
")















 medicine
medicineSimilar presentations:


")







Наследственные ферментопатии обмена металлов у новорожденных детей
1. Наследственные ферментопатии обмена металлов у новорожденных детей
АО «Медицинский университет Астана»НАСЛЕДСТВЕННЫЕ
ФЕРМЕНТОПАТИИ
ОБМЕНА МЕТАЛЛОВ
У НОВОРОЖДЕННЫХ
ДЕТЕЙ
Подготовила: 756 гр. педиатрия
Проверила: Чехович Г.И
2. БОЛЕЗНЬ МЕНКЕСА (НАРУШЕНИЯ ТРАНСПОРТА МЕДИ, БОЛЕЗНЬ СТАЛЬНЫХ ВОЛОС, БОЛЕЗНЬ КУРЧАВЫХ ВОЛОС, СИНДРОМ ЗАТЫЛОЧНЫХ РОГОВ)
3.
БолезньМенкеса
(болезнь
курчавых
волос)
—
прогрессирующее заболевание с Х-сцепленным рецессивным
типом наследования. Ген, мутации которого приводят к
развитию болезни Менкеса, кодирует АТФазу Р-типа,
переносящую медь. Мутации в гене, кодирующем этот
протеин, ассоциируются с низким уровнем меди и
церулоплазмина, а также с дефектами абсорбции и транспорта
меди в кишечнике.
Генетика: мутациии гена, кодирующие
Cu(2+)-транспортиующую ATФ-азу,
полипептид альфа (ATP7A; MIM *300011).
Ген картирован на длинном плече Хромосомы (локус Хq12-q13).
Тип наследования:: Х-сцепленный
рецессивный
Эпидемиология:частота в мире 1:40 0001:350 000 живых новорожденных
мальчиков.
4.
Патогенез:в результате мутаций гена ATP7A, кодирующего одиниз полипептидов трансмембанного белка происходит
нарушение транспорта ионов меди. Патогенетические
механизмы, приводящие к заболеванию, обусловлены
сочетанной недостаточностью медь-содержащих белков,
участвующих в окислительном фосфорилировании, продукции
меланина, процессинге коллагена и кератина, дезинтоксикации
свободных радикалов, продукции дофамина.
Концентрация медьсвязывающих белков- церулоплазмина в
сыворотке и активность некоторых медьсодержащих ферментов
(например,
аминооксидаз
соединительной
ткани
)
снижены. Волосы курчавые . Нарушение синтеза коллагена и
эластина приводит к образованию расслаивающих аневризм,
разрыву миокарда, эмфиземе легких и остеопорозу . Анемия
отсутствует. Смерть обычно наступает в первые 5 лет жизни.
5.
Клинические проявления:обычно заболевание манифестирует в неонатальныйпериод. К числу ранних клинических симптомов относятся: гипотеpмия,
гипеpбилиpубинемия, задержка физического развития. В неонатальный период
волосы как правило, не изменены, но у части больных встречается образование
узелков на волосах (trichorexis nodosa) и веретенообразные волосы (monilethrix).
Иногда у таких больных наблюдается себорейный дерматит. К 2-3 месяцам жизни
становится очевидным отставание роста, задержка психомоторного развития и
прогрессирующие неврологические расстройства с потерей ранее приобретенных
навыков, появляются различные типы эпилептических приступов (фокальные,
генерализованные, миоклонические). Позднее формируется спастический
тетрапарез. С течением времени трихополидистрофия становится очевидной:
волосы спутанные, тусклые, жесткие, седые или волосы цвета "слоновой кости".
Появляются осложнения со стороны сосудистой системы в виде субдуральных
кровоизлияний, разрывов артерий и тромботической болезни. При ангиографии
выявляются удлиненные, извитые, варьирующего калибра артерии с
чередованием областей расширения и сужения - в головном мозге, внутренних
органах, конечностях. При контрастной рентгенографии выявляются дивертикулы
мочевого тракта, которые могут привести к разрывам и предрасполагают к
рекуррентным инфекциям мочевого тракта, что становится большой проблемой
для пациентов с более длительным сроком жизни. Часто наблюдаются
патологические переломы pебеp, гиперрастяжимость кожи, диффузная
гипопигментация кожи, гиперподвижность
6.
