










")























")



















")























 medicine
medicineSimilar presentations:










Наследственные заболевания нервной системы у детей
1. Наследственные заболевания нервной системы у детей
Кафедра нервных болезней и нейрохирургии КГМУд.м.н. Заболотских Наталья Владимировна
2. Общая классификация наследственных заболеваний нервной системы
• Заболевания, при которых поражениенервной системы составляет ведущий
симптомокомплекс, «ядро» клинической
картины.
• Заболевания с системными наследственными
дефектами метаболизма , при которых,
наряду с поражением различных органов и
тканей, в качестве одного из проявлений
может наблюдаться вовлечение нервной
системы
3. Классификация наследственных болезней нервной системы в соответствии с уровнем поражения.
• Наследственные нервно- мышечные болезни:прогрессирующие мышечные дистрофии;
врождённые непрогрессирующие миопатии ( структурные миопатии)
наследственные болезни мотонейрона ( спинальные амиотрофии )
наследственные невропатии
наследственные миотонические синдромы
наследственные миастенические синдромы
• Наследственные болезни центральной нервной системы:
заболевания с преимущественным вовлечением экстрапирамидной системы
заболевания с преимущественным вовлечением пирамидной системы
заболевания с преимущественным вовлечением координаторных систем
заболевания с преимущественным вовлечением когнитивной сферы
наследственные формы эпилепсии
другие формы наследственных заболеваний ЦНС
• Наследственные болезни нервной системы с мультиорганными
проявлениями:
заболевания, характеризующиеся нарушением клеточной пролиферации
наследственные болезни обмена с вовлечением обмена нервной системы
4. Проблемы классификации наследственных заболеваний нервной системы
Для наследственных заболеваний нервной системыхарактерен исключительно выраженный меж- и
внутрисемейный полиморфизм.
Полиморфизм морфологической картины, отражающий
в первую очередь вариабельный характер и тяжесть
мутаций у конкретных больных.
Патогенез большинства форм наследственных
неврологических заболеваний изучен недостаточно.
5. Основные типы наследования
• Аутосомно-доминантный• Аутосомно-рецессивный
• Х- сцепленный доминантный и
рецессивный
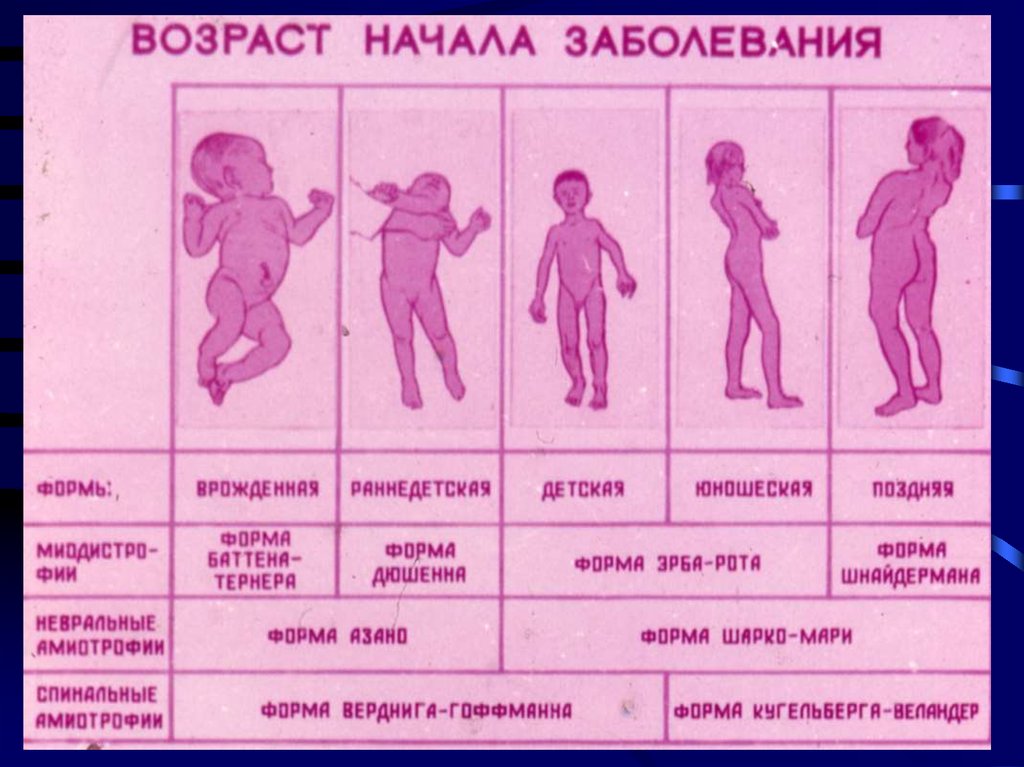
• Митохондриальный или материнский
6.
Аутосомно- доминантный тип наследования1. Патологический ген является
доминирующим, распределение аллелей
Аа×аа
2. Вертикальная передача болезни (от
родителей к детям)
3. Риск возникновения болезни -50%
(соотношение больного и здорового
потомства 50%)
4. Мужчины и женщины поражаются в
равной степени
7.
