



































 47XX, 47XY")









 medicine
medicineSimilar presentations:










Наследственная патология
1. Наследственная патология
НАСЛЕДСТВЕННАЯПАТОЛОГИЯ
ПРАКТИЧЕСКОЕ ЗАНЯТИЕ
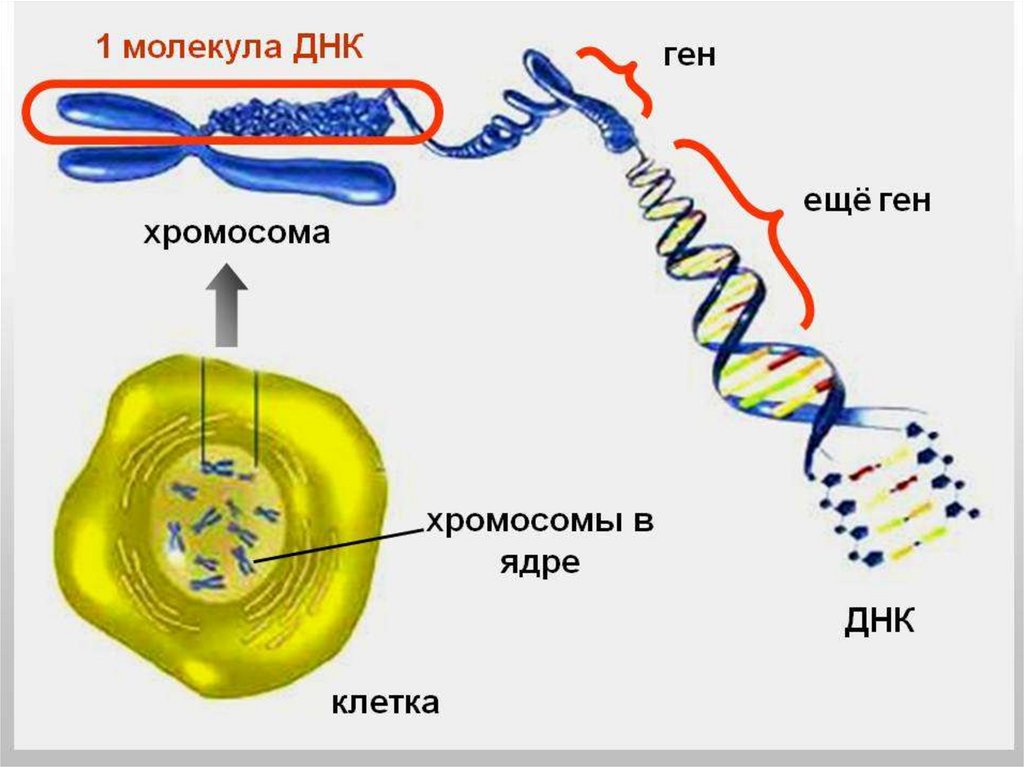
2. О геноме
О ГЕНОМЕ• 20-25 тыс. генов благодаря альтернативному
сплайсингу обеспечивают образование более 100
тыс. белков
Генетика изучает отдельные гены
Геномика изучает геном в целом
Протеомика изучает все белки, экспрессируемые в
клетке или ткани
У двух любых человек совпадает 99,5%
последовательностей ДНК.
Уникальность каждого индивида – 0,5% генома (15 млн.
пар нуклеотидов)
3. Гены и болезни человека
ГЕНЫ И БОЛЕЗНИ ЧЕЛОВЕКА• 670 генетических заболеваний приходится на
1000 человек
• Каждый человек – носитель 5-8 повреждённых
генов
4.
5.
6. Причины увеличения частоты наследственных форм патологий.
ПРИЧИНЫ УВЕЛИЧЕНИЯ ЧАСТОТЫ НАСЛЕДСТВЕННЫХФОРМ ПАТОЛОГИЙ.
1. Ликвидация и уменьшение частоты ряда инфекционные и
алиментарных заболеваний.
2. Увеличение средней продолжительности жизни человека.
3. Рост числа и разнообразия мутагенных факторов в
окружающей среде.
4. Совершенствование методов диагностики наследственных
форм патологий.
7. Основная причина наследственных заболеваний - мутация.
