

































 medicine
medicineSimilar presentations:










Наследственность и патология. Тема 4
1.
ТЕМА 4.НАСЛЕДСТВЕННОСТЬ И
ПАТОЛОГИЯ
• Наследственные болезни и их классификация.
• Понятие о моногенных и хромосомных заболеваниях.
• Понятие о мультифакториальных (полигенных)
заболеваниях, их особенности, профилактика.
• Синдромы с числовыми аномалиями аутосом
• Синдромы с числовыми аномалиями половых хромосом
• Моногенные заболевания
2.
Наследственные патологииделят на:
Генные болезни - это заболевания, вызываемые генными мутациями. Они
передаются из поколения в поколение и наследуются по законам Менделя.
Хромосомные болезни - это заболевания, возникающие в результате
хромосомных и геномных мутаций.
Мультифакториальные болезни - болезни с наследственной
предрасположенностью - это заболевания, возникающие в результате
соответствующей генетической предрасположенности и наличия определенных
факторов внешней среды.
Генетические соматические болезни - группа генетических болезней,
возникающих в результате мутаций в соматических клетках. К ней относятся
некоторые опухоли, отдельные пороки развития, аутоиммунные заболевания.
Болезни генетической несовместимости матери и плода. Они
развиваются в результате иммунологической реакции матери на антиген плода.
3.
Характерные особенности наследственныхпатологии:
1. Ранняя манифестация. Около 25% наследственных заболеваний
проявляются непосредственно после рождения
ребенка (врожденные); около 70% - к трем годам жизни, а к концу
пубертатного периода - 90%.
2. Хроническое прогредиентное течение. Прогредиентным
называется течение заболевания с постоянным ухудшением общего
состояния и с нарастанием негативных симптомов у пациента. Хронический
характер течения наследственных болезней определяется постоянным
функционированием мутантного гена.
3. Относительная резистентность к терапии.
4.
Характерные особенности наследственныхпатологии:
4. Множественность поражения. Известно, что более чем при 60%
наследственных заболеваний в патологический процесс вовлекается более
одной системы органов.
5. Семейный характер заболевания.
6. Клинический полиморфизм
5.
Генные болезниЭто разнообразная по клинической картине группа заболеваний,
обусловленная мутациями единичных генов.
Количество известных в настоящее время моногенных наследственных
заболеваний составляет около 4000-5000 нозологических форм. Встречаются
эти заболевания с частотой 1:500-1:100 000 и реже.
Особенности наследования генных заболеваний определяются законами
Г. Менделя
Начало патогенеза любой генной болезни связано с первичным эффектом
мутантного аллеля. Он может проявляться в следующих вариантах:
• отсутствие синтеза белка;
• синтез аномального по первичной структуре белка;
• количественно избыточный синтез белка;
• количественно недостаточный синтез белка.
6.
Классификация генных заболеванийпо клиническому принципу
Т.е. на отнесении болезни к той или иной группе в
зависимости от системы органов, наиболее вовлеченной в
патологический процесс:
-моногенные заболевания нервной системы,
- моногенные заболевания дыхательной системы,
- моногенные заболевания сердечно-сосудистой систем,
- моногенные заболевания кожи,
- моногенные заболевания органов зрения,
- моногенные психические заболевания,
- моногенные эндокринные заболевания и так далее.
7.
Классификация генных заболеванийпо патогенетическому принципу
• наследственные болезни обмена веществ:
-наследственные нарушения аминокислотного обмена,
- нарушения обмена углеводов,
- нарушения липидного обмена,
- нарушения стероидного обмена и т.д.
• моногенные синдромы множественных
врожденных пороков развития (синдром Холт-Орама)
•комбинированные формы
8.
Классификация генных заболеванийпо генетическому принципу
(по типам наследования )
• аутосомно-доминантные,
• аутосомно-рецессивные,
• Х-сцепленные доминантные,
• Х-сцепленные рецессивные,
• У-сцепленные (голандрические)
• митохондриальные.
9.