Аутосомно- рецессивный тип наследования• Мутантный ген находится на обеих
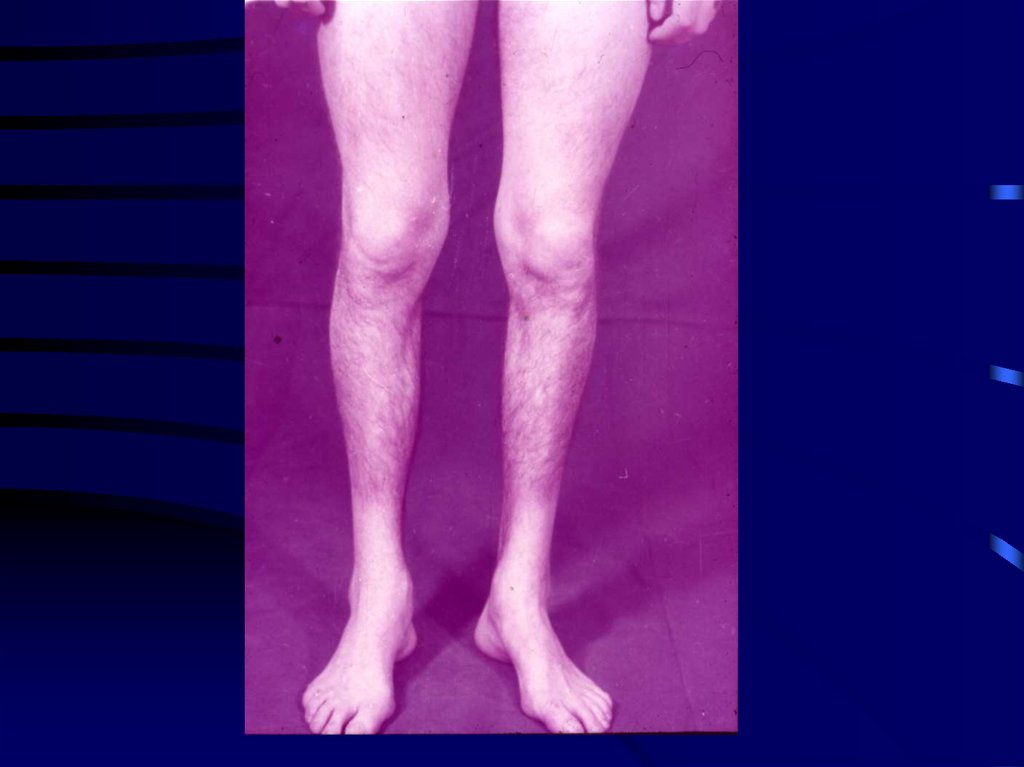
гомологичных хромосомах, унаследованных
от родителей (Аа ×Аа)-а-рецессивный
мутантный ген)
• «Горизонтальная передача болезни» болезнь проявляется в одном поколении
среди сибсов, родители при этом клинически
здоровы
• Доля пораженных сибсов – 25%
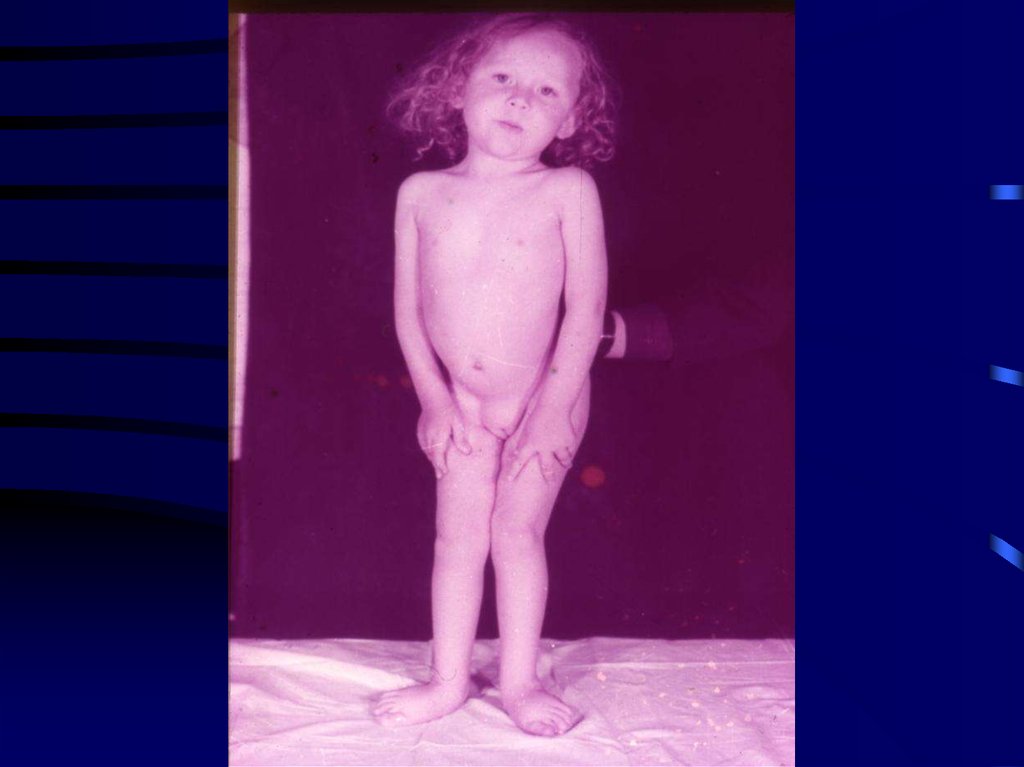
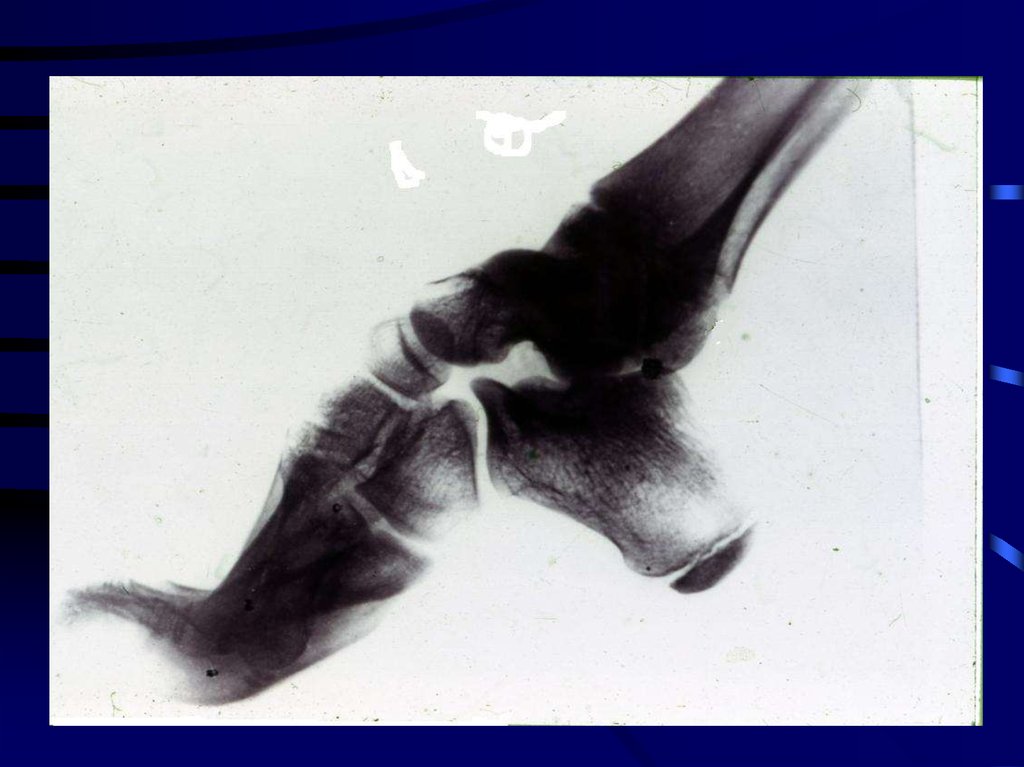
• У родителей больных часто
кровнородственный брак
• Мужчины и женщины поражаются в равной
степени
8.
