









")

.")







")


































")



 medicine
medicineSimilar presentations:



")






Дегенеративные, демиелинизирующие, нейромышечные заболевания нервной системы
1. Дегенеративные, демиелинизирующие, нервно-мышечные заболевания нервной системы
ДЕГЕНЕРАТИВНЫЕ,ДЕМИЕЛИНИЗИРУЮЩИЕ, НЕРВНОМЫШЕЧНЫЕ ЗАБОЛЕВАНИЯ
НЕРВНОЙ СИСТЕМЫ
Евсевьева О.М.
2. Нейродегенеративные заболевания
НЕЙРОДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯГруппа медленно прогрессирующих,
приобретенных или наследственнообусловленных заболеваний.
В основе прогрессирующая гибель нервных
клеток (нейродегенерация), ведущая к
различным неврологическим симптомам —
прежде всего, к деменции и нарушению
движений. Заболевания могут наступить в
различном возрасте, протекают
генерализированно, гистологически
определяется специфический тип изменений.
3.
К нейродегенеративным заболеваниям относятсяТаупатии:
Болезнь Альцгеймера
Прогрессирующий супрануклеарный парез взора
Кортикобазальная дегенерация
Болезнь серебряного зерна
Фронтотемпоральная деменция и паркинсонизм 17
хромосомы (FTDP-17)
Болезнь Пика
Патоморфологическая основа этой группы НДЗ чрезмерное фосфорилирование белка, называемого
таупротеином и его фиксация в клетках головного мозга
в виде своеобразных патологических структур. Этот
процесс нарушает жизнедеятельность нейрона.
4.
Синуклеопатии:Болезнь Паркинсона
Деменция с тельцами Леви
Мультисистемная атрофия
Для этой группы характерно накопление в клетках
мозга α-синуклеина, представляющего комплекс из
35 аминокислот. Из этого белка образуются
различные структуры, приводящие к гибели клеток.
Первыми симптомами обычно бывают
двигательные расстройства, позднее развивается
деменция. У некоторых больных отмечается наряду
с накоплением α-синуклеина, отмечается
отложение и β-амилоида, типичного для таупатий.
5.
Тринуклеотидные заболевания:Хорея Хантингтона
Спинобульбарная мышечная атрофия, тип Кеннеди
Атаксия Фридрейха
Спиноцеребеллярная атаксия
Дендаторубро-паллидолуизальная атрофия (DRPLA)
Наследственные болезни, характерное свойство которых
нарастающее количество повторов цепочек аминокислот
тринуклеотидов. Достижение определенного критического
количества повторов тринуклеотидов определяет
вероятность заболеть равной 100%. Это носит название
динамической мутации. В последующих поколениях число
тринуклеотидных повторов неуклонно нарастает, что
приводит к утяжелению течения заболевания. Это
называется генетическая антиципация.
6.
Прионные заболевания:Болезнь Крейтцфельдта — Якоба
Синдром Герстмана — Штраусслера — Шейнкера
Фатальная семейная бессонница
Куру
Заболевания мотонейрона
Боковой амиотрофический склероз
Первичный боковой склероз
Спинальная мышечная атрофия
Нейроаксональные дистрофии
Инфантильная нейроаксональная дистрофия
Нейродегенерация с отложением железа в мозге
Неклассифицированные заболевания
Дегенерация фронтотемпоральных долей
7. Болезнь Крейтцфельда-Якоба
БОЛЕЗНЬ КРЕЙТЦФЕЛЬДА-ЯКОБАПрионовые болезни –накопление в клетках ЦНС
прионовых белков медленные инфекции
Аутосомно-доминатный тип наследования
С 50 лет резкая слабость, изменения психики,
делирий, зрительные агнозии, мозжечковая
атаксия, кокровая миоклония, паркиносонизм,
эпилептические припадки
МРТ головного мозга-атрофия коры б.п,
Смерть в течении года
8. Боковой амиотрофический склероз
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗБолезнь двигательных нейронов
Хроническое прогрессирующее заболевание
н.с с избирательным поражением
центральных и периферических
мотонейронов и характеризуется
нарастающей слабостью бульбарных мышц,
мышц плечевого и тазового пояса туловища,
мышц живота.
