
























 medicine
medicineSimilar presentations:










Наследственная патология
1. Наследственная патология
2.
Общая характеристика хромосомныхзаболеваний:
1. Поражение двух и более систем;
2. Полиморфизм внешних дефектов,
нарушение пропорций тела, дисплазия
развития (заячья губа);
3. Если поражены аутосомы, то обязательно
поражаются ССС, НС, костно-мышечная
система;
4. В 99% случаев хромосомные болезни
наследственно не отягощены, т.е. не
унаследованы;
5. Характерна умственная отсталость;
3.
Фенотипическое проявление хромосомных болезней.I. Хромосомные болезни по половым хромосомам, их
фенотипическое проявление.
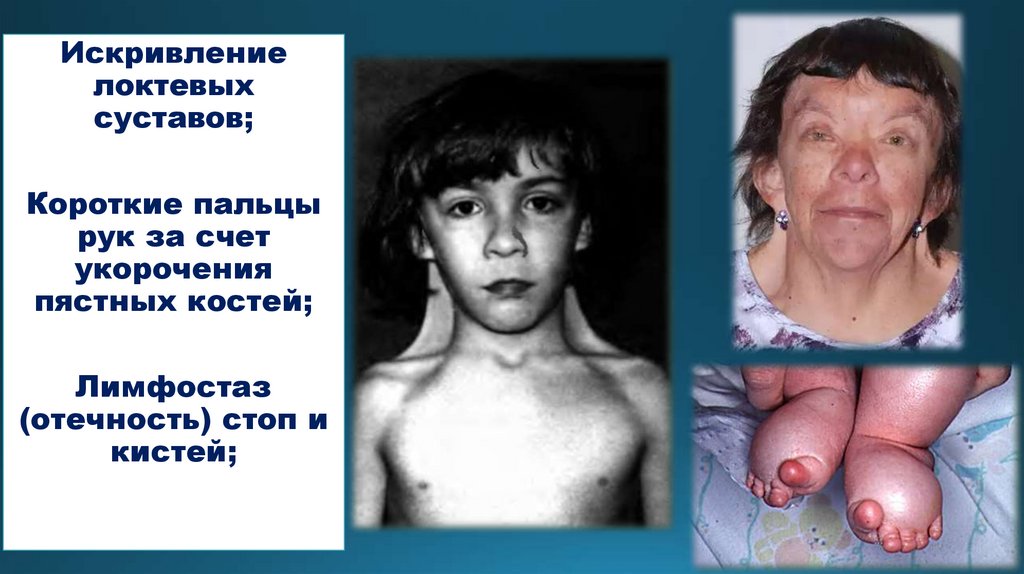
1. Синдром Шерешевского-Тернера 45(44+Х0)
Частота рождения 1 из 3000.
Рождаются девочки со сниженной массой тела, с
отеком стоп и голеней. На боковой поверхности шеи
складка – птеригиум шейный.
Впоследствии не развиваются вторичные половые
признаки, больные бесплодны. Характерна детская
фигура, голос, отсутствует менструальный цикл, могут
быть умственно отсталыми.
4.
Искривлениелоктевых
суставов;
Короткие пальцы
рук за счет
укорочения
пястных костей;