Х-сцепленный рецессивный тип наследования• Заболевание проявляется только у мужчин,
унаследовавших от матери мутантную хромосому
• Заболевание передается клинически здоровыми
женщинами половине сыновей с мутантной Ххромосомой
• Отсутствие прямой передачи болезни от мужчин их
сыновьям (наследуют от отца нормальнуюY-хромосому)
Х-сцепленный доминантный тип наследования (очень
редкий)
• Ген, локализованный на Х- хромосоме, определяет
развитие доминантного признака
• Все дочери отца наследуют заболевание
• Передача заболевания от отца к сыну невозможна
(сыновья наследуют здоровую Y-хромосому)
• Вероятность рождения больного ребенка любого пола от
больной матери равна 50%
• В каждой родословной число больных женщин в 2 раза
больше, ем больных мужчин
9.
Митохондриальный или материнский типнаследования
Набор митохондрий в клетках организма
имеет исключительно материнское
происхождение
• Заболевание передается от больной матери
ко всем ее детям
• Сыновья и дочери (мужчины и женщины)
поражаются в равной степени
• Передача болезни по мужской линии
невозможна
10.
Миопатии – группа болезней или синдромовпри которых основные патологические,
биохимические, электрофизиологические
изменения происходят в скелетных мышцах. Эти
изменения не связаны с первичным поражением
периферической или центральной нервной
системы
11.
В мышце могут повреждаться:• нити актина и миозина
• митохондрии
• лизосомы
• плазматические мембраны эндоплазматического
ретикулума и сарколеммы
• межмышечная соединительная ткань
• нервно-мышечный синапс
• постсинаптическая мембрана
• двигательная концевая пластинка
12. Заболевания, связанные с патологией мышц (миопатии)
Прогрессирующиемышечные
дистрофии
Дюшена, Бекера, плече-лопаточно-лицевая ЛандузиДежерина, конечностно-поясная миодистрофия ЭрбаРота, окулофарингеальная миодистрофия, дистальные
миодистрофии, скапуло-перонеальная, глазная и др.
Врожденные
Болезнь центрального стержня, миотубулярная
непрогрессирующие миопатия, немалиновая миопатия и др.
(структурные)
миопатии
Метаболические
миопатии
Гликогензависимые миопатии, митохондриальные
энцефаломиопатии
Миотонические
миопатии
Дистрофическая миотония Куршманна-ШтейнертаБаттена, врожденная миотония Томсена. Врожденная
миотония Эйленбурга (или парамиотония).Миотония
Беккера
Семейные
периодические
параличи
Гипокалиемический, гиперкалиемический,
нормокалиемические параксизмальные миоплегии
13.
14.
Прогрессирующие мышечные дистрофии –наиболее обширная группа заболеваний.
Клинически миопатии в отличие от других
поражений моторной единицы проявляются
прогрессирующей мышечной слабостью,
атрофией мышц
15.
Мышечная дистрофия Дюшена и Беккера относится ксамым частым формам мышечных дистрофий
• Сцепленные с Х-хромосомой рецессивные
заболевания
• Проявляются у мальчиков в возрасте 2-6 лет
• Матери детей являются носителями рецессивного
гена
• Миопатия Дюшена связана с делецией в локусе
Хр21 (на коротком плече Х-хромосомы)
• Данный ген является самым большим из известных
на сегодня генов человека и кодикует белок
«дистрофин»
• Этот белок является важной составной частью
цитоскелета и обеспечивает связь между
сократительным аппаратом мышечного волокна и
сарколеммой
• При отсутствии синтеза дистрофина проявления
заболевания соответствуют ПМД Дюшена
• При снижении количества и изменениях размера
молекулы дистрофина - ПМД Беккера
16.
• Генная мутация ведет к полному нарушению синтезадистрофина – структурного мышечного белка → гибель
миофибрилл → поражение скелетных и гладких мышц
• Форма Дюшена - наиболее злокачественная форма
мышечных дистрофий
• Начинается в первые годы жизни с быстрым
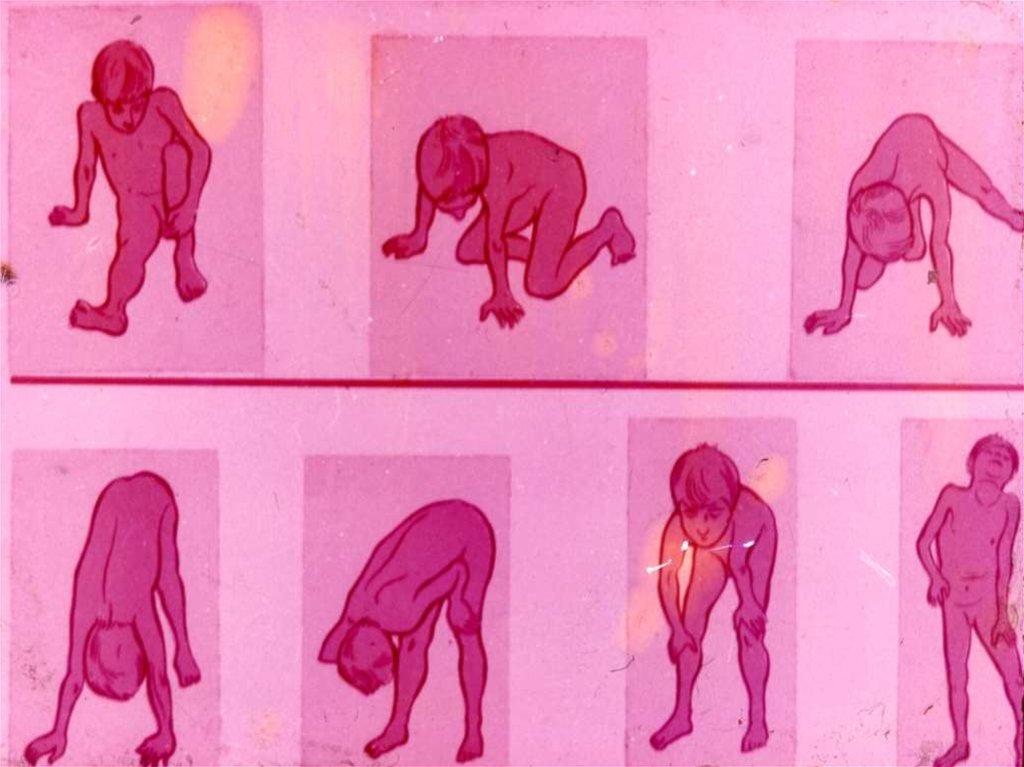
прогрессированием
• Отмечается отставание детей в моторном развитии (с
задержкой начинают садиться, вставать, ходить)
• Двигательная неловкость, неустойчивость, частые
спотыкания, падения, двигательная пассивность, нежелание
детей ходить.