9.
От 50 лет, чаще мужчиныВирусы по типу медленной инфекции
Слабость дистальных отделов конечностей,
затруднение речи, атрофии и парезы мелких
мышц, фасцикуляции. Затем парезы и атрофии
прогрессируют распрастраняются на мышцы
плечевого пояса, спины, грудной клетки.
Псевдобульбарный синдром, слабость мышц шеи
из-за поражения ядер IX X XI XII гр.ЧМН
Формы: бульбарная, шейно-грудная, поясничнокрестцовая
Диагностика: МРТ ШОП, миелография, ЭНМГ, ЭМГ,
Лечение симптоматическое: витамины гр Б, АТФ,
рилузол.
Прогоноз неблагоприятен
10.
Экстрапирамидные нарушения (поражениеполосатого тела, бледного шара, черной субстанции).
Два синдрома: гипертонически-гипокинетический
(синдром Паркинсона поражение черной субстанции,
бледного шара), гипотонически-гиперкинетический
синдром (поражение полосатого тела).
11. Болезнь Паркинсона (дрожательный паралич)
БОЛЕЗНЬ ПАРКИНСОНА (ДРОЖАТЕЛЬНЫЙПАРАЛИЧ)
Идиопатическое медленно прогрессирующее
дегенеративное заболевание ЦНС,
характеризующееся замедленностью
движений, ригидность мышц, тремором в
покое и нарушением позных рефлексов.
В основе-поражение дофаминергических
нейронов черной субстанции и других
дофаминергических структур ядер ствола
головного мозга
12.
13. Б. Паркинсона (гипертонически-гипокинетический синдром).
Б. ПАРКИНСОНА (ГИПЕРТОНИЧЕСКИГИПОКИНЕТИЧЕСКИЙ СИНДРОМ).Характерны:
-общая скованность;
-поза с согнутой головой и туловищем, полусогнутыми и
приведёнными руками;
-замедленность и маловыразительность движенийсимптом «восковой куклы»;
-мышечная ригидность, симптом «зубчатого колеса»
-скудность мимики;
-замедленная мелкими шажками походка;
-монотонная тихая речь;
-мелкий статический тремор в дистальных отделах
конечностей, чаще рук-по типу «скатывания пилюль»,
реже нижней челюсти.
14.
15.
16.
17.
18.
19.
20.
21. Болезнь Вильсона-Коновалова (гепатоцеребральная атрофия)
БОЛЕЗНЬ ВИЛЬСОНА-КОНОВАЛОВА(ГЕПАТОЦЕРЕБРАЛЬНАЯ АТРОФИЯ)
-хроническое прогрессирующее наследственнодегенеративное заболевание,
характеризующееся сочетанным поражением
подкорковых узлов головного мозга и печени.
Аутосомно-рецессивный тип наследования
-нарушение синтеза белка церулоплазмина,
транспортирующего медь, в результате высокая
концентрация меди в крови и она откладывается
в печени, мозге, роговице, почках.
Медь токсична
22.
мышечная ригидность-нарушение походки,глотания, речи
Цирроз печени
Гиперкинезы разнообразные
Псевдобульбарный симптомы
Интелектуальное снижение
Кольца Кайзера-Флейшера по периферии
роговицы
23.
24.
Диагностика:-кровь, мочу на содержание меди
-кровь на церулоплазмин
Повышение печеночных, почечных проб
Лечение: унитиол, полисорб
25. Хорея-Хантингтона
ХОРЕЯ-ХАНТИНГТОНАНаследственное нейродегенеративное
заболевание, постепенное начало в возрасте
35-50 лет, сочетание прогрессирующих
хореических гиперкинезов и психических
расстройств (деменцией).
Аутосомно-доминатный тип наследования
В основе- гибель нервных клеток, снижение
содержания нейромедиаторов, нарушение
обмена, нарушение контроля и мышечного
тонуса. Атрофия головного мозга
26.