Лимфостаз
(отечность) стоп и
кистей;
5.
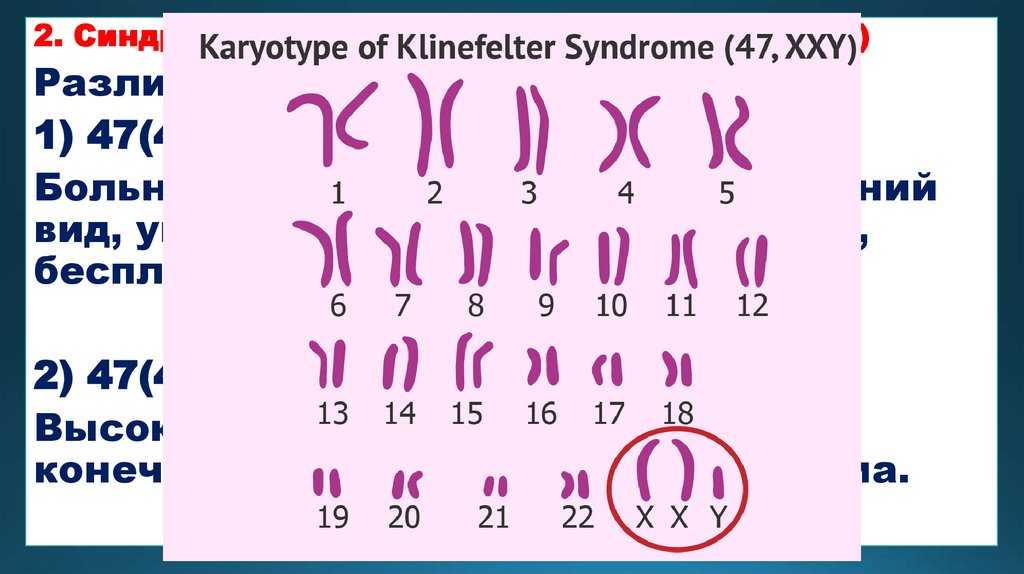
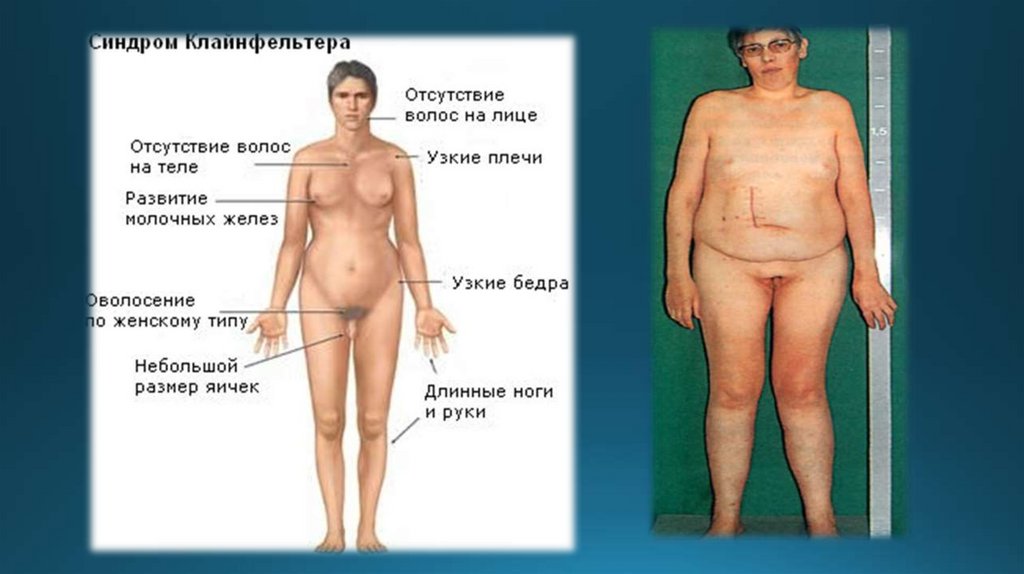
2. Синдром Клайнфельтера (мужская трисомия)Различают два вида трисомий:
1) 47(44+ХХУ)
Больные имеют женоподобный внешний
вид, увеличенные молочные железы,
бесплодие, умственно-отсталые.
2) 47(44+ХУУ)
Высокие, худые, с длинными
конечностями, агрессивны до садизма.
6.
7.
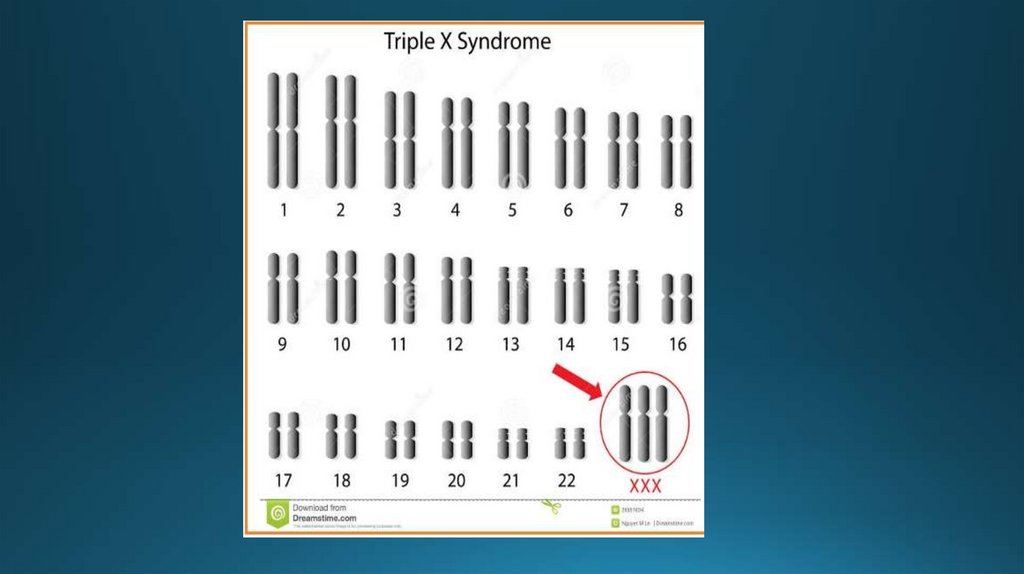
3. Женская трисомия 47(44+ХХХ)Больные маленького роста, длинные
конечности, маленькая голова,
недоразвитые уши, поражение
нервной системы в виде умственной
отсталости различной степени,
нарушение процессов овогенеза.
Больные чаще бесплодные, иногда
имеют детей, половина из которых
нормальные.
8.
9.
II. Хромосомные болезни по аутосомам.1.Синдром Дауна47(45+ХУ)
трисомия 21 пары хромосом 1:300-500
Больные имеют характерный внешний вид
Башенный
череп,
маленькая
голова,
недоразвитые,
низкорасположенные
уши,
монголоидный разрез глаз, широкий плоский
нос, большой язык, который не помещается во
рту. Короткая шея, обезьянья складка на
ладони, поражение кровеносной и костномышечной
систем.
