





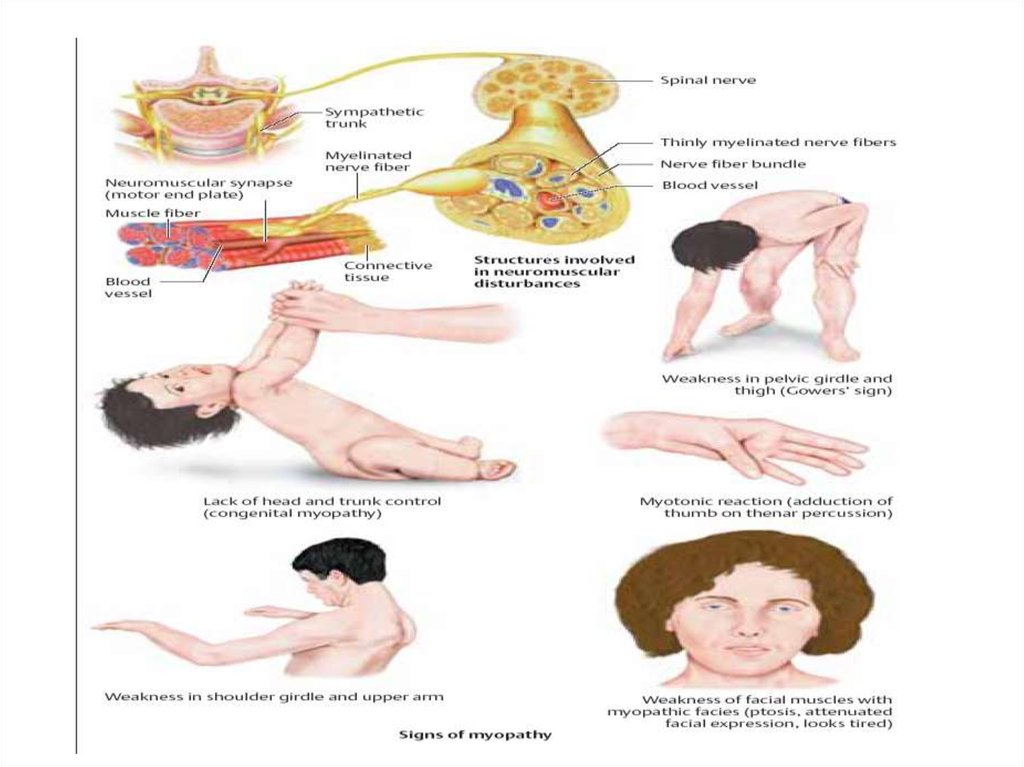

 - это группа наследственно-мышечных заболеваний, проявляющихся мышечной слабостью и атрофией мышц")

















































 с медуллярно-клеточной дифференцировкой, эпидермоидный тип роста")








 medicine
medicineSimilar presentations:










Нервно-мышечные заболевания
1. Медицина в изобразительном искусстве
Гиппократ - одни из самыхизвестных древнегреческих
врачевателей. Изображен
неизвестным византийским
художником начала XIV века
(Национальная библиотека. Париж)
Богиня Исида. Рельеф из храма
Исиды на о. Фале. Песчанник I век
до н.э. Исида была богиней
плодородия, воды и ветра,
символом женственности и
семейной верности, богиней
мореплавания и врачевания. В
руке у Исиды посох, на голове
змея, вокруг сосуда - элементы
будущего символа медицины, в
левой руке богини находится анк
или крестообразная петля,
олицетворяющая физическое и
психическое здоровье.
Урок анатомии доктора Тюльпа. 1632
год, Рембрандт Гарменс ван Рейн
2. Скоро уж четыре столетия, как Мона Лиза лишает здравого рассудка всех, кто, вдоволь насмотревшись, начинает толковать о ней.
Грюйе, конец XIX векаПортре́т госпожи́ Ли́зы дель
Джоко́ндо, итал. Ritratto di Monna Lisa
del Giocondo) — картина Леонардо
да Винчи, находящаяся в Лувре
(Париж, Франция), одно из самых
известных произведений живописи в
мире, которое, как считается, является
портретом Лизы Герардини, супруги
торговца шёлком из Флоренции
Франческо дель Джокондо, написанным
около 1503—1505 года.
3. НЕРВНО-МЫШЕЧНЫЕ ЗАБОЛЕВАНИЯ
НЕРВНОМЫШЕЧНЫЕЗАБОЛЕВАНИЯ
к.м.н., доцент КНиП МИ СВФУ
Попова Т.Е.