Интелектуальные расстройстваХореические гиперкинезы быстрые,
неритмичные, беспорядочные в различных
мышечных группах. Тяжело выполнить
произвольные движения, гримасничание,
нарушение речи, снижение мышечного
тонуса.
27.
Диагностика :ЭЭГ, МРТ головного мозгаатрофия корыЛечение симптоматическое
28. Торсионная дистония
ТОРСИОННАЯ ДИСТОНИЯХроническое пргрессирующее заболевание
нервной системы, клинически
проявляющееся изменением мышечного
тонуса и непроизвольными, тоническими
сокращениями мышц туловища и
конечностей.
Семейная форма –аутосомно-доминантная,
аутосомно-рецессивная
Нарушение обмена допамина
29.
Развивается до 15 лет нарушение походки,спастическая кривошея, тонические спазмы
мышц туловища, головы.
Гиперкинезы усиливаются при стрессе
Спастическая кривошея, писчий спазм,
блефароспазм, хореоатетоз.
Лечение симптоматическое препаратами
леводопы, холинолитики, седативные
препараты.
30.
Демиелинизирующие заболевания —это заболевания нервной системы при
которых
повреждается миелиновая оболочка нейроно
в
В результате нарушается проведение
сигналов нервам, это приводит к нарушениям
чувствительности, движений, когнитивных
функций, и других функций в зависимости от
того, какие затронуты нервы.
Рассеянный склероз
Рассеянный энцефаломиелит
31. Рассеянный склероз
РАССЕЯННЫЙ СКЛЕРОЗХроническое демиелинизирующее
заболевания н.с., характеризующееся
многочаговыми поражениями, протекающие
с обострениями и ремиссиями, либо
прогродиентно.
2-7 на 10 000 населения
Северные регионы, женщины болеют в 2
раза чаще
32.
Полиэтиологично, несколько теорийнаследственная, вирусная, бактериальнаяинфекция, аутоиммунная теория, нехватка
вит. Д.
Возраст 15-50 лет, женщины в 2 раза
чаще
Симптоматика волноообразная
Рассеянная симптоматика
Начало чаще всего--ретробульбарный
неврит, косоглазие, диплопия.
33.
Сначала болезни минимальнаясимптоматика, затем она накапливается, и
в каждое обострение появляется что-то
новое-прогрессирующее течение.
Пелена перед глазами, онемение
половины тела, парестезии, ощущение
прохождения электрического тока вдоль
позвоночника-синдром Лермита,
нарушение координации, гемипарез,
асимметрия лица, головокружение,
нарушение речи, НФТО, нарушение
когнитивных функций, эмоциональная
неустойчивость.
34.
Ухудшение состояния после горячей ванны,физической нагрузки
Диагностика: МРТ головного, спинного мозга
с контрастом (критерии Макдоналда 2005изменение в пространстве и времени)-очаги
демиелинизации около боковых желудочков,
мозолистое тело, мозжечок, ствол, спинной
мозг. Многоочаговое поражение белого
вещества.
ЦСЖ-лимфоцитарный плеоцитоз (5-50 кл,
белок не более 1 г\л), повышение
иммуноглобулинов G, олигоклональные АТ
35.
ЛечениеИзбегать жарких помещений, повышенной
физической нагрузки, отказ от алкоголя и
курения, минимум вакцинации,
При обострении--ГЛК-метилпреднизолон 1-2 г.
пульс-терапия в\в 5-10 суток
Плазмаферез
Иммуномодуляторы-ПИТРС
Препараты бетаинтерферона-Авонекс, Ребиф
Глатирамат ацетат-копаксон
Иммуносупрессоры-метотрексат,
циклофосфамид, азатиоприн
36. Рассеянный энцефаломиелит
РАССЕЯННЫЙ ЭНЦЕФАЛОМИЕЛИТОстрое аутоиммунное заболевание ЦНС
после перенесенной вирусной или
бактериальной инфекции, после вакцинации.
Корь, краснуха, опоясывающий герпес,
прививка от бешенства.