• В 2-3 года появляется мышечная слабость, проявляющаяся
при физической нагрузке – длительной ходьбе, подъеме по
лестнице
• Развивается атрофия мышц сначала тазового, а затем и
плечевого пояса
• Это сопровождается слабостью проксимальных групп мышц,
что приводит к переваливающейся («утиной») походке,
затруднениям при вставании и подъеме по лестнице,
затруднению при попытке сесть из положения лежа на спине
• Атрофии всегда симметричны
17.
18.
• Вначале атрофии мышц локализуются в мышцахтазового пояса, бедер, а через 1-3 года быстро
распространяются на плечевой пояс, мышцы
спины
• Вследствие атрофий углубляется поясничный
лордоз, формируются крыловидные лопатки,
«осиная» талия
• Одним из наиболее характерных симптомов
является псевдогипертрофии различных мышц
(наиболее часто икроножных, дельтовидных,
ягодичных мышц) - атлетический вид мышц
голеней сопровождается снижением их силы
• Исчезают коленные, позднее – разгибательно- и
сгибательно-локтевые рефлексы
• Рефлекс с ахиллова сухожилия длительное время
остается сохранным
19. Типичное расположение псевдогипертрофий при форме Дюшенна
20. Внешний вид больного миопатией Дюшенна
21.
22.
23.
24.
25.
26.
27.
28.
29.
При миодистрофии Дюшена имеется сочетание с патологиейкостно-суставной системы и внутренних органов (сердечнососудистой и эндокринной).
Костно-суставные нарушения характеризуются
• деформацией позвоночника (сколиоз, поясничный лордоз)
• уплощением и деформацией грудной клетки («килевидная
грудь», «ладьевидная грудь»)
• высоким сводом стопы
• эквиноварусная деформация стоп
• контрактуры в крупных суставах
Сердечно-сосудистые расстройства проявляются
гипертрофической или дилатационной кардиомиопатией:
• тахикардия, аритмия, блокадой ножек пучка Гиса и т.д.
• сердечная недостаточность, гипертрофия левого желудочка,
снижение сократительной способности миокарда,
• лабильностью пульса, артериального давления
• расширением границ сердца
30.
• К 7-10 годам возникают грубыедвигательные расстройства - выраженные
изменения походки, снижение мышечной
силы, значительно ограничение
самостоятельного передвижения
• К 14-15 годам наступает обездвиженность,
развитие контрактур, дыхательные
нарушения
• Гибель больных наступает на 2 - 3
десятилетии жизни.
31.
Диагноз ставится на основании• данных генеалогического анализа (рецесивный
сцепленный с Х-хромосомой тип наследования)
• раннего начало в 1-3 года,
• клинических данных (симметричные атрофии
проксимальных групп мышц, развивающиеся в
восходящем направлении, псевдогипертрофии
икроножных мышц, грубые соматические и
нейроэндокринные расстройства
• снижение интеллекта
• быстрое злокачественное течение болезни
• данных биохимических исследований (типично
раннее, с 5-го дня жизни ребенка, увеличение
активности КФК – в 30-50 раз выше нормы),
• игольчатой ЭМГ миогенный тип поражения
• бипсии мышц, выявляющих первичномышечный тип поражения
32.
Форма Беккера• рассматривается как «мягкий» клинический вариант
ПМД Дюшена с более поздним началом (в 12-15 лет),
относительно доброкачественным течением и
сохранностью способности к самостоятельной ходьбе на
протяжении 15-20 лет от момента появления первых
симптомов
• Умеренные соматические расстройства, медленное течение.
• Костно- суставные деформации возможны
преимущественно в поздние сроки заболевания.
• В крови повышена активность КФК
Имеется большее разнообразие клинических проявлений:
У ряда больных доминирует картина кардиомиопатии при
малой степени вовлечения в патологический процесс
скелетных мышц
у других имеет место медленное прогрессирование
симптомов мышечной слабости, они сохраняют способность
к самостоятельной ходьбе до 60 лет
Относительно часто встречается лишь «полая стопа»
33. Внешний вид больного миопатией Беккера
34.
Для подтверждения клинического диагнозадистрофинопатии используют:
• молекулярно-генетические (обнаружение
мутантного гена)
• иммунобиохимические или
иммуногистохимические (обнаружение
уменьшения или отсутствия дистрофина)
• методы исследования мышечных
биоптатов с помощью АТ к дистрофину
35.
Прогрессирующая мышечная дистрофия Эрба–Рота(конечностно-поясная форма прогрессирующей мышечной
дистрофии, лопаточно-бедренный тип Эрба (тип II А))
• При этой форме на сегодняшний день обнаружено
около 10 различных генетических дефектов и
различают как аутосомно-доминантные, так и
аутосомно-рецессивные формы
• тип Эрба - аутосомно-рецессивная форма
Заболевание чаще начинается в 14-16 лет, крайне
редко – в 5-10 лет
• Мышечная слабость преобладает в
проксимальных отделах верхних конечностей и
плечевом поясе
• Иногда миодистрофический процесс одновременно
поражает мышцы тазового и плечевого пояса
• Мимическая мускулатура интактна
36.
37.
• Довольно значительно страдают мышцы спины и живота• Вследствие атрофий возникает лордоз, «крыловидные
лопатки», «осиная» талия, «утиная» походка
• При вставании больные применяют вспомогательные
приемы – вставание «лесенкой».
• Псевдогипертрофии мышц, контрактуры суставов,
сухожильные ретракции малохарактерны, если и
наблюдаются, то выражены умеренно.