Вероятность
рождения
ребенка с синдромом связана с возрастом
матери,
чем
старше
мать,
тем
больше
вероятность.
10.
11.
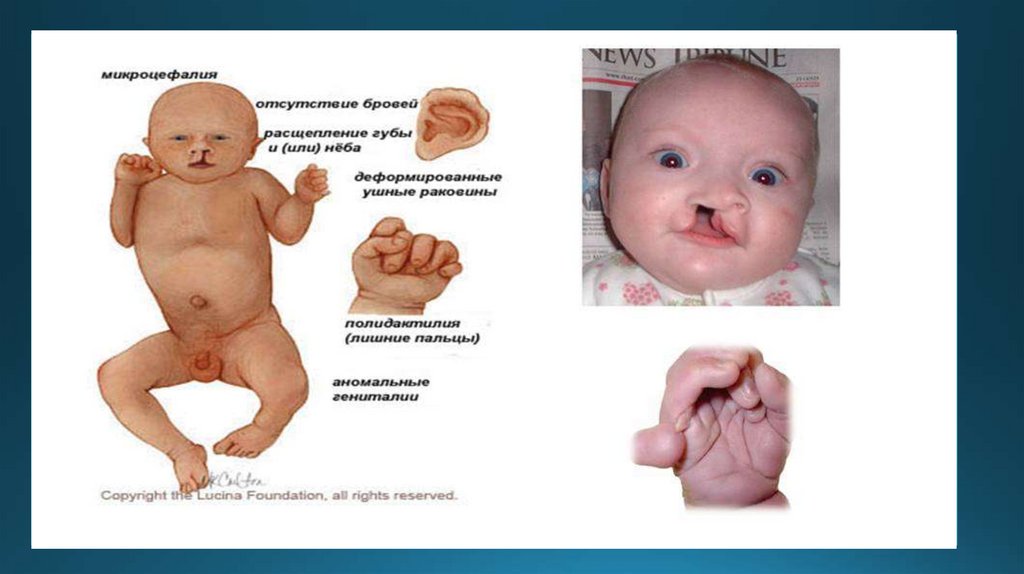
2.Синдром Патау(1:7000-1:14000)Трисомия в 13 паре хромосом
Окружность черепа уменьшена,
низкий лоб, узкие глазные
щели, дисплазия губы и неба,
помутнение роговицы,
полидактилия(шестипалость).
Умирают в течение года после
рождения.
12.
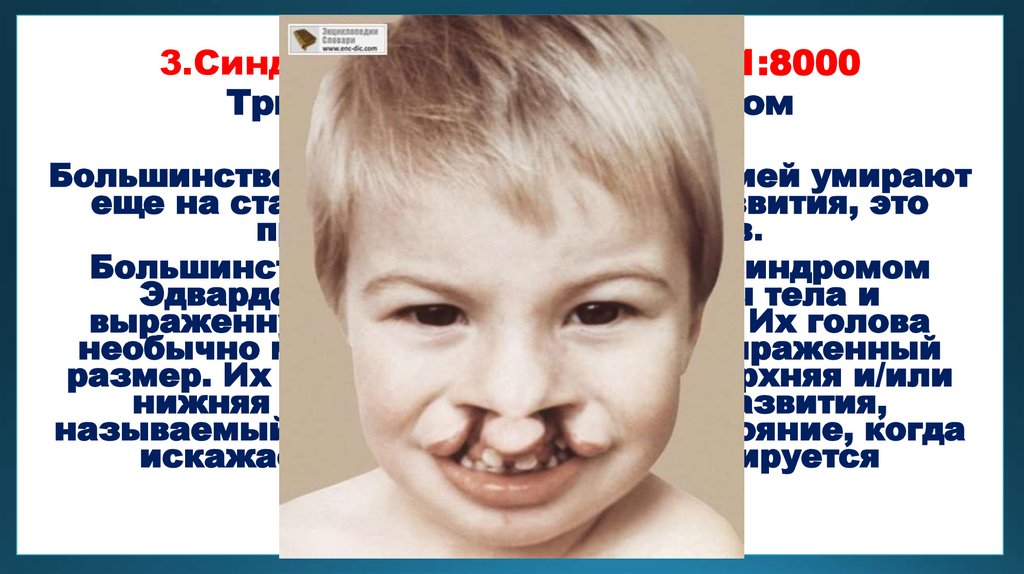
3.Синдром Эдвардса 1:3000—1:8000Трисомия 18-пары хромосом
Большинство детей с данной патологией умирают
еще на стадии эмбрионального развития, это
происходит в 60 % случаев.
Большинство детей, рожденных с синдромом
Эдвардса, имеют дефицит массы тела и
выраженную задержку в развитии. Их голова
необычно мала, а затылок имеет выраженный
размер. Их уши низко посажены, верхняя и/или
нижняя челюсть имеет дефект развития,
называемый микрогнатией. Это состояние, когда
искажается форма лица и формируется
неправильный прикус.
13.
4.Синдром «кошачий крик»Связан с делецией короткого
плеча 5-ой хромосомы.
Плач новорожденного похож на
крик кошки, что связано с
аномалиями развития гортани и
голосовых связок, дети плохо
растут, отстают в психическом
развитии. Умирают в детском
возрасте.