Варианты:
острый геморрагический лейкоэнцефалит
Поперечный миелит
Неврит зрительного нерва
Оптикомиелит
Церебеллит
Стволовой энцефалит
37.
Клиника острая: повышение температуры,общемозговая симптоматика,
менингеальные симптомы,
эпилептический припадок, очаговые
симптомы.
В клин.ан.кр-лейкоцитоз, повышение СОЭ
Исход заболевания м.б.различен
МРТ очаги демиелинизации
ЦСЖ-лимфоцитарный плеоцитоз более 100
кл, повышение белка более 1 г\л.
38.
Лечение:ГЛК
Плазмаферез
циклофосфамид
39. Острая воспалительная полирадикуломиелоневропатия
ОСТРАЯ ВОСПАЛИТЕЛЬНАЯПОЛИРАДИКУЛОМИЕЛОНЕВРОПАТИЯ
Синдром Гийена-Барре-Штроля
Аутоиммунная природа
Острая демиелинизация корешков
спинномозговых и черепных нервов
Прогрессирующая мышечная слабость с
расстройствами дыхания, отсутствия глубоких
рефлексов
2 на 100 000 человек
Возраст 30-50 лет, чаще у мужчин
40.
После 3-х недель после инфекциивирусной, бактериальной,
микоплазменной
Парестезии, боли в дистальных отделах рук
и ног, иногда вокруг рта и в языке, общая
слабость, повышение температуры до
субфебрильных цифр, слабость лицевых и
бульбарных мышц, двигательные
нарушения, нервные стволы болезненны
при пальпации, симптомы натяжения
положительны.
Акроцианоз, похолодание конечностей
41.
ЦСЖ-белково-клеточная диссоциация (белок3-5 г\л)
Восходящий паралич Ландри –
распрастранение параличей на мышцы
туловища, дыхательные, бульбарную
мускулатуру, может привести к смерти.
Лечение:
Плазмаферрез
Глк-преднизолон 1-2 мг\кг\сут
Иммуноглобулины в\в
Вит.гр.Б
Прозерин 1 мл. п\к
-ИВЛ
42. Нервно-мышечные заболевания
НЕРВНО-МЫШЕЧНЫЕ ЗАБОЛЕВАНИЯМиастения-заболевание характеризуется
нарушением нерво-мышечной передачи и
проявляется слабостью и патологической
утомляемостью поперечно-полосатых мышц.
Преимущественное поражение мышц лица,
губ, глаз, языка, глотки, шеи.
В молодом возрасте, женщины болеют чаще.
43.
Аутоиммунное заболевание, м.б.семейным
При миастении повышение фермента
холинэстеразы, ацетилхолин
перерабатывается, блокируются
антителами ацетилхолиновые рецепторы, в
результате дальнейшая передача
импульсов страдает.
М.б. при гиперплазии вилочковой железы,
раке легкого, молочной железы, яичника,
предстательной железы
44.