• Кардиомиодистрофии, снижение интеллекта отсутствуют
• К 30-40 годам наблюдается глубокая физическая
инвалидизация, к 20 годам у 50% больных наблюдается
утрата самостоятельной ходьбы
• В ранних стадиях заболевания в крови активность КФК
значительно повышена, но по мере прогрессирования
заболевания она может снижаться до нормальных величин.
• На ЭМГ – первично-мышечный тип поражения
38. Лицелопаточно-плечевая миодистрофия (тип Ландузи-Дежерина)
• Заболевание наследуется по аутосомно-доминантному типу свысокой пенетрантностью и вариабельной экспрессивностью
• Начинается в возрасте около 20 лет, иногда позже.
• Мышечная слабость и атрофии вначале появляются в
мышцах плечевого пояса (лопатки, плечи) с последующим
распространением на лицо
• Это приводит к возникновению симптомов свободных
надплечий, «крыловидных» лопаток, уплощению грудной
клетки в передне-заднем направлении и ротацией внутрь
плечевых суставов
• Постепенно процесс распространяется на мышца
проксимальных отделов рук, а затем и на нижние конечности
• Имеется преимущественное атрофия двуглавой и трехглавой
мышц плеча, большой грудной, передней зубчатой,
трапецивидной мышц.
• Имеется своеобразие поражения мышц: вначале появляется
поражение разгибателей стоп, что приводит в «степажу», а
лищь позже поражаются мышцы проксимальных отделов.
39.
40.
41.
42.
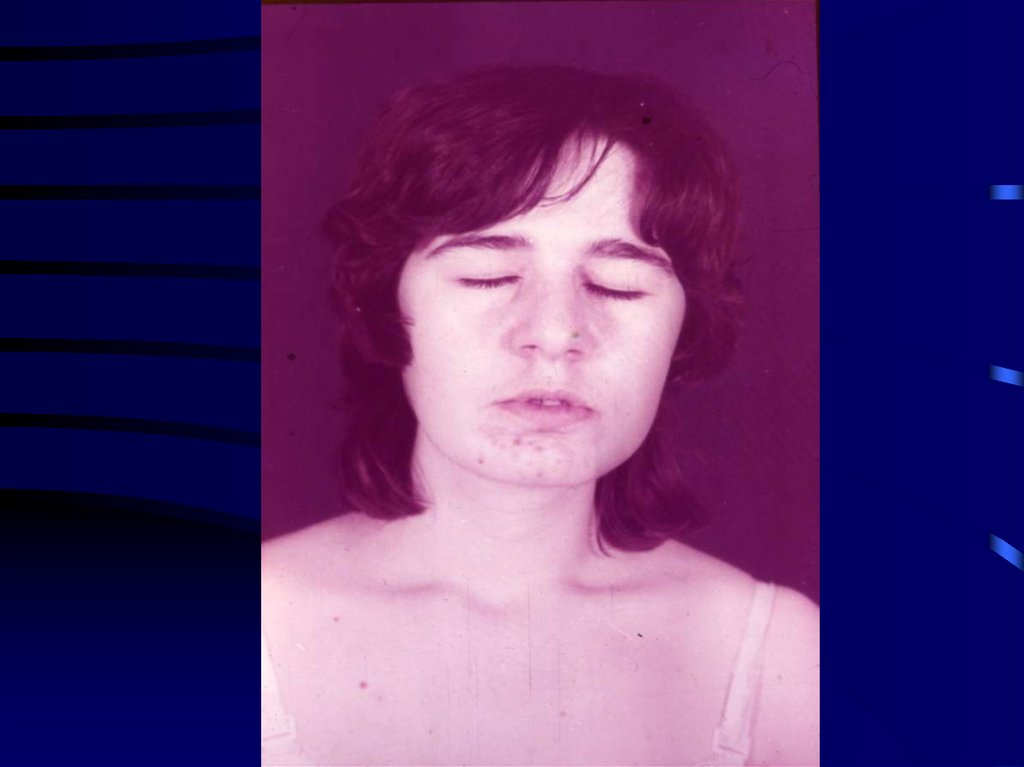
• Лицо становится гипомимичным (лицомиопата). Грубо страдают круговая мышца
глаз и рта
• Типичны «полированный» лоб, лагофтальм,
«поперечная» улыбка (улыбка Джаконды),
толстые, иногда вывороченные губы,
протрузия верхней губы («губы тапира»).
• Нередко имеется асимметрия поражения
мышц, псевдогипертрофия икроножных,
дельтовидных мышц
43.
44.
45.
• Сухожильные рефлексы сниженыпреимущественно с двуглавой и трехглавой мышц
плеча
• Кардиопатии практически не выявляются
• Интелект не страдает
• Имеются аномалии сосудов сетчатки, что приводит
к снижению зрения, отек и отслойка сетчатки.
Телеангиэктазии на глазном дне
• Может наблюдаться снижение слуха
• Активность КФК не увеличена (в некоторых
случаях повышается в 5 раз)
• Болезнь медленно прогрессирует
• Больные длительное время сохраняют
трудоспособность, продолжительность жизни в
большинстве случаев не снижается.
46.
Имеются варианты распределения атрофий исоответственно выделяют
• лопаточно-плечеягодично-бедренный
• лицелопаточно-плечеягодично-бедренный
• лицелопаточно-плечеперонеальный
• лопаточно-плечебедренный
• лицелопаточно-плечеягодично-бедренноперонеальный варианты и др.
47. Спинальные амиотрофии детского возраста
Это группа наследственных заболеваний, при которыхмышцы страдают вторично при поражении передних рогов
спинного мозга или двигательных ядер ствола мозга.
Процесс обусловлен дефектами программируемой клеточной
гибели – апоптоза
• недоразвитие клеток передних рогов спинного мозга,
двигательных ядер V, VI, VII, IX, X, XI, XII черепных
нервов
• В скелетных мышцах выявляются нейрогенные изменения
– вялые параличи
• Центральный мотонейрон не страдает
• Нет чувствительных нарушений
• Наследуются по аутосомно-рецессивному типу
• Различают врожденную форму, раннюю детскую форму
(второе полугодие жизни) и позднюю форму (1.5- 2.5 лет).
48.
Различают• Острую злокачественную инфантильную
СА Вернига-Гофмана (или СА I типа)
(врожденная форм –первые 6 мес)
• Хроническая инфантильная СА (СА II
типа) (ранняя детская форма (второе
полугодие жизни – 2 года)
• Ювенильная СА (СА III типа) (поздняя
форма (2 - 15 лет) (болезнь Кугельберга –
Веландера)
49.
