
































 biology
biologySimilar presentations:
")









Поиск схожих последовательностей в базах данных. Локальное выравнивание. Семейство программ серии BLAST
1.
Поиск схожих последовательностей вбазах данных. Локальное
выравнивание. Семейство программ
серии BLAST.
2.
Выравнивание последовательностей —размещениедвух или более последовательностей ДНК, РНК или
белков друг под другом таким образом, чтобы легко
увидеть сходные участки в этих последовательностях.
Сходство первичных структур двух молекул может
отражать их функциональные, структурные или
эволюционные взаимосвязи.
Все выравнивания принято делить на глобальные
(последовательности сравниваются полностью) и
локальные (сравниваются только определённые
участки последовательностей).
3.
Цель выравнивания – добиться максимальногоколичества совпадений. Что лучше?
• Просто написать последовательности друг под
другом
• Двигать друг относительно друга
• Вставлять пробелы
4.
Алгоритм локального выравнивания был предложен Т.Ф. Смитом и М. Уотерменом в 1981 г.
В обоих случаях число совпадающих нуклеотидов - 8
5.
Система оценкиЦена изменения данной позиции в выравнивании
последовательностей
– Стоимость замены, вcтавки, делеции
Значительно чаще 1 длинная делеция, чем много
коротких => штраф за удлинение делеции
– Бонус за совпадение букв
6.
Score – общая мера сходства:AACGTTTCCAGTCCAAATAGCTAGGC
===--===