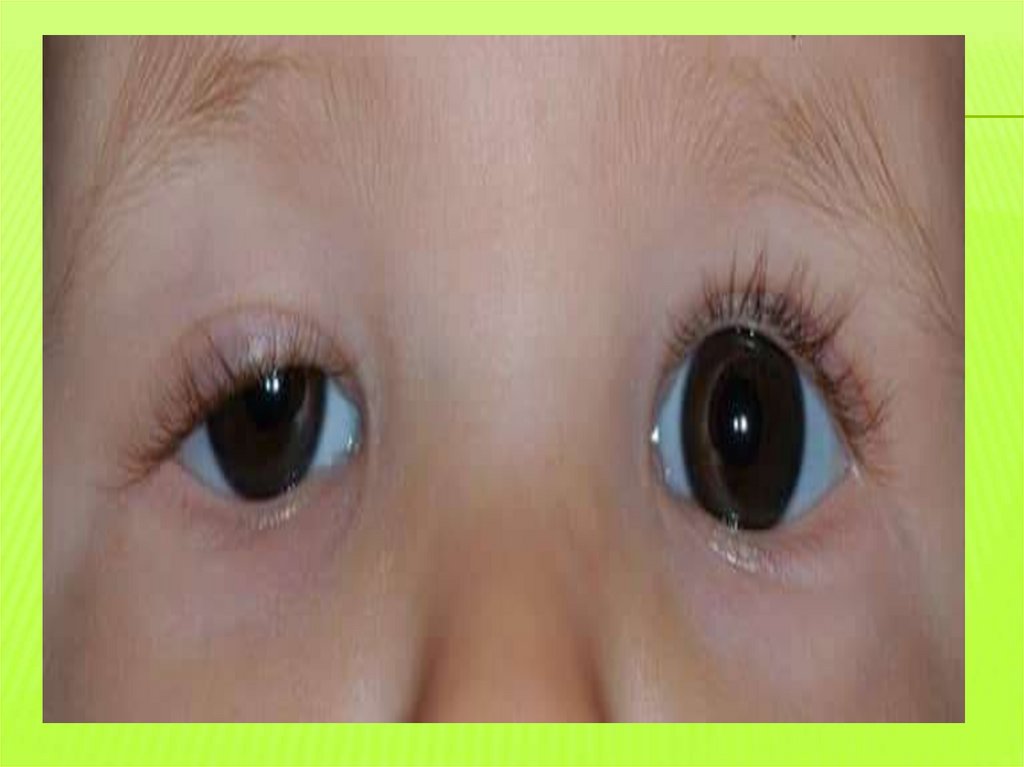
Мышечная слабостьДиплопия, птоз, косоглазие, трудность речи
и глотания после длительного разговора,
повышенная утомляемость, плохая
переносимость физ. нагрузок
Восстановление после короткого отдыха
45.
46.
Формы:-глазная
-генерализованная
-бульбарная
Диагностика: ЭНМГ, кровь на АТ к ацетихолиновым
рецепторам, КТ средостения, исследование
щитовидной железы, кровь на ревмофактор, онкопоиск
Лечение: антихолинэстеразные препараты-уменьшить
холинэстеразу, увеличить выработку АЦХ, уменьшить
блок антителами ацх-х рецепоторов.
прозерин, калимин-антихолинэстеразный препарат
иногда с атропином, нейромедин, ГЛК курсами,
тимэктомия, плазмаферрез, иммуноглобулины (Игвен),
иммуносупрессоры (циклофосфамид, азатиоприн)
47. Миастенический криз
МИАСТЕНИЧЕСКИЙ КРИЗНеотложное состояние!!!!!
При недостаточности антихолинэстеразных
средств, при запущенных формах миастениипрогрессирующая слабость скелетной
мускулатуры, дыхательных и бульбарных мышц
(одышка, не способность сглатывать слюну,
держать голову, ослабление голоса) -прозерин 1
мл.0,05% в\в + атропин 0,5 в\м, в\м по 2-3 мл.
через 2 часа, интубация, трахеостомия, ИВЛ,
баланс жидкости и электролитов.
48. Холинэргический криз
ХОЛИНЭРГИЧЕСКИЙ КРИЗНеотложное состояние!!!!!
Передозировка ацетилхолинэстеразных
препаратов (блокируется нервно-мышечная
передача)-узкие зрачки, парез аккомодации,
мышечные подергивания, гиперсекреция
бронхиальной слизи, слюны, рвота,
брадикардия, гиотензия, диарея.
Отмена ацетилхолинэстеразных препаратов и
введение атропина 0,5 мл. в\в или п\к
49. Миопатии
МИОПАТИИЗаболевания с нарушением сократительной
способности мышечных волокон и
проявляющихся слабостью мышц,
снижением их тонуса, атрофией (иногда
псевдогипертофей), уменьшением объема
активных движений.
50. Прогрессирующая мышечная дистрофия Дюшенна
ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯДИСТРОФИЯ ДЮШЕННА
Начало в раннем возрасте
Симметричная атрофия мышц в сочетании с
сердечно-сосудистыми, костно-суставными,
психическими нарушениями.
Рецессивное, Х-сцепленным наследованием,
чаще мальчики
Перерождение мышечной ткани на жировую
и соединительную, некроз отдельных волокон
51.
