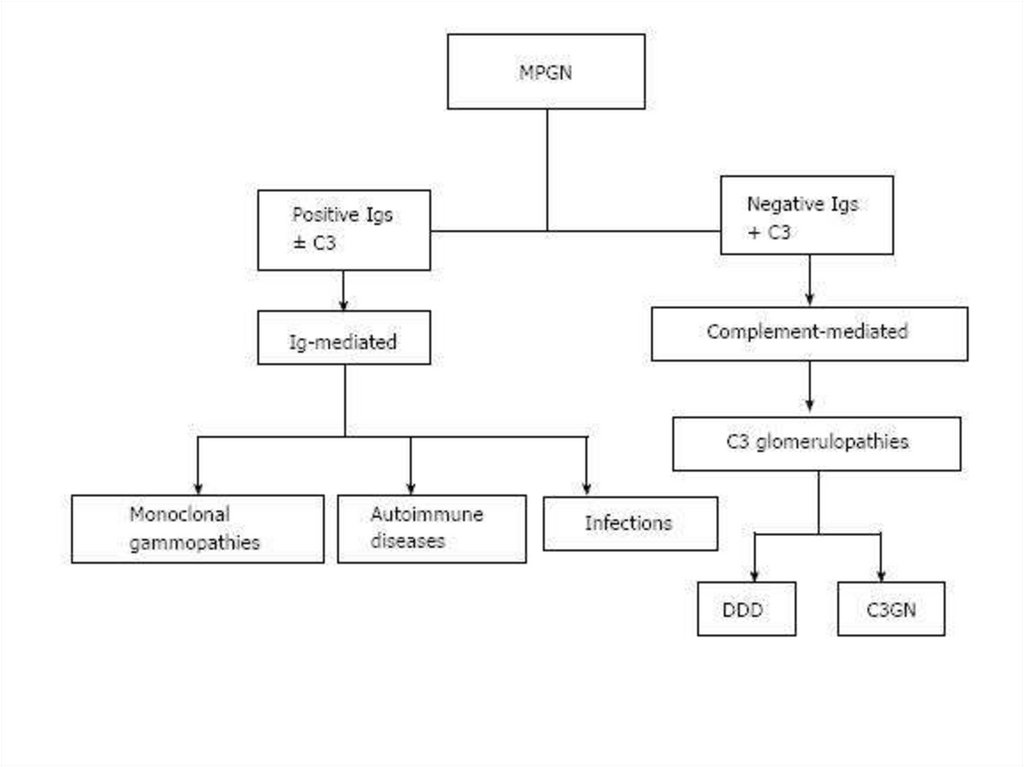
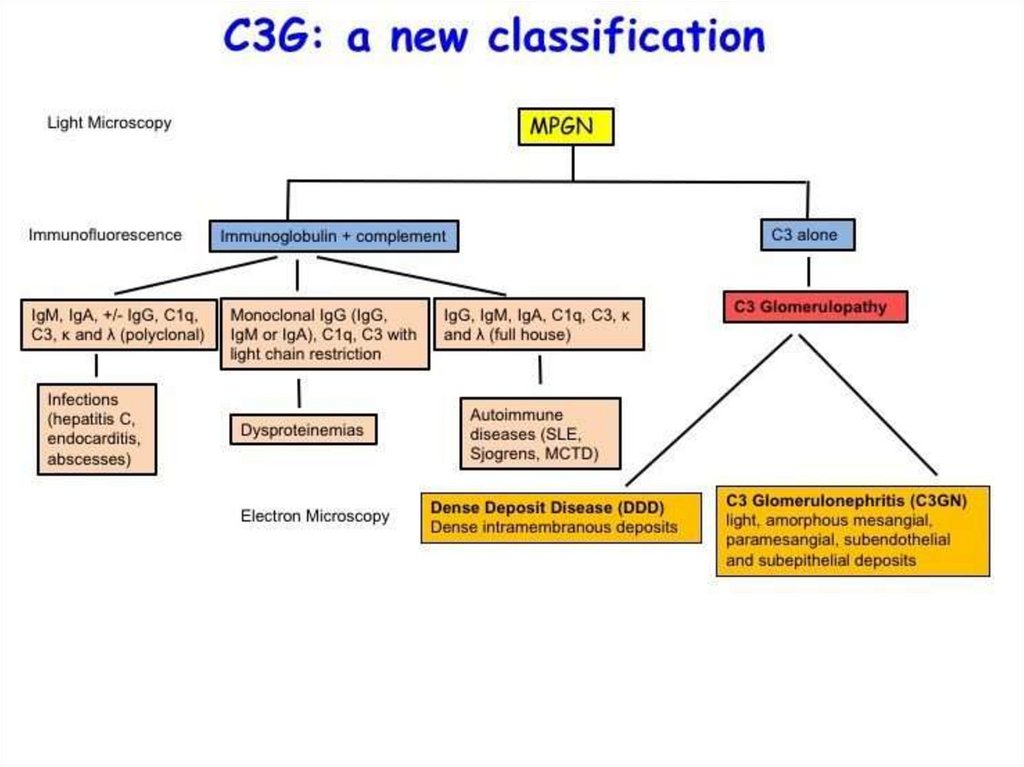
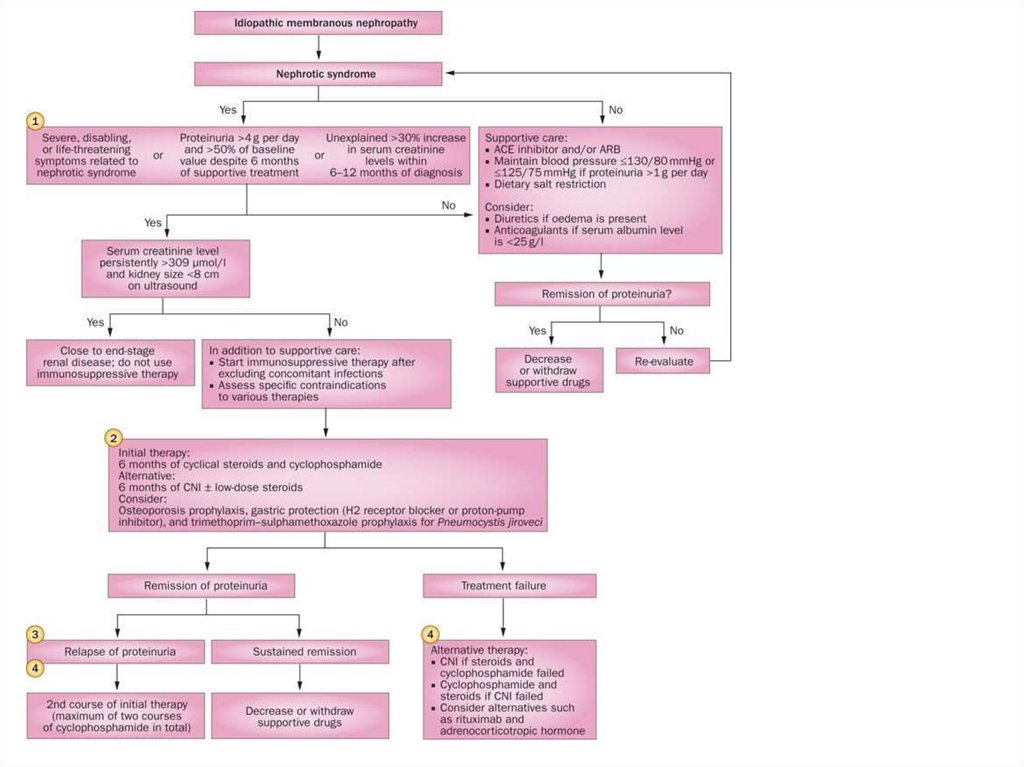
Differential diagnosis of nephrotic and nephritic syndrome
1.
Differential diagnosis of nephroticand nephritic syndrome
2.
3.
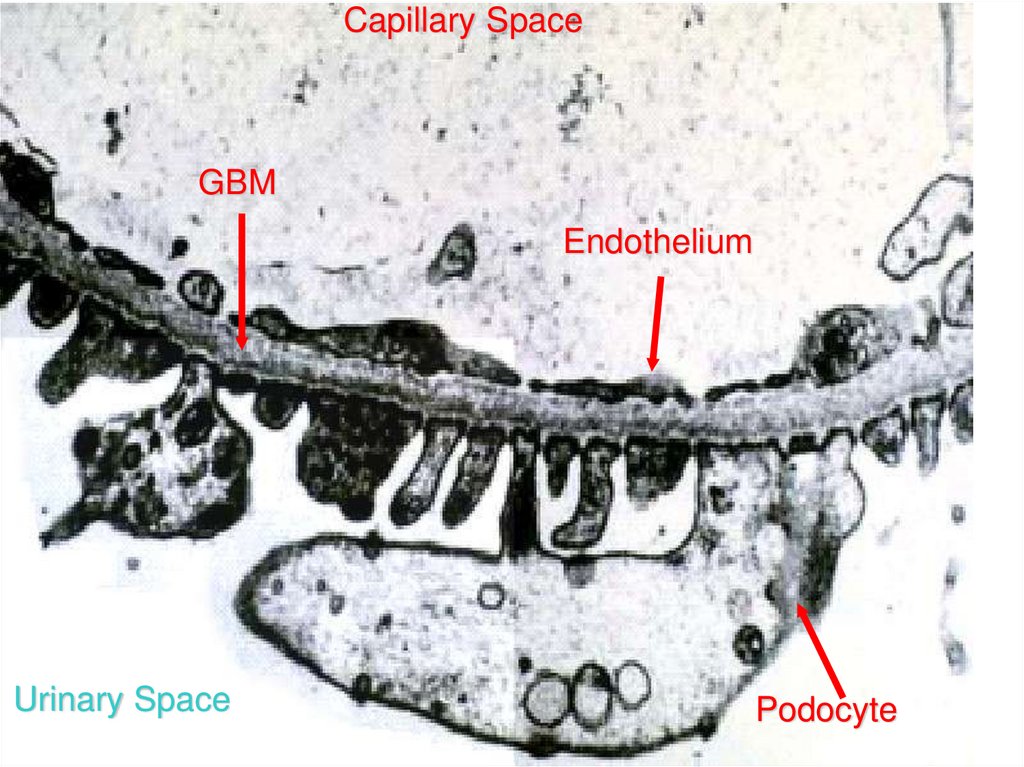
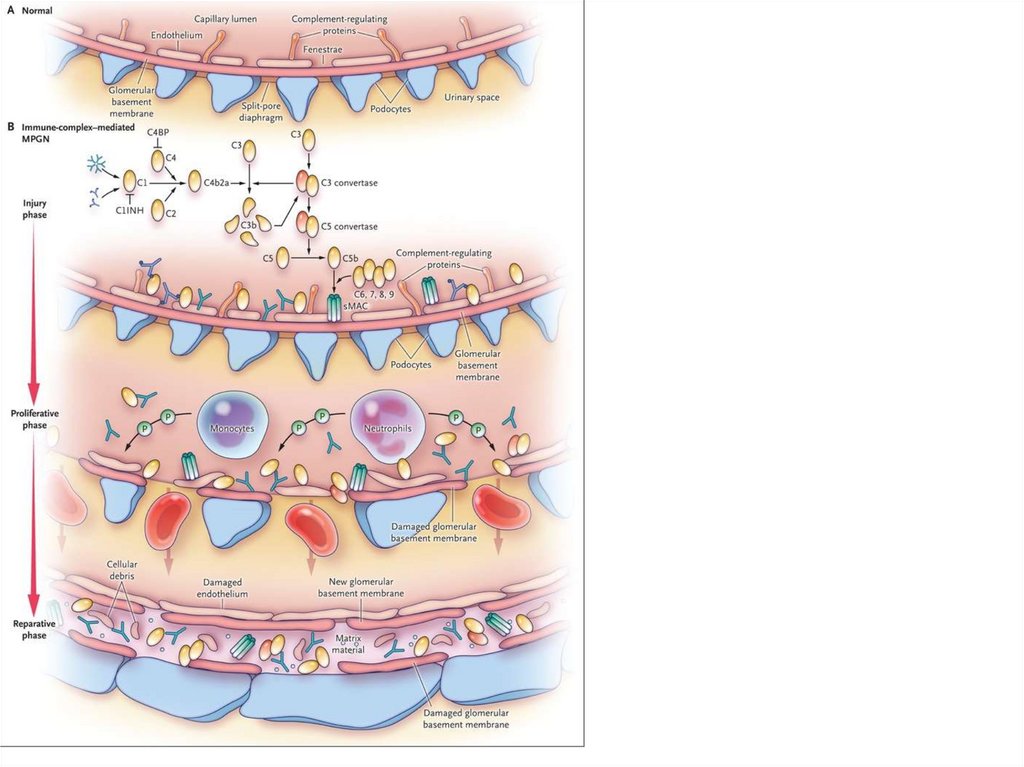
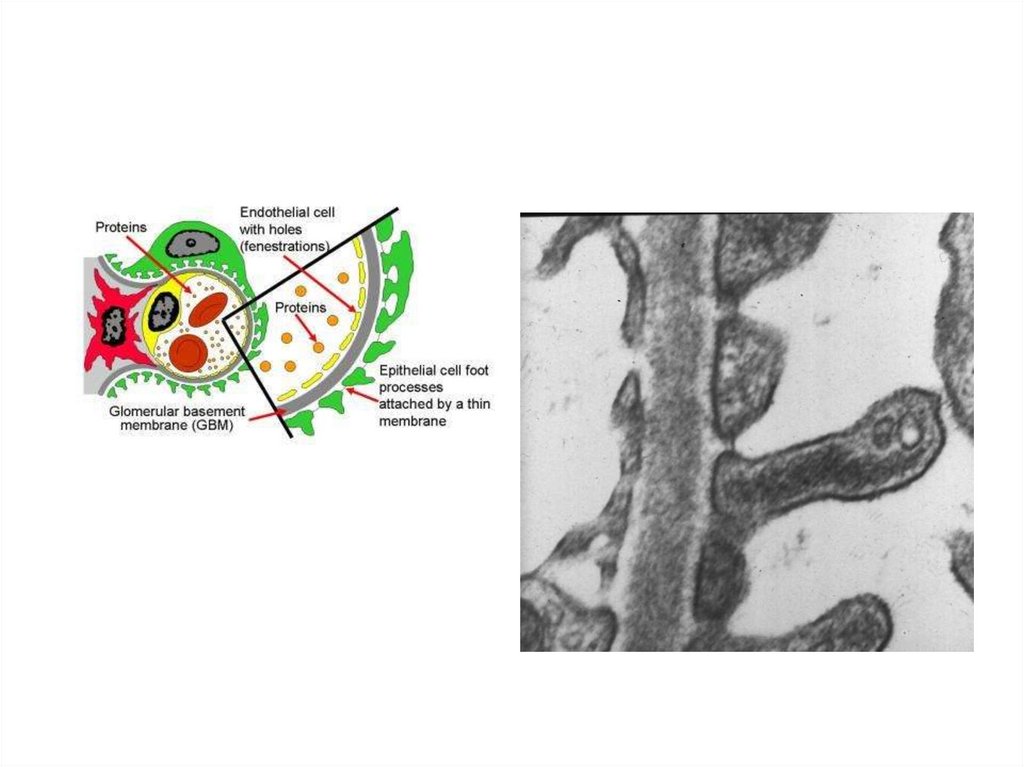
Capillary SpaceGBM
Endothelium
Urinary Space
Podocyte
4.