4. План лекции
• 1. Нервно-мышечные заболевания: классификация• 2. Миодистрофии с Х-сцепленным типом
наследования (Дюшена, Беккера, Эмери-Дрейфуса)
• 3. Миодистрофии с аутосомным типом наследования
(Ландузи-Дежерина, Эрба-Рота, ОФМД)
• 4. Невральная амиотрофия Шарко-Мари-Тут
• 5. Спинальные амиотрофии (Верднига-Гоффмана,
Кугельберга-Веландера)
• 6. Миотонии
• 7. Миастения
5.
• Наследственные нервно-мышечныезаболевания – большая гетерогенная
группа болезней, в основе которых
лежит генетически детерминированное
поражение периферических нервов,
передних рогов спинного мозга и
скелетных мышц.
6.
• Первичные миопатии с рецессивным Хсцепленным наследованием• Первичные миопатии с аутосомным
наследованием
• Конгенитальные (врожденные) миопатии
• Спинальная мышечная атрофия
(амиотрофия)
• Миотонии
• Нейропатии
• Миастения
• Воспалительные миопатии
7.
8.
1.2.
3.
В Якутии, по данным медико-генетической
консультации, в структуре наследственных
заболеваний ведущее место занимают болезни
нервной системы и, в первую очередь, группа
нервно-мышечной патологии.
Многими формами заболеваний начинают страдать
с детства и симптоматика прогрессирует на
протяжении всей жизни.
Важнейшим является ранняя диагностика
заболеваний, определение клинической формы,
типа наследования. Правильная диагностика
позволяет определить тактику патогенетической
терапии, прогноз и дать обоснованное заключение
при медико-генетическом консультировании.
9. МИОПАТИИ (миодистрофии) - это группа наследственно-мышечных заболеваний, проявляющихся мышечной слабостью и атрофией мышц
МИОПАТИИ (миодистрофии) - это группа наследственномышечных заболеваний, проявляющихся мышечнойслабостью и атрофией мышц
• Мышечная слабость не сопровождается болью, хотя
возможны мышечные спазмы – крампи
• При большинстве ПМД слабость раньше проявляется в
проксимальных отделах, плечевой и тазовый пояс
• Сухожильные рефлексы снижаются пропорционально
выраженности мышечной слабости
• Расстройства чувствительности встречаются нечасто,
обычно при невральной амиотрофии
• Заболевание не влияет на функции тазовых органов
10.
Наследственные нервно – мышечныезаболевания у детей в Якутии
Распространенность
наследственных
нервно – мышечных
заболеваний
составила 13,3 на
100 000 детского
населения РС(Я).
• Инвалидность
составила 100%.
11. Первичная заболеваемость ННМЗ в Якутии у детей до 14 лет
0,030,025
0,025
0,021
первичная
заболеваемость в
среднем составила
0,01
0,02
0,015
0,012
0,01
0,008
0,005
‰
0
2006 г.
2007 г.
2008 г.
2009 г.
12. Х-сцепленный с полом тип наследования
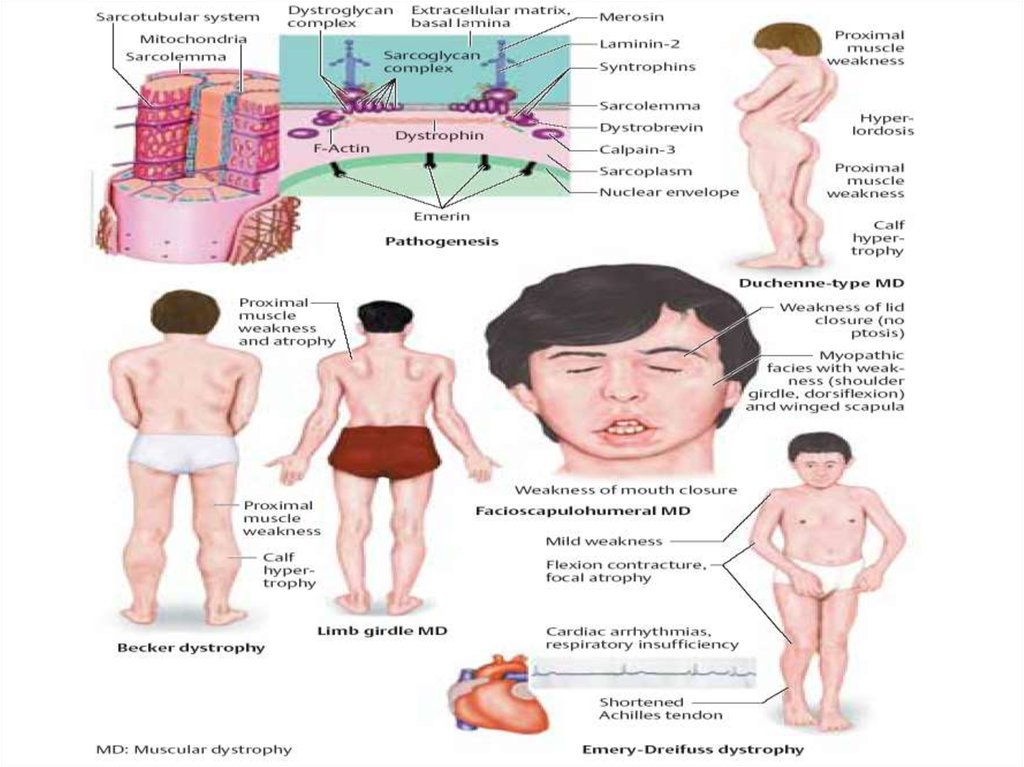
13. Миодистрофия Дюшенна
Наиболее злокачественная форма, передается порецессивному сцепленному с Х-хромосомой типу,
отсутствует продукт данного гена – структурный
белок дистрофин, что ведет к гибели мифибрилл.