АУТОСОМНО-ДОМИНАНТНЫЕ ЗАБОЛЕВАНИЯ• Синдром Марфана
• Нейрофибороматоз I
• Синдром Холт-Орама
10.
Синдром МорфанаНаследственное заболевание соединительной ткани,
вызываемое множественными мутациями генов ,
проявляющееся изменениями скелета:
• высоким ростом с относительно коротким
туловищем
• длинными паукообразными пальцами
(арахнодактилия)
• разболтанностью суставов
• часто сколиозом , кифозом
• деформациями грудной клетки ( ямка или киль)
• аркообразным небом.
Характерны также поражения глаз – вывих
хрутсалика.
11.
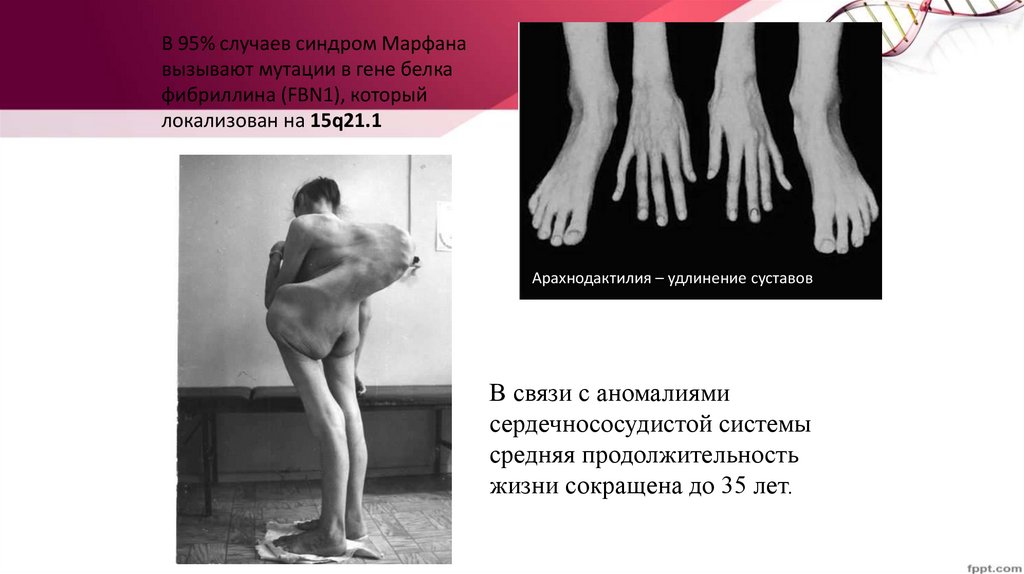
В 95% случаев синдром Марфанавызывают мутации в гене белка
фибриллина (FBN1), который
локализован на 15q21.1
Арахнодактилия – удлинение суставов
В связи с аномалиями
сердечнососудистой системы
средняя продолжительность
жизни сокращена до 35 лет.
12.
Нейрофиброматоз типа I (Болезнь Реклингхаузена)• Характеризуется нейрофибромами –
доброкачественными опухолями
нервов, состоящими из шванновских
клеток и фибробластов, а также
очаговой гиперпигментацией кожи –
так называемыми пятнами цвета кофе
с молоком.
• Причиной болезни служит мутация
гена NF1, локализующегося на 17-й
хромосоме и кодирующего белок
нейрофибромин .
• частота не реже 1:3000 — 1:4000
населения
13.
НФ-1 илипериферический
нейрофиброматоз
Нейрофибромы представляют собой
мягкие узелки, при надавливании как
бы проваливающиеся в кожу, симптом «кнопки звонка».
• Подкожные узелки располагаются по ходу нервных стволов (округлые
бусинки диаметром 1-2 см, подвижные, не прикрепленные к коже).
Помимо этого, у некоторых больных развиваются массивные диффузные
опухолевидные образования.
• Наблюдаются изменения костной системы - кифоз, сколиоз,
псевдоартрозы, локальный гигантизм, неспецифические черепно-лицевые
аномалии.