14.
Диагностика хромосомныхболезней определяется
цитогенетическим методом,
при котором исследуется
клетка и определяется
кариотип.
15.
Генные болезни16.
Генные болезни – это группазаболеваний, обусловленная
мутацией единичных генов.
17.
Генные болезни встречаются чаще, чемхромосомные.
I.Аутосомно- доминантные
II.Аутосомно-рецессивные
III.Сцепленные с полом
18.
I.Аутосомно- доминантные19. 1. Аномалии конечностей
А) Полидактилиямногопалость, полноеили частичное
развитие добавочных
пальцев(от 6 –до 9).
Полидактилия чаще
наследуется по
аутосомнодоминантному типу, но
иногда это
анатомическое
отклонение
наследуются по
аутосомнорецессивному типу.
20.
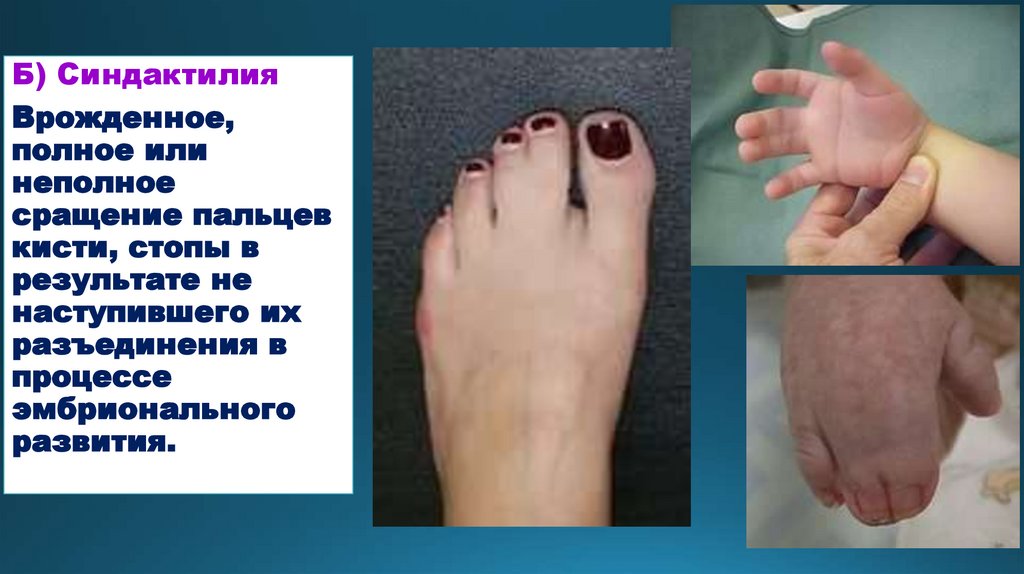
Б) СиндактилияВрожденное,
полное или
неполное
сращение пальцев
кисти, стопы в
результате не
наступившего их
разъединения в
процессе
эмбрионального
развития.
21.
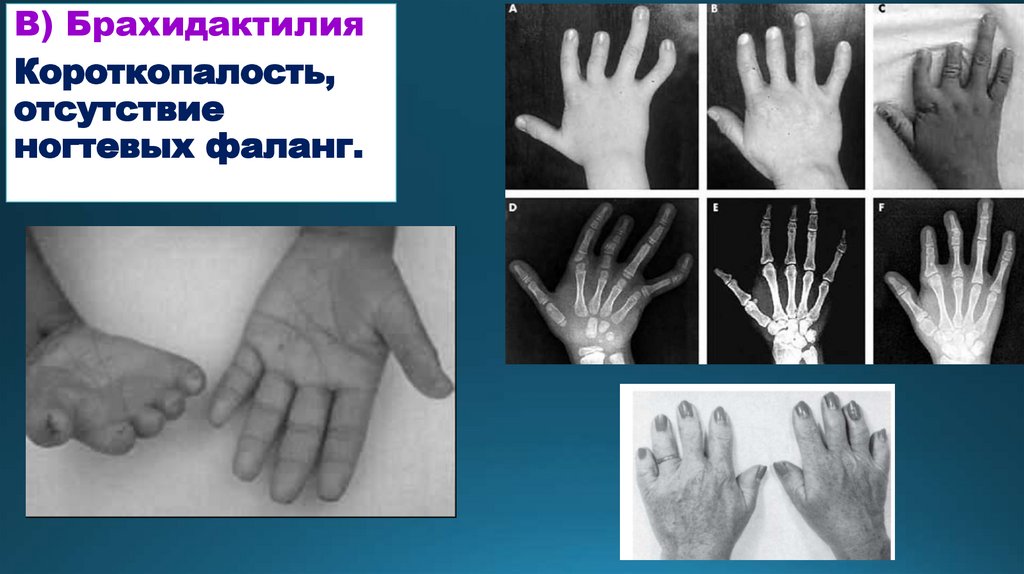
В) БрахидактилияКороткопалость,
отсутствие
ногтевых фаланг.
22.