Classification• Glomerular
- Glomerulonephritis
- Amyloidosis
- Diabetes mellitus
• Tubular and interstitial
- Pyelonephritis
- Tubulointerstitial nephritis
- Nephrolythiasis
• Tumors
- Benign
- Malignant
• Development abnormalities
• Other diseases (parasites, abscess, embolism etc)
5.
Glomerular diseasesNephritic syndrome
(glomerular
inflammation)
- Arterial hypertension
- Oedemas
- Urinary syndrome: microand macrohematuria,
moderate proteinuria
(about1 g /24 hrs); RBC
casts
Nephrotic syndrome
(GBM affection)
- Heavy proteinuria (> 3.5
g/daily)
- Hypoproteinemia
(< 60g/l)
- Hypoalbuminemia(<30
g/l)
- Hyperlipidemia (> 7,5 g/l)
- Oedemas (severe)
6.
Differential diagnosisGllomerular
Tubular
Protein > 1 g daily
< 1 g daily
Casts
absent
WBC – only in nephrotic Present
syndrome
Bacteria - absent
At exacerbation and RF
- olyguria
In pyelonephritis; in TIN
– sterile WBC-uria
May be polyuria + RF
(TIN)
7.
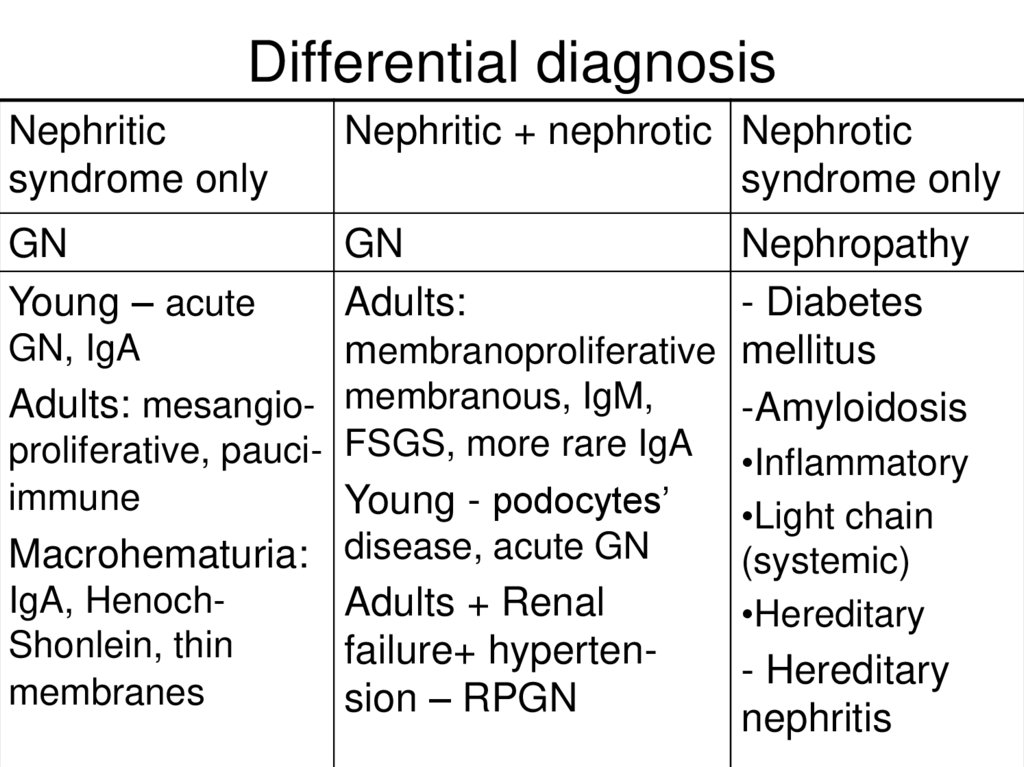
Differential diagnosisNephritic
syndrome only
Nephritic + nephrotic Nephrotic
syndrome only
GN
Young – acute
GN
Nephropathy
Adults:
- Diabetes
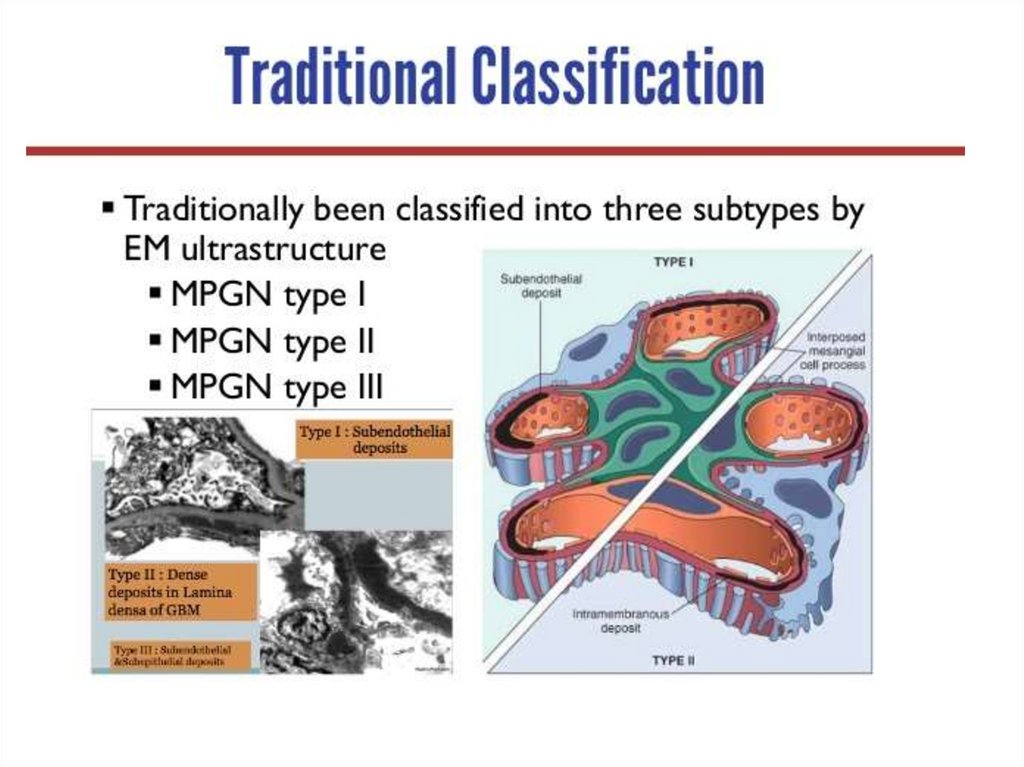
GN, IgA
membranoproliferative mellitus
Adults: mesangio- membranous, IgM,
-Amyloidosis
proliferative, pauci- FSGS, more rare IgA
immune
Young - podocytes’
Macrohematuria: disease, acute GN
IgA, HenochAdults + Renal
Shonlein, thin
failure+ hypertenmembranes
sion – RPGN
•Inflammatory
•Light chain
(systemic)
•Hereditary
- Hereditary
nephritis
8.
Acute nephritis syndromeHeart failure
1st day – 100%; normalizing 1-2 weeks
Usually moderate 150/180/90-100 mm Hg
Very rare (3%)
Bradycardia
33-50% of cases
Diuresis decrease
Thurst
(oliguria; anuria); Most early; if more than 1
week – prolonged course
Oedemas or NS
Early
Back pain
Early
Headache
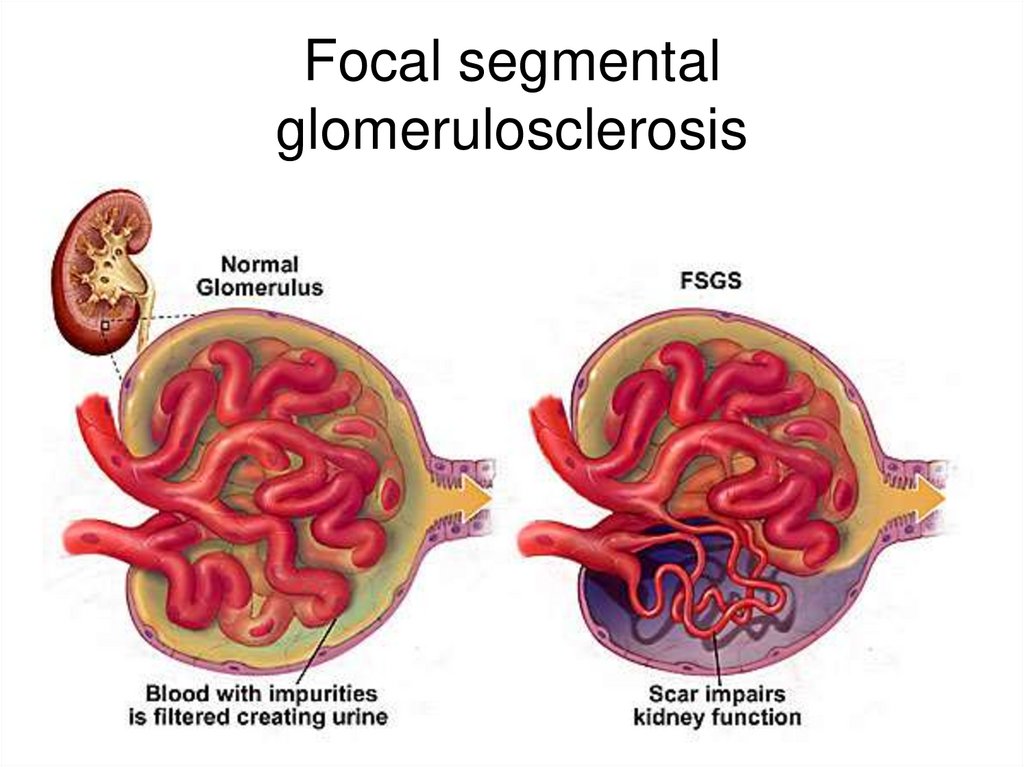
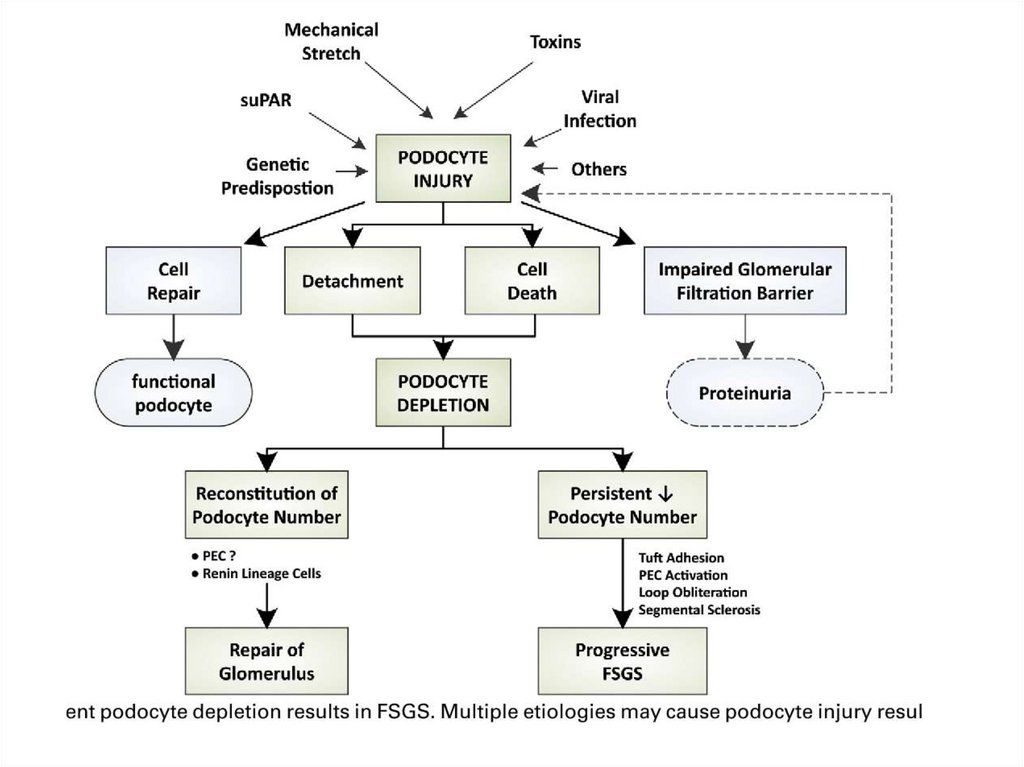
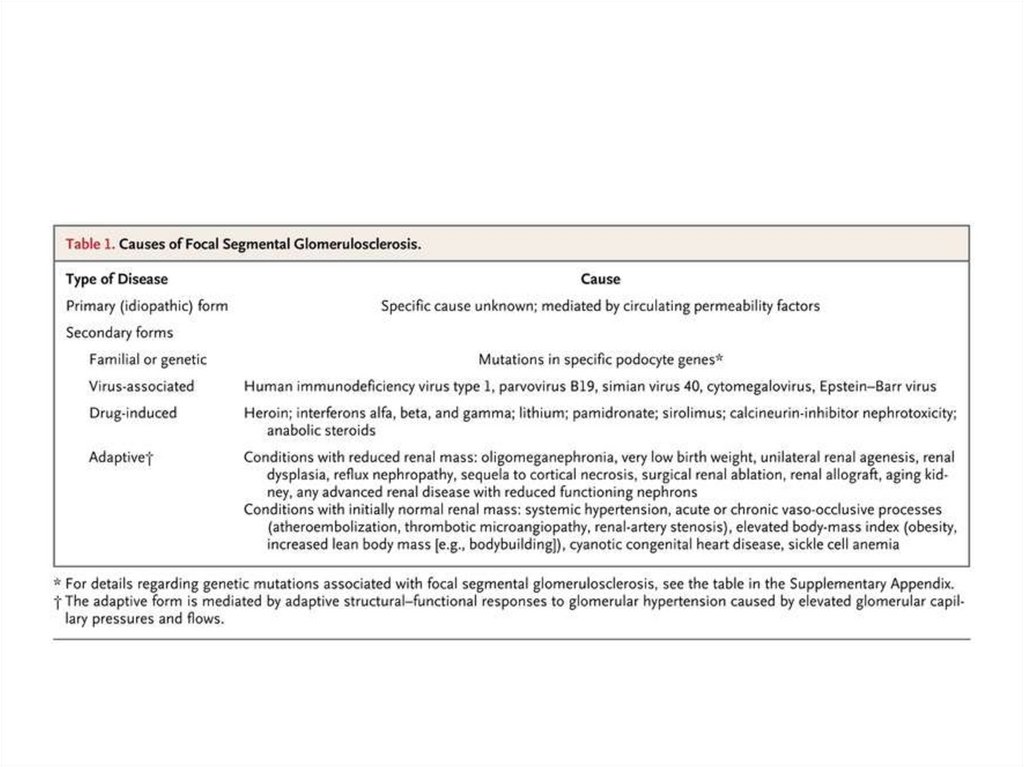
early
Eclampsy
Very rare
Hypertension
9.