• При локализации в средостении, в брюшной полости, в глазнице они приводят
к нарушению функций прилегающих органов. – Плексиформная
нейрофиброма
• Затруднения в обучении наблюдаются у 30% больных. Умственная
14.
Синдром Холт-Орама(синдром рукасердце).
• моногенный синдром множественных
врожденных пороков развития.
• Аномалии верхних конечностей и
врожденные пороки сердца.
• Пороки развития руки варьируют от гипо или
аплазии 1-го пальца кисти, трехфалангового 1
пальца кисти до гипо- и аплазии лучевой кости
(лучевая косорукость). Левая рука поражается
чаще.
15.
Синдром Холт-Орама(синдром рукасердце).
• Врожденные пороки сердца
неспецифичны и проявляются в
виде дефектов межпредсердной и
межжелудочковой перегородок,
открытого аортального протока,
коарктации аорты, тетрады Фалло,
стеноза легочной артерии, пролапса
митрального клапана и др.
• Прогноз жизни зависит от тяжести
поражения сердца.
16.
АУТОСОМНО-РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ• Муковисцидоз
•Фенилкетонурия
•Адреногенитальный синдром
•Галактоземия
17.
Муковисцидоз (кистофиброзподжелудочной железы)
• Это заболевание обусловлено генерализованным поражением
экзокринных желез.
• Частота муковисцидоза (МВ) среди новорожденных в
европейской популяции составляет 1:2500.
• Ген МВ локализован на 7-й хромосоме (белок муковисцидозный трансмембранный регулятор проводимости)
• Нарушается транспорт хлоридов в эпителиальных клетках.
• Гиперсекреция густой слизи в клетках экзокринной части
поджелудочной железы, эпителии бронхов, слизистой оболочке
желудочно-кишечного тракта.
• Ферменты поджелудочной железы не поступают в просвет
кишечника.
• Закупорка мелких бронхов и последующему присоединению
инфекции. Подобные процессы развиваются в придаточных
пазухах носа и в канальцах семенников.
18.
Различают следующие клинические формы МВ:• смешанная (легочно-кишечная - 65-75% от всех больных);
• преимущественно легочная (15-20%);
• преимущественно кишечная (5-10%);
• мекониальный илеус (не более 1%);
• стертые и абортивные формы (незначительная доля).
Интеллектуальное развитие у детей не страдает.
Прогноз жизни при всех формах, кроме атипичных,
неблагоприятный. В настоящее время пациенты со
смешанными формами редко живут более 40 лет.
19.
Фенилкетонурия (фенилпировинограднаяЧастота в европейских странах составляет
олигофрения).
1:10000
• Это одна из самых частых форм наследственных дефектов
обмена аминокислот.
• Дефицит фенилаланина - 4-гидроксилазы, фермента,
контролирующего превращение фенилаланина в тирозин.
• Ген, кодирующий данный фермент, картирован на 12-й
хромосоме.
• Концентрация фенилаланина увеличивается в организме в
десятки раз. Часть его выводится с мочой, а остальное
количество превращается в фенилпировиноградную,
фенилуксусную, фенилмолочные кислоты и другие фенилкетоновые производные,
• Нарушение формирования миелиновой оболочки вокруг
аксонов в центральной нервной системе.
20.
ФКУ• Ребенок рождается без каких-либо симптомов
• В первые месяцы жизни беспокойство или, наоборот,
вялость, сонливость.
• Отмечается гипопигментация кожи, волос, радужной
оболочки (результат снижения уровня тирозина).
• Своеобразный «мышиный запах».
• Неврологические симптомы - мышечную гипотонию или,
наоборот, гипертонию, повышение рефлексов.
• К 6-ти месяцам выявляется задержка психомоторного
развития.
• После 3-х лет клиническая картина характеризуется
умственной отсталостью (в 95% случаев это имбецильность
или идиотия), нарушением поведения (чаще возбуждение,
расторможенность, психотические расстройства),
судорожным синдромом.
21.
Адреногенитальный синдром (врожденнаягиперплазия коры надпочечников).
• Относится к группе наследственных
нарушений биосинтеза стероидных гормонов.
• Распространенность всех форм АГС равна 1:12
000 новорожденных.