Диагностика:приМРТ/КТ
головного
мозга
обнаруживают кортикальную и субкортикальную
атрофию, фокальные очаги некроза в веществе
головного мозга и атрофию мозжечка. При КЭЭГ
регистрируют мультифокальные спайки и медленноволновую активность. Микроскопическое исследование
волос также помогает в диагностике данного
заболевания. Основными методами диагностики
является снижение уровня меди и церулоплазмина в
сыворотке крови. При биопсии печени обнаруживают
низкое содержание меди, в слизистой кишечника и
культуре клеток кожных фибробластов - высокое
содержание.
7.
Лечение:применяют введение гистидинатамеди 0,2-0,45 мг/день внутривенно, что
обеспечивает восстановление нормального
содержания ионов меди, медь-зависимых
белков (церулоплазмин) в сыворотке, печени и
ЦСЖ. Для лечения тяжелых неврологических
нарушений лечение малоэффективно.
Прогноз
Неблагоприятный.
8. Гемохроматоз, или бронзовый диабет, гемосидероз, пигментный цирроз, гемомеланоз, сидерофилия, синдром Труазье-Ано-Шоффера
Гемохроматоз, или бронзовый диабет, гемосидероз, пигментныйцирроз, гемомеланоз, сидерофилия, синдром Труазье-АноШоффера
9.
Гемохроматозноворожденных
—
редкая
форма
фульминантного поражения печени неизвестной причины с
диффузным отложением железа в печени, поджелудочной
железе, сердце и эндокринных железах без повышенного
поступления железа в организм (с пищей или в виде
трансфузий). В некоторых случаях отмечено наследование по
аутосомно-рецессивному
типу.
Дети
могут
родиться
недоношенными, либо у них отмечается внутриутробная
задержка развития. При беременности определяется большая
плацента; быстрое прогрессирование заболевания со
смертельным
исходом
характеризуется
развитием
гепатомегалии,
гипогликемии,
гипопротромбинемии,
гипоальбуминемии и гипербилирубинемии.
10.
ФормыПервичный (идиопатический) гемохроматоз – наследственное
заболевание, связанное с мутацией (нарушением структуры,
повреждением) гена, отвечающего за обмен железа в организме.
Неонатальный гемохроматоз (избыточное содержание железа в
организме у новорожденных) – быстропрогрессирующее редкое
заболевание новорожденных. Причины его неизвестны.
Также
выделяют
следующие
стадии
гемохроматоза:
1 стадия - гемохроматоз без перегрузки железом (обмен железа в
организме нарушен, но содержание железа в организме еще не
превышает границы нормы);
2 стадия - перегрузка железом без клинических проявлений (избыток
железа в организме);
3 стадия - наблюдают клинические проявления (общие симптомы:
гиперпигментация, нарушения функций печени, почек, сердца,
поджелудочной железы).
11.
У небольшого процента людей, гомозиготных по генудефицита основного ингибитора сывороточных протеаз,
а-антитрипсина, отмечается холестаз новорожденных с
последующим развитием цирроза в детском возрасте.
На долю а—антитрипсина, синтезируемого в печени
ингибитора протеаз, приходится до 80 % сывороточной
фракции
а-глуболина.
Синтез
а,-антитрипсина
регулируется 20 кодоминантными аллелями, некоторые
из которых определяют синтез дефектных ингибиторов
протеаз. Самый распространенный аллель системы
ингибиторов протеаз (Pi) назван М; дети с нормальным
фенотипом имеют формулу PiMM.
12.
Этиология и встречаемость наследственного гемохроматоза.Наследственный гемохроматоз (MIM №235200) — болезнь накопления
железа, встречающаяся у некоторых лиц с гомозиготными или
компаундными гетерозиготными мутациями в гене HFE. Большинство
пациентов (90-95%) с наследственным гемохроматозом гомозиготны
по мутации Cys282Tyr; оставшиеся 5-10% больных — компаундные
гетерозиготы по Cys282Tyr и другой мутации, His63Asp. Гомозиготность
по мутации His63Asp не ведет к клиническому гемохроматозу, если нет
дополнительных причин перегрузки железом.
Наследственный гемохроматоз первоначально считался простым
моногенным заболеванием. В настоящее время по генному дефекту и
клинической картине выделяют 4 формы ПГХ:
классический аутосомно-рецессивный HFE-1;
ювенильный HFE-2;
HFE-3, связанный с мутацией в трансферриновом рецепторе 2-го типа;
аутосомно-доминантный гемохроматоз HFE-4.