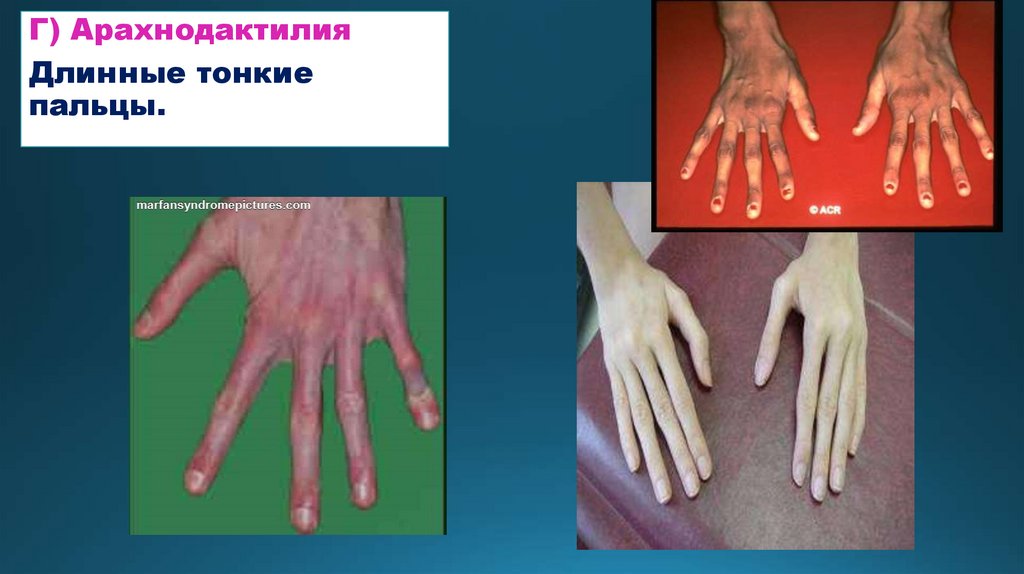
Г) АрахнодактилияДлинные тонкие
пальцы.
23.
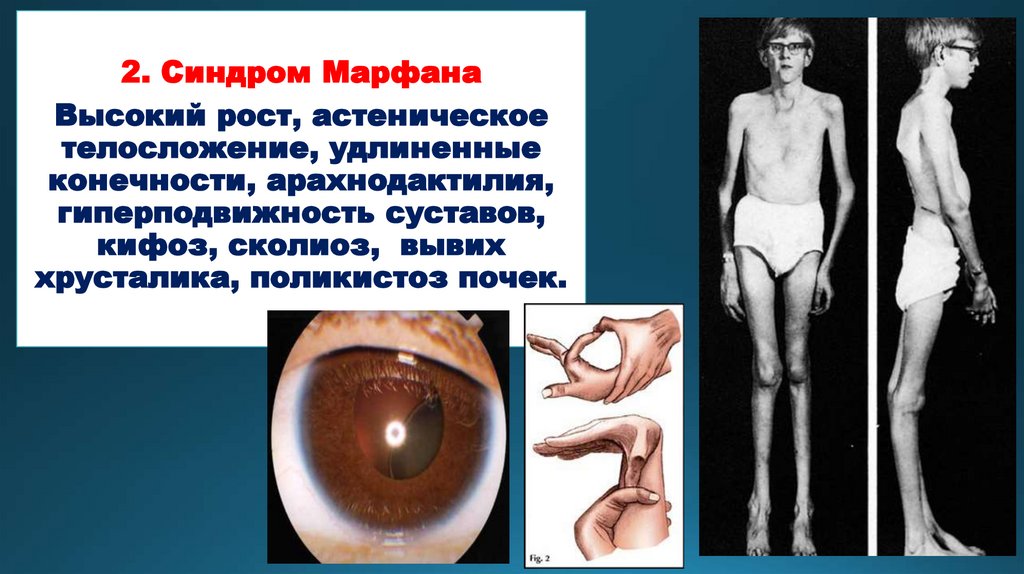
2. Синдром МарфанаВысокий рост, астеническое
телосложение, удлиненные
конечности, арахнодактилия,
гиперподвижность суставов,
кифоз, сколиоз, вывих
хрусталика, поликистоз почек.
24.
3. Нейрофиброматоз (Болезнь Реклингхаузена)Ген НФ полностью расшифрован, расположен в
17 хромосоме, в нем обнаружено более 100
мутаций.
Появляется с рождения или в первом
десятилетии жизни образованием на коже
пигментных пятен типа «кофе с молоком». С
возрастом они превращаются в мелкие опухоли,
число опухолей доходит до 1000, могут
перерождаться в злокачественные
новообразования.
25.
26.