Различают несколько форм заболевания:
• вирильную,
• солетеряющую,
• поздняя форма,
• латентная форма.
22.
ГалактоземияОтносится к группе наследственных болезней обмена углеводов (3
генетических варианта).
Обусловлено недостаточностью основных ферментов, участвующих
в превращении галактозы в клетках печени в глюкозу
Токсическое действие на мозг, печень, кишечник и почки галактозы
и галактозо-1-фосфата, накапливающихся в организме.
на 1-2 неделе жизни у ребенка отмечаются частые срыгивания,
плохая прибавка массы тела, нарушения стула. Затем
присоединяются симптомы поражения печени
У 70% детей, не получающих соответствующего лечения,
формируется катаракта в первый месяц жизни.
Без соответствующего лечения дети погибают в течение первого
полугодия жизни от острой печеночной недостаточности.
23.
Х-СЦЕПЛЕННЫЕ ЗАБОЛЕВАНИЯ• Мышечная дистония Дюшенна
• Синдром Мартина-Белл
• Гемофилия А
24.
Ппсевдогипертрофическая мышечнаядистония Дюшенна.
Частота ее составляет 1:3000 - 1:5000 мальчиков.
Это одна из самых частых форм рецессивных
наследственных нервно-мышечных заболеваний.
Мышечные дистрофии характеризуются дегенеративными
изменениями в поперечно-полосатой мускулатуре
Заболевание обусловлено нарушением синтеза белка
дистрофина. Ген дистрофина локализован в коротком плече Ххромосомы.
Признаки заболевания проявляются у детей трех-пяти лет.
Примерно у 50% детей отмечается снижение интеллекта - от
пограничных состояний до выраженной дебильности.
Острая сердечная недостаточность - наиболее частая причина
смерти. огибают больные, как правило, на третьем
десятилетии жизни, а к 14-15 годам они, обычно,
обездвижены.
25.
Синдром умственной отсталости сломкой Х-хромосомой
(синдром Мартина-Белл).
• Одна из наиболее часто встречающихся форм
Х-сцепленной умственной отсталости.
• Популяционная частота СМБ составляет
1:2000-5000 живорожденных, больных мальчиков в 2-3
раза больше, чем девочек.
• Признаки: высокий рост; крупные кисти и стопы; высокий
выступающий лоб; удлиненное лицо, с уплощенной
срединной частью; прогнатия; толстые губы (нижняя
часто вывернута); клювовидный нос; высокое
арковидное небо; «оттопыренные», увеличенные в
размерах уши; гиперэластичность кожных покровов;
переразгибаемость суставов, плоскостопие
• Умственная отсталость, типичная для этого синдрома,
оценивается как умеренная или выраженная
26.
ГемофилияГемофилия — наследственное заболевание,
характеризующееся снижением или нарушением синтеза
факторов свертывания крови. Обычно болезнью страдают
мужчины, женщины же выступают как носительницы
гемофилии.
Гемофилия появляется из-за изменения одного гена в хромосоме X.
Гемофилия А (рецессивная мутация в X-хромосоме) вызвана
генетическим дефектом, отсутствием в крови необходимого белка —
так называемого фактора VIII (антигемофильного глобулина). Такая
гемофилия считается классической, она встречается наиболее часто, у
80-85 % больных гемофилией. Тяжёлые кровотечения при травмах и
операциях наблюдаются при уровне VIII фактора — 5-20 %.
Гемофилия B вызвана дефектным фактором крови IX (рецессивная
мутация в X-хромосоме). Нарушено образование вторичной
коагуляционной пробки.
27.
Хромосомные болезниЭто группа врожденных наследственных заболеваний, которые клинически
характеризуются наличием множественных пороков развития.
• Этиологической основой их являются численные или структурные аномалии
хромосом.
Механизмы, лежащие в основе хромосомных болезней, обусловленных
изменением числа хромосом:
1) Нерасхождение хромосом. Может произойти во время 1 или 2-го
мейотического деления. При этом в анафазе гомологичные хромосомы или
сестринские хроматиды отходят к одному полюсу. Нерасхождение
хромосом в зиготе или на ранних стадиях ее дробления приводит к развитию
организма с клетками разной хромосомной конституции (два типа и более).