13.
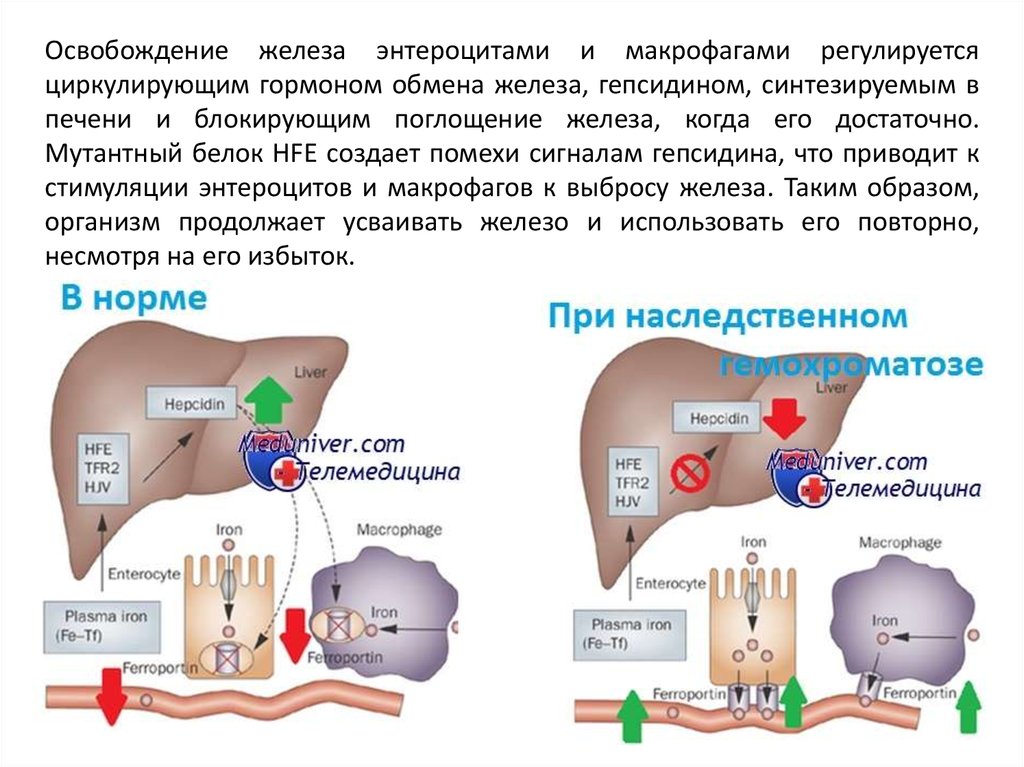
Освобождение железа энтероцитами и макрофагами регулируетсяциркулирующим гормоном обмена железа, гепсидином, синтезируемым в
печени и блокирующим поглощение железа, когда его достаточно.
Мутантный белок HFE создает помехи сигналам гепсидина, что приводит к
стимуляции энтероцитов и макрофагов к выбросу железа. Таким образом,
организм продолжает усваивать железо и использовать его повторно,
несмотря на его избыток.
14.
Течение заболевания может существенно различаться. В первую неделю жизниобычно уже присутствуют желтуха, ахолический кал и гепатомегалия, но ко 2-4-му
месяцу желтуха исчезает. Возможно полное разрешение заболевания, стойкого
поражения печени или даже цирроза. У детей старшего возраста развивается
картина хронического поражения печени или цирроза с портальной
гипертензией.
Диагностика основана на определении генотипа Pi и гистологическом
исследовании биоптатов печени. В перипортальных гепатоцитах обнаруживают
цитоплазматические ШИК-позитивные гранулы. Иммунофлюоресцентное и
иммуноцитохимические исследования показали, что эти гранулы содержат
материал, генетически сходный с а—антитрипсином. Высказано предположение,
что нарушение биосинтеза белка или его гликозилирования может влиять на
экскрецию продукта из шероховатого эндоплазматического ретикулума во
внеклеточное пространство. При электронной микроскопии находят
бесформенные отложения гликопротеина в расширенном шероховатом
эндоплазматическом ретикулуме. Поражение печени у новорожденных
варьирует в широких пределах: гигантоклеточная трансформация печеночных
клеток, минимальное воспаление, стаз желчных кислот, возможен портальный
фиброз различной степени с разрастанием желчных протоков. Трансплантация
печени может привести к излечению; другой эффективной терапии пока не
предложено, хотя существует перспектива генной терапии
15. Семейный периодический паралич
16.