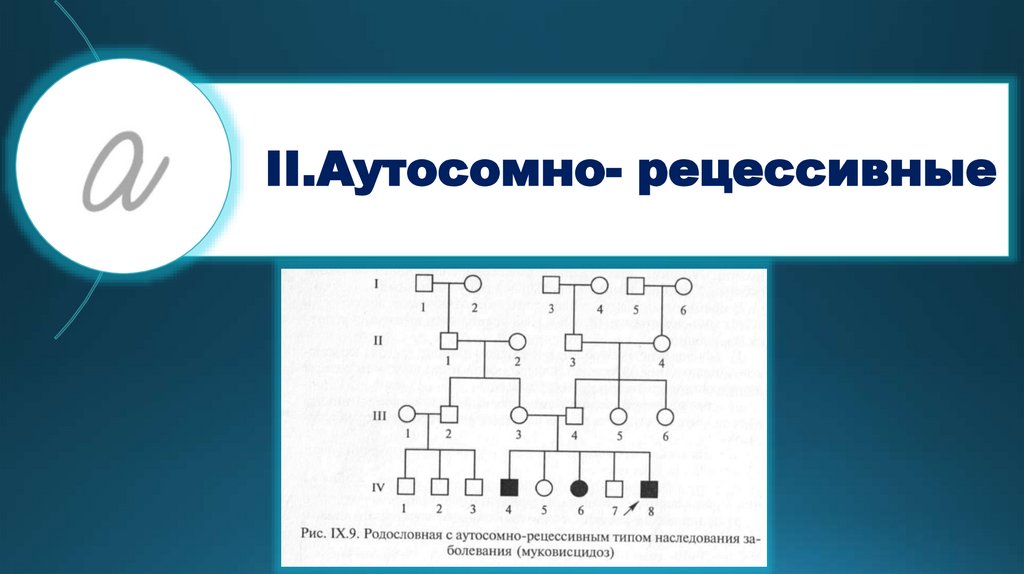
II.Аутосомно- рецессивные27.
Тяжелые болезни обменавеществ, больные дети
рождаются от здоровых
гетерозиготных родителей.
28.
1.ФенилкетонурияНарушение белкового обмена(аминокислотного)
У таких больных аминокислота фенилаланин не
превращается в тирозин и накапливается в организме,
она является ядом.
У больных отмечается рвота, понос, повышение
мышечного тонуса, судороги, цирроз печени.
Кожа, волосы, моча обладают характерным мышиным
запахом, нервная система отстает в развитии. Все
симптомы развиваются постепенно, к 3-5 годам ребенок
умирает.
29.
Лечение – диетотерапия с первых днейжизни и до 8 лет из рациона
исключаются продукты содержащие
фенилаланин.
Диагностика
В роддоме, на смоченную мочой пеленку
капают реактив FeCl3 , при повышенном
содержании аминокислот окрашивание
сине-зеленое. Анализ крови на
ферменты, контролирующие
превращение фенилаланина в тирозин.
30.
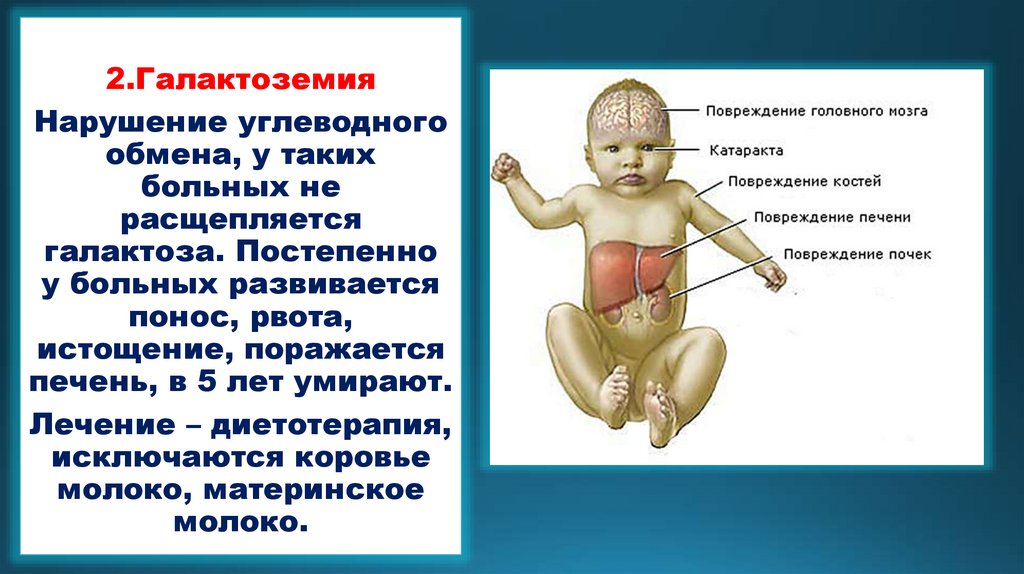
2.ГалактоземияНарушение углеводного
обмена, у таких
больных не
расщепляется
галактоза. Постепенно
у больных развивается
понос, рвота,
истощение, поражается
печень, в 5 лет умирают.
Лечение – диетотерапия,
исключаются коровье
молоко, материнское
молоко.
31.