ОСНОВНАЯ ПРИЧИНА НАСЛЕДСТВЕННЫХЗАБОЛЕВАНИЙ - МУТАЦИЯ.
Мутация - внезапное
скачкообразное стойкое изменение
наследственности.
Мутагены:
1. Физические:
ионизирующая радиация
R-излучение
ИФ-излучение
2. Химические:
фенол, ксилол, пестициды,
лекарственные средства.
3. Биохимические:
вирусы (краснухи, оспы)
8. мутации
МУТАЦИИ• Мутация – стойкое изменение в ДНК.
Мутации бывают
1) Генные (изменение структуры ДНК)
2) Хромосомные (изменение структуры
хромосом)
3) Геномные (изменение числа хромосом)
9. Генные мутации
ГЕННЫЕ МУТАЦИИ• Точечные мутации в кодирующих
последовательностях
I. Замена одной АК на другую
Изменение значения последовательности –
миссенс-мутация (бессмысленная)
Консервативная
(нет значимых
нарушений
функции белка)
Неконсервативная –
значительное нарушение
функции белка. Пример:
CTC -> CAC при серповидноклеточной анемии
10. Генные Мутации
ГЕННЫЕ МУТАЦИИII. Превращение кодирующей АК в стоп-кодон
Преждевременная остановка трансляции гена
нонсенс-мутация
(CAG -> UAG (стоп-кодон) в β-глобине приводит к
дефициту цепей β-глобина – развитие βталассемии)
11. Генные мутации
ГЕННЫЕ МУТАЦИИ• Мутации в некодирующих последовательностях
(некоторые формы анемии) – не затрагивают
экзоны, но возникают в промоторной и
энхансерной зонах, в результате чего снижается
или прекращается транскрипция. Если возникают
в интронах, то нарушается формирование мРНК –
продукт гена не синтезируется.
12. Генные мутации
ГЕННЫЕ МУТАЦИИ13. Генные мутации
ГЕННЫЕ МУТАЦИИ• Делеции
• Вставки (инсерции)
Мутации со сдвигом рамки
считывания
Кроме того, выделяют тринуклеотидные повторы
(пример: многочисленные повторы CGG в гене
FMR1 приводят к врождённному слабоумию)
14. Хромосомные мутации
ХРОМОСОМНЫЕ МУТАЦИИ15. Геномные мутации
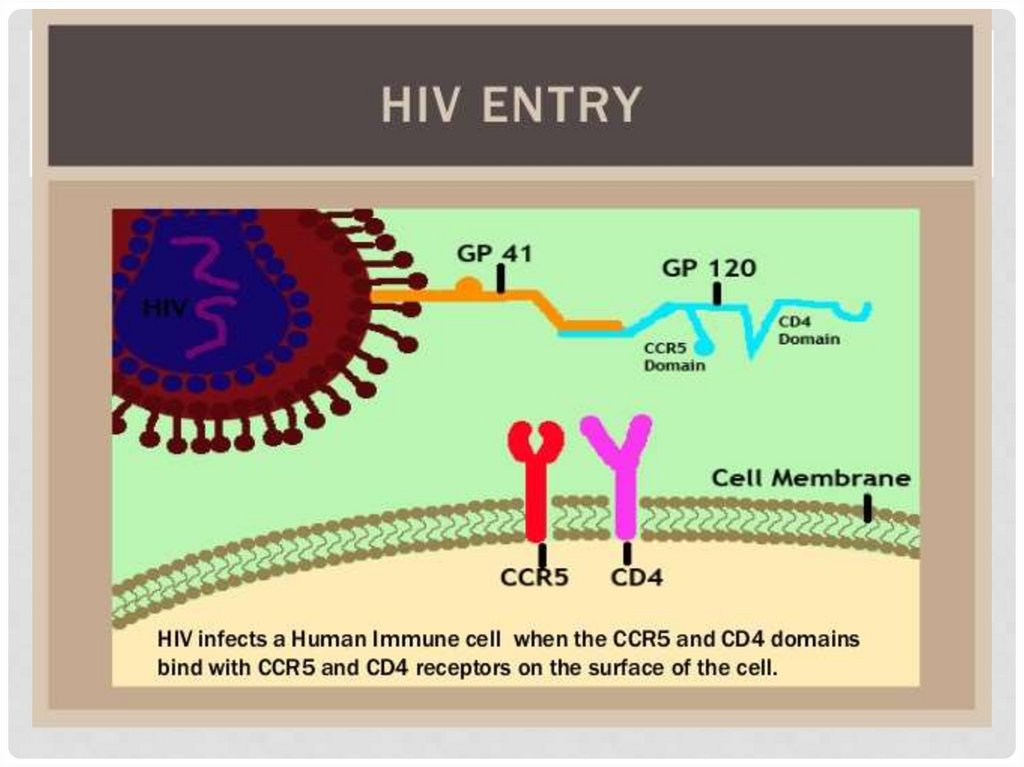
ГЕНОМНЫЕ МУТАЦИИ16. Мутации – только вред?