Такие формы называются мозаичными.
2) Утрата отдельной хромосомы вследствие «анафазного отставания».
3) Полиплоидизация - увеличение числа хромосом, кратное гаплоидному
набору. Причинами могут являться либо двойное оплодотворение, либо
диплоидная гамета одного из мейотических делений.
28.
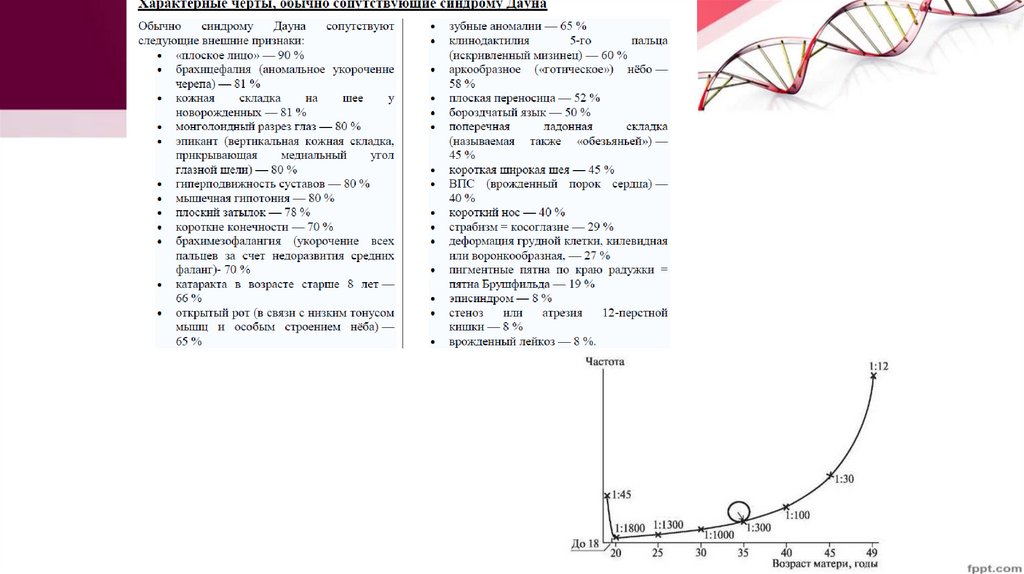
Синдром Даунатрисомия 21Был описан в 1866 году английским
педиатром Л. Дауном, но только в 1959 году
французским генетиком и врачом Дж.
Леженом с соавторами было доказано, что
это заболевание хромосомной природы
Частота этого синдрома составляет 1:700800 новорожденных.
29.
30.
Синдром Патау, трисомия 13Генетическая природа была
расшифрована в 1960 г.
американским генетиком К. Патау.
Частота данного заболевания
составляет 1:6000 рождений,
занимая второе место по частоте
встречаемости (после синдрома
Дауна) среди полных аутосомных
трисомий.
Мальчики и девочки страдают
этим заболеванием с одинаковой
частотой.
31.
Трисомия 13 – синдром Патау• аномалии черепа и лица - микроцефалия,
выраженная тригоноцефалия, скошенный
лоб, узкие глазные щели, запавшее
переносье, низкорасположенные и
деформированные ушные раковины.
• дефекты скальпа овальной или округлой
формы, до 1 см
• расщелина губы и неба
• полидактилия
• врожденные пороки сердца,
пищеварительного тракта, почек , органов
зрения.
• Центральная нервная система поражается в 100% случаев.
• Продолжительность жизни у детей с синдромом Патау резко снижена. На первом
году жизни умирает 95% больных, причем 60-65% в перинатальном периоде.
• В возрасте старше 3-х лет остаются в живых единицы.
• Все дети с синдромом Патау имеют тяжелую умственную отсталость (глубокая
идиотия).
32.
Трисомия 18 – синдром ЭдвардсаОписан в 1960 г. Д. Эдвардсом
Частота синдрома Эдвардса
среди новорожденных
составляет 1:7000 . Девочки
примерно в 3 раза чаще.