=-===-==-======
AACCGTTC
TACAATTACCTAGGC
Hits(+1): 18
Misses (-2): 5
Gaps (existence -2, extension -1): 1 Length: 3
Score = 18 * 1 + 5 * (-2) – 2 – 2 = 6
7.
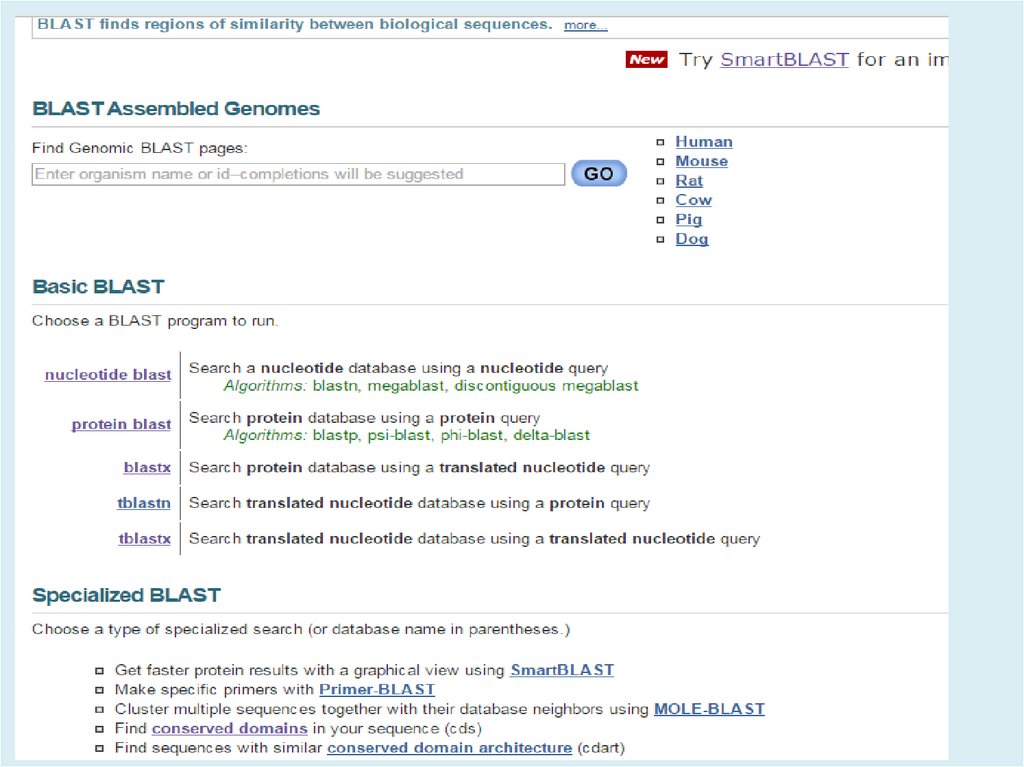
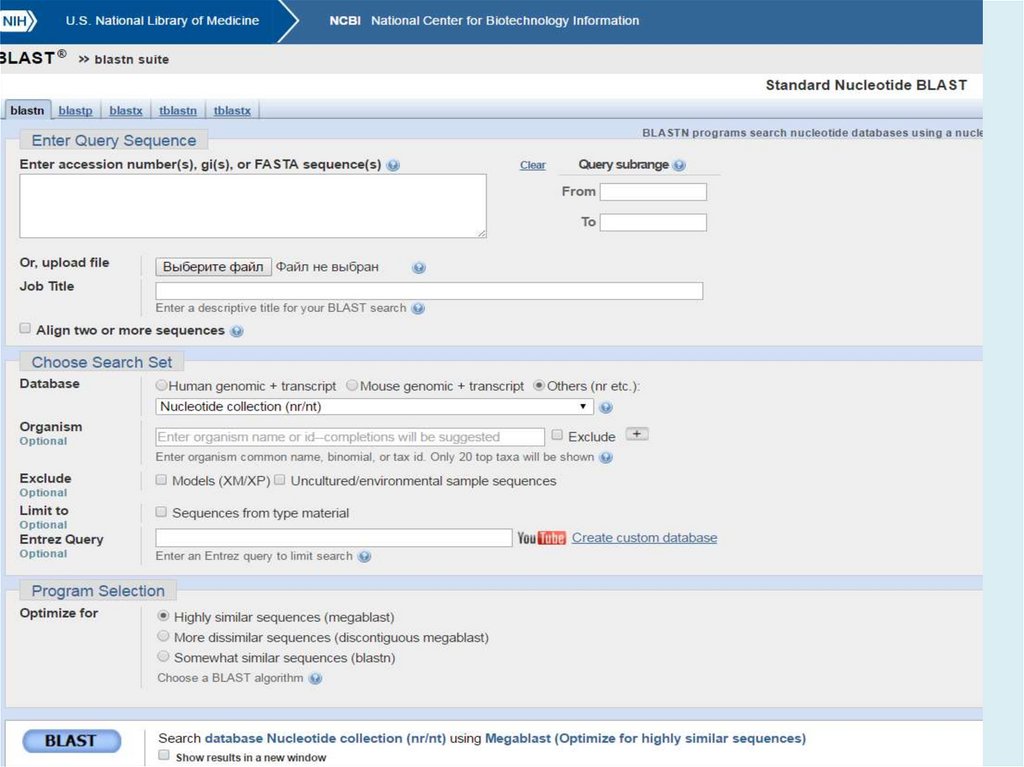
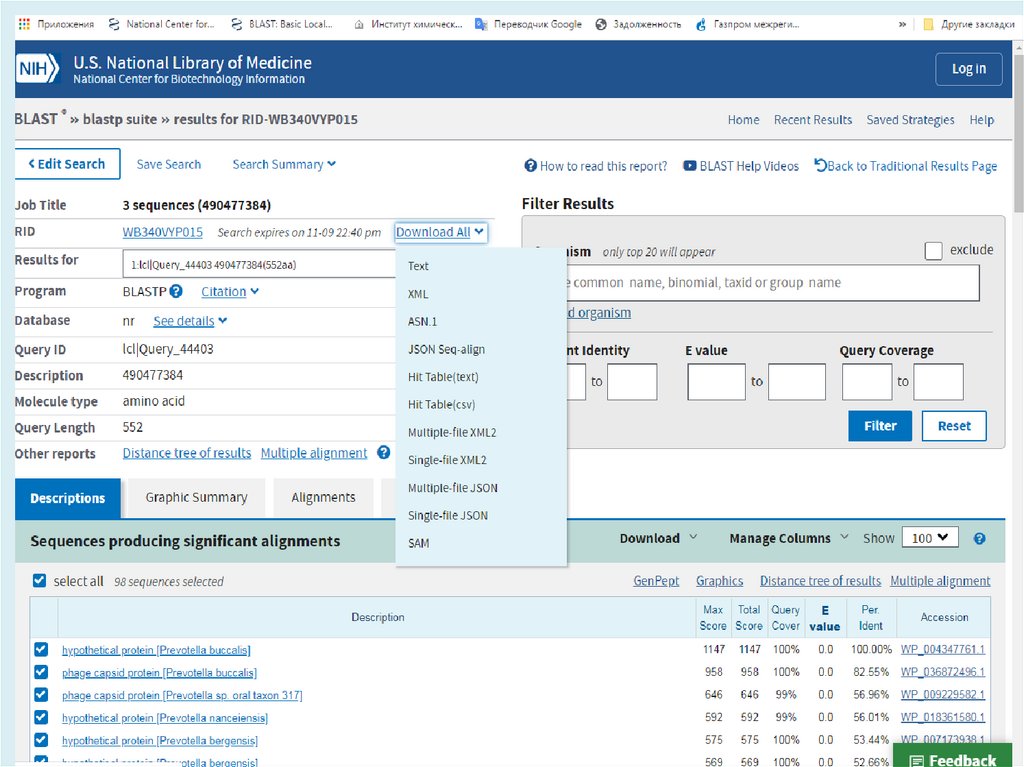
http://blast.ncbi.nlm.nih.gov/Blast.cgiBLAST (Basic Local Alignment Search Tool) — семейство
компьютерных программ, служащих для поиска гомологов
белков или нуклеиновых кислот, для которых известна
первичная структура или её фрагмент.
GenBank – экспоненциальный рост объема данных,
следовательно нужна быстрая (даже в ущерб точности)
программа
Главная задача – поиск похожих последовательностей в
базах данных (=> главное достоинство – скорость)
8.
Используя BLAST, можно сравнить имеющуюсяпоследовательность с последовательностями из базы
данных и найти последовательности предполагаемых
гомологов. Является важнейшим инструментом для
молекулярных биологов.
Программа BLAST была разработана S. Altschul, W. Gish, W.
Miller, E. Myers, и D.J. Lipman в 1990-1993 г.г.
Основана на алгоритме Смита — Ватермана , и сравнивает
отрезки всех возможных длин и оптимизирует меру сходства
по всем отрезкам и всем выравниваниям этих отрезков.
Несмотря на снижение сходства родственных
последовательностей с течение времени при их эволюции,
мы можем рассчитывать обнаружить короткие участки
высокого сходства, не затронутые мутациями.
9.