Possible in following diseases• Idiopathic
- Acute diffuse endocapillary proliferative GN
(postinfectious)
- Membranoproliferative
- Ig A nephropathy
- Minimal changes disease
- Ig M nephropathy
- More rare – FSGS, rapidly progressing
• Secondary
- Henoch-Shonlein
- SLE (lupus-nephritis)
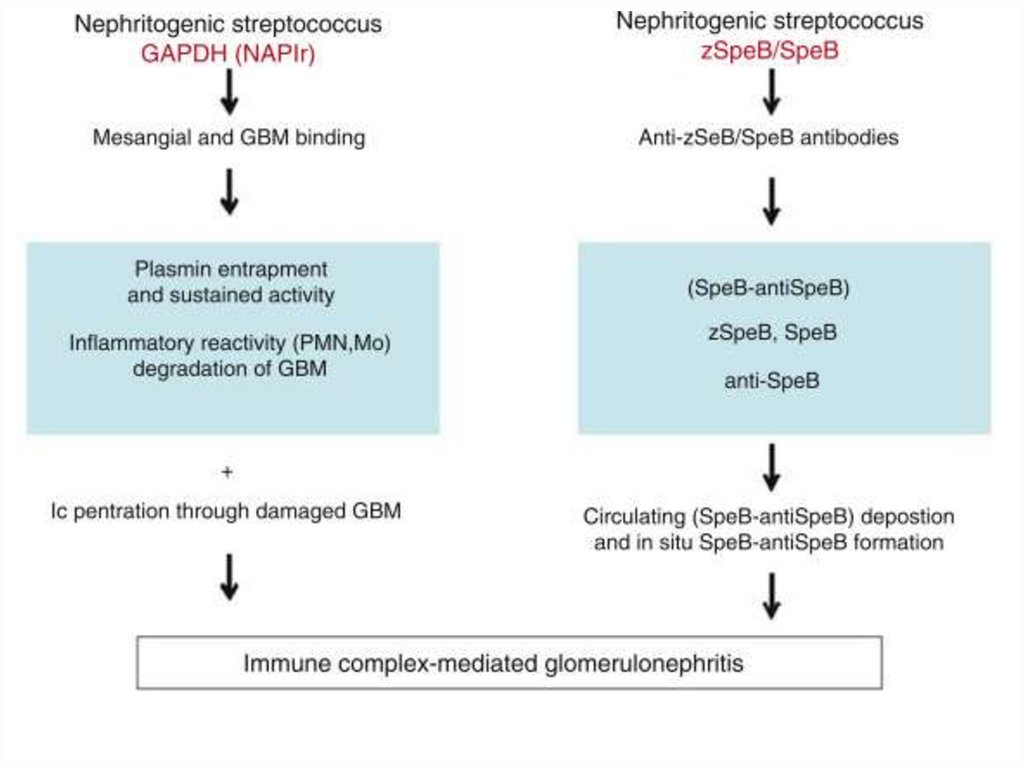
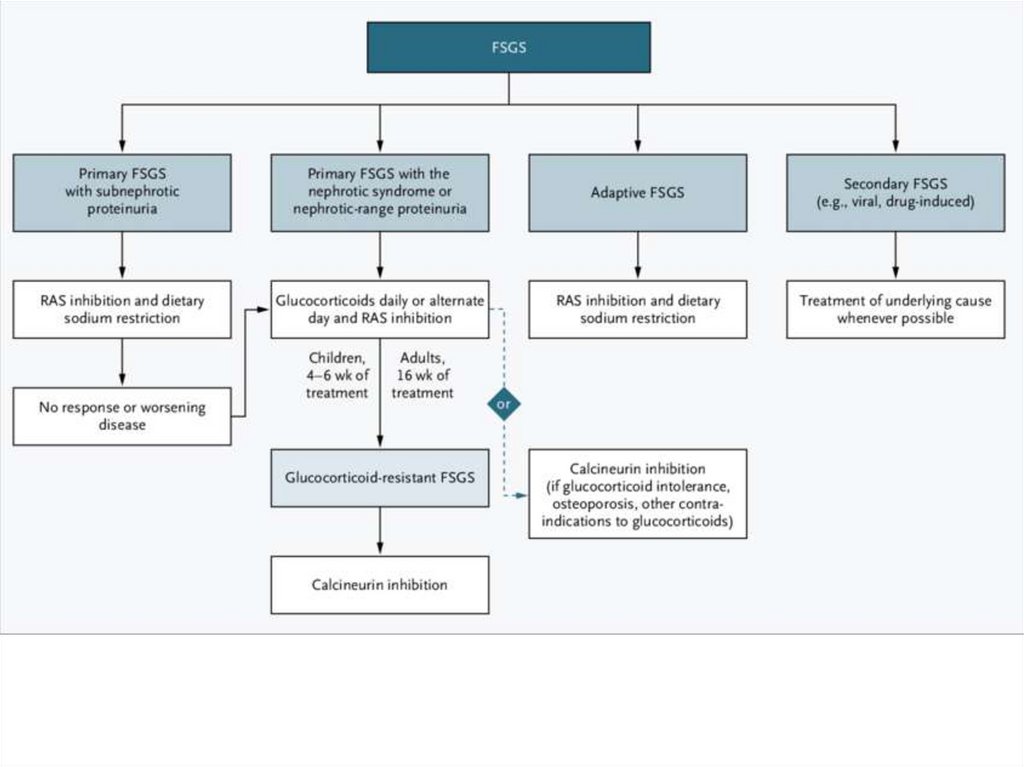
10.
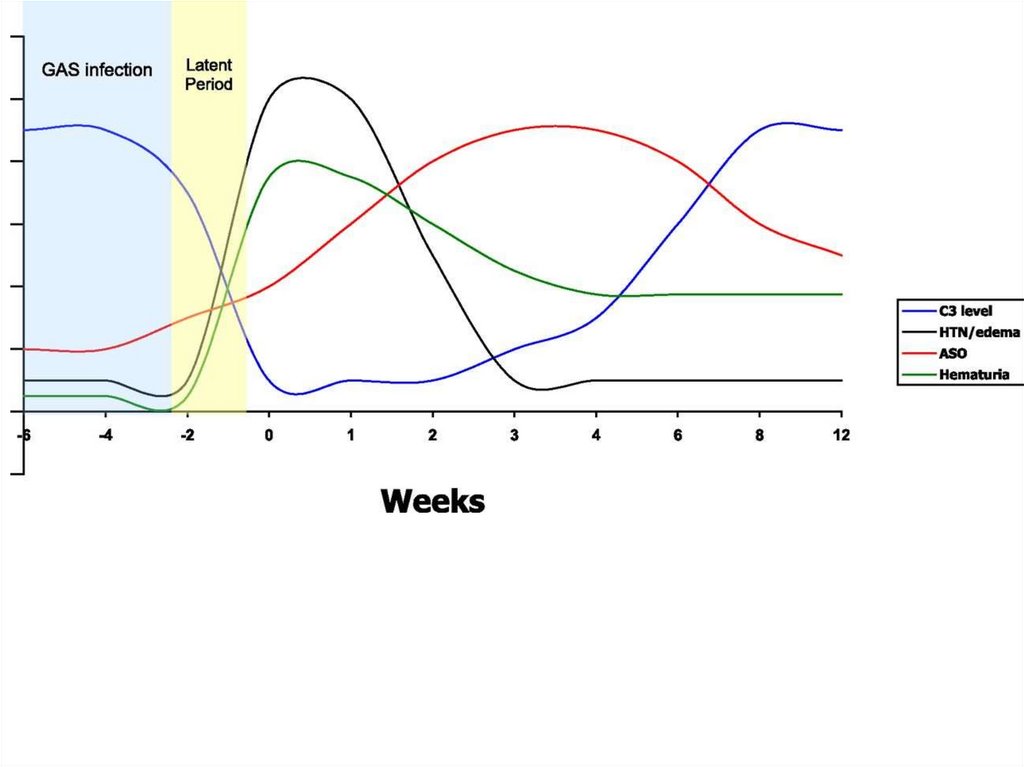
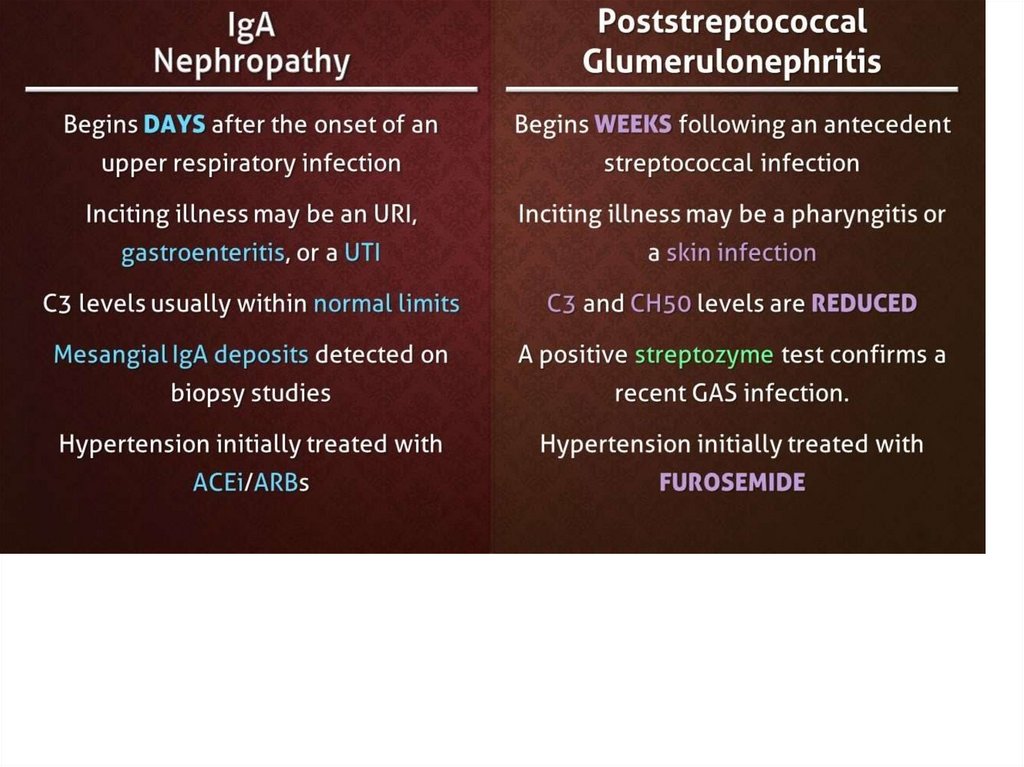
Postinfectious3-4 weeks before onset – usually infection
episode
Aethiological factors
• Streptococci: group A (β-hemolytic); types 1, 3,
4, 12, 49 (more often 12) - 15-20% cases;
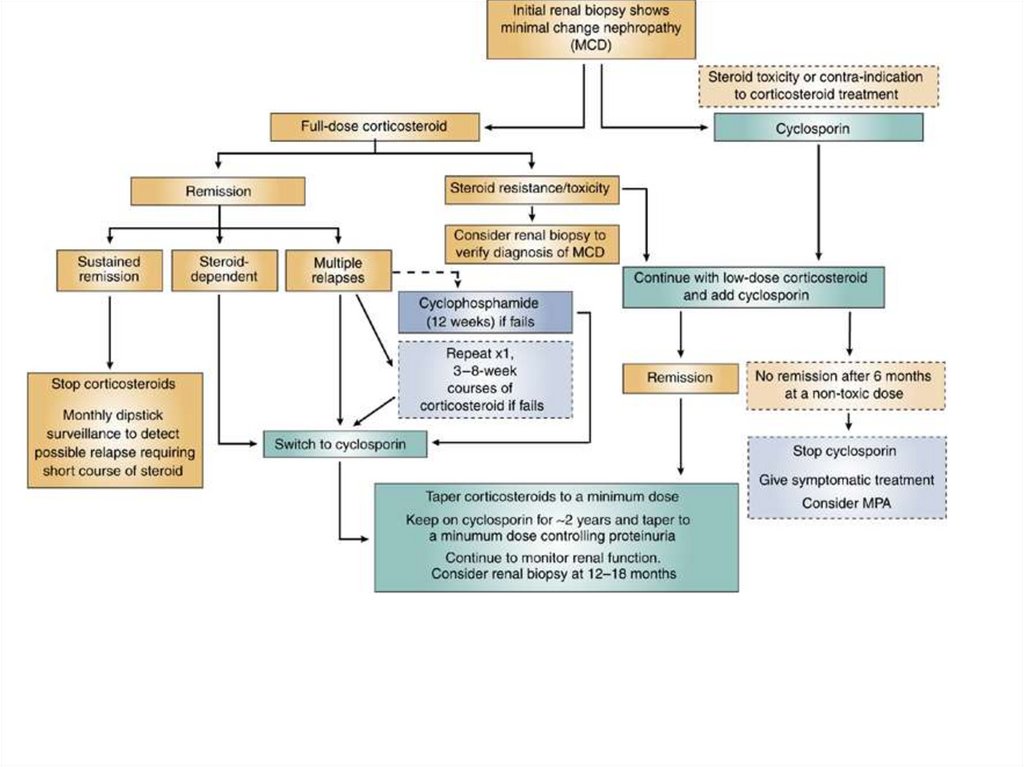
more common – pharyngitis, impetigo
• Other bacterial and viral agents
• Vaccination
11.
12.
13.
14.
15.
Phase of pathological processExcudative: hemorrhagic excudate in
glomeruli (clinically - hematuria)
Excudative-proliferative
Proliferative
16.