Дети с этим синдромом
рождаются с низкой массой
тела, в среднем - 2180 г
Череп долихоцефалической
формы, нижняя челюсть и
отверстие рта маленькие,
глазные щели узкие и короткие,
ушные раковины маленькие,
низкорасположенные, наружный
слуховой проход сужен, иногда
отсутствует.
33.
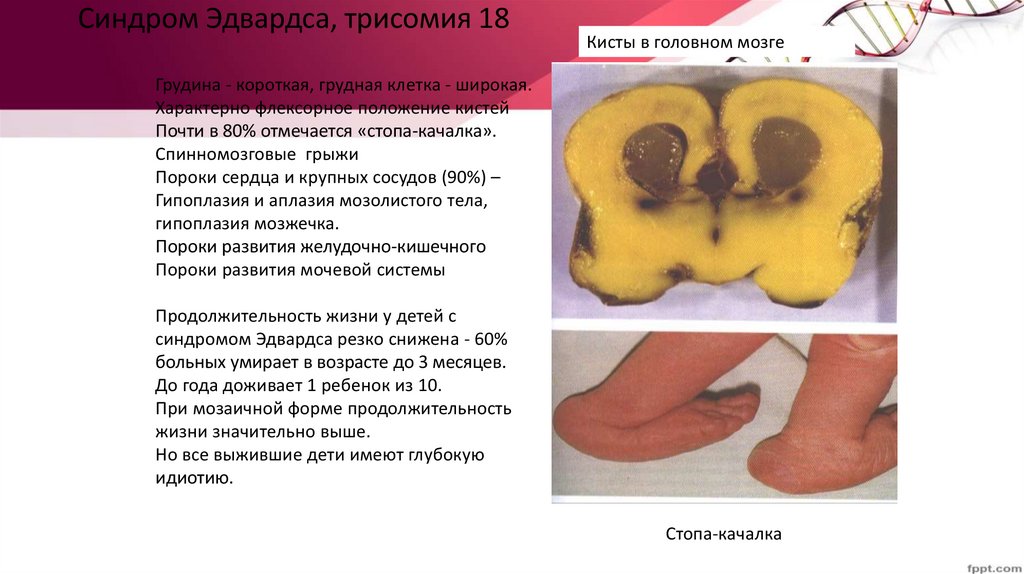
Синдром Эдвардса, трисомия 18Кисты в головном мозге
Грудина - короткая, грудная клетка - широкая.
Характерно флексорное положение кистей
Почти в 80% отмечается «стопа-качалка».
Спинномозговые грыжи
Пороки сердца и крупных сосудов (90%) –
Гипоплазия и аплазия мозолистого тела,
гипоплазия мозжечка.
Пороки развития желудочно-кишечного
Пороки развития мочевой системы
Продолжительность жизни у детей с
синдромом Эдвардса резко снижена - 60%
больных умирает в возрасте до 3 месяцев.
До года доживает 1 ребенок из 10.
При мозаичной форме продолжительность
жизни значительно выше.
Но все выжившие дети имеют глубокую
идиотию.
Стопа-качалка
34.
Анеуплоидии по половым хромосомамНе приводят к тяжелым нарушениям развития благодаря
способности Х хромосомы образовывать тельце Барра
Общая частота полисомий по Х- и У-хромосомам составляет
1,5-2,0:1000 новорожденных.
35.
Синдром трисомии Хкариотип ХХХ
Впервые цитогенетика этого синдрома была описана в 1959 г. Р. Джекобсом.
Частота этого синдрома составляет 1:1000-1:2000 новорожденных девочек.
Как правило, физическое и психическое развитие у женщин с этим синдромом не
имеет отклонений от нормы.
Умственное развитие также обычно нормально, иногда на нижних границах
нормы.
Лишь у некоторых женщин отмечаются нарушения со стороны репродуктивной
функции (различные нарушения цикла, вторичная аменорея, ранняя менопауза).