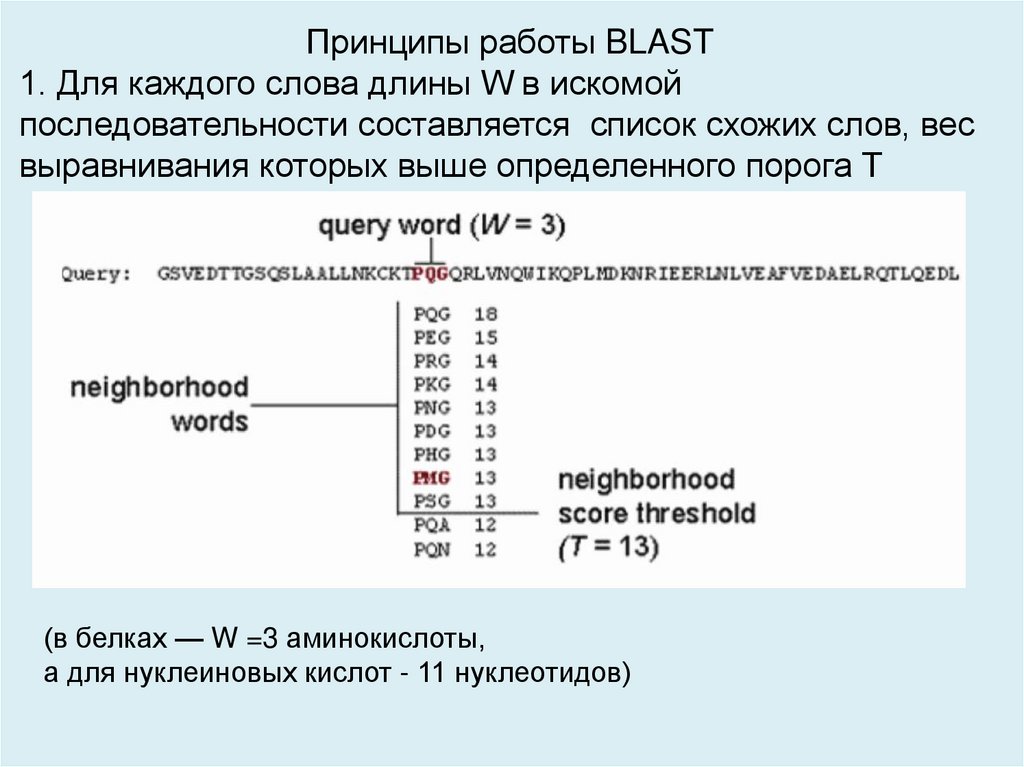
Принципы работы BLAST1. Для каждого слова длины W в искомой
последовательности составляется список схожих слов, вес
выравнивания которых выше определенного порога T
(в белках — W =3 аминокислоты,
а для нуклеиновых кислот - 11 нуклеотидов)
10.
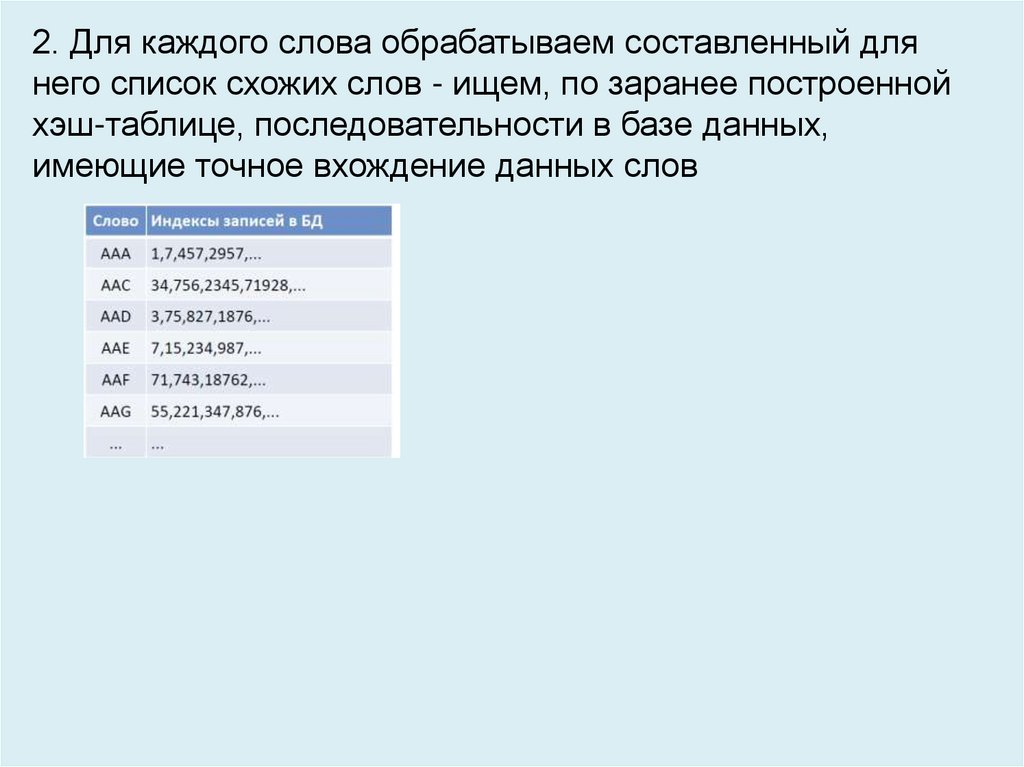
2. Для каждого слова обрабатываем составленный длянего список схожих слов - ищем, по заранее построенной
хэш-таблице, последовательности в базе данных,
имеющие точное вхождение данных слов
11.
3. Расширяем выравнивание вправо и влево от найденных“затравок” используя алгоритм динамического
программирования сначала без гэпов (пробелов), а затем с
их использованием.
пары сегментов с максимальным сходством
12.