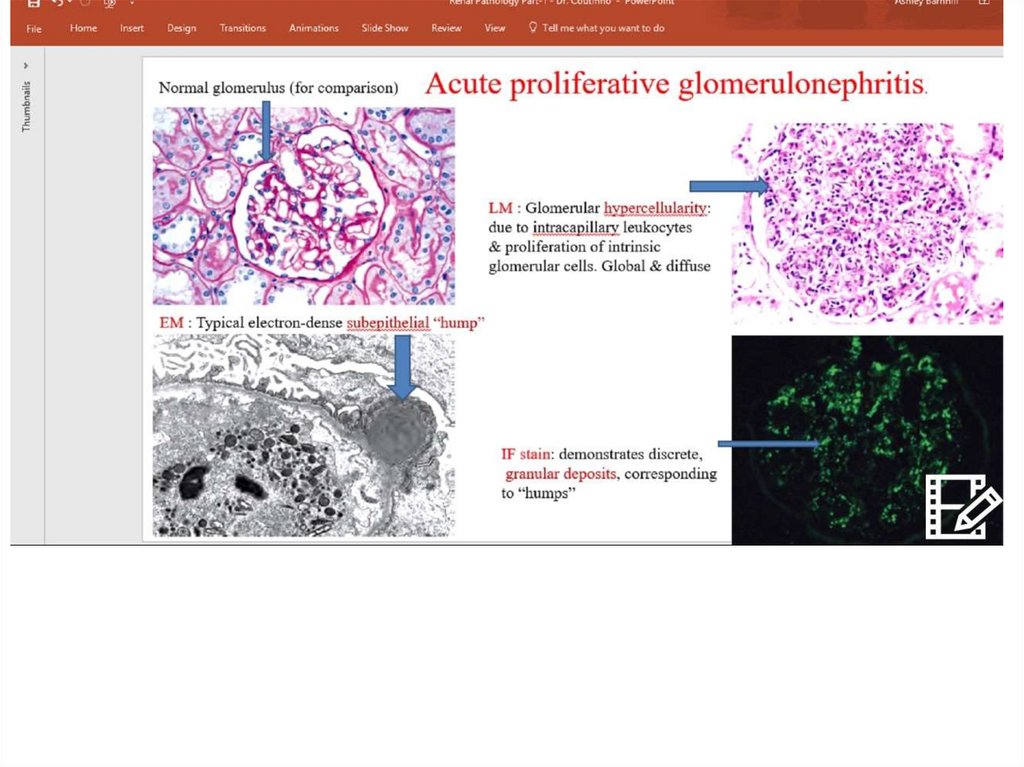
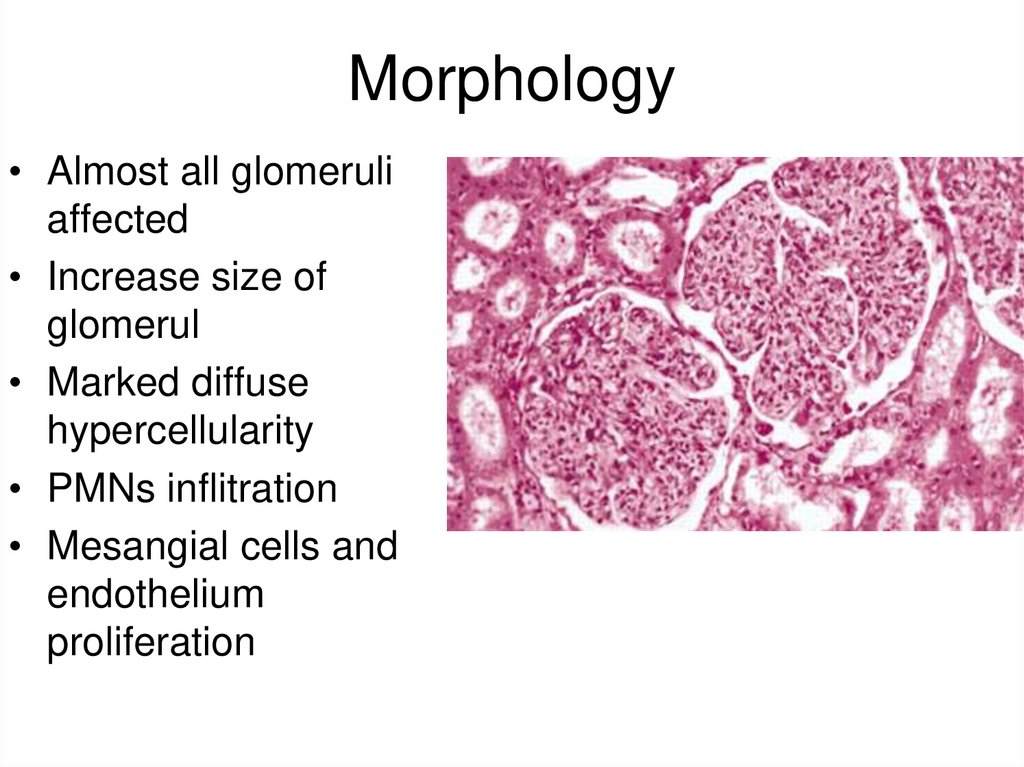
Morphology• Almost all glomeruli
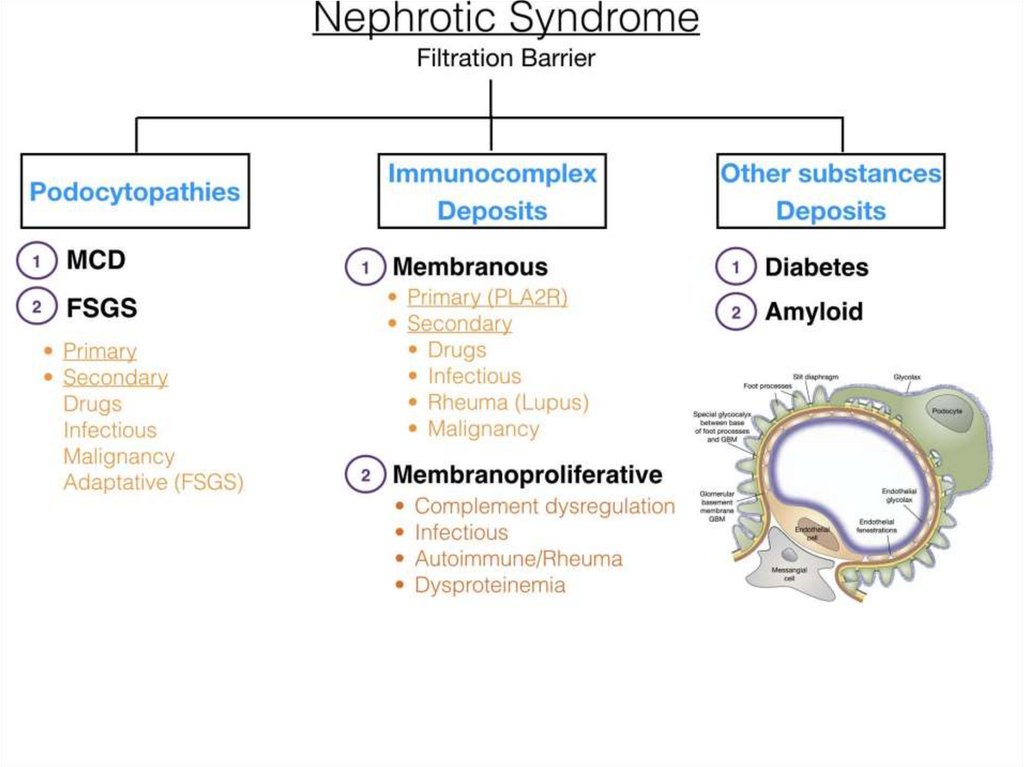
affected
• Increase size of
glomerul
• Marked diffuse
hypercellularity
• PMNs inflitration
• Mesangial cells and
endothelium
proliferation
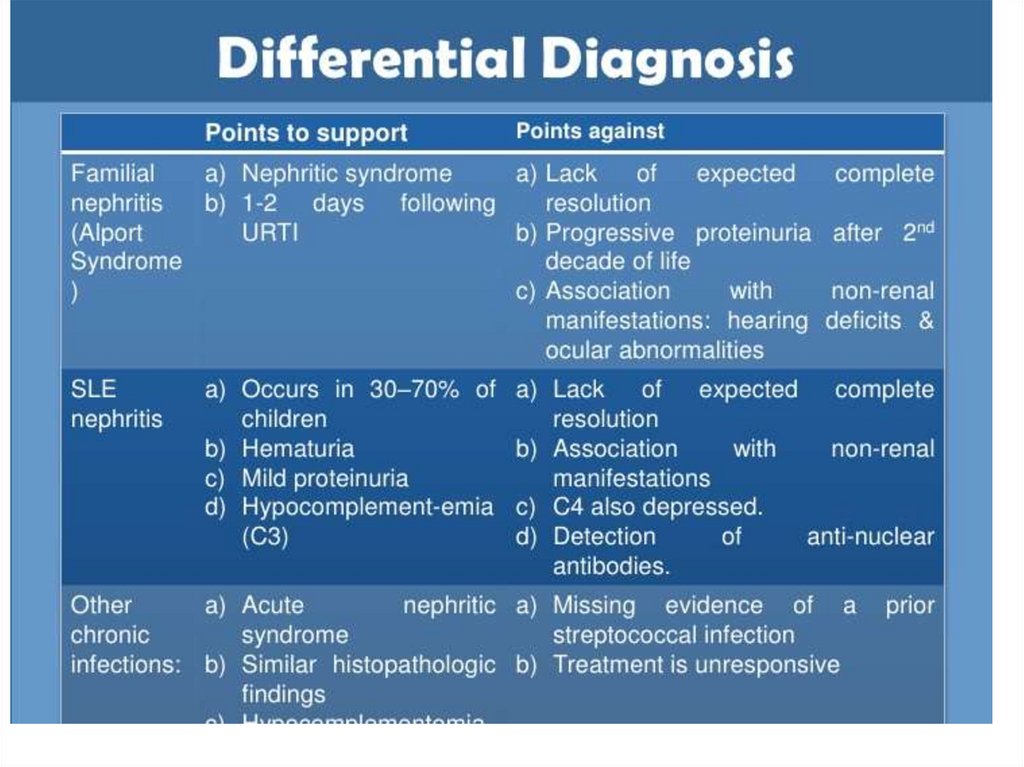
17.
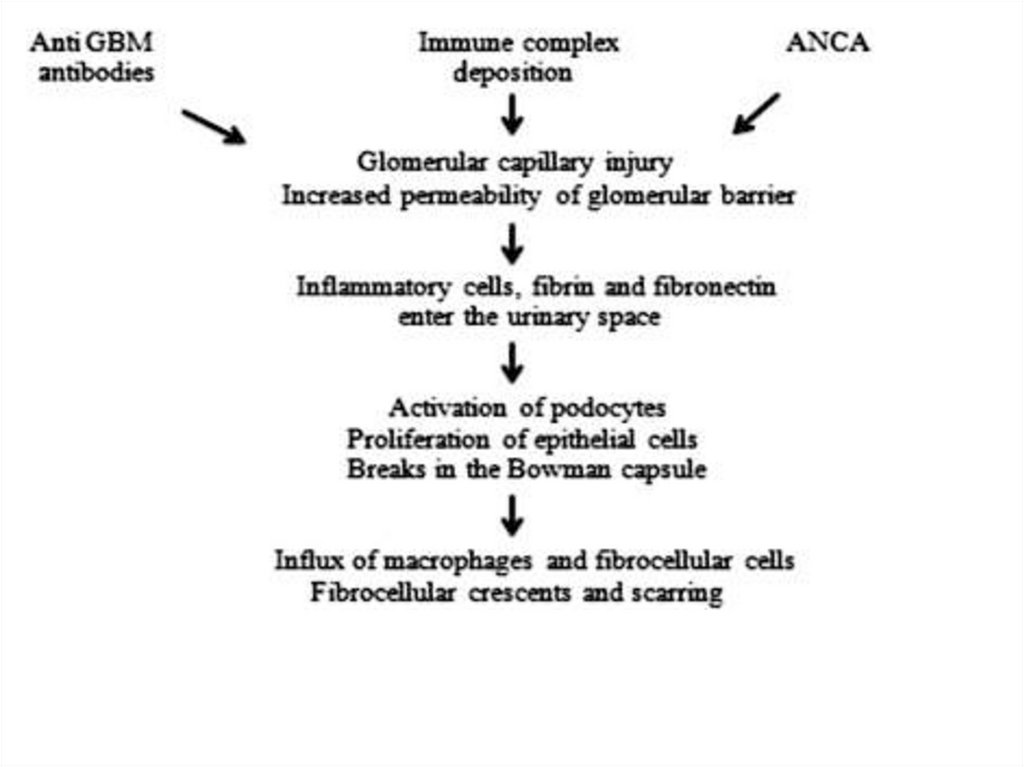
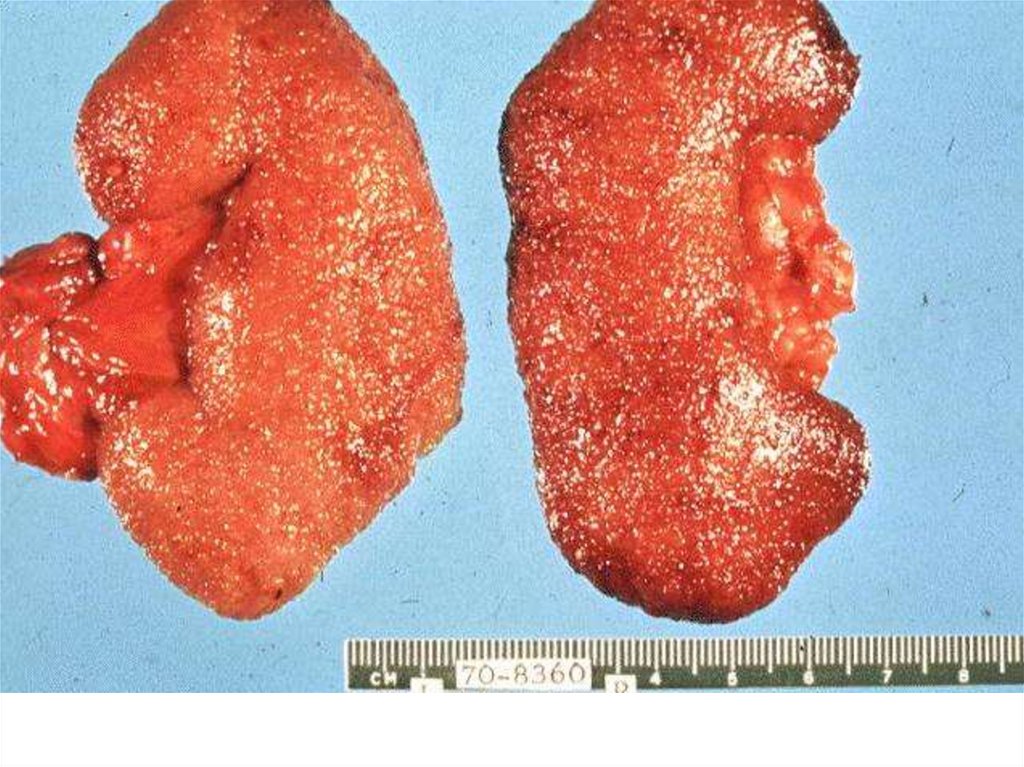
Immune fluorescence and electronmicroscopy
• Granular small deposits