Острую злокачественную инфантильную СА ВернигаГофмана (или СА I типа)Эта форма встречается с частотой 1:25 000 новорожденных.
Клиническая картина
• Возраст дебюта — с рождения ребенка до 6 мес жизни.
• Снижение двигательной активности плода может быть
отмечено еще внутриутробно по вялому шевелению
• Обнаруживаются генерализованная слабость,
превалирующая в проксимальных мышечных группах
• Гипотония и сухожильная арефлексия
• В положении на спине - «поза лягушки» с разведением и
наружной ротацией бедер
• Мимическая мускулатура сохранна, глазодвигательные
мышцы не вовлечены
• Могут быть выявлены атрофия и фасцикуляции в языке,
фасцикулярный тремор кистей
• В тех случаях, когда развивается бульбарный синдром и
исчезает глоточный рефлекс, значительно затрудняется
кормление, что может привести к гибели ребенка от
аспирационной пневмонии
50.
• Часто формируется деформация грудной клетки• Если мышечная слабость выявляется сразу же
после рождения, то смерть обычно наступает
приблизительно в 6-месячном возрасте
• При появлении первых симптомов после З мес
жизни срок выживаемости может составлять
около 2 лет.
• Основная причина смерти — дыхательная
недостаточность на фоне интеркуррентных
респираторных заболеваний
• Концентрация КФК обычно в норме, но у детей с
быстро прогрессируюшей слабостью она может
быть несколько повышена
• ЭМГ обнаруживает нейрональный тип поражения
мышц
51.
СА 1 типа необходимо дифференцировать от другихсостояний, вызывающих синдром «вялого
ребенка»
• врожденные миодистрофии и невропатии,
• структурные миопатии
• врожденная или неонатальная миастения
• метаболические миопатии
• внутриутробный полиомиелит
• Ботулизм
• хромосомная патология
• атоническая форма церебрального паралича
• синдром Марфана
52.
Хроническая инфантильная спинальная амиотрофия(СА II типа)
• Выявляется мышечная слабость между 6-м и 24-м месяцами
жизни
• Если слабость возникает между З-м и 6-м месяцами, то тип
течения болезни более злокачественный
• Первоначальные проявления слабости обычно
симметричны и наблюдаются в проксимальных мышечных
группах конечностей
• Слабость мышц бедер — наиболее заметный симптом
• Сухожильные рефлексы с пораженных мышц угнетены или
исчезают
• Все больные способны сидеть, большинство —
самостоятельно стоять, а некоторые даже могут ходить
• Мимическая мускулатура и наружные мышцы глаза на
ранних этапах болезни не поражаются
• Мышечная слабость прогрессирует медленно
• В отдельных случаях она остается стабильной многие годы,
а затем прогрессирование возобновляется
• Предполагается выживание больных вплоть до зрелого
возраста
53.
• Формируются контрактуры• Часто наблюдаются псевдогипертрофии
икроножных и ягодичных мышц, что может
вызывать мысль о миодистрофии Дюшенна.
• Стопы постепенно приобретают эквиноварусную
позицию
• У детей часто обнаруживаются тремор кистей,
фасцикуляции в языке и конечностях,
деформации позвоночника и грудной клетки,
врожденный вывих тазобедренных суставов
• Концентрация КФК в норме
• Результаты генетического анализа и данные
ЭМГ идентичны таковым при острой
инфантильной форме
54.
55.
56.
57.
Ювенильная СА (СА III типа) (поздняя формаболезнь Кугельберга – Веландера)
• Частота - 1,2 на 100 000
• Дебют между 2 и 15-м годом жизни, в большинстве
случаев до возраста 5 лет и всегда после 18 мес
жизни. Описаны случаи начала заболевания в 15-30
лет.
• В начале болезни наблюдается патологическая
мышечная утомляемость в ногах при длительной
физической нагрузке (ходьба, бег), подергивания в
мышцах
• Увеличиваются икроножные мышцы.
• Атрофии первоначально локализуются в
проксимальных группах мышц нижних
конечностей, тазового пояса, бедер и всегда
симметричны -затруднение при подъеме по
лестнице, вставании с горизонтальной плоскости,
«утиная» походка.
58.
• Через несколько лет появляются атрофии впроксимальных мышцах верхних конечностей,
выявляются «крыловидные» лопатки, ограничивается
объем движений в проксимальных отделах рук, плечевом
поясе
• Мышечный тонус снижается в проксимальных группах
мышц
• Сухожильные рефлексы угасают сначала на ногах, затем
на руках
• Выявляются фасцикулярные подергивания в мышцах,
фибриляции языка, мелкий тремор пальцев
• Костно-суставные нарушения не выражены или выражены
минимально
• Болезнь очень медленно прогрессирует, больные
длительное время сохраняют возможность
самообслуживания
• По клиническим симптомам заболевание напоминает
миодистрофию Эрба
• Активность КФК может быть умеренно повышена.
59.
Врожденная миотония – миотония Томсена• Относится к наследственным нервно-мышечным
заболеваниям, при которых первичный молекулярный
дефект связан с каналопатией, обусловленной мутацией
генов ионных каналов - мутация гена хлорного канала на
хромосоме 7q35
• Это приводит к нарушению механизма расслабления мышц
после их сокращения в связи со снижением проводимости
мышечной мембраны для ионов хлора (в норме ионы хлора
обеспечивают стабилизацию мембранного потенциала
покоя после акта сокращения)
• Тип наследования – аутосомно-доминантный
• Проявляется сразу после рождения или в детстве
• Первыми признаками миотонии у ребенка - изменение
голоса при плаче, он начинает задыхаться, а после плача
лицо очень медленно расслабляется
• Заболевание обычно протекает мягко и не прогрессирует.
60.
• Первые признаки болезни могут появляться в 8-15 лет ввиде миотонических спазмов
• Миотонические феномены локализуются чаще в мышцах
кисти, ног, жевательных мышцах и круговых мышцах
глаз.
• Чаще поражаются ноги, чем руки
• Больные жалуются на локальное повышение мышечного
тонуса в начале движения, уменьшающееся при его
продолжении
• Миотонический феномен легко вызывается при
перкуссии
• С возрастом развивается генерализованная мышечная
гипертрофия (атлетизм), являющаяся классическим
признаком миотонии Томсена
• При пальпации мышцы плотные, твердые, однако
объективно мышечная сила в них снижена
• Сухожильные рефлексы нормальные
• С возрастом миотонические проявления становятся менее
выраженными
• Продолжительность жизни не меняется
• Уровень КФК не повышен.