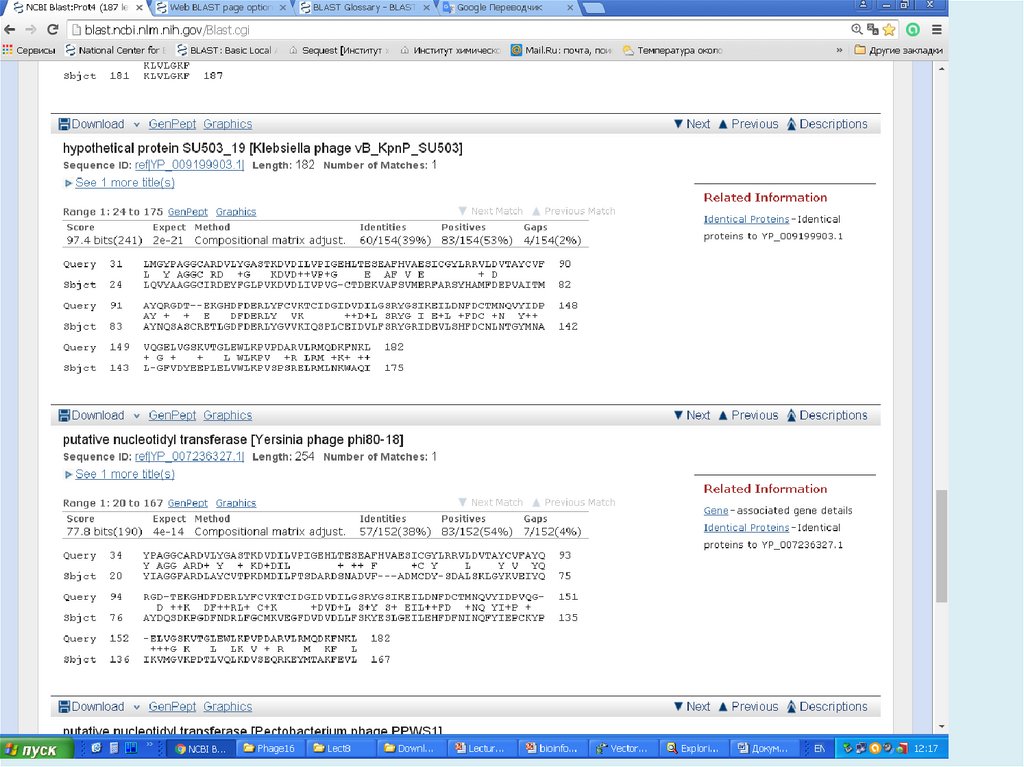
13.
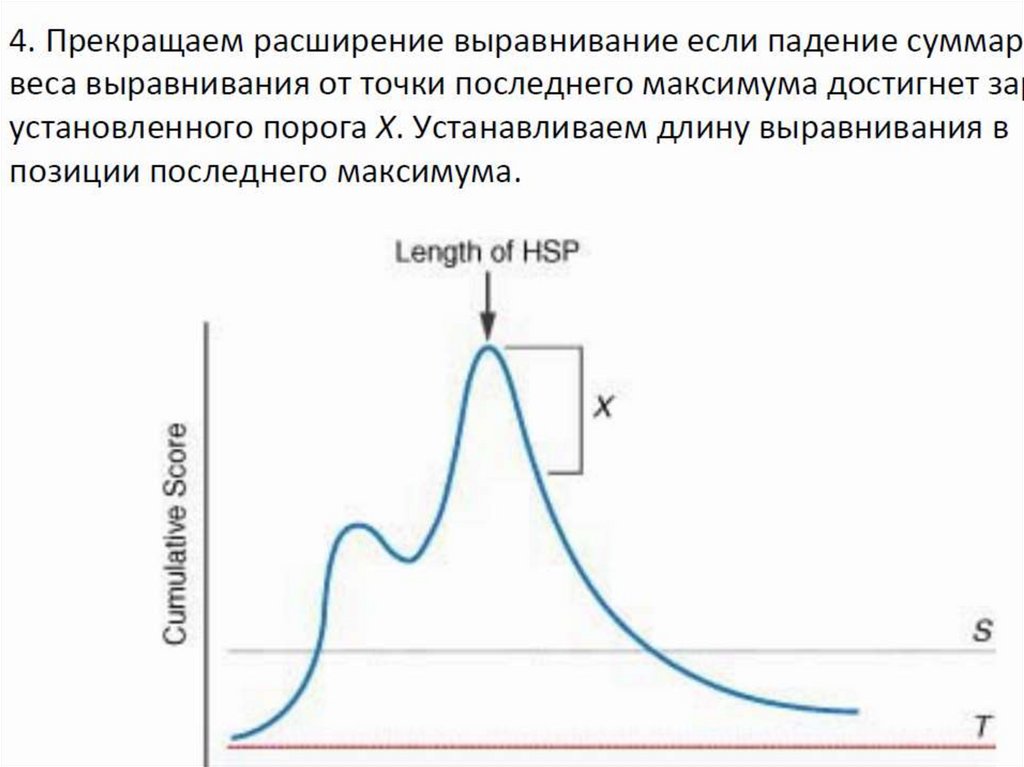
После максимального продления размеров всехвозможных «слов» изучаемой последовательности,
определяются выравнивания с максимальным
количеством совпадений для каждой пары запрос —
последовательность базы данных, и полученная
информация фиксируется.
Теоретически локальное выравнивание может начинаться
с любой пары нуклеотидов или аминокислот выровненных
последовательностей. Однако HSP, как правило, не
начинаются близко к краю (началу или концу)
последовательностей.
14.
15.
16.