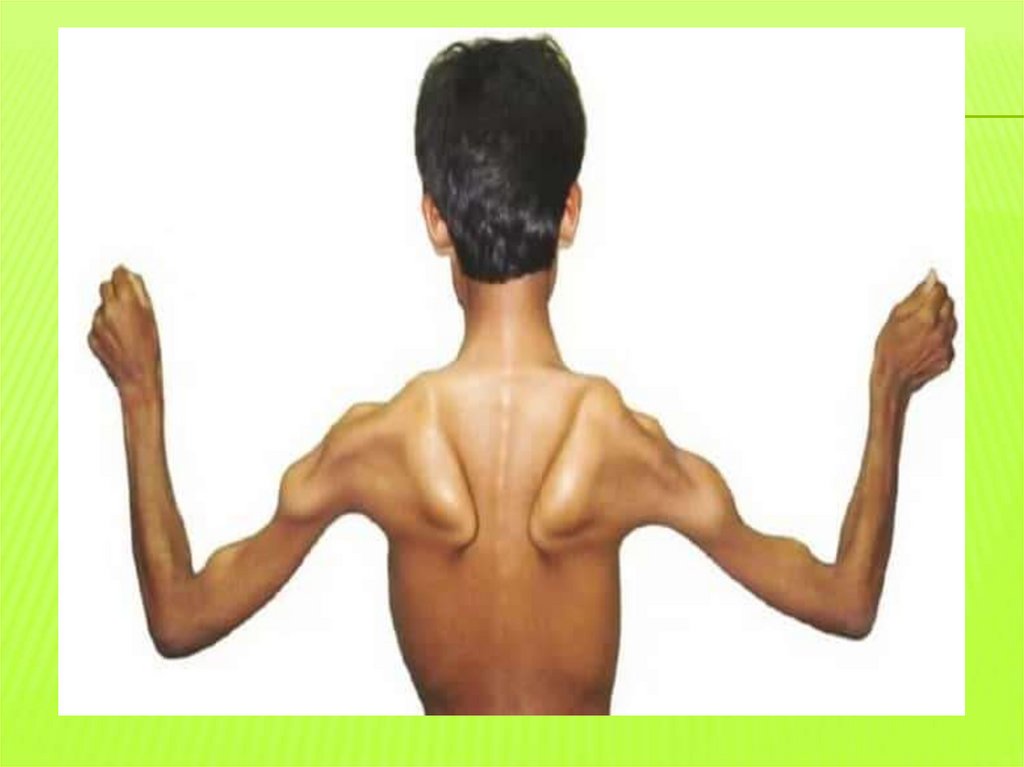
Проявляется в 1-3 года жизни слабость мышцтазового пояса, отставание в моторном развитии, с
задержкой начинают садиться, вставать, ходить.
В 2-3 года мышечная слабость, патологическая
мышечная утомляемость, проявляющаяся при физ.
нагрузке - «утиная» походка, «взбирание по самому
себе»=взбирание лесенкой при вставании.
Атрофии в проксимальных группах мышц сначала
нижних конечностей, затем и верхних
Лордоз, «крыловидные лопатки», «осиная» талия
Злокачественное заболевание к 7-10 годам
выраженные двигательные расстройства
К 14-15 годам обездвиженность
52.
53.
54.
55.
56.
Диагностика:Осмотр генетика
Б\х ан.кр- КФК, АЛТ, АСТ
ЭМГ
Сосудистые, нейроэндокринные заболевания
-биопсия мышцы
Лечение : ГЛК-преднизолон, симптоматическоевит. гр Б, Е, глютаминовая к-та, АТФ в\м,
средства для нейромышечной передачипрозерин, нейромедин
57.
58. Пароксизмальные миоплегии
ПАРОКСИЗМАЛЬНЫЕ МИОПЛЕГИИНаследственные нервно-мышечные
заболевания характеризующиеся внезапной
мышечной слабостью и параличами.