61. Наследственные моторно-сенсорные невропатии (НМСН)
Наследственные моторносенсорные невропатии (НМСН)Старые названия:
Невральная амиотрофия Шарко-Мари-Тута
Перонеальная мышечная атрофии.
62.
НМСН тип IРазличают аутосомно-доминантную и аутосомнорецессивную формы НМСН типа I.
• Заболевание начинается на 1-ом и реже на 2-ом десятилетии
жизни.
• Рецессивные случаи протекают намного тяжелее, начинают
проявляться раньше (до 10 лет), СПИ эфф. ниже 20 м\с
• Изменение нервов происходит по типу сегментарной
демиелинизации, сопровождающейся гипертрофическими
изменениями шванновских клеток с образованием
разрастаний миелиновой оболочки по типу «луковичных
головок»
• Вначале больные испытывают трудности при ходьбе или
беге, появляется деформация стоп.
• В связи с ранним вовлечением в процесс мышц стопы за счет
их денервации возникает ее деформация по типу «конской
стопы», «полой стопы», молоточкообразных пальцев.
• Часто такие больные лечатся у ортопедов.
63.
64.
65.
• Постепенно нарастает слабость и развиваются атрофиипреимущественно перонеальной группы мышц
• Ноги приобретают форму «перевернутых бутылок»
• В дальнейшем присоединяются слабость и атрофии мышц
верхних конечностей, особенно кистей и пальцев.
• Сухожильные рефлексы изменяются неравномерно:
ахилловы выпадают всегда, коленные – в 50% случаев,
разгибательно- и сгибательно локтевой рефлексы – только
при выраженном поражении верхних конечностей
• Наблюдается снижение поверхностной и глубокой
чувствительности
• Атаксия и дрожание обнаруживается приблизительно у 30
% больных НСМН типа I.
• Пациенты приспосабливаются к своему дефекту и
способность к самостоятельному передвижению
длительное время.
• Способность к самостоятельному передвижению
утрачивается после 50-летнего возраста.
• Болезнь прогрессирует очень медленно и нередко по
прошествии периода роста переходит в стабильное
течение.
66.
• У 10% больных никогда не возникаютсимптомы заболевания и диагноз
устанавливается только при клиническом
обследовании.
• Скорость проведения по двигательным
нервам рук при НМСН типа I обычно
составляет от 10 до 35 м\с.
• Мелкие мышцы стоп часто денервированы.
• Потенциал чувствительного нерва не
удается зарегистрировать.
67. НСМН тип II
Аутосомно-доминантная НМСН типа IIИзменение нервов происходит по типу аксональной дегенерации
наряду с минимальными проявлениями сегментарной
демиелинизации.
• Пик возникновения болезни приходится на 2-е десятилетие
жизни, но многие случаи протекают бессимптомно до более
позднего возраста, иногда вплоть до 60 лет и более.
• Заболевание по своим проявлениям очень похоже на НМСН
типа I и их невозможно достаточно четко различить
клинически.
• Приблизительно у 50% больных отсутствуют клинические
признаки нарушения чувствительности
• СПИэфф. в верхних конечностях может сохраняться в
нормальных пределах, но обычно составляет 38-58 м\с.
• В нижних конечностях СПИ эфф. замедленная в большей
степени
• СПИ афф. нарушено во всех случаях, но ПДЕ
чувствительного нерва иногда удается зарегистрировать.
68.
Аутосомно-рецессивная НМСН типа II• в возрасте до 5 лет и очень редко - после 20
лет.
• Слабость мышц при рецессивной форме
выражена в большей степени и может
охватывать проксимальные группы мышц.
• СПИ эфф. колеблется в пределах от 35 м\с до
верхней границе нормы.
• Мелкие мышцы стоп могут быть
денервированы
• ПД чувствительных нервов резко снижены
или их вообще не удается записать.
69. НМСН тип III
НМСН тип III (болезнь Дежерина –Сотта или гипертрофическийполиневрит детей) –аутосомно-рецессивный синдром
• В основе болезни лежит врожденная гипомиелинизация в сочетании с
утолщением периферических нервов, демиелинизацией,
пролиферацией шванновских клеток с образованием луковичных
утолщений.
• Начало болезни – врожденное или с первых лет жизни.
• Моторное развитие детей задерживается и никогда не достигает
нормального уровня.
• Наблюдается выраженный сколиоз.
• Клинические проявления такие же, как и при НМСН.
• Атрофии мышц и легкие нарушения чувствительности локализуются
в дистальных отделах конечностей, реже в мышцах лица,
• наблюдаются фасцикулярные подергивания.
• Отмечаются утолщение периферических нервных стволов, которые
могут определяться даже визуально.
• Скорость проведения по двигательным волокнам нервов обычно ниже
10 м\с.
• Течение заболевания тяжелое, медленно прогрессирующее.
70.
71.
72.
73.
Гепатоцеребральная дистрофия(гепатолентикулярная дегенерация, болезнь
Вильсона, болезнь Вильсона— Коновалова)
• Системное заболевание, возникающее вследствие
нарушения метаболизма меди, передающееся
аутосомно-рецессивным путем
• Характеризуется сочетанием цирроза печени с
дегенеративными изменениями базальных
ганглиев (преимущественно чечевицеобразных
ядер).
• Распространенность болезни в США составляет 1
случай на 50 000—100 000 (точных данных по РФ
нет)
74.
• Ген болезни расположен на длинном плече 13-й хромосомы• Он кодирует медьтранспортирующую АТФазу,
участвующую в превращениях церуллоплазмина.
• Для развития заболевания ребенок должен рецессивно
унаследовать 2 патологических гена (один от матери,
другой от отца), т.е. типичная клиническая картина
разворачивается только у гомозигот
• Для гетерозигот (при одном нормальном гене и одном
патологическом) характерно субклиническое течение.
• Гетерозиготы являются носителями патологического гена
и могут передать его своим детям
• для ранних дебютов характерны большие делеции и
полное разрушение гена; при менее тяжелых формах
транспорт меди страдает частично
• Генетические мутации вызывают необратимые
нарушения обмена меди: страдает экскреция металла с
желчью и нарушается образование церуллоплазмина.
75.
• Медь начинает откладываться в печени• Ионы меди проникают в кровь и разносятся по
всем тканям организма, включая мозг.