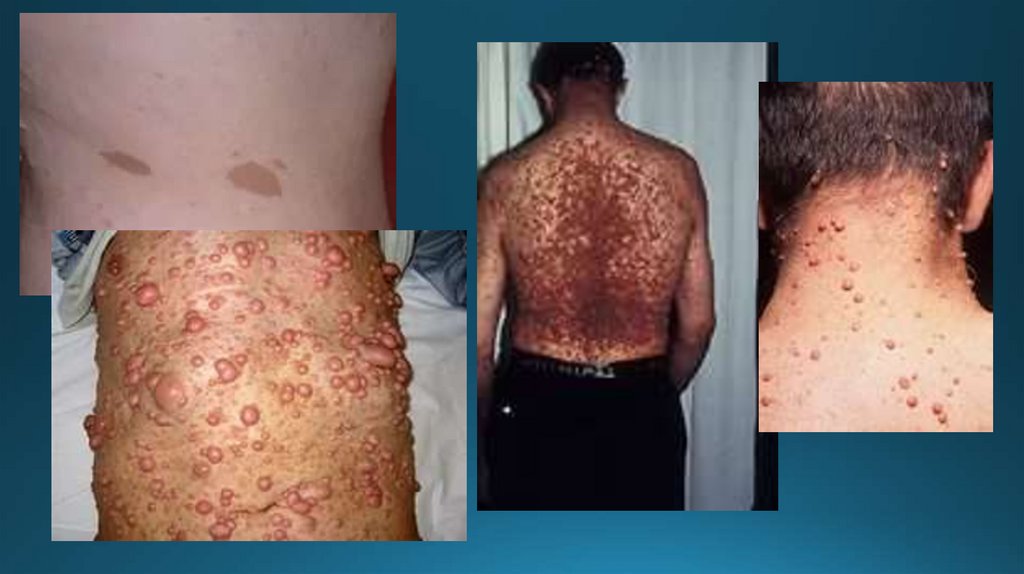
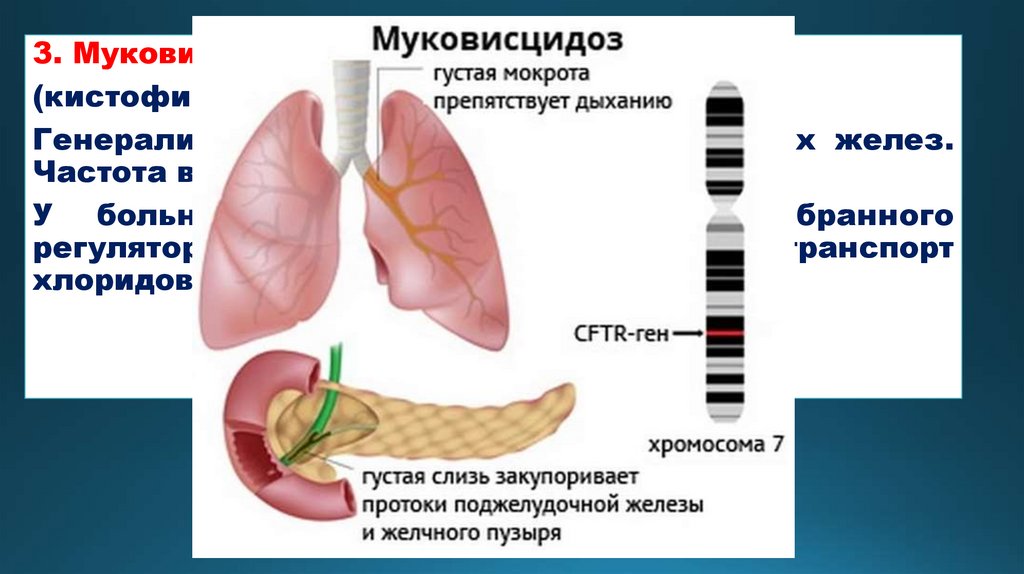
3. Муковисцидоз(кистофиброз поджелудочной железы)
Генерализованное поражение экзокринных желез.
Частота встречаемости 1: 2500
У больных отсутствует ген трансмембранного
регулятора, вследствие чего нарушается транспорт
хлоридов в эпителиальных тканях.
32.
III.Заболевания, сцепленные с полом.33.
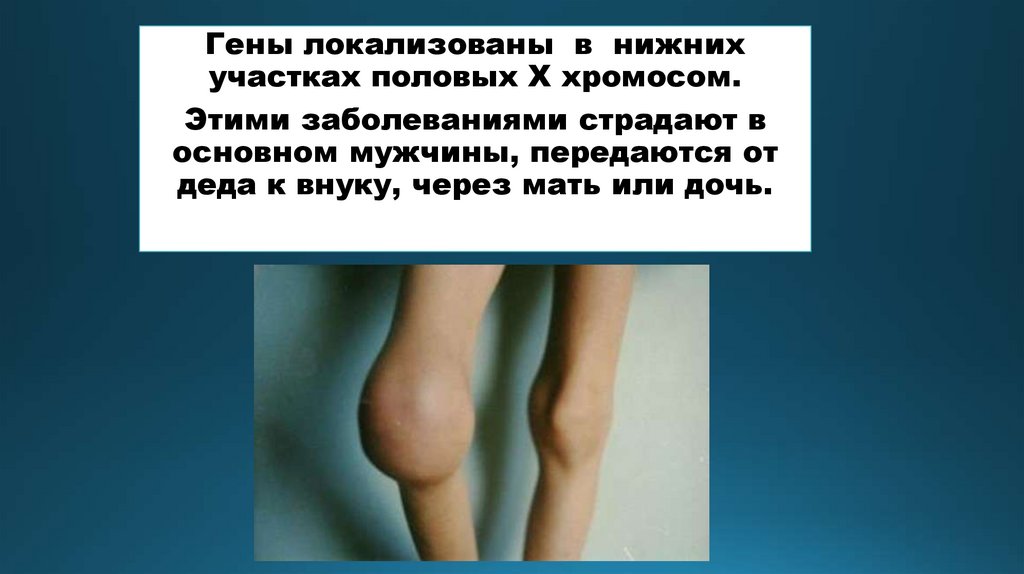
Гены локализованы в нижнихучастках половых Х хромосом.