МУТАЦИИ – ТОЛЬКО ВРЕД?CCR5 – белок, кодируемый одноименным геном
на коротком плече 3 хромосомы. Находится на
поверхности Т-лимфоцитов. При делеции 32 пар
оснований в гене возникает мутация CCR5-D32
(CCR5 delta 32). У гетерозигот по данной мутации
снижен риск инфицирования ВИЧ, у гомозигот
инфицирование невозможно.
17.
18. Наследственное, семейное или врождённое заболевание?
НАСЛЕДСТВЕННОЕ, СЕМЕЙНОЕ ИЛИВРОЖДЁННОЕ ЗАБОЛЕВАНИЕ?
• Наследственные нарушения происходят от
одного из родителей, передаются через гаметы
от поколения к поколению, следовательно,
являются семейными.
• Врождённое заболевание = присутствующее
при рождении.
НЕ ВСЕ врождённые заболевания генетически
обусловлены (врождённый сифилис)
НЕ ВСЕ генетические заболевания врождённые
(симптомы при болезни Хантингтона появляются в
после 30-40 лет)
19. Моногенность и полигенность
МОНОГЕННОСТЬ И ПОЛИГЕННОСТЬ• Моногенные заболевания наследуются по
законам Менделя. Типы наследования:
аутосомно-доминантный, аутосомнорецессивный, сцепленный с X-хромосомой.
• Полигенные заболевания (многофакторные) –
взаимодействие определенных комбинаций
аллелей и экзогенных факторов
(предрасположенность с СД, ИБС, ГБ,
атеросклерозу и т.д.)
20. Аутосомно-рецессивные заболевания
АУТОСОМНО-РЕЦЕССИВНЫЕЗАБОЛЕВАНИЯ
Система
Заболевание
Метаболическая
Кистозный фиброз
Фенилкетонурия
Галактоземия
Гомоцистинурия
Болезнь Вильсона
Гемохроматоз
Болезни накопления гликогена
Кроветворения
Серповидно-клеточная анемия
Талассемии
Эндокринная
Врожденная гиперплазия надпочечников
Опорно-двигательная
Алкаптонурия
Нервная
Нейрогенная мышечная атрофия
Атаксия Фридрейха
Спинальная мышечная атрофия
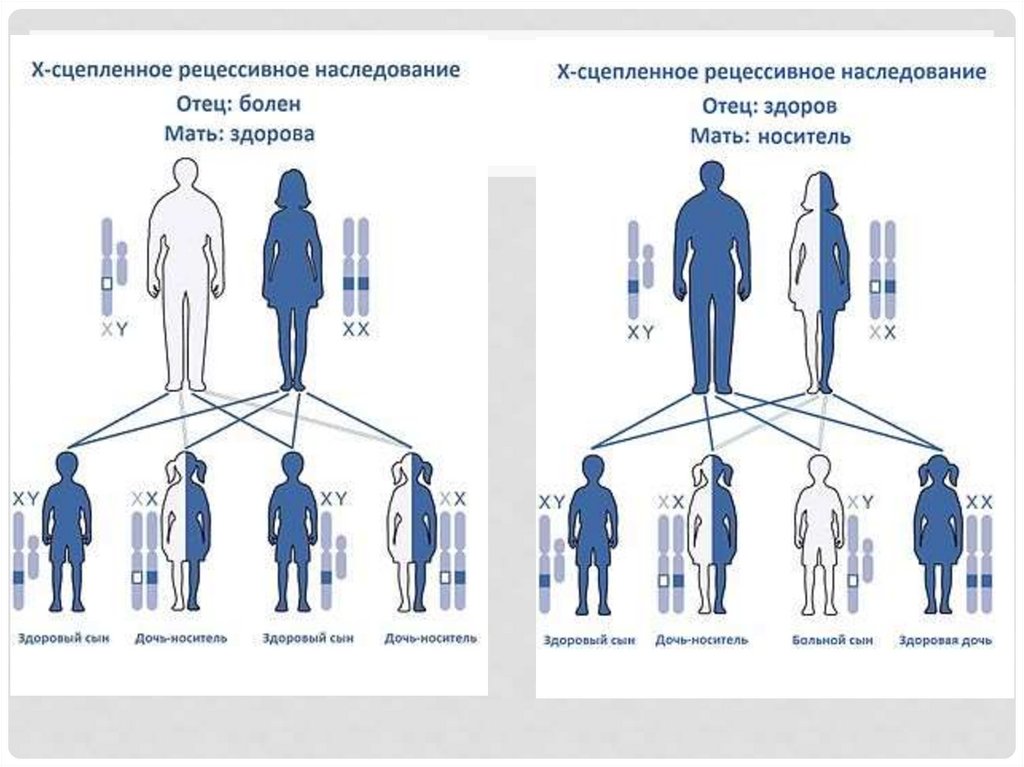
21. Заболевания, сцепленные с Х-хромосомой
ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С ХХРОМОСОМОЙ• Больной мужчина не передает заболевания
сыновьям. 50% сыновей гетерозиготной женщины
имеют шанс унаследовать мутантный ген
• Гетерозиготные женщины редко имеют все
фенотипические признаки заболевания, т.к. у них
есть парный нормальный аллель.
• Практически все заболевания рецессивные.