• Поражение печени с распадом ее ткани и
снижением барьерной функции ведет к
аутоинтоксикации продуктами гепатолиза и
чужеродным белком
• Медь откладывается в почках, селезенке. В мозге
– в базальных , хвостатом теле, бледном шаре,
глубоких слоях коры, зубчатых ядрах, мозжечке,
субталамических ядрах
• Медь откладывается также в роговице с
образованием колец Кайзера-Флейшера. Их цвет
может варьировать между желтым, зеленым и
коричневым.
76.
77.
Клиническая картина• Заболевание начинается в детском или молодом возрасте и
имеет хроническое прогрессирующее течение
• Появлению симптомов поражения НС обычно задолго
предшествуют висцеральные расстройства в виде
нарушения деятельности печени и желудочно-кишечных
расстройств (желтуха, боли в правом подреберье,
диспепсические явления)
• Возникает развернутый гепатолиенальный синдром
• Чем сильнее страдает печень, тем раньше начинается и
быстрее течет заболевание; 90 % больных умирают до 30
лет
• Со стороны НС на первый план выступают
экстрапирамидные симптомы в виде мышечной
ригидности, гиперкинезов и расстройства психики
• Пирамидные симптомы могут быть, но чаще отсутствуют.
• Чувствительность обычно не расстроена
78.
По Н.В. Коновалову, выделяют 5 форм гепатолентикулярнойдегенерации:
1. Брюшная форма (тяжелое заболевание печени,
приводящее к смерти раньше появления симптомов со
стороны нервной системы)
2. Ригидно - аритмогиперкинетическая, или ранняя, форма
• Отличается быстрым течением (2-З года), начинается в
детском возрасте
• Преобладают мышечная ригидность, приводящая к
контрактурам, бедность и замедленность движений,
• Хореоатетозные или торсионные насильственные
движения
• Лицо амимично, часто искажено застывшей тримасой
• Дизартрия и дисфагия
• Нередки эпилептические приступы
• Аффективные расстройства и умеренное снижение
интеллекта
79.
3. Дрожательно-ригидная форма• Встречается чаще других
• Начинается в юношеском возрасте
• Течет медленнее (в среднем 5-6 лет), с ремиссиями
и внезапными ухудшениями, сопровождающимися
субфебрильной температурой
• Характеризуется одновременным развитием
тяжелой ригидности и типичного ритмичного (2-8
дрожаний в 1 с) тремора, который резко
усиливается при статическом напряжении мышц,
движениях и волнении (в покое и во сне он
исчезает), захватывает конечности, голову и
туловище
• Примешиваются атетоидные и хореиформные
насильственные движения
• Наблюдается также дисфагия и дизартрия
80.
4. Дрожательная форма• Начинается в возрасте 20-30 лет, течет медленно (10 лет и
больше)
• В неврологическом статусе преобладает тремор
• Ригидность появляется лишь в конце болезни
• Не редко - гипотония, амимия, медленная, монотонная речь
(брадилалия), брадикинезия
• Тяжелые изменения психики: часты аффективньте
вспышки.
• Эпилептические приступы
5. Экстрапирамидно-корковая форма
• Встречается реже других форм, длится 6-8 лет
• Начинается обычно как одна из описанных выше форм
• Типичны экстрапирамидные нарушения
• В дальнейшем присоединяются пирамидные парезы,
эпилептические приступы и развивается деменция
81.
При всех формах типичным симптомом болезни является• кольцо Кайзера-Флейшера — отложение зеленоватобурого пигмента, содержащего медь, по периферии
роговой оболочки
• Иногда - желтовато-коричневая пигментация кожи
туловища и лица
• Часты геморрагические явления (кровоточивость десен,
носовые кровотечения, положительная проба жгуга),
мраморность кожи, акроцианоз
• Капилляроскопия обнаруживает атонию капилляров и
застойность кровотока
• Отмечаются суставные боли, профузные поты, остеопороз,
ломкость костей
• Обычны лейкопения и тромбоцитопения, гипохромная
анемия, явления геморрагического диатеза, купрурия
(более 100 мг меди в суточной моче)
• В крови снижен уровень церуллоплазмина
• Из-за большого количества мутаций пренатальная
диагностика не разработана
82.
• Фильм83.
Лечение• Направлено на профилактику отложения меди,
является пожизненным
• Д-пеницилламин (купримин, делен) назначают в
стартовой дозе 600-3000 мг в день (образует с медью
прочное соединение, которое экскретируется
почками
• Ацетат цинка (50 мг З раза в день) уменьшает
всасывание меди в желудочно-кишечном тракте
• Исключение из рациона богатой медью пищи какао, шоколад, грибы, орехи
• Через 12-24 мес от начала лечения уменьшаются
неврологические симптомы
• через 6-8 мес исчезает кольцо Кайзера-Флейшера.
• Прогноз в отношении выздоровления остается
тяжелым
84.
Фенилкетонурия• Наследственное нарушение обмена
• Наследование — аутосомно-рецессивное
• Ген локализован на 12q24.1
В основе патогенеза заболевания лежит дефицит
фермента печени фенилаланин-4-гидроксилазы
Нарушается гидроксилирование фенилаланина в
тирозин.
Накопление фенилаланина и его дериватов
(фенилпировиноградная, фенилуксусная,
фенилмолочная кислоты и др.) приводит к
вторичным нарушениям, в том числе накоплению
в ткани мозга недоокисленных продуктов обмена,
блокирующих нормальный метаболизм
85.
Клинические симптомы• Задержка психического развития,
обнаруживаемая в возрасте 4-6 мес
• Психические расстройства касаются
интеллектуальной сферы, поведения
• Уже в первые месяцы жизни отмечается
повышенное беспокойство в виде
беспричинного крика, нарушения сна,
реже вялость, сонливость
• У более старших детей - возбуждение с
двигательными стереотипами, аутизм,
психотические расстройства
• Судорожный синдром (у 50% больных)
86.
Неврологическая симптоматика:• вторичная микроцефалия
• изменение мышечного тонуса по
гипотоническому или гипертоническому типу
• гиперрефлексия, клонус стоп, пирамидные знаки
• гиперкинезы
• В развернутой стадии заболевания характерен
своеобразный затхлый, «мышиный» запах,
связанный с присутствием в моче фенилуксусной
кислоты
• Выявление гомозигот по патогенному гену в
периоде новорожденности в рамках
скринирующих программ и последующая диета,
обедненная фенилаланином, позволяют избежать
вторичных осложнений, характерных для
фенилкетонурии