Этими заболеваниями страдают в
основном мужчины, передаются от
деда к внуку, через мать или дочь.
34.
1.ГемофилияЗаболевание обусловлено резкой недостаточностью
факторов свертывающей системы крови.
Различают 2 основные формы гемофилии.
При гемофилии А, имеется дефицит VIII фактора
свертывания (антигемофильного глобулина).
Гемофилия В характеризуется дефицитом IX
фактора (плазменного компонента тромбопластина)
Гемофилия А встречается примерно в 5 раз чаще,
чем гемофилия В.
35.
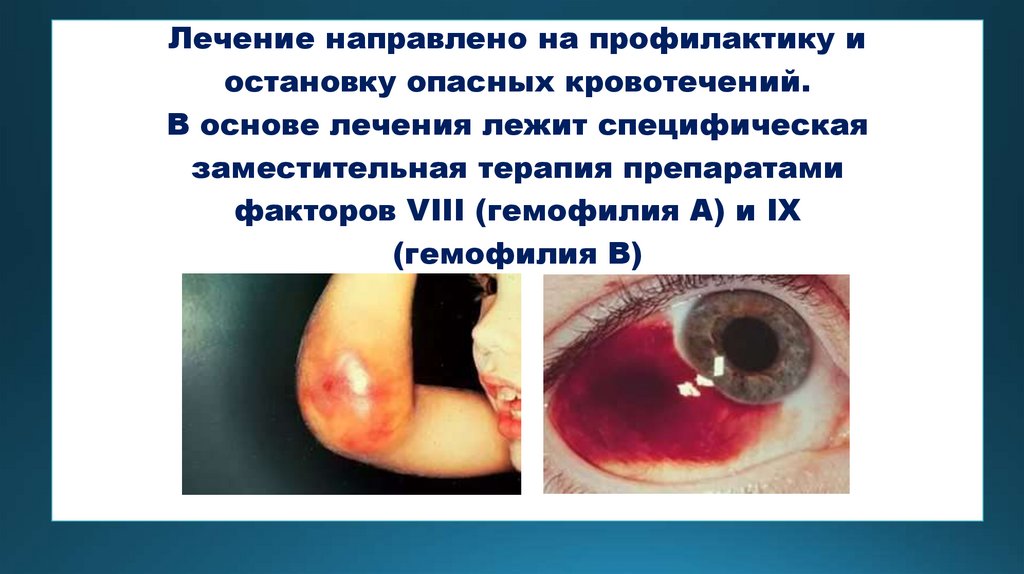
Лечение направлено на профилактику иостановку опасных кровотечений.
В основе лечения лежит специфическая
заместительная терапия препаратами
факторов VIII (гемофилия А) и IX
(гемофилия В)
36.
мультифакториальные37.
МУФЗаболевания, которые проявляются
при наличии наследственной
патологии и определенных факторов
внешней среды.
Различают моногенные МУФ и
полигенные МУФ
38.
Моногенные МУФРазвиваются при наличии одного гена и строго
определенного фактора внешней среды.
Пример: непереносимость коровьего молока.
39.
Полигенные МУФРазвиваются при наличии нескольких генов и
определенных
факторов
внешней
среды:
гипертоническая болезнь, язвенная болезнь,
сахарный диабет, экзема, болезни сердца,
миопия.
40. Лечение наследственных заболеваний
Существует 4 принципа лечения наследственныхзаболеваний:
1. симптоматическое – (лечение по симптомам) –
пластические операции.
41.
2. Патологическое – заместительная терапия:диетотерапия, инсулинотерапия, витаминотерапия.
42.
3.Этиологическое–
(происхождение)
–
направление
будущего,
основано
на
коррекции
мутаций
благодаря
генной
инженерии.
43.
4. адаптивное – адаптация детей к жизни вобществе
(дом ребенка, школы для глухих, слепых,
умственно отсталых)
44.
Медико-генетическое консультированиеЦель: предупреждение рождения больного ребенка.
Задачи:
1.Определение
прогноза
здоровья
для
будущего
потомства в семьях, где был, есть и предполагается
больной с наследственной патологией.
2.Объяснение родителям в доступной форме смысла
генетического риска и помощи им в принятии решения.
3.Помощь врача в постановке диагноза наследственных
заболеваний с проведением специальных методов
диагностики.
4.Проведение диспансеризации и выявление группы
повышенного риска среди родственников пробанда.
5.Пропаганда
медико–генетических
знаний
среди
населения.
45.
Выделяют три этапа МГК1. уточнение диагноза. Диагноз уточняется врачом –
генетиком и врачом – специалистом в данной области.
2. выявление генетического риска, используют метод
родословных.
3. заключение и рекомендация врача, в письменной
форме.