(«stars in the sky»)
• Mesangial type –IgG C3
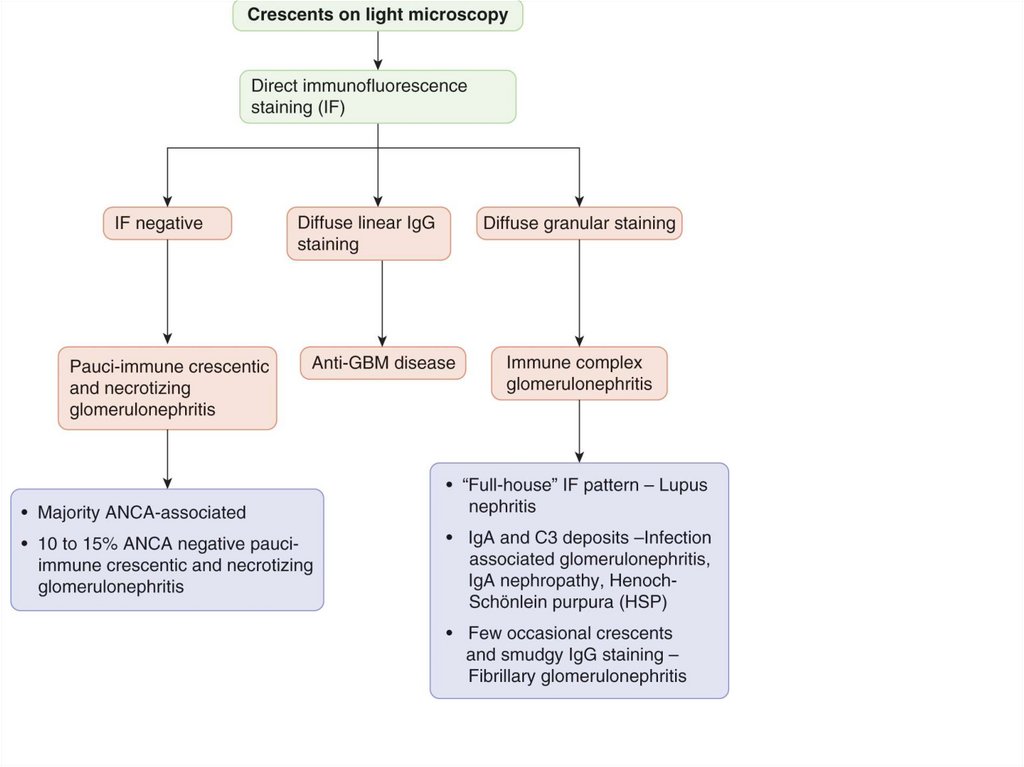
deposits in mesangium
• Large dense
subendothelial deposits
(«humps» - less
favourable - photo)
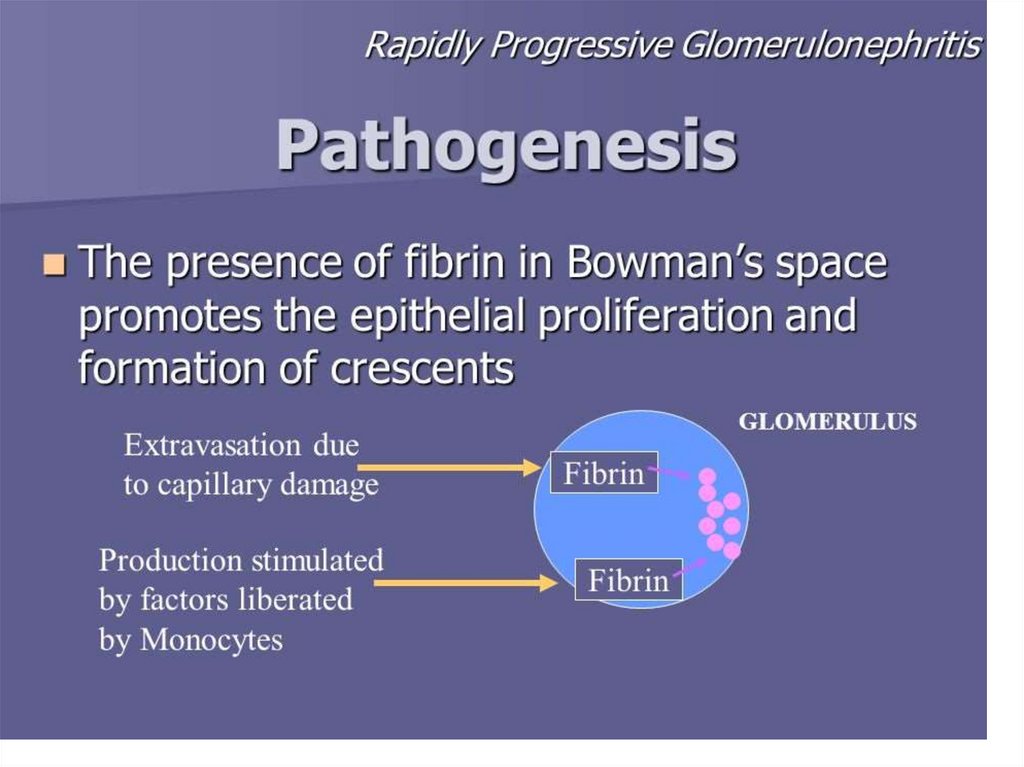
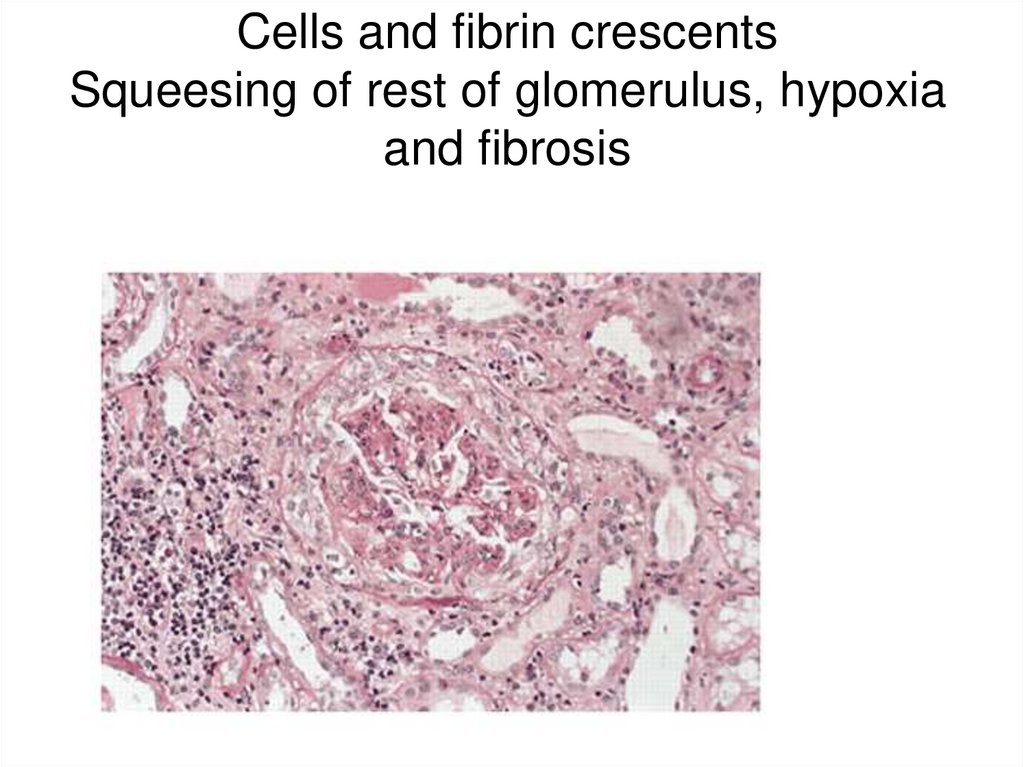
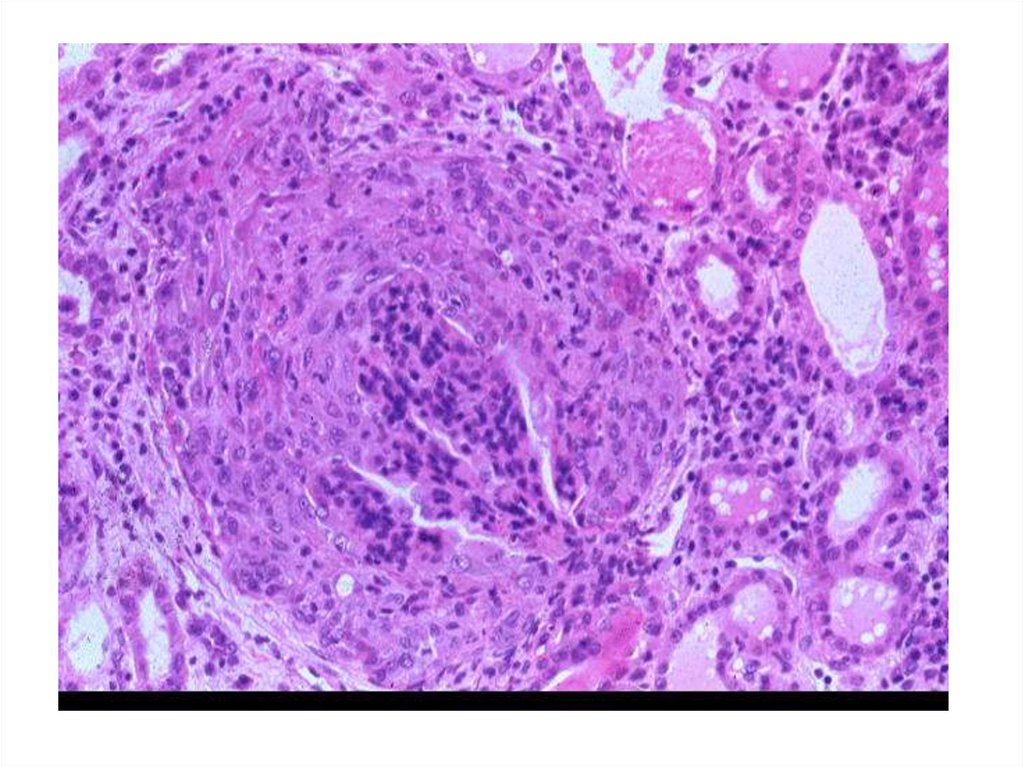
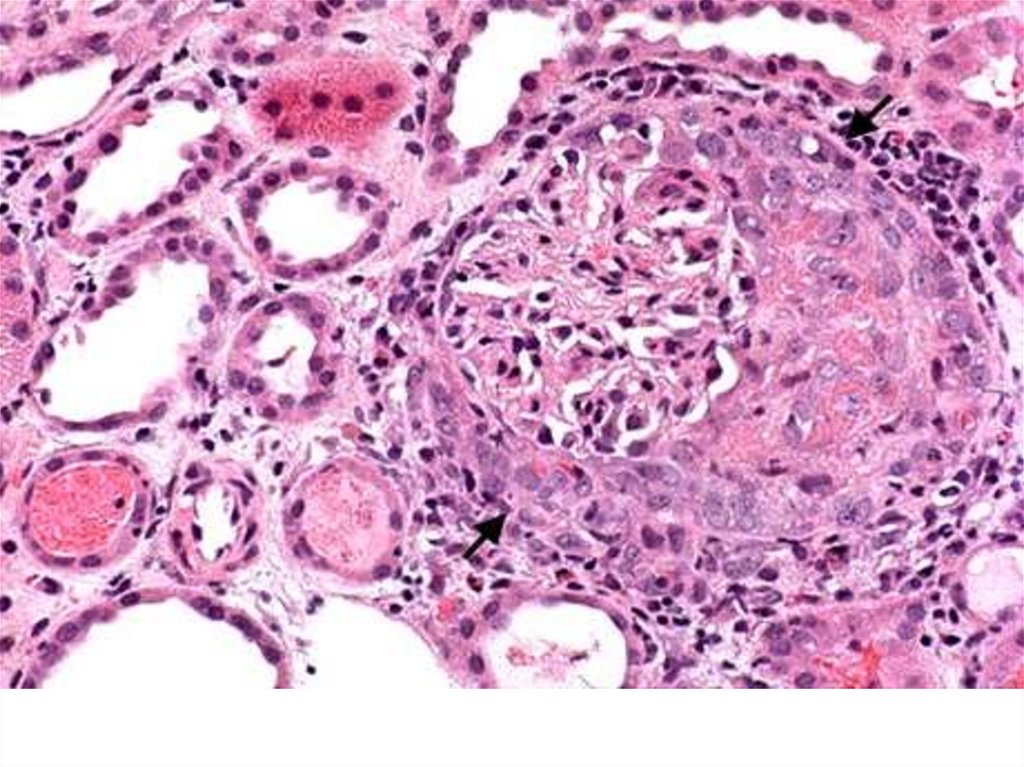
• Immune negative type–
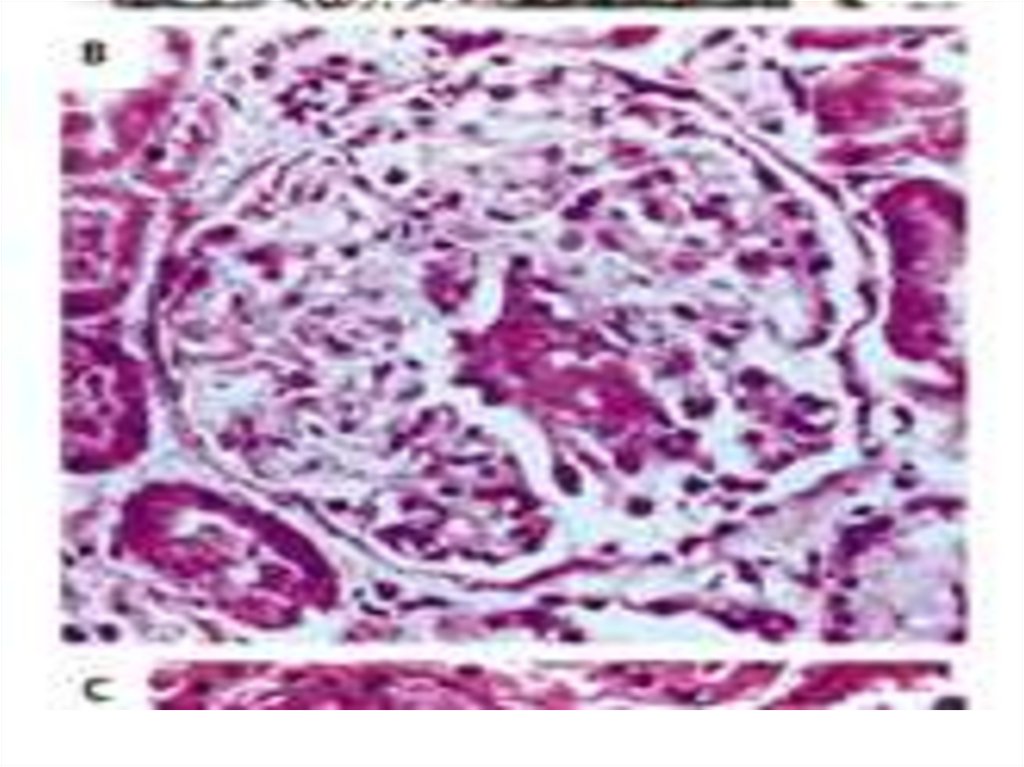
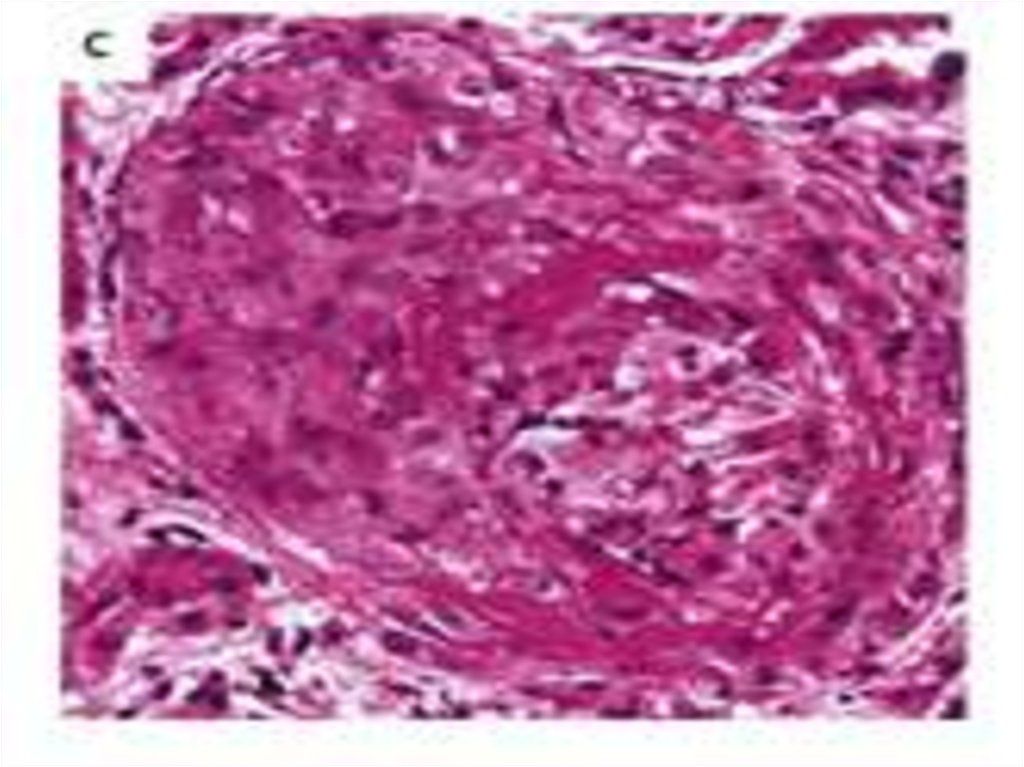
absence of IgG and
complement deposits
18.