22. Заболевания, сцепленные с Х-хромосомой
ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С ХХРОМОСОМОЙСистема
Заболевания
Опорно-двигательная
Миодистрофия Дюшенна
Кроветворная
Гемофилия А и B
Хроническая гранулематозная болезнь
Дефицит глюкозо-6фосфатдегидрогеназы
Иммунная
Агаммаглобулинемия
Синдром Вискотта-Олдрича
Метаболическая
Несахарный диабет
Синдром Леша-Нихана
Нервная
Синдром ломкой Х-хломосомы
23.
24.
25. Основы моногенных заболеваний
ОСНОВЫ МОНОГЕННЫХ ЗАБОЛЕВАНИЙМутация в гене
Аномальный белок или его отсутствие
I. Дефекты
мембранных
рецепторов и
транспортных
систем (семейная
гиперхолестеринемия)
II. Дефекты
ферментов
III. Нарушения
структуры,
функции или
количества
неферментных
белков
(альбинизм,
галактоземия,
гликогенозы,
мукополисахаридозы)
Накапливаются
непереработанные
(талассемия)
вещества
IV. Генетически
детерминированные
побочные реакции
на ЛС (антималярийный
перпарат примахин при
дефиците глюкозо-6фосфатдегидрогеназы
вызывает гемолитическую
анемию)
26. Семейная гиперхолестеринемия
I. Дефекты мембранныхрецепторов
СЕМЕЙНАЯ ГИПЕРХОЛЕСТЕРИНЕМИЯ
Метаболизм ЛПНП
Мутация в гене, кодирующем
рецептор к ЛПНП (описано более
900 мутаций в этом гене,
отнесенных к 5 группам)
1:500 человек
Уровень холестерина с рождения
повышен в 2-3 раза у гетерозитот и в
5-6 раз – у гомозигот -> более
ранние клинические проявления АС
27. Регуляция метаболизма холестерина
РЕГУЛЯЦИЯ МЕТАБОЛИЗМАХОЛЕСТЕРИНА
28. Лизосомные болезни накопления
II. Дефектыферментов
ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
Кислые гидролазы (ферменты лизосом)
перерабатывают внутриклеточные органеллы
(аутофагия) или внеклеточные субстраты
(гетерофагия). При недостаточности фермента
катаболизм субстрата остается незавершенным, он
накапливается в лизосомах.
Лизосом особенно много в фагоцитах
увеличение органов, где много фагоцитов
гепато- и спленомегалия
29. Лизосомные болезни накопления
II. Дефектыферментов
ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
30. БолеЗнь тея-сакса
Лизосомные болезни накопленияБОЛЕЗНЬ ТЕЯ-САКСА
• Ганглиозидоз, обусловленный дефицитом
фермента гексоаминидазы А.