Болеют мальчики. Начало с конца 1 года, к 6-10
годам с трудом передвигаются. Развивается
кардиомиопатия, гипоплазия надпочечников,
остеопороз.
Неврологи из университета штата Вашингтон доказали, что с
помощью генно-инженерных методов можно полностью
излечить миодистрофия Дюшенна.
14. Миопатия Дюшенна
Мышечная слабость проявляетсясимметрично и прогрессирует постепенно
Утиная походка
«икры гнома» - псевдогипертрофия
икроножных мышц
«осиная талия», «лягушачий» живот,
крыловидные лопатки
лицо «сфинкса», губы «тапира»,
«поперечная» улыбка Джоконды
вставание лесенкой,
синдром «вялого ребенка»
сухожильные рефлексы снижаются
15.
• Через 1 – 2 года от началазаболевания:
• Мышечная
слабость,
патологическая
утомляемость,
изменение походки по
типу «утиной»
отмечается у 100 %
больных детей.
• Симптом
«взбирание
лесенкой» или
«взбирание по самому
себе» отмечается у
57,14% больных.
Рис.№1. Вставание с корточек «лесенкой»симптом Говерса
16.
• Через 3 – 5 лет от начала заболевания:• Атрофия мышц спины и
конечностей
А)
Рис. №2.
лордоз, «крыловидные»
лопатки, «осиная талия» выявлены
у 85,71% больных.
А) лордоз,
«крыловидные»
лопатки, «осиная
талия».
• «Классический» симптом –
псевдогипертрофия икроножных
мышц у 85,71% больных.
Б) Характерная
поза. Общая
мышечная
гипотония,
атрофия мыши
плечевого и
тазового поясов.
• Мышечный тонус снижен у
100% детей.
Сухожильные рефлексы:
Не изменены –14,29%;
Снижены с рук – 14,29%;
Снижены коленные – 28,57%;
Ахилловые снижены –14,29%;
Живые – 14,29%;
Отсутствуют – 57,14%.
в)
псевдогипертрофия
икроножных мышц.
Б)
В)
17.
18.
АБ
Больной А., 4 года.
Псевдогипертрофия
икроножных мышц.
Больной Д., 10 лет.
А) Псевдогипертрофия
икроножных мышц.
Б) Контрактура голеностопных
суставов.
19. Миопатия Беккера
• Начинается в раннем детском возрасте(в первый год жизни) и проявляется
атрофией мышц конечностей, спины, а
потом всего туловища, быстро
прогрессирует, исход —
неблагоприятный. Бывают и другие
редкие разновидности
прогрессирующих мышечных
дистрофий.
20. Форма Эмери-Дрейфуса
• Возраст начала – позднее детство,юношество, возможно у взрослых
• Основные симптомы – поражение мышц
тазового пояса, трудности при подъеме по
лестнице, беге, ригидность мышц шеи и
спины, деформация стопы по типу конской,
сердечные нарушения
• Прогноз – медленное течение, прогноз
определяют сердечные нарушения
• У девочек и женщин м.б. АД наследованя
21.
22.
23. Миопатия Ландузи-Дежерина
Плече-лопаточно-лицевая форма миодистрофии,которая может быть в возрасте от 6-ти до 52-х лет
(чаще в 10-15 лет) и характеризуется поражением
мышц лица с постепенной последующей атрофией
мышц плечевого пояса, туловища и конечностей.
Ранними признаками болезни являются плохо
смыкающиеся и незакрывающиеся веки, полиостью
не смыкающиеся губы что создает нечеткую речь и
невозможность надуть щеки. Заболевание протекает
медленно.