Course• Monosymptomous (mostly – urinary
syndrome, in first days – moderate
hypertension)
• Full complex: oedemas, hypertension,
urinary syndrome
• Nephrotic: nephrotic syndrome,
hypertension
In 60-80 – monosymptomous course
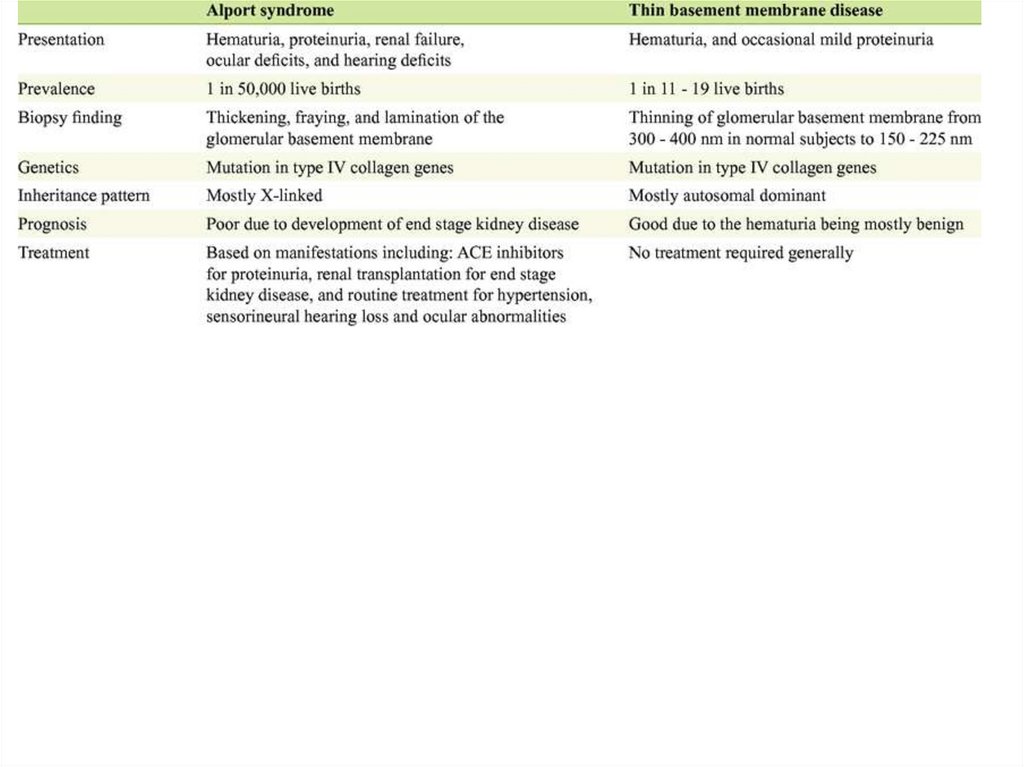
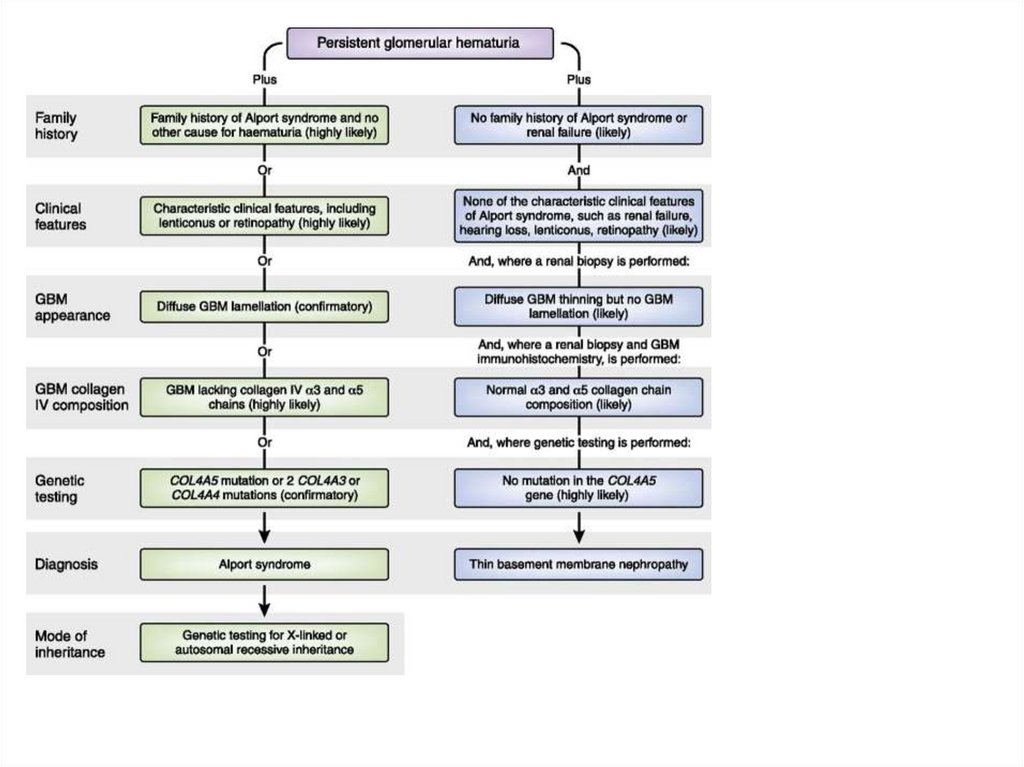
dominated, now – full complex
19.
Differential diagnosis• In acute endocapillary proliferative GN –
cyclic course of the disease
• Early (1 week) – diuresis increase,
oedemas disappear
• BP – 1st week
• Urine analysis – normal in 6 month (more
rare – 12)
• Kidney biopsy
20.
21.
22.
23.
24.
Treatment: drugs• Pathogenetic
- ACE inhibitors – decrease blood pressure in
glomeruli (better – monopril) – from 1st day
(dose depend on BP)
- Antiinflammatory – if symptoms persist after 2
days of symptomatic treatment
• Symptomatic
- Diuretics: Furosemid 40-80 daily up to edemas
disappearing; in severe cases i.v. 80-300 mg
daily every 2nd day
- Antihypertensive: ACE inhibitors
25.
26.
renal biopsy• patients with rapidly progressive renal failure
• If evidence of crescentic glomerulonephritis with
more than 30% of the glomeruli involved, a short
course of intravenous pulse steroid therapy is
recommended (500 mg to 1 g/1.73 m2 of
methylprednisone qd for 3-5 d)
• Long-term treatment with steroids or
immunosuppressives is not recommended.
27.
Chronic GN• With macrohematuria syndrome (IgАnephropathy; Henoch-Shonlein syndrome, thin
membranes disease)
• With neprhotic syndrome:
- GBM affection (membranous,
mebranoproliferative, minimal changes, focal
segmental glomerulosclerosis, IgMnephropathy)
• Rapidly progressive (crescentic)
• Mesangioproliferative idiopathic
(including focal)
28.
Isolated urinary syndrome• Focal mesangioproliferative GN
• Mesangioproliferative GN
• Acute endocapillary proliferative GN rare
29.
Idiopathic mesangioproliferativeglomerulonephritis
• Prevalence: The most common variant of
the disease (70% of all cases of
glomerulonephritis)
• Prevalence: more often in young males
• Aethiology: more often unknown
(revealed during preventional medical
examinations, when occasionally urine
changes are found); in women may
manifest as nephropathia of pregnant.
30.
Morphology Light microscopy• Focal and diffuse variants
• Diffuse variant
• diffuse proliferation of mesangial cells;
increased mesangial matrix
• thinned basement membrane
• sclerosis (globular or segmental)
• inosculations between glomerular loops
and capsule
31.
Immunofluorescent investigation:
In immunopositive forms – focal or linear
Ig deposits
Electron microscopy:
Increase of number and size of
mesangial cells, thinnering of basement
membrane; electron-dense deposits of
immune complexes.
32.
Clinical manifestations:• Urinary syndrome: for the long time – the
only syndrome of the diseases
• Oedemas and hypertension: appear
when the sclerotic changes in glomeruli
develop; oedemas are usually mild and
located in suborbital regions.
• Prognosis: chronic renal failure - 15-20
years after the onset of the disease.
33.
Treatment:Regimen: to avoid cold
Antiinflammatory: only in exacerbations
Prednisolone
1 month - 60 mg daily with subsequent
gradual reducing of dose up to 20 mg daily
• up to 1 year – 20 mg subsequent intake
• Antyhypertensive: ACE inhibitors when
hypertension develops
34.
Focal mesangioproliferativeglomerulonephritis
• only some glomeruli involved
• segmental (some of the glomerular loops)
mesangial cells proliferation
• dystrophy and atrophy of some tubules
35.
• Due to focal affection – minimal clinicalmanifestations.
• Oedemas – absent
• BP – normal
• Urinary syndrome: moderate proteinuria up to
1 g daily
• Course: very slow progression.
• Treatment: only regimen (avoid cold) and diet
(moderate restriction of salt and proteins)
36.
Urinary syndrome + hypertension• Mesangioproliferative GN
• Acute endocapillary proliferative GN rare
• Ig A nephropathy
• Membranous nephropathy (early
phase)
37.
Macrohematuria + back pain• Isolated (accompanied by proteinuria and
casts)
- Ig A nephropathy
- Thin membranes disease
• Systemic manifestations
- Skin, intestine, joints – Henoch-Shonlein
- Lungs, progressive RF - Goodpasture
• Non-GN: urolythiasis, diseases of
gallbladder, renal cancer; more rare – TIN,
PN
38.
Ig A nephropathy• Primary IgA nephropathy is an immunecomplex–mediated glomerulonephritis
defined immunohistologically by the
presence of glomerular IgA deposits
accompanied by a variety of
histopathologic lesions
• most common throughout the world
39.
Causes of Ig A neprhopathy-
Primary
Ig A nephropathy
Henoch-Sholein purpura
Secondary
Autoimmune diseases of liver, hepatitis B,
schistosomatosis
Autoimmune diseases of intestine
Hepatitis herpetiformis, psoriasis
Lungs: bronchiolitis obliterans, hemosiderosi, sarcoidosis,
cystic fibrosis
Neoplasms: pancreas, lung, larynx
Infections: HIV, leprosy
Autoimmune: SLE, RA, cryoglobulinemia, seronegative
arthrites, Goodpasture, Becchet, Wegener, ANCAvasculitis
40.
Predisposition:• To IgA (including abnormal) synthesis
by mucosal and blood cells
• Increase production of abnormal IgA
as a response to mucosal exposure of
infectious antigen
• IgA-containing deposits in kidneys
• If liver elimination fails – systemic
disease (Shonlein-Henoch)
41.
42.
IgA nephropathy• Prevalence: highest in Asia (Singapore,
Japan, and Hong Kong), Australia, Finland,
and southern Europe (20 to 40 percent). US
– 2-10
• 20-30 y.o.
• More common in whites and Asians
• No consistent genetic abnormalities; in
familial cases – association with genes
responsible for Ig A production
• Re-occuring in kidney transplant
43.