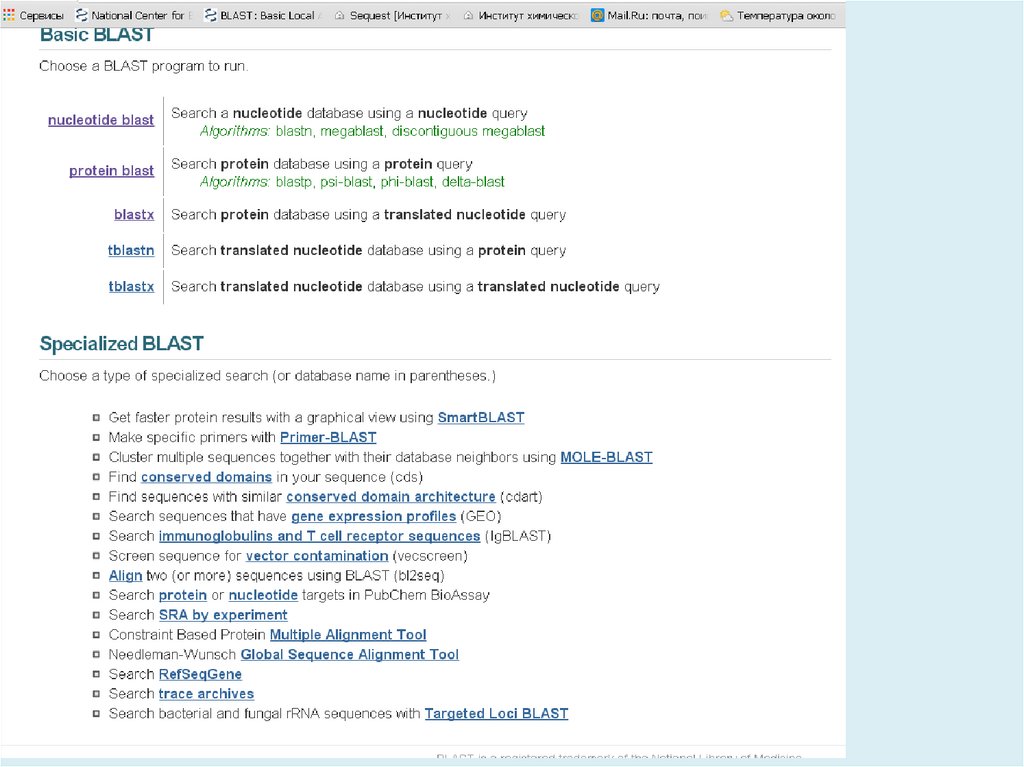
ПрограммаЗапрос
Тип БД
Сравнивает
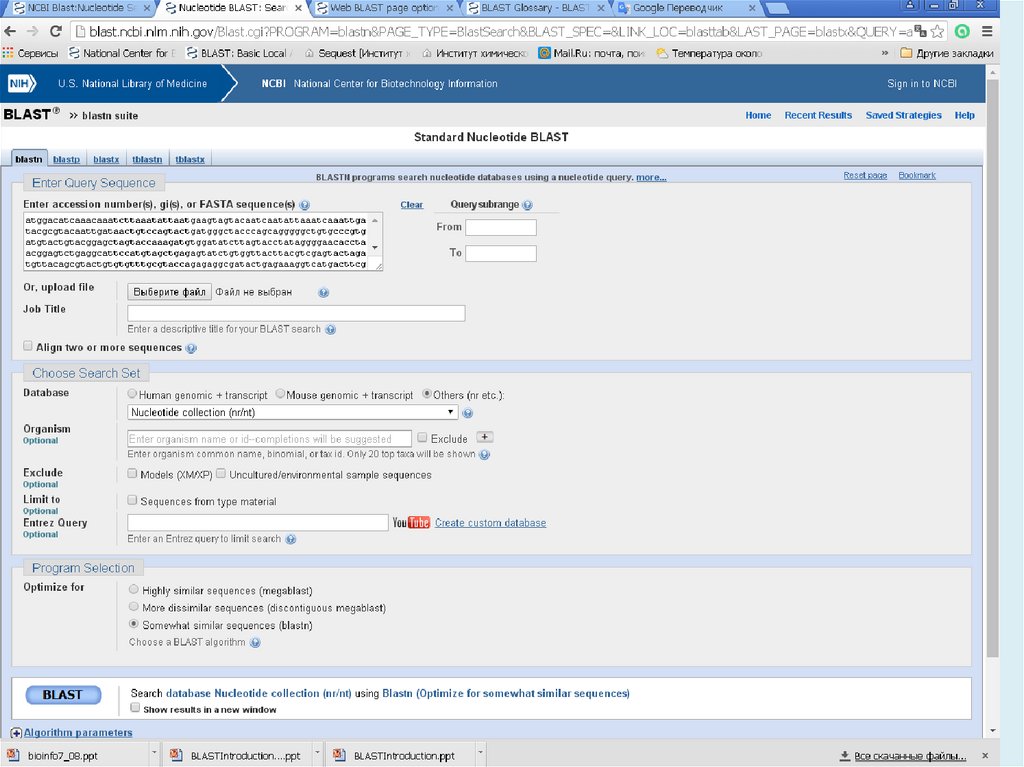
Blastn
ДНК
ДНК
ДНК
Blastp
белок
белок
белки
Blastx
ДНК
белок
белки
Tblastn
белок
ДНК
белки
Tblastx
ДНК
ДНК
белки
17.
18.
ДНК:megaBLAST – другой алгоритм для сравнения ДНК.
Оптимизирован для длинных похожих
последовательностей. Оптимален для поиска
совпадений в родном геноме или очень близких видах
Discontiguous megaBLAST – аналогично, параметры
подобраны для более далеких видов
19.
20.