Чаще всего гипо-нормо- и
гиперкалиемические формы
Дефект сарколеммы нарушающий
проницаемость ионов натрия и калия
59. Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля)
ГИПОКАЛИЕМИЧЕСКАЯ ФОРМАПАРОКСИЗМАЛЬНОЙ МИОПЛЕГИИ (БОЛЕЗНЬ
ВЕСТФАЛЯ)
С 6-15 лет, внезапно ночью или утром пароксизмы
мышечной слабости, обездвиженности, снижением
мышечного тонуса и глубоких рефлексов,
вегетативными расстройствами-лабильность
пульса, АД
-парциальные и генерализованные
Нарушения сердечно-сосудистой системы
Продолжительность-несколько часов-несколько
суток
Содержание калия в крови ниже 2 ммоль\л
Диета богатая калием, р-р хлорида калия в\в,
панангин
60. Миотонии
МИОТОНИИГруппа нервно-мышечных заболеваний,
характеризующаяся неспособностью
произвольной мускулатуры к расслаблению
после сокращения.
61. Врожденная миотония Лейдена-Томсона
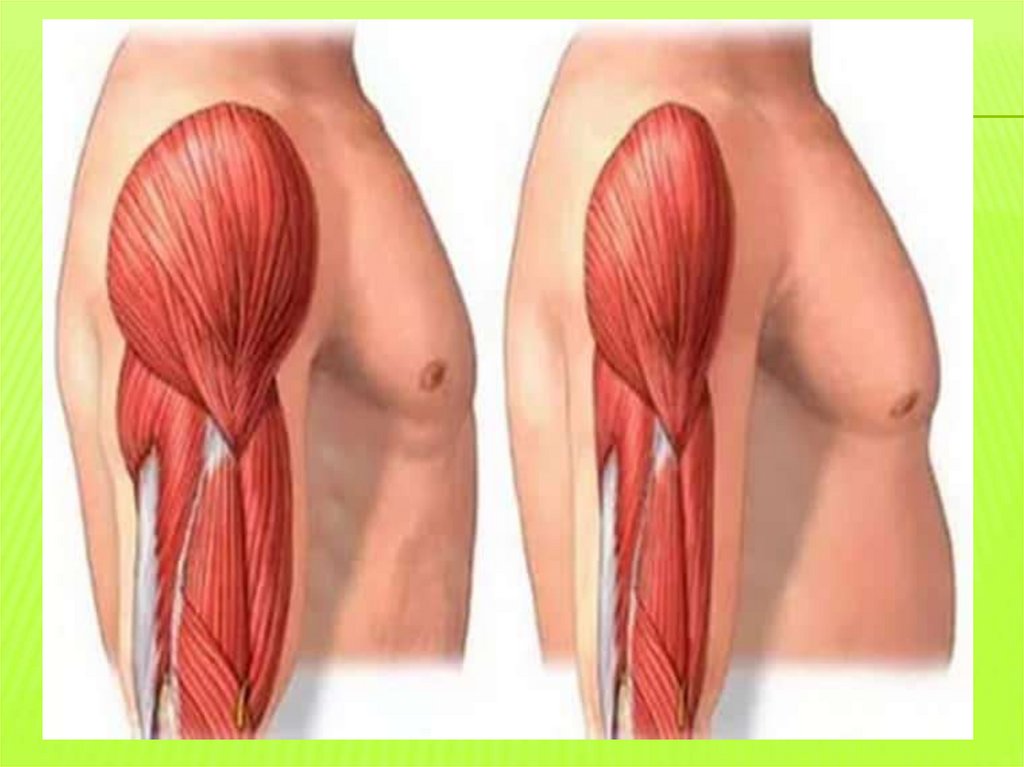
ВРОЖДЕННАЯ МИОТОНИЯ ЛЕЙДЕНА-ТОМСОНААутосомно-доминантный тип наследования
С 8-15 лет миотонические спазмызатруднение рассслабления мышц после
активного напряжения
Диффузные гипертрофии мышц напоминают
спортивных атлетов
Диагностика: генетика, ЭМГ, биопсия мышцы.
Лечение : дифенин-тормозящее действие на
нервно-мышечное проведение в ЦНС,
диакарб-изменение проницаемости
мембраны