FamilialUp to 15% of cases of the common primary glomerulonephritis IgA nephropathy
(IgAN) are classified as 'familial', indicating an underlying genetic component
Familial IgAN is probably underdiagnosed, as urinary abnormalities that cause clinical
suspicion can occur intermittently
Analysis of extended kindreds has shown that some patients with 'sporadic' forms of
IgAN share common ancestors
Families affected by IgAN are often affected by other glomerular diseases such as
IgM nephropathy, Henoch-Schönlein purpura nephritis and focal segmental
glomerulosclerosis
Autosomal dominant inheritance with incomplete penetrance is a likely form of
transmission in families with IgAN
44.
45.
46.
47.
48.
49.
Main clinical features• Associated with current respiratory tract
infection (not after, but during infection)
• Macroscopic hematuria + unilateral back
pain
• More rare – isolated US or nephrotic
syndrome
• Up to 20% - asymptomatic undiagnosed
course and presentation at late stage by
RF
50.
Prognosis• 15-40% - end-stage RF, develops
after prolonged course of disease
• In most of patients – RF in about
15 or more years
51.
52.
53.
Treatment• ACE
• Steroids (1 g i.v. per day for three days at the
beginning of months 1, 3, and 5 + 0.5 mg/kg)
• n–3 polyunsaturated fatty acids (fish-oil
supplements) may involve mechanisms
reducing inflammation and glomerulosclerosis
• Tonsillectomy – used in some countries
• Transplantation – in end-stage (10% of all cases
of transplantation)
54.
55.
56.
57.
Thin membranes disease• Hereditary condition
• Inflammatory changes absent or minimal
• Included to the group of minimal changes
nephritis
• Thickness of GBM is about 240 nm (normal
– 350)
• Clinical manifestations – macrohematuria,
unilateral back pain, disuria
• Renal failure and hypertension – not
reported
58.
59.
60.
61.
62.
63.
64.
GNs with nephrotic syndrome• Membranous
• Membranoproliferative
• Mesangioproliferative with IgМ
deposits
• Focal segmental glomerulosclerosis
• Minimal changes disease (podocytes
disease)
65.
66.
Clinical course (general)• Acute onset – all variants
• Hypertension at onset, renal failure in
5-7 years in 4 diseases except
minimal changes
• Development of chronic renal failure
in minimal changes diseases is not
reported
67.
Treatment (general)• All diseases require steroids from the
onset
• In all variants except minimal
changes cytotoxic drugs are usually
added
• FSGS is usually steroid resistant
68.
Membranoproliferative• Other name - mesangiocapillary
glomerulonephritis or MCGN
• mesangial cells proliferation and basement
membrane changes
• Usually below 30, with equal rate of men and
women.
• revealed in 20-30% of glomerulonephritis cases
and in respectively 8% (in children) 14% (in
adults) of nephrotic syndrome cases.
69.
• Aethiology: in most cases idiopathic,may be in infectious endocarditis,
hepatitis B and C infections, systemic
lupus erythematosus.
• Pathogenesis: Complement
abnormalities, indicating predominant
activation of the alternative pathway of
complement
70.
71.
72.
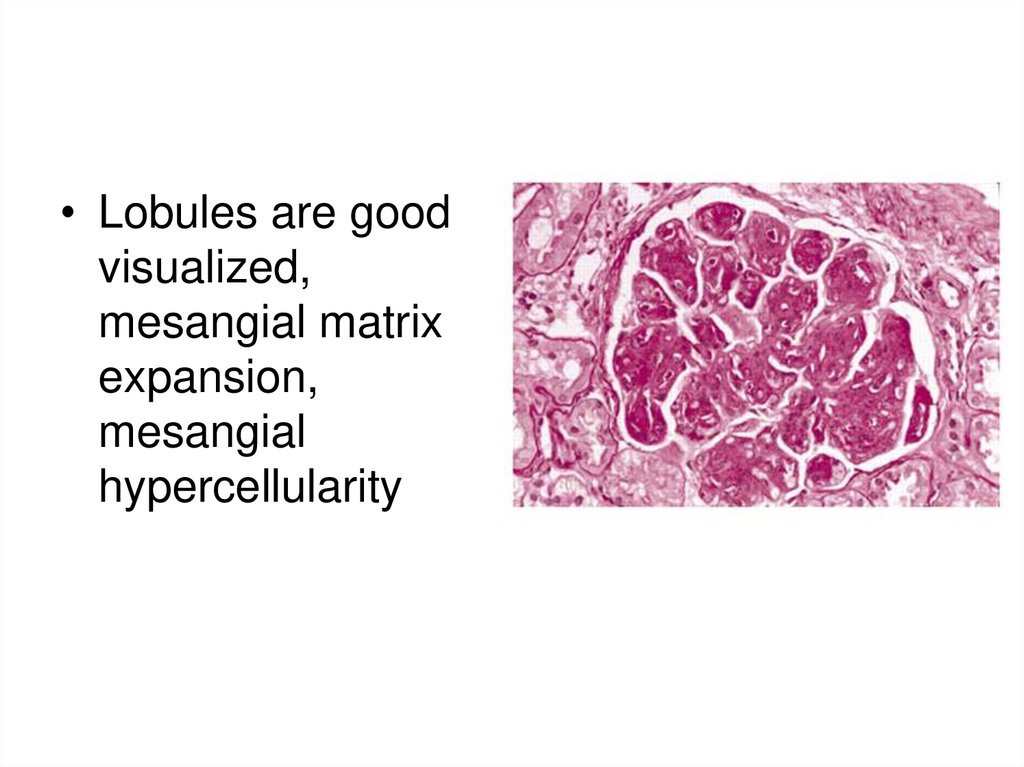
• Lobules are goodvisualized,
mesangial matrix
expansion,
mesangial
hypercellularity
73.
- Basement membrane affection –3 variants:
• I type – subendothelial deposition of immune
complexes
• II type – dense deposits inside the basement
membrane (disease of dense deposits)
• III type- severe increase of deposits number
causing splitting of the basement membrane and
contacts between intramembraneous and
subendothelial deposits; sometimes with
duplicated appearance of the glomerular
basement membrane.
74.
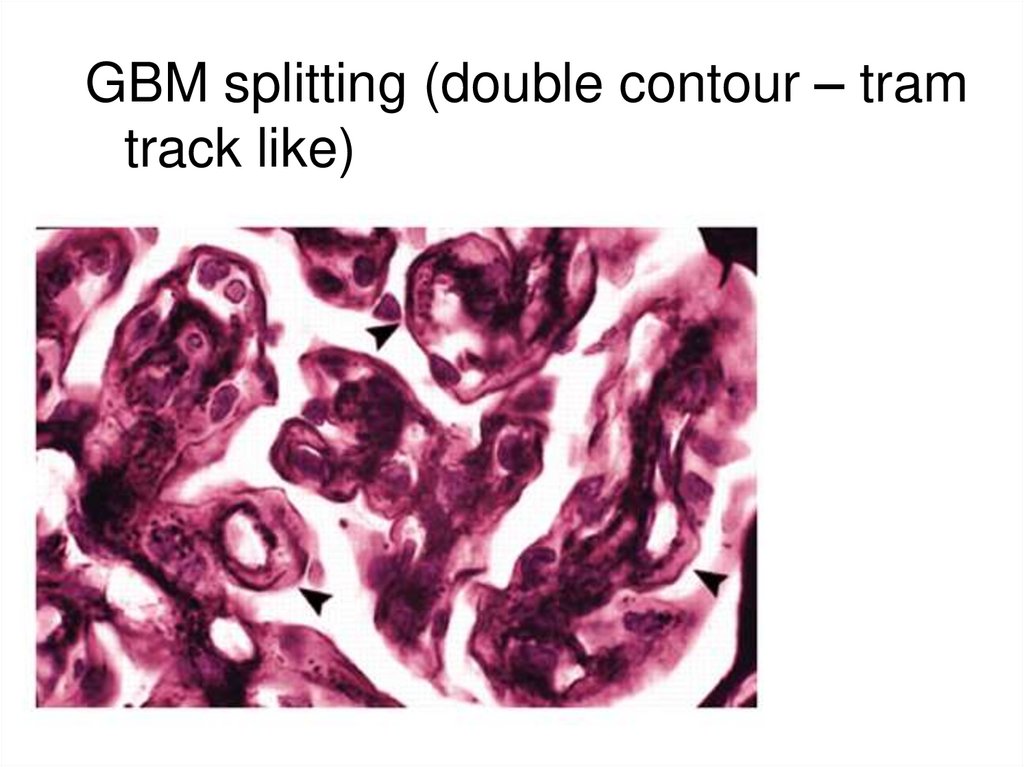
GBM splitting (double contour – tramtrack like)
75.
Electron microscopy: GBM splitting76.
Marked mesangial hypercellularity77.
78.
79.
80.
81.
82.
83.
84.
85.
Clinical manifestationsAcute onset of the disease with simultaneous
appearance of all the 3 main syndromes
Urinary syndrome (clinical): macroscopic
haematuria in 20% of cases
Hypertension: high BP from the 1st days of
the disease in 60% of cases
Oedemas: usually marked, in case of
nephrotic syndrome hardly respond to
treatment and are associated with high activity
of the disease
Nephrotic syndrome: in 50% of patients
86.
Diagnostic formula• Membranoproliferative
glomerulonephritis, I type, immune
positive with IgA, M and G deposits in
glomeruli and interstitium, exacerbation.
Nephrotic syndrome. Secondary
hypertension (date), III type of course.
Initial disturbances of renal function.
87.
88.
89.
90.
91.
92.
Membranous• predominant affection of basement
membrane without mesangial cells
proliferation
• 3-10% of all glomerulonephritis types and no
less than 30% of all cases with nephrotic
syndrome (2-5% of these in children). More
often middle-aged and aged (most commonly in
the fifth and sixth decades), more often men.
• usually in patients with chronic infections
93.
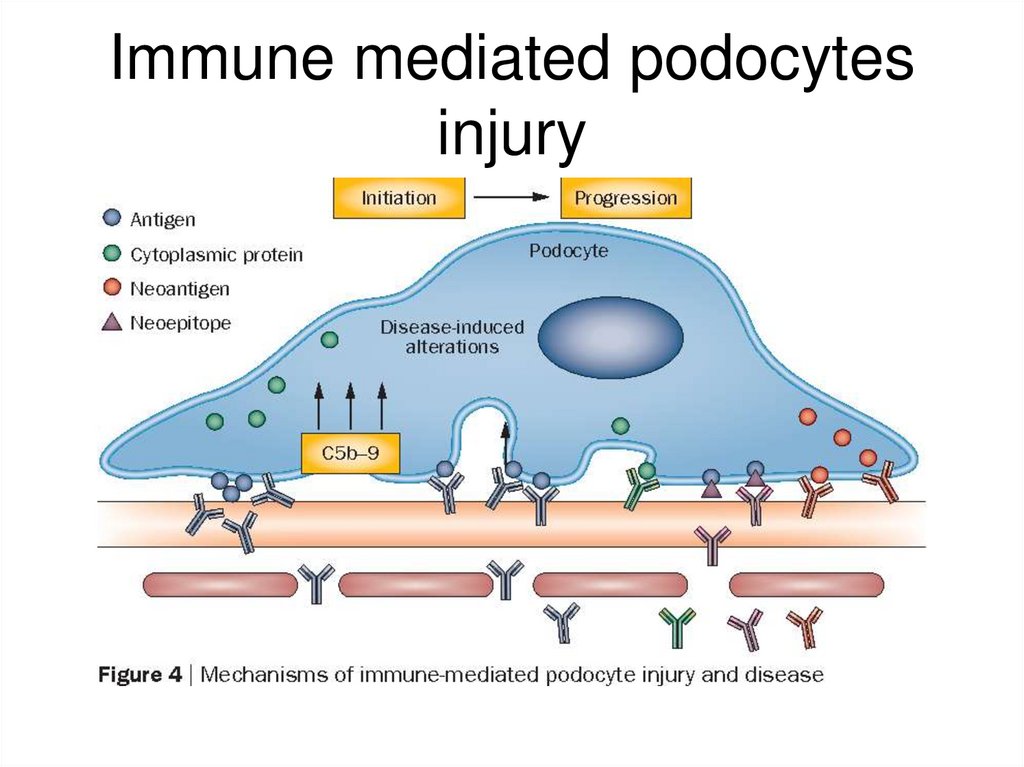
• auto–anti-PLA2R (podocytes antigen)antibodies
• deposition of immune complexes
• In situ formation of immune complexes
through the reaction of circulating
autoantibody to a native glomerular
(podocyte) antigen.
• formation of immune complexes with a
nonnative (extrinsic) antigen artificially
bound to the capillary wall.
94.
Immune mediated podocytesinjury
95.
96.
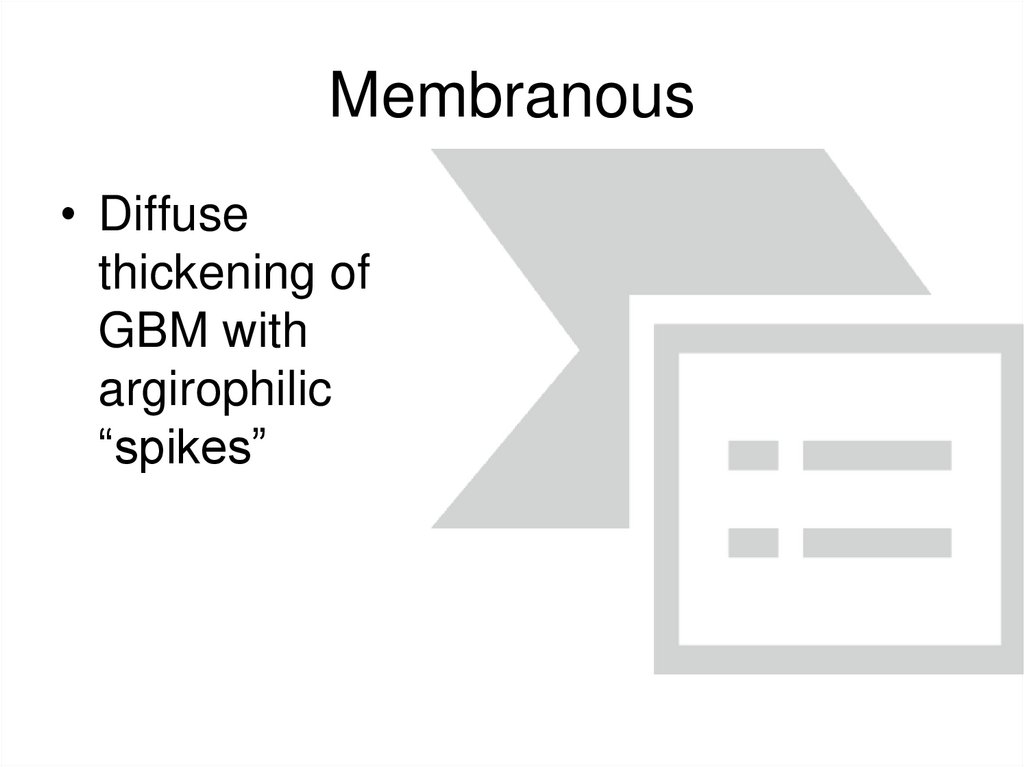
Membranous• Diffuse
thickening of
GBM with
argirophilic
“spikes”
97.