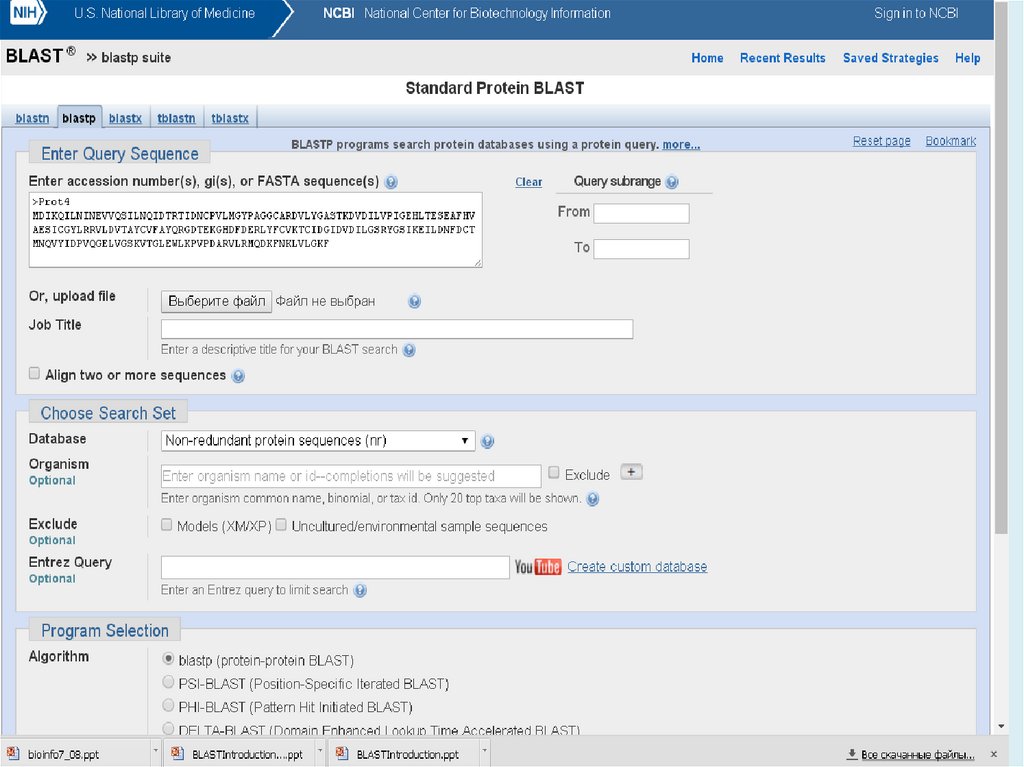
Пример последовательности в формате FASTA :>OVAX_CHICK GENE X PROTEIN
QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNV
ATLPAEKMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVY
Пустые строки в середине не допускаются
Последовательности должны быть представлены в стандартных IUB / IUPAC
кодах аминокислот и нуклеиновых кислот, со следующими исключениями:
строчные буквы принимаются; один дефис или тире может быть использованы
для представления пробела неопределенной длины. Перед отправкой запроса
любые цифры в последовательности запроса должны быть либо удалены либо
заменены соответствующими буквенными кодами (например, N для
неизвестного остатка нуклеиновой кислоты или Х для неизвестного остатка
аминокислоты). Поддерживаемые коды нуклеиновых кислот:
A
adenosine C
cytidine
G
guanine
T
thymidine N
A/G/C/T (any)U
uridine
K
G/T (keto) S
G/C (strong) Y
T/C (pyrimidine)
M
A/C (amino)W
A/T (weak)
R
G/A (purine)
B
G/T/C
D
G/A/T
H
A/C/T
V
G/C/A
-
gap of indeterminate length
21.
Вырожденные нуклеотидные коды (красный цвет),рассматриваются как «mismatches» нуклеотидного
выравнивания.
Для тех программ, которые используют аминокислотные
последовательности (BLASTP и TBLASTN), принятые коды
аминокислот :
A alanine P proline B aspartate/asparagine Q glutamine
C cystine R arginine D aspartate S serine E glutamate
T threonine F phenylalanine U selenocysteine G glycine
V valine H histidine W tryptophan I isoleucine Y tyrosine
K lysine Z glutamate/glutamine L leucine X any
M methionine * translation stop N asparagine - gap
Для аминокислот, U заменяется X перед началом поиска
blast не будет принимать «-» в запросе. Для представления
пробелов, используйте N или X
22.
Nucleotide Sequence DatabasesNr All GenBank + RefSeq Nucleotides + EMBL + DDBJ + PDB sequences (excluding
HTGS0,1,2, EST, GSS, STS, PAT, WGS).
refseq_rna RNA entries from NCBI's Reference Sequence project
refseq_genomic Genomic entries from NCBI's Reference Sequence project
Est Database of GenBank + EMBL + DDBJ sequences from EST Divisions
est_human Human subset of est.
est_mouse Mouse subset.
est_others Non-Mouse, non-Human subset of est.
Gss Genome Survey Sequence, includes single-pass genomic data, exon-trapped
sequences, and Alu PCR sequences.
Htgs Unfinished High Throughput Genomic Sequences: phases 0, 1 and 2 (finished,
phase 3 HTG sequences are in nr)
Pat Nucleotides from the Patent division of GenBank.
Pdb Sequences derived from the 3-dimensional structure from Brookhaven Protein
Data Bank
Month All new or revised GenBank + EMBL + DDBJ + PDB sequences released in the
last 30 days.
Dbsts Database of GenBank+EMBL+DDBJ sequences from STS Divisions .
Chromosome A database with complete genomes and chromosomes from the NCBI
Reference Sequence project.
Wgs A database for whole genome shotgun sequence entries.
env_nt Nucleotide sequences from environmental samples
23.
Peptide Sequence DatabasesNr All non-redundant GenBank CDS translations + RefSeq Proteins + PDB + SwissProt +
PIR + PRF
Refseq RefSeq protein sequences from NCBI's Reference Sequence Project.
Swissprot Last major release of the SWISS-PROT protein sequence database (no
updates).
Pat Proteins from the Patent division of GenPept.
pdb Sequences derived from the 3-dimensional structure from Brookhaven Protein
Data Bank.
Month All new or revised GenBank CDS translation+PDB+SwissProt+PIR+PRF released
in the last 30 days.
env_nr Protein sequences from environmental samples.
24.