Заболевание наиболее распространено среди
евреев, частота 1:30.
Повреждение нейронов (ЦНС,
включая сетчатку)
Клиническая картина с 6 месяцев –
ухудшение умственного и
психомоторного развития,
дискоординации.
За 1-2 года развивается полное
вегетативное состояние.
Ds – ферментативный анализ
Лизосомы с множеством
слоев мембраны, похожие
на луковичную кожуру
31. БолеЗнь тея-сакса
Лизосомные болезни накопленияБОЛЕЗНЬ ТЕЯ-САКСА
Вишнево-красное пятно на макуле – симптом многих
болезней накопления, поражающих нервную систему
32. Болезнь нимана-пика
Лизосомные болезни накопленияБОЛЕЗНЬ НИМАНА-ПИКА
Типы А и В – нет сфингомиелиназы -> накопление
сфингомиелина – обязательного компонента клеточных
мембран –> липиды накапливаются в лизосомах.
Спленомегалия до 10 раз больше нормы. Характерный признак
– выпирание живота из-за гепатомегалии. Лимфаденопатия.
• Тип А: Обширное поражение ЦНС, прогрессирующее
истощение организма, смерть в течение первых 3 лет жизни.
• Тип В: органомегалия, симптоматики со стороны ЦНС обычно
нет. Доживают до взрослого возраста.
Диагностика – биохимический анализ активности
сфингомиелиназы в биоптате печени или костного мозга
Тип С – нарушение транспорта липидов, накапливаются в
клетке. Клиника гетерогенна.
33. Болезнь нимана-пика
Лизосомные болезни накопленияБОЛЕЗНЬ НИМАНА-ПИКА
Зеброподобные тельца –
Переполненные лизосомы
Пенистый вид цитоплазмы
гепатоцитов – следствие
отложения липидов
34. Болезнь гоше
Лизосомные болезни накопленияБОЛЕЗНЬ ГОШЕ
Накопление глюкоцереброзида (компонент клеточных
мембран стареющих клеток) в результате мутации в
гене, кодирующем глюкоцереброзидазу.
Гиперспленизм, панцитопения, патологические
переломы, лимфаденопатия.
Тип I (ненейропатический) – спленомегалия,
поражение ОДА без вовлечения ЦНС. Начало в
молодом возрасте. Продолжительность жизни немного
снижена.
Тип II (острая нейропатическая форма) –
прогрессирует поражение ЦНС (конвульсии,
умственная отсталость), смерть в молодом возрасте.
Тип III – промежуточный, системные поражения,
начало в молодом возрасте.
35.
Группыгликогенозов
Специфичес
кий тип и
пример
Дефектный
фермент
Морфологические
изменения
Клинические признаки
печеночная
Гепаторенальн
ый гликогеноз –
болезнь Гирке
(тип I)
Глюкозо-6фосфатаза
Гепатомегалия:
интрацитоплазматическ
ое накопление
гликогена и небольшого
количества липидов,
реномегалия:
интрацитоплазматическ
ое накопление
гликогена в эпителии
канальцев коркового
вещества
Без лечения: нарушение нормального
развития, остановка роста, гепато- и
реномегалия.
Гипогликемия вследствие нарушения
мобилизации глюкозы, что приводит к
судорогам.
Гиперлипидемия и гиперурикемия в
результате нарушенного метаболизма
глюкозы, у многих подагра и ксантомы
Склонность к кровотечениям из-за
дисфункции тромбоцитов.
При лечении большинство выживают, но
развиваются поздние осложнения
мышечная
Болезнь
МакАрдла (тип
V)
Мышечная
фосфорилаза
Отложение гликогена
происходит только в
скелетных мышцах,
преимущественно под
плазматической
мембраной мышечного
волокна
Проявляется после 20 лет. Болезненные
судороги после физ.нагрузки, которая не
повышает уровень лактата в венозной
крови. В 50% случаев выявляют
миоглобинурию. Продолжительность
жизни сравнима с нормальной.
смешанная
Генерализован
ный гликогеноз
– болезнь
Помпе (тип II)
Лизосомная
глюкозидаза
Гепатомегалия легкой
степени: вздутие
лизосом за счет
гликогена с
формированием
кружевного
цитоплазматического
узора. Гликоген внутри
саркоплазмы и в
мембраносвязанном
состоянии. Скелетные
мышцы: аналогично
сердцу
Массивная кардиомегалия, мышечная
гипотония, кардиореспираторная
недостаточность в течение 2 лет. При
более мягкой форме во взрослом
возрасте выявляются поражение только
скелетных мышц и хроническая
миопатия.