98.
4 degrees of basement membraneaffection
• I degree: basement membrane thickening and
increase of density, no spikes are present
• II degree: large subepithelial deposits separated
by spikes of basement membrane
• III degree: deposits incorporated into a
thickened basement membrane with many
spikes
• IV degree: a very thick irregular basement
membrane with no spikes and resorbed
deposits. Some sites of basement membrane
arte thickened, others are wrinkled.
99.
Deposits in GBM100.
Clinical manifestations• 1. Onset: gradual, from mild/moderate
proteinuria (I morphological degree of basement
membrane affection) appearing and
disappearing; becoming more and more marked;
stable marked level is reached in several years
(quicker in secondary glomerulonephritis); by
that time symptoms appear
• 2. Oedemas: more and more marked with
disease progression up to nephrotic syndrome
• 3. Nephrotic syndrome: usually appears at IIIIV morphological degree
• 4. Hypertension: at III-IV degree
• 5. Urinary syndrome: macroscopic haematuria
in 10-20% of children, but rare in adults
101.
102.
Complications• Strokes, angina and congestive heart
failure may occur (hypertension, age,
atherosclerosis progression in patients
with nephrotic syndrome)
• 7. Chronic renal failure
• 8. Renal vein thrombosis: 5-10%; due to
hypercoagulable state of the nephrotic
syndrome and not a primary cause of
membranous nephropathy
103.
Diangosis formula• 1. Membranous glomerulonephritis, I
degree wih Ig G and M deposits. Isolated
urinary syndrome with moderate
proteinuria (date). Course: I type, without
renal function impairment
104.
105.
106.
107.
FSGS• glomerulonephritis with focal and segmental
(affection of some capillars of glomerulus)
nephrone affection.
• Prevalence: rather rare (0.5-1% of all
glomerulonephritis types; in adults – 10% of all
glomerulonephritis types, presenting with
nephrotic syndrome, in children – 7% of these).
Predominantly in young people, in children peak
age is 6-8 years old; in adults – 20-30 years old
(but rarely occur in age over 70).
108.
Morphology• - focal and segmental affection (only some
loops of glomerular capillars)
• - hyaline masses in glomeruli, with the
progressing of the disease hyaline masses
are replaced by sclerosis
• - focal obsolescence of glomeruli,
mesangial matrix increase, moderal
hypercellularity of mesangium, focal
canals changes
109.
110.
111.
• Electron microscopy• Absence of diffuse foot process
effacement
• Immunofluorescent microscopy:
• deposits of IgM, G and C3 may be seen in
the sclerotic areas.
112.
Focal segmentalglomerulosclerosis
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
Clinical manifestations• 1. Acute onset with marked symptoms from the
beginning of the disease
• 2. Oedemas: marked
• 3. Nephrotic syndrome
• 4. Hypertension: high BP
• 5. Urinary syndrome: see laboratory
investigations
• 6. Heart failure, tachycardia
• 7. Pallor
123.
Treatment• Steroid response – 20-25%
• Cyclophosphamide
• Cyclosporin A
124.
125.
Mesangioproliferativeglomerulonephritis, immunopositive
with IgM deposits (IgM-nephropathy)
• Prevalence: very rare, 1% of all
glomerulonephritis cases. Males under
40 dominate.
• Aethiology: unknown
• Morphology: typical picture of
mesangioproliferative
glomerulonephritis
• with IgM deposits.
126.
Clinical manifestations:• 1. Active onset and course of the disease
• 2. Oedemas – from the first day of the
disease, marked; with subsequent
• 3. Nephrotic syndrome development
• 4. Arterial hypertension: present,
appears sometimes later than oedemas
127.
128.
Treatment• Prednizolone
• Cyclophosphan=Endoxan=Cyclophospha
mide (cytotoxic drug - alkiling agent): -first
3 days – i.v. 200-400 mg; than – same
dose per os with subsequent lowering the
dose
129.
Minimal change disease=LipoidNephrosis=Nil Disease
• Glomerulonephrtits with minimal changes is a
disease with marked clinical manifestations but
without significant morphological changes.
• Prevalence: Most common in children 2-5 years
old (under 6 years old – 80%); sometimes may
be revealed in adults (middle-aged, even aged).
In adults revealed in 0-3% of glomerulonephritis
cases (25% of these with nephritic syndrome);
men: women ratio is 2:1.
130.
• A. Idiopathic: aethiology is unknown, but disease occurfollowing:
• -viral upper respiratory tract infections
• - immunizations,
• - bee stings.
• B. Secondary:
• - Hodgkin's disease (paraneoplastic)
• - Gold preparations use in rheumatoid arthritis patients
• - Lithium preparations use
• - Antibiotics
• - NSAIDs (non-steroid anti-inflammatory drugs)
• - narcotics (heroin)
131.
132.
Morphology• absence of changes
• - focal obsolescence of glomeruli
• - minimal increase of mesangial matrix
without changes of mesangial cells
number
• - focal changes in canals
133.
2. Electron microscopy:• characteristic "fusion" of the epithelial foot
processes, which is not a specific finding
for this disorder but occurs in all
glomerular diseases associated with
significant proteinuria
134.
Immunofluorescence:• - negative
• - in some patients, diffuse mesangial
deposits of IgM and C3. These individuals
tend to have more marked hematuria and
hypertension than these with “pure”
disease
135.
136.
Minimal changes disease• Loss of charge by
foot processes of
podocytes without
morphological
changes of GBM
137.
138.
Clinical manifestations• 1. Acute onset with marked symptoms
• 2. Oedemas: rapidly progressing up to nephrotic
syndrome
• 3. Nephrotic syndrome with ascitis, hydrothorzx,
hydropericardium
• 4. Hypertension: infrequent (9%); may be revealed at
the late stages (10 and more years after the onset)
• 5. Urinary syndrome: see laboratory investigations
• .
139.
complications• - High susceptibility to infections with
gram-positive microorganisms (due to
severe hypoproteinemia), in children - in
particular cellulitis and pneumococcal
peritonitis
• - thromboembolic events
• - severe hyperlipidemia
• - protein malnutrition
140.
Treatment• A. Steroids – long-time treatment (for several
years)
• - In adults with nephrotic syndrome – i.v.
Prednoizolone 300-500 mg daily – 3-5 days
• - then – per os 60-80 mg daily - 1 month
• - then – lowering the dose gradually up to
minimally possible to avoid proteinuria (usually
10-20 g daily)
• Cytotoxic agents (Cyclophosphane) in patients
with contraindications to steroids, frequent
relapses and steroid-dependent individuals;
beginning with i.v. administration, then – per os.
141.
142.
143.
Non-GN causes of NS• Isolated proteinuria
• WBC may be in heavy proteinuria
(passive penetration)
• No casts
• Main causes: diabetic neprhopathy,
amyloidosis
144.
Diabetic nephropathy• Main cause of RF and NS in US and
Europe (46% - of all end-stage RF cases)
• More common in 2nd type (41%)
• Pathogenesis: GBM hyalinosis
• Starts with gradually increasing
proteinuria, than NS and renal failure
• The course and prognosis depends on
efficacy of diabetes control
145.
Main features• Consider in all cases of isolated
proteinuria and NS in diabetes
patients (especially type II)
• Presence of DN doesn’t exclude
GN!!! If nephritic syndrome is present,
diagnosis should be based on biopsy
results
• Treatment – control of diabetes, in
end-stage – treatment of CKD
146.
Amyloidosis• Main manifestation in SAA and
familial renal variants of amyloidosis
• In mediterranian countries – outcome
of familiar mediterranian fever (FMF)
• In other variants (AL, AH, ATTR) –
coexisting with other clinical
manifestations
147.
Main features• Consider in cases of isolated proteinuria in
patients with chronic inflammations, tumors
and microbial infections
• The first cause of proteinuria in
Mediterranian region in patients with periodic
chest and joint pain and fever (FMF)
• Isolated proteinuria and NS in patients
without any inflammatory reactions –
consider familial renal variants (renal biopsy
with Congo Red staining)
148.
• In systemic autoimmune diseases –kidney biopsy for differential
diagnosis of SAA and GN (or coexisting)
• Treatment
- SAA – treatment of main disease (incl
anticytokines)
- FMF - Colchicin
- Familial renal – kidney transplantation
149.
Nephrotic syndrome in early age• GN – minimal change disease
• Hereditary GN
150.
Hereditary nephritis: malignant• Congenital Nephrotic Syndrome of the
Finnish Type – NS after bifth, steroid-resistant
• Corticosteroid-Resistant Nephrotic
Syndrome – a-dominant, early childhood,
resistant to immunosuppressive therapy,
progression to FSGS
• Pierson's Syndrome - a-recessive; diffuse
mesangial sclerosis and distinctive ocular
anomalies characterized by microcoria – death
in 2 months
151.
More benign syndromes• Nail–Patella Syndrome – a-dominant: symmetric
abnormalities of the nails, skeleton, eyes, and kidneys.
thickening with splitting and fibrillar collagen deposits;
vary from RF in infancy to onset in late adult
• Denys–Drash Syndrome and Frasier's Syndrome
male pseudohermaphroditism, gonades tumors and
progressive glomerulopathy. Denys–Drash – NS from
infancy, end-stage – 3 years; Frasier's – start as
FSGS late childhood; end-stage – 20-30 y.o.
• Autosomal Dominant Focal Segmental
Glomerulosclerosis onset of mild proteinuria during
adolescence or early adulthood, with slow progression
to segmental glomerulosclerosis and, ultimately, to
end-stage renal disease
152.
Nephritic syndrome + RF• Outcome of any other variant of GN
• Most malignant course – FSGS,
membranoproliferative GN, Ig M
nephropathy
• In case of rapid symptoms progression –
suspect RPGN
• In case of very young people or children –
Alport’s syndrome
153.
Rapidly progressiveglomerulonephritis
• Extracapillary diffuse proliferative
glomerulonephritis==diffuse crescentic
glomerulonephritis
• progress from onset to endstage renal
failure within weeks or months. The
characteristic histological appearance
is of a focal necrotizing
glomerulonephritis with crescent
formation
154.
155.
• Prevalence: usually in young, up to30-40 years old, both men and
women (equal). Rate is about 1% of
all glomerulonephritis types.
156.
157.
Aethiology:• Idiopathic (some authors suspect it to be
a renal-limited form of systemic vasculitis)
• Antiglomerular basement membrane
disease (antibodies to 4th type of collagen)
• Primary systemic vasculitis: Wegener's
granulomatosis,
• Other systemic diseases: systemic lupus
erythematosus
• Often cold or tonsillitis precedes the
development of the disease
158.
159.
160.
161.
Variants• With immune complexes deposits
• Anti-GBM antibodies
• Pauci-u\immune (antineutrophilic
antibodies – ANCA)
162.
Morphology:• Light microscopy
• marked diffuse cells proliferation; cells are located
extracapillary and form crescents (present in no less
than 50% of glomeruli, usually – in 60-80%).
Crescents are situated extracapillary inside the
Boumen’s capsule. Crescents consist of proliferating
capsule cells, monocytes and lymphocytes,
sometimes fibrin is revealed.
• Crescents squeeze loops and initial part of proximal
part of Henle loop
• Gradual increase of fibrin amount in crescents –
further squeezing and glomerular obsolescence.
• As a result – glomerular capillary loops necroses
• Further mesangial cells proliferation
163.
Cells and fibrin crescentsSqueesing of rest of glomerulus, hypoxia
and fibrosis
164.
165.
166.
167.
168.
Clinical manifestations:– Acute onset with marked symptoms
– Urinary syndrome (clinical): severe decrease of
diuresis up to olyguria
– Oedemas: frequent increase (mass increase – 6-10 kg
during 1st week), localized on face and lower
extremities; ascitis, hydrothorax, hydropericardium.
– Hypertension: high BP (malign AH may be present)
– Nephrotic syndrome: rare
– Heart failure symptoms: may be present in aged
patients
– Other symptoms: headache (water retention,
intoxication); thirst, absence of appetite; pallor
169.
170.
Prognosis• rapid progression; most of patients die
in first months of the disease, in spite of
active treatment, due to chronic renal
failure or complications of malign
arterial hypertension (stroke, myocardial
infarction).
• Chronic renal failure: 1st months of
the disease
171.
Treatment• Plasma exchange therapy: 3-5 sessions
with removing of 500-600 ml of blood;
RBC are returned to patient and plasma is
removed. The efficacy of treatment is due
to possible removal of P-proteins , which
block cells receptors. In case of nephrotic
syndrome with hypoproteinemia removed
volume of plasma can be replaced by
solutions (albumin)
172.
Steroids:2. Prednizolone 0.05
- initial dose – 500-800 mg i.v. 3-5 days
- then per os 80 -100 mg daily (2 mg/kg daily) – 1 month
- 2-times lowering the dose (up to 40-50 mg), the dose is
lowered ½ tab once 2 days
- 40 mg dose – 6 months
- 2-times lowering the dose for 1 year
- The further therapy is planned afterwards if the patient
is still alive.
3. Instead of Prednizolone Cyclophosphan can be
administered:
- 200 mg daily i.v. – 1 month
- per os 100 mg - 6 months
- if treatment is effective – 2-times lowering of dose and
1-year treatment course.
173.
Main method• Kidney transplant
174.
Alport syndrome• COL4A1, COL4A2, COL4A3, COL4A4,
COL4A5, and COL4A6 genes mutation
• Most common - X-linked inheritance of
mutations in the COL4A5
• More rare - autosomal dominant
inheritance of dominant negative
mutations in the COL4A3 or COL4A4
gene
175.
Morphology• thin glomerular basement membranes thicken
over time into multilamellations surrounding
lucent areas that often contain granules of
varying density — the so-called split basement
membrane
• areas of thinning and splitting glomerular
basement membrane
• tubules drop out, segmental glomerular scars
progress, and the kidneys eventually fail
because of interstitial fibrosis
176.
177.
178.
179.
180.
Clinical manifestationsHematuria
Non-nephrotic proteinuria
progressive renal failure
sensorineural deafness
Lenticonus of the anterior lens capsule (positive
"oil droplet sign")
• retinopathy (dot and fleck reflections)
• Rare- mental retardation
• Rare - leiomyomatosis
181.
Course• juvenile form: renal failure and
sensorineural deafness generally develop
by 30 years of age (nonsense or missense
mutations, reading-frame shifts, or large
deletions)
• adult form: symptoms start after 30 years
of age; deafness is mild or late-onset
(splice variants, exon-skipping mutations,
or missense mutations of glycines in the
collagen helix)