При тетрасомиях ХХХХ встречается высокий рост, телосложение по мужскому
типу, эпикант, гипертелоризм, уплощенное переносье, высокое небо,
аномальный рост зубов, деформированные и аномально расположенные ушные
раковины. У этих женщин описаны различные нарушения менструального цикла,
бесплодие, преждевременный климакс.
Снижение интеллекта - от пограничной умственной отсталости до различных
степеней олигофрении, описано у двух третей больных.
36.
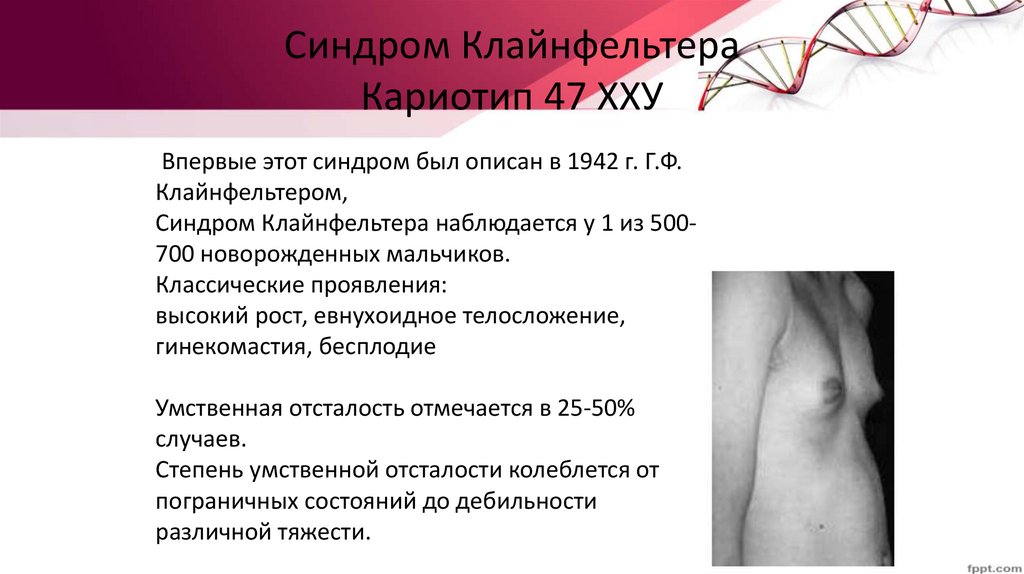
Синдром КлайнфельтераКариотип 47 ХХУ
Впервые этот синдром был описан в 1942 г. Г.Ф.
Клайнфельтером,
Синдром Клайнфельтера наблюдается у 1 из 500700 новорожденных мальчиков.
Классические проявления:
высокий рост, евнухоидное телосложение,
гинекомастия, бесплодие
Умственная отсталость отмечается в 25-50%
случаев.
Степень умственной отсталости колеблется от
пограничных состояний до дебильности
различной тяжести.
37.
Синдром Шерешевского-Тернера, 45,ХОСиндром был описан в 1925 г. русским врачом Н.А.
Шерешевским. В 1938 г Ц. Тернером.
Частота этого заболевания составляет 1:2000-5000
новорожденных девочек.
короткая с кожными складками шея (шейный птеригиум) и
лимфатический отек кистей и стоп
низкорасположенные ушные раковины, короткая шея,
низкий рост волос на шее, широкая грудная клетка.
В пубертатном периоде наблюдается отсутствие
формирования вторичных половых признаков
Пороки развития кровеносной системы, патология почек
Интеллект больных с синдромом Шерешевского-Тернера
сохранен
38.
Мультифакториальные болезни можно разделить на:• врожденные пороки развития (расщелина губы и
неба, спинно-мозговая грыжа, стеноз привратника,
аненцефалия и черепно-мозговая грыжа, вывих
бедра, гидроцефалия, врожденная косолапость)
• распространенные психические и нервные болезни
(шизофрения, эпилепсия, рассеянный склероз)
• Распространенные соматические болезни
«среднего» возраста (псориаз, бронхиальная астма,
ревматизм, сахарный диабет, ИБС, атеросклероз,
язвенная болезнь, желчекаменная и мочекаменная
болезнь, ревматоидный артрит, многие формы рака)