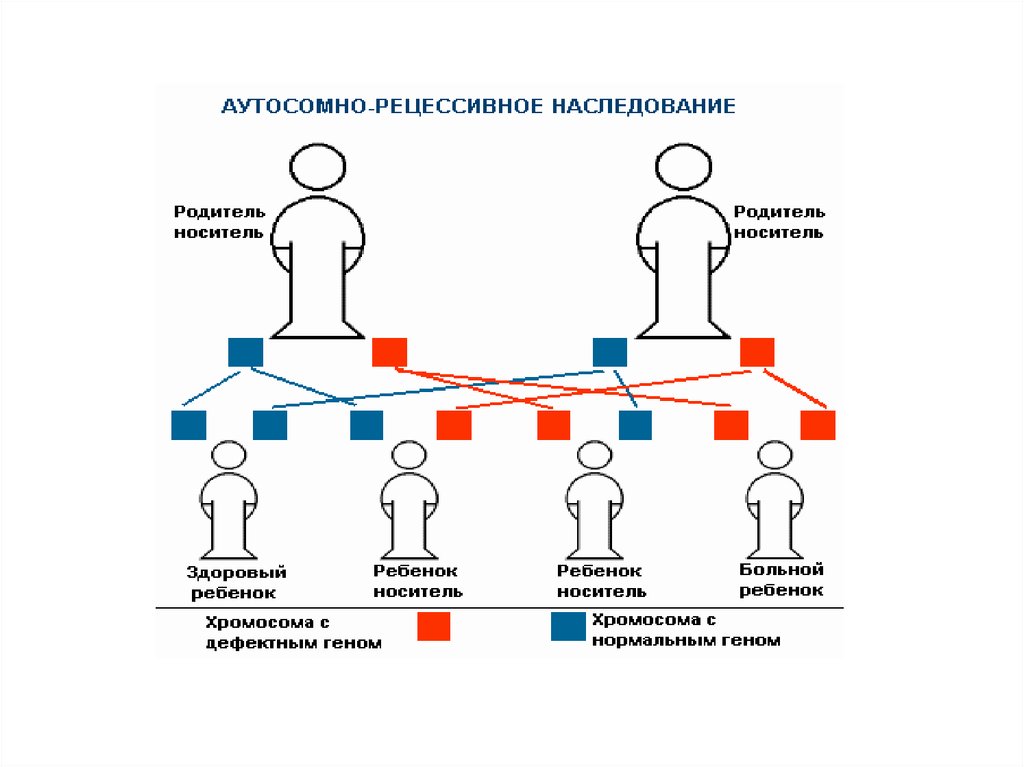
24. Ювенильная миодистрофия Эрба-Рота
Конечностно-поясная мышечнаядистрофия – развивается в детском и
юношестком возрасте (чаще в 14-16
лет).
Глубокая инвалидизация через 10-20
лет. Передается по аутосомнорецессивному типу. В настоящий
момент известно 15 генов,
ответственных за возникновение ЮМ.
25.
При миодистрофии Эрбастрадают мышцы бедра и
плечевого пояса (т.е.
проксимальные мышцы ближайшие к центру тела;
дистальные мышцы удаленные от центра тела
могут иногда тоже
поражаться, но на поздних
стадиях).
26.
• Первым симптомом появившейся болезниможет быть изменение походки. Из-за
слабости проксимальных мышц ног
человек начинает ходить вперевалку.
Такую походку еще называют "утиной".
• К походке добавляются трудность при
вставании со стула и поднимание по
лестницам.
27.
• Из-за слабости мышц плечевого поясачеловеку может быть очень тяжело (или
невозможно) держать руки над головой
(например, при расчесывании волос
или попытке дотянуться до высокой
полки), вытянутыми перед собой и
переносить тяжелые вещи. Некоторым
людям трудно набирать текст на
клавиатуре компьютера и
самостоятельно принимать пищу.
28. Окулофарингеальная форма
29. Диагностика
• КФК-креатинфосфокиназа - этофермент, повышенное содержание
которого в крови может
сигнализировать о повреждении мышц.
При МД уровень КФК может быть в
десятки, сотни и тысячи раз выше
нормы.
• ЭНМГ - позволяет определить,
является ли слабость пациента
следствием проблем в самих мышцах
или в двигательных нервах,
контролирующих мышцы.
30. Диагностика
• Биопсия небольшого образцамышечной ткани определяет
конкретный тип МД. Белки
являются неотъемлемой частью
мышц. Когда белок неполноценен,
когда его количество в мышцах
очень низкое или его вообще нет,
мышцы начинают разрушаться.