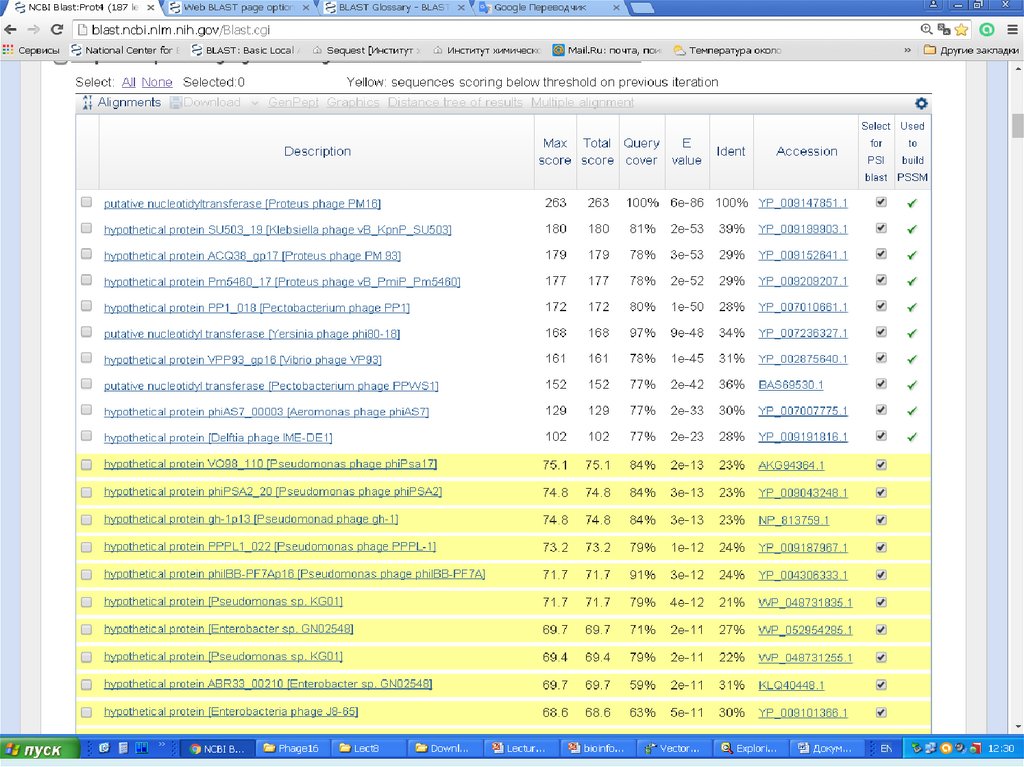
25.
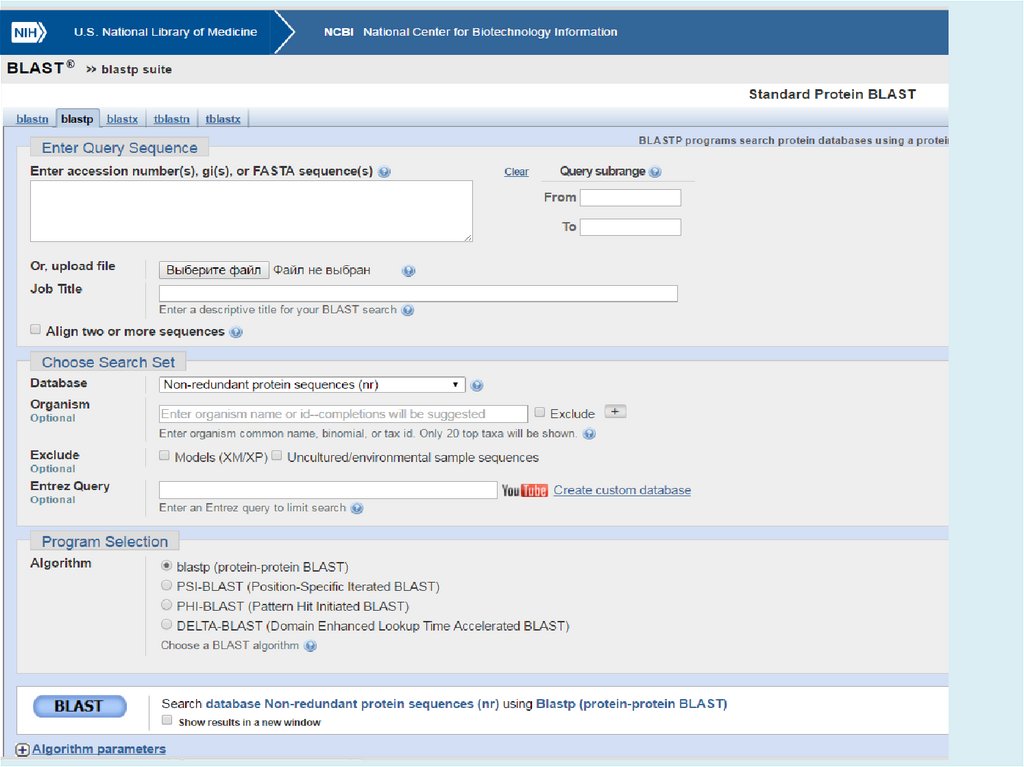
Белок:PSI-BLAST (Position-Specific Iterated -BLAST) поиск удаленных
белковых гомологов с использованием PSSM (position-specific
scoring matrix)
PHI-BLAST (Pattern-Hit Initiated -BLAST) ищет гомологичные
белки, удовлетворяющие заданному паттерну
26.
27.
При определении сходства ключевым элементомявляется матрица замен, так как она определяет
показатели сходства для любой возможной пары
нуклеотидов или аминокислот. В большинстве
программ серии BLAST используется матрица
BLOSUM62 (Blocks Substitution matrix 62 % identity,
блоковая матрица замен с 62 % идентичностью).
28.