36. Хромосомные болезни
ХРОМОСОМНЫЕ БОЛЕЗНИ• 6:1000. Геномная или хромосомная мутация
37. Чаще встречаются:
ЧАЩЕ ВСТРЕЧАЮТСЯ:Трисомии:
- 1. По 21-й хромосоме - болезнь Дауна
нарушение лицевого черепа и мозга
сердце, ЖКТ, легкие, мозг, аномалии кистей, ног. Частота: 1
ребенок на 500 (800) новорожденных.
- 2. По 13-й хромосоме - синдром Патау.
нарушение мозгового и пищевого черепа
полидактилия, брахидактилия
незавершенный поворот кишечника
незаращение перегородки сердца
Частота: 1 ребенок на 5-7 тыс. новорожденных.
- 3. По 18-й хромосоме - синдром Эдвардс.
Частота : 1 на 7 тыс. новорожденных.
дефекты сердца, кишечника, конечностей.
• - 4. YXX - набор половых хромосом. Синдром Кляйнфельтера.
Обнаруживается половой хроматин.
Частота: 1 на 800 (1000) новорожденных мальчиков.
- 5. YYX - повышена агрессивность.
- 6. Трисомия Х - женский организм, первичная аменорея 2 половых
хроматина
38. Стигмы дизэмбриогенеза
СТИГМЫ ДИЗЭМБРИОГЕНЕЗА• Анатомическое отклонение от строения органа или части
тела, проявляется с новорожденного периода. Это
результат единичных мутаций, но наличие у ребенка
нескольких стигм часто обусловлено серьёзной
хромосомной аномалией.
Примеры: аномалии развития мозгового черепа
(долихоцефалия, брахицефалия), низкое расположение
ушных раковин, эпикантус, запавшее переносье, микро- и
макростомия, «карпий» рот, готическое нёбо, «заячья» губа,
разнообразие форм грудной клетки, удлинение или
укорочение как всей конечности, так и пальцев,
полидактилия, плоская стопа, гипогонадизм, гипоплазия
наружных половых органов и т.д.
39.
Долихоцефалия,плоский и широкий
нос, низкое
расположение
ушных раковин
Полидактилия
Готическое нёбо
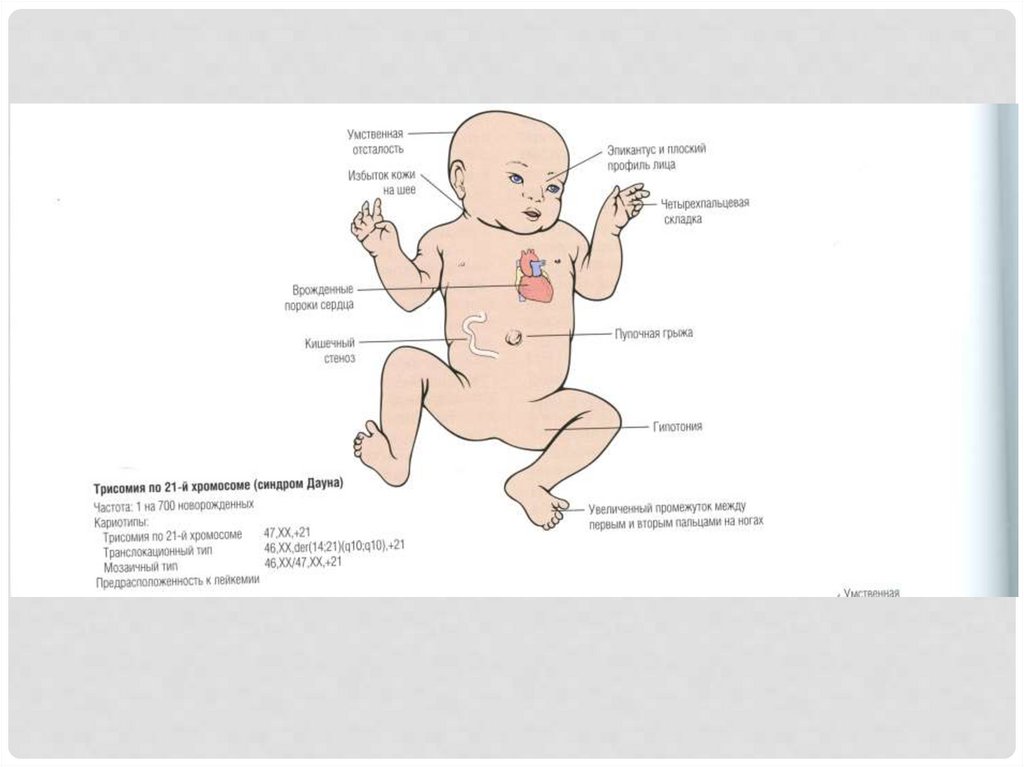
40. Трисомия по 21-й хромосоме (синдром дауна) 47XX, 47XY
Геномные заболеванияТРИСОМИЯ ПО 21-Й ХРОМОСОМЕ
(СИНДРОМ ДАУНА) 47XX, 47XY
Родители здоровы. Причина – нерасхождение
хромосомы в мейозе.