• ДНК-диагностика (ПЦР, ПДРФ)
31. Лечение
• При легких формах – достаточно повторных(каждые 6-12 мес.) исследований
двигательных функций
• В тяжелых случаях, разъяснить суть и
прогноз заболевания родственникам
• Преднизолон (0,75 мг/кг/сут) способствует
увеличению мышечной силы
• Аминокислоты, эссенциале, вит.Е
• ЛФК
• Профилактика МГК
32. Невральная амиотрофия Шарко-Мари-Тут
медленно прогрессирующеенаследственно-дегенеративное
заболевание с преимущественным
поражением периферического
двигательного нейрона, основным
признаком которого является атрофия
мышц в дистальных отделах нижних
конечностей.
33.
• Морфологическую основу болезни составляютдегенеративные изменения, главным образом в
периферических нервах и нервных корешках,
касающиеся как осевых цилиндров, так и миелиновой
оболочки.
• Изменения в мышцах носят преимущественно
неврогенный характер в виде «пучковой» атрофии
мышечных волокон. В ряде случаев отмечаются
изменения и в спинном мозге. Они складываются из
атрофии клеток передних рогов, главным образом, в
поясничной и шейной части спинного мозга и
различной степени поражения проводниковых
систем.
34.
• Мужчины болеют несколько чаще, чемженщины. Большая часть случаев (80 %)
наследуется по аутосомно-доминантному
типу.
• Болезнь нередко обнаруживается у когонибудь из родителей больного, но
встречаются и спорадические случаи.
• Начало заболевания относится большей
частью ко второй половине первого или к
первой половине второго десятилетия жизни,
но может наблюдаться и раньше, и в
значительно более позднем возрасте.
Максимум заболеваемости падает на возраст
5 — 10 лет.
35. Основными клиническими симптомами заболевания являются
• амиотрофии, которые начинаются симметрично сдистальных отделов нижних конечностей. В первую
очередь поражаются разгибатели и абдукторы стопы,
в результате чего стопа свисает, появляется
характерная походка — степпаж.
• Атрофия мышц стопы приводит к когтевидной
установке пальцев и деформации ее.
Амиотрофический процесс постепенно
распространяется на более проксимальные отделы.
Однако в подавляющем большинстве случаев
проксимальные отделы конечностей остаются
сохранными; процесс не распространяется также на
мышцы туловища, шеи и головы.
36.
Больной Н., 11летА) дистальная атрофия мышц
нижних конечностей.
37. Спинальная амиотрофия Верднига-Гоффмана
• Заболевание описаноВерднигом в 1891 г. и
Гофманом в 1893 г.
Наследуется по
аутосомнорецессивному типу.
• Частота: 1 на 100
000 населения, 7 на
100 000
новорожденных.
38. Патоморфология
• Обнаруживают недоразвитие клеток передних роговспинного мозга, демиелинизацию передних
корешков. Часто имеются аналогичные изменения в
двигательных ядрах и корешках V, VI, VII, IX, X, XI и
XII черепных нервов. В скелетных мышцах
нейрогенные изменения характеризуются «пучковой
атрофией», чередованием атрофированных и
сохранных пучков мышечных волокон, а также
нарушениями, типичными для первичных миопатий
(гиалиноз, гипертрофия отдельных мышечных
волокон, гиперплазия соединительной ткани).
39. Клиническая картина
Различают три формы заболевания:
врожденную,
раннюю детскую и
позднюю, отличающуюся временем
проявления первых клинических
симптомов и темпом течения
миодистрофического процесса.
40. При врожденной форме
• дети рождаются с вялыми парезами. С первых дней жизнивыражены генерализованная мышечная гипотония и снижение
либо отсутствие глубоких рефлексов.
• Рано определяются бульбарные расстройства, проявляющиеся
вялым сосанием, слабым криком, фасцикуляциями языка,
снижением глоточного рефлекса. Выявляется парез
диафрагмы.
• Заболевание сочетается с костно-суставными деформациями:
сколиозом, воронкообразной или «куриной» грудной клеткой,
контрактурами суставов. Развитие статических и локомоторных
функций резко замедлено. Лишь у ограниченного числа детей с
большим опозданием формируется способность держать
голову и самостоятельно садиться. Однако приобретенные
двигательные навыки быстро регрессируют. У многих детей с
врожденной формой болезни снижен интеллект.
41. Ранняя детская форма
• первые признаки болезни возникают, как правило, на второмполугодии жизни. Моторное развитие в течение первых месяцев
удовлетворительное. Дети своевременно начинают держать
голову, сидеть, иногда стоять.