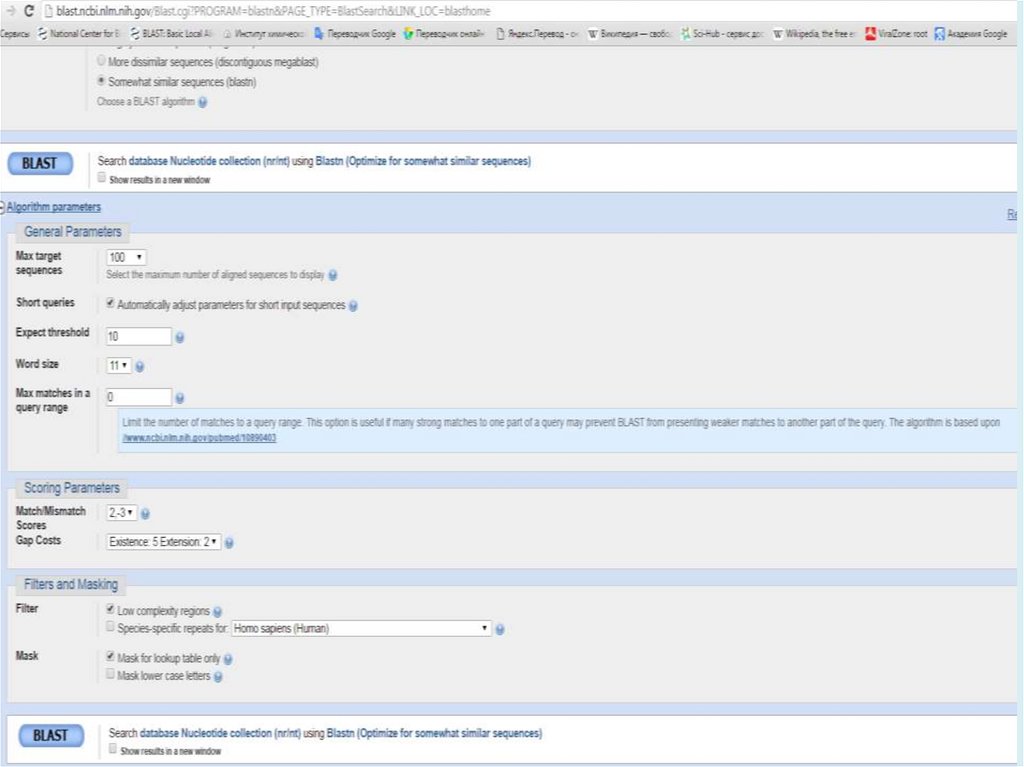
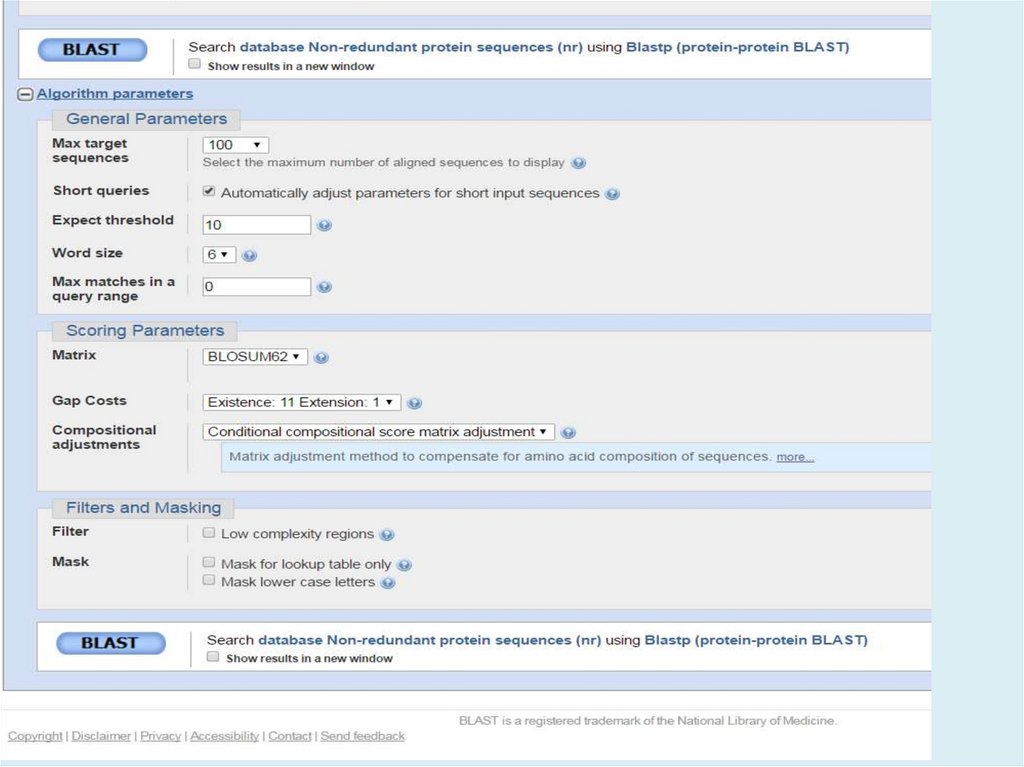
Выбор параметровМеняйте параметры только, если по умолчанию не
работает (параметры по умолчанию подобраны хорошо
для большинства ситуаций)
Для того, чтобы выбрать более подходящие параметры
надо очень ТОЧНО сформулировать задачу
Фильтрация
Low-complexity region. Если Ваш белок содержит большой
регион низкой сложности – попробуйте использовать
BLAST без соответствующей фильтрации
Если Ваш белок содержит очень часто встречающиеся
домены, их тоже можно отфильтровать – в ручную
29.
Выбор параметровМатрица:BLOSUM для локального выравнивания обычно
лучше, чем PAM
Чем выше номер BLOSUM – тем строже выравнивание
(BLOSUM80 вместо BLOSUM45 – более короткие
выравнивания)
РАМ – чем ниже, тем строже
Штрафы за делеции:
Чем больше штраф за внесение, тем короче выравнивания
Штраф за удлинение ~10 раз ниже, чем за внесение
Если