Синдром Дауна – ведущая причина умственной
отсталости, около 80% имеют IQ 25-50, пациенты
отличаются мягким характером, дружелюбны.
Около 40% имеют пороки сердца – причина
большинства летальных исходов.
Риск заболевания лейкемией в 10-20 раз выше.
Продолжительность жизни в среднем 47 лет.
После 40 лет нейропатологические болезни, схожи с
болезнью Альцгеймера.
Молекулярные основы заболевания до сих пор не
изучены.
41.
42.
43.
44. Заболевания, вызванные поражением половых хромосом
ЗАБОЛЕВАНИЯ, ВЫЗВАННЫЕПОРАЖЕНИЕМ ПОЛОВЫХ ХРОМОСОМ
• Синдром Клайнфельтера – мужской гипогонадизм,
47XXY (90%). 1:660 мальчиков.
• Синдром Тернера – первичный гипогонадизм у
фенотипических женщин. 1:2000 девочек. 45Х (57%),
структурные аномалии Х-хромосомы (14%),
мозаицизм (29%).
• Гермафродитизм и псевдогермафродитизм.
Истинный гермафродитизм – наличие овариальной
и тестикулярной ткани, 46ХХ или 46ХХ/46XY
(мозаицизм). Ложный (псевдогермафродитизм) –
несоответствие фенотипического пола гонадному,
46XX.
45.
46. Молекулярная диагностика генетических заболеваний
МОЛЕКУЛЯРНАЯ ДИАГНОСТИКАГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ
47. Показания к пренатальной диагностике наследственных заболеваний
ПОКАЗАНИЯ К ПРЕНАТАЛЬНОЙ ДИАГНОСТИКЕНАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
• Возраст матери >35 лет
• Один из родителей является носителем мутантного
гена
• У одного из родителей уже есть ребенок с
хромосомной аномалией
• Выявленные на УЗИ нарушения развития плода
• Один из родителей носитель сцепленного с Ххромосомой заболевания, важно определить пол
плода
• Аномальные уровни -фетопротеина, βхорионического гонадотропина человека и
эстрадиола при трехкратном повторении теста
48. Показания к постнатальной диагностике наследственных заболеваний
ПОКАЗАНИЯ К ПОСТНАТАЛЬНОЙ ДИАГНОСТИКЕНАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
• Множественные врожденные пороки
• Не объясненная другими причинами умственная
отсталость и/или задержка развития
• Подозрение на анеуплоидию (признаки
синдрома Дауна)
• Подозрение на аномалии половых хромосом
(синдром Тернера)
• Подозрение на синдром ломкой Х-хромосомы
• Бесплодие
• Многократные самопроизвольные аборты
49. Методы молекулярной диагностики геномных изменений
МЕТОДЫ МОЛЕКУЛЯРНОЙ ДИАГНОСТИКИГЕНОМНЫХ ИЗМЕНЕНИЙ
ПЦР
Секвенирование ДНК
Расширенный геномный анализ
Саузерн-блоттинг
Метод флуоресцентной гибридизации in situ
(FISH)
Метод сравнительной геномной гибридизации
Анализ РНК
50.
FISH-метод, сверху слевахромосома с перестройкой
ПЦР. Слева и справа – слоты с
маркерной ДНК, между ними продукты
ПЦР