• Заболевание развивается подостро, нередко после инфекции,
пищевой интоксикации. Вялые парезы первоначально
локализуются в ногах, затем быстро распространяются на
мышцы туловища и руки. Диффузные мышечные атрофии
сочетаются с фасцикуляциями языка, мелким тремором
пальцев, сухожильными контрактурами. Мышечный тонус,
глубокие рефлексы снижаются. В поздних стадиях возникают
генерализованная мышечная гипотония, явления бульбарного
паралича.
• Течение злокачественное, хотя и мягче по сравнению с
врожденной формой. Летальный исход наступает к 14-15 годам
жизни
42. Поздняя форма
• первые признаки болезни возникают в 1,5-2,5 года. К этомувозрасту у детей полностью завершено формирование
статических и локомоторных функций. Большинство детей
самостоятельно ходят и бегают. Заболевание начинается
незаметно.
• Движения становятся неловкими, неуверенными. Дети часто
спотыкаются, падают. Изменяется походка - они ходят, сгибая
ноги в коленях (походка «заводной куклы»).
• Вялые парезы первоначально локализуются в проксимальных
группах мышц нижних конечностей, в дальнейшем
сравнительно медленно переходят на проксимальные группы
мышц верхних конечностей, мышцы туловища; атрофии мышц
обычно малозаметны вследствие хорошо развитого подкожного
жирового слоя.
43. Амиотрофия Кугельберга-Веландера
Заболевание развивается после 18 месяцевжизни с пиком манифестации клинических
проявлений в 5-17 лет. Первым симптомом
является слабость мышц проксимальных
отделов нижних конечностей. Дети не могут
бегать, часто падают, испытывают трудности
при ходьбе по лестнице и при вставании. В
дальнейшем, присоединяются мышечные
атрофии, снижение сухожильных рефлексов, в
первую очередь с двуглавой, трехглавой мышц
плеча и коленных. Особенностью течения
спинальной амиотрофии Кугельберга Веландера является относительная
доброкачественность. В течение многих лет
болезнь не вызывает глубокой инвалидизации.
44. Миотонии
Миотонии (греч, myos мышца + tonosнапряжение) — группа наследственно
обусловленных нервно-мышечных
заболеваний; характеризуется тоническим
спазмом мышц, возникающим в начале
активных движений.
45.
Различные формы М. отличаются разнымтипом наследования, вариабельностью
возраста проявления заболевания, его
течения и другими признаками.
К наиболее часто встречающимся формам
относятся врожденная (неатрофическая)
миотония Томсена и атрофическая
(дистрофическая) миотония Гоффманна —
Россолимо — Штейнерта — Куршманна.
Тип наследования аутосомно-доминантный,
изредка аутосомно-рецессивный, при
котором чаще заболевают мужчины, и
течение болезни бывает тяжелым.
46.
Врожденная (неатрофическая) миотонияТомсена развивается вначале незаметно,
постепенно прогрессируя, достигает
клинически определяемой степени.
Одинаково часто заболевают лица обоего
пола. Дети с М. отстают от сверстников в
играх, испытывают затруднения при беге,
прыжках.
47.
Часто обращают внимание на неуклюжесть
ребенка. Характерны жалобы на затруднения
в самом начале активных движений,
особенно если движения совершаются
быстро и со значительным усилием.
• Застывание, скованность, «скрючивание»
исчезают после повторных однотипных
движений. Отмечается утомляемость при
физических нагрузках. Ходьба, особенно
после длительного покоя, затруднена,
первые шаги совершаются с большим
усилием (особенно первые шаги по
лестнице).
48. Миотонические проявления
49. Миотонический ровик на языке
Миотонические спазмыусиливаются под влиянием
холода и отрицательных эмоций,
в тепле и под действием
алкоголя они ослабевают;
проявляются как в мышцах рук
и ног, так и в мускулатуре лица,
языка, гортани, в жевательных
мышцах, мышцах шеи и
туловища.
•Повышение механической
возбудимости выявляют при
ударе по мышце
неврологическим молоточком. У
больного М. на месте удара
образуется углубление,
удерживающееся несколько
секунд, миотонический ровик
(перкуссионная миотония).
Наиболее постоянен
миотонический ровик на языке
50. ЭНМГ
Электровозбудимость мышц также нарушена.После раздражения мышцы фарадическим или
гальваническим током наблюдается ее
замедленное расслабление (электрическая
миотоническая реакция).
На электромиограммах выявляются
миотонические нарушения: после прекращения
активного движения (например, сжимание
кисти в кулак) в фазе расслабления
обнаруживают биоэлектрическую активность
не только в мышцах-разгибателях, но и
мышцах-сгибателях.
51. ЛЕЧЕНИЕ
• Фенитоин – 5 мг/кг/сут внутрь,эффективен и в наименьшей степени
влияет на сердце
• Кортикостероиды показаны в тяжелых
случаях
52. Миастения
• Миастения является классическим аутоиммуннымзаболеванием человека. Основным клиническим
проявлением миастении является синдром
патологической мышечной утомляемости (усиление
проявлений миастении после физической нагрузки и
уменьшение их после отдыха).
• Впервые заболевание было описано Томасом
Уиллисом в 1672 году. Антитела при миастении
впервые были выделены Strauss в 1960 году.
Окончательно аутоиммунное происхождение болезни
было доказано Patrick и Lindstrom в опыте на
кроликах, иммунизированных Torpedo californica
AChR, у которых появились признаки миастении.
53.
• Последнее время заболеваемостьмиастенией растет, на сегодняшний момент
распространенность составляет
приблизительно 5-10 человек на 100 000
населения.
• Заболевание может начаться в любом
возрасте, однако наибольший пик
заболеваемости встречается среди молодых
женщин 20-40 лет.
54.
• По данным Миастенического центраженщины болеют чаще мужчин 3 : 1. В
пожилом возрасте соотношение
выравнивается, мужчины болеют также
часто, как и женщины, 1 : 1.
• Детская миастения встречается достаточно
редко, максимум 1-3% от всех заболевших.
55. Прогноз
Ранее миастения была тяжелымзаболеванием с высокой летальностью - 3040%. Однако при современных методах
диагностики и лечения летальность стала
минимальной – менее 1%, около 80% на
фоне лечения достигают полной/неполной
ремиссии. Но все-таки заболевание является
хроническим, серьезным и требует
тщательного наблюдения и лечения
56.
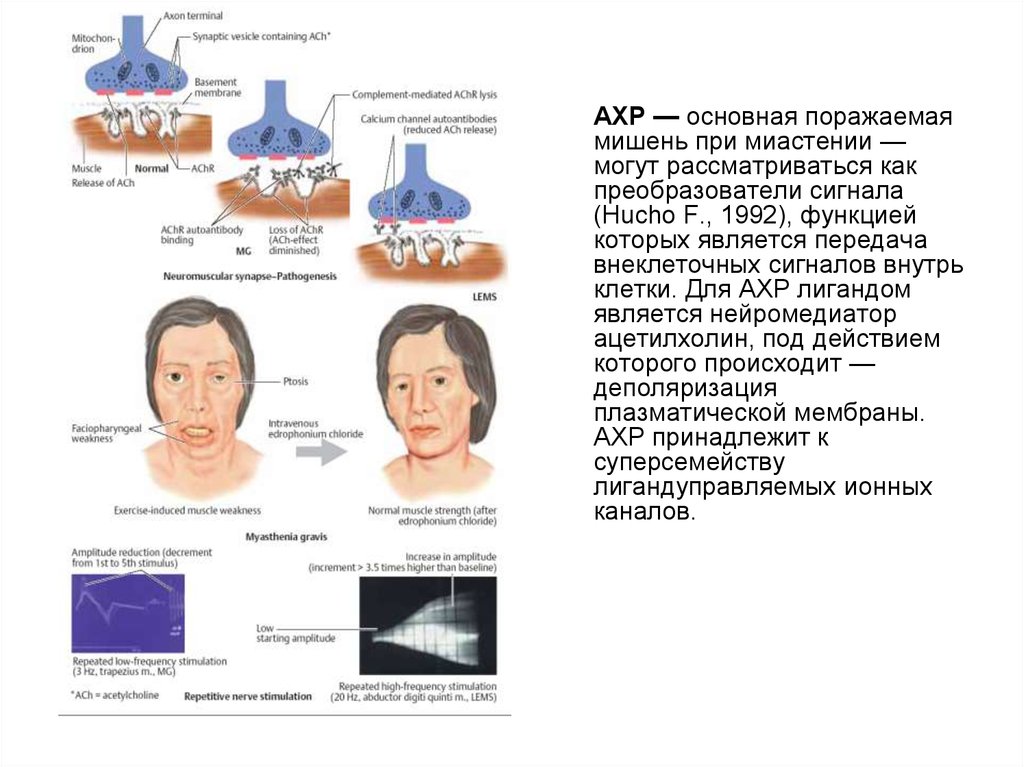
АХР — основная поражаемаямишень при миастении —
могут рассматриваться как
преобразователи сигнала
(Hucho F., 1992), функцией
которых является передача
внеклеточных сигналов внутрь
клетки. Для АХР лигандом
является нейромедиатор
ацетилхолин, под действием
которого происходит —
деполяризация
плазматической мембраны.
АХР принадлежит к
суперсемейству
лигандуправляемых ионных
каналов.
57. Субъединица ацетилхолинового рецептора
58. Формы миастении
• Глазная миастения (14%)• Легкая генерализованная миастения с
глазными симптомами
• Умеренная генерализованная миастения с
легкими бульбарными и глазными
симптомами
• Острая тяжелая миастения с бульбарными и
дыхательными нарушениями
• Поздняя тяжелая миастения (конечная
стадия других форм миастении, развивается
в течение двух лет от начала болезни)
59. Глазная форма миастении
60. Кризы
• Миастенический криз – нарастание общей слабостиобусловлено нарушением всасывания препарата или
прогрессированием заболевания, необходимо
увеличить дозу АХЭ
• Холинергический криз – ингибиторы АХЭ могут
соединяться с холинорецепторами, поэтому при их
передозировке происходит блокада нервно-мышечной
передачи. Нарастает мышечная слабость,
сопровождающаяся признаками побочных эффектов
АХЭ (повышенная перистальтика ЖКТ, урчание,
слюнотечение, фасцикулярные подергивания мышц,
судороги, брадикардия, потливость, боль в животе,
страх смерти, ступор). При передозировке АХЭ
необходимо введение атропина 0,1% по 0,5-1,0 мл
через 10-15 минут до прекращения приступа
61. ДИАГНОСТИКА
1. Клинический осмотр и выяснение истории болезни.2. Функциональная проба на выявление синдрома
патологической мышечной утомляемости.
3. Электромиографическое исследование
4. Прозериновая проба
5. Клинический осмотр для выявления обратимости
миастенических изменений на фоне прозерина
6. Анализ крови на антитела к ацетилхолиновым
рецепторам и антитела к титину
7. Компьютерная томография органов переднего
средостения (вилочковой железы, синоним: тимуса).
62. Тимома (thymoma) с медуллярно-клеточной дифференцировкой, эпидермоидный тип роста
63. ЭНМГ
Описание методикиЭНМГ
Пациент находится в положении
сидя или лежа, на кожу
накладываются поверхностные
электроды. Стимулирующий
поверхностный электрод
располагается по ходу нерва.
Пациент ощущает покалывание в
месте стимуляции и ритмичное
сокращение исследуемой мышцы.
Ощущения могут быть
болезненными. Исследование
одной мышцы длится около 5-15
минут. При проведении декременттеста часто проводятся
фармакологические пробы с
повторным исследованием на фоне
действия препарата (чаще всего
прозерина).
64. Лечение
• Удаление тимомы• Антихолинэстеразные препараты (АХЭП) прозерин 0,05%1,0, калимин 0,06, оксазил 0,005
• При неоперабельной опухоли или сохранении симптомов
назначают гуанидин (20-25 мг/кг/сут в 3-4 приема).
• Плазмаферез
• Пульс-терапия метилпреднизолоном
• Цитостатики (азатиоприн, метотрексат)
• Иммуностимуляторы (тималин, тимопентин)
Противопоказаны: миорелаксанты, йодсодержащие
контрастные средства, нейролептики, антидепрессанты,
барбитураты, карбамазепин и др.
65. Синдром Итона-Ламберта
• Встречается при злокачественныхновообразованиях, чаще всего при
одноклеточном раке легких.
• Отличается от миастении уменьшением
слабости при физической нагрузке, частыми
вегетативными нарушениями (сухость во рту)
и признаками нейропатии (отсутствие
сухожильных рефлексов) и ЭНМГ-картиной.
• В основе – выработка аутоантител против
пресинаптических окончаний мотонейронов.
66. ЛЕЧЕНИЕ
В случаях легкой впервыевыявленной миастении и глазной
формы в лечении применяется
только калимин 0,06 и препараты
калия 1 г..
67.
• В случаях выраженной мышечнойслабости или наличии бульбарных
нарушений применяется
глюкокортикоидная терапия:
преднизолон (метипред) в дозе 1 мг/кг
веса строго через день в утренние часы
(обычные дозы составляют 60-80 мг в
сутки, минимально эффективные дозы
составляют 50 мг в сутки через день).
68.
• В первые 1-2 года от началазаболевания при генерализованной
форме миастении проводится
оперативное вмешательство по
удалению вилочковой железы
(тимэктомия). Эффект от тимэктомии
развивается в интервале 1-12 месяцев
с момента тимэктомии, оценка
эффективности тимэктомии
производится через 1 год.
69.
• При обострении миастении допустимо иоправданно проведение плазмафереза и
введение внутривенного иммуноглобулина.
Плазмаферез целесообразно проводить по
500 мл через день N5-7 с замещением
плазмой или альбумином.
70. Хирургическое лечение
• Удаление тимомы• Примерно в 90% случаев длительность
устойчивого улучшения или ремиссии
после тимэктомии у таких больных
превышает 5 лет.