Семейный периодический паралич - это редкоеаутосомное состояние, характеризующееся эпизодами
вялого паралича с потерей глубоких сухожильных
рефлексов и отсутствием ответа мышц на электрическую
стимуляцию. Существуют 3 формы: гиперкалиемическая,
гипокалиемическая
и
нормокалиемическая.
Гипокалиемическая
форма
семейного периодического паралича развивается
вследствие мутации в гене кальциевого канала,
ассоциированного с рецептором дигидропиридина.
Гиперкалиемическая форма является следствием мутаций
гена, кодирующего альфа-субъединицу натриевых
каналов в скелетной мускулатуре (SCN4A). Причина
нормокалиемической формы остается неясной; при
некоторых обстоятельствах она может быть следствием
мутации гена, кодирующего натриевые каналы.
17.
При гипокалиемической форме эпизоды обычно появляются в возрастедо 16 лет. На следующий день после активных занятии, упражнении
пациент часто просыпается со слабостью, которая может быть легкой и
ограниченной определенными группами мышц или может охватывать
все 4 конечности. Эпизоды провоцируются пищей, богатой углеводами.
Щадятся глазодвигательные, мышцы, иннервируемые бульбарной
группой черепных нервов, и дыхательные мышцы. Сознание не
страдает. Уровень калия в крови и моче снижен. Слабость сохраняется
вплоть до 24 часов.
При гиперкалиемической форме эпизоды часто начинают появляться в
более раннем возрасте и обычно являются более короткими, более
частыми и менее тяжелыми. Эпизоды провоцируются физическими
упражнениями после еды или голоданием. Часто отмечается миотония
(позднее наступление расслабления после мышечного сокращения).
Миотония век может быть единственным симптомом.
При нормокалиемической форме пораженные пациенты чувствительны
к приему калия с пищей, и у них отмечаются эпизоды легкой мышечной
слабости при нормальном уровне калия в сыворотке крови.
18.
Лучшим диагностическим показателем является анамнез - типичные эпизоды. Приизмерении во время эпизода уровень калия в сыворотке крови может быть
измененным. Семейный периодический паралич иногда может провоцироваться
введением глюкозы и инсулина (гипокалиемическая форма) или хлорида калия
(гиперкалиемическая форма), однако только опытные врачи должны проводить
эти процедуры, так как при спровоцированном эпизоде могут развиться паралич
дыхательной мускулатуры или нарушение внутрисердечной проводимости.
Эпизоды гипокалиемического паралича лечат назначением хлорида калия в
растворе для приема внутрь (без добавления сахара) или внутривенным
введением калия. Показана диета с низким содержанием углеводов и натрия;
исключение деятельности, требующей мышечного напряжения, а также алкоголя
после периодов отдыха; прием ацетазоламида внутрь один раз в день может
способствовать предотвращению развития гипокалиемических эпизодов.
Начинающийся легкий эпизод гиперкалиемического паралича можно прервать
легкими физическими упражнениями или приемом углеводов из расчета 2 г/кг.
Развившийся эпизод требует тиазидов, ацетазоламида или ингаляционных бетаагонистов. Тяжелые приступы требуют внутривенного введения глюконата кальция
или глюкозы с инсулином. Регулярный прием углеводов, низкое содержание калия
в пище и избегание голодания и деятельности, требующей мышечного
напряжения, после еды, а также нахождения на холоде помогают предотвратить
гипокалиемические эпизоды.
19.
Сисок использованной литературы:1. http://meduniver.com/Medical/Neurology/bolezn_menkesakurchavix_volos.html MedUniver
2. http://www.rare-diseases.ru/rare-diseases/encyclopediadiseases/123-2010-07-0318-28-23
3. http://meduniver.com/Medical/genetika/nasledstvennii_gemoxromatoz.html MedUni
ver
4. https://detstrana.ru/service/disease/newborns/gemohromatoz/
5. http://dommedika.com/phisiology/gemoxromatoz_i_deficit_antitripsina.html Domme
dika
6. http://ilive.com.ua/health/semeynyy-periodicheskiy-paralich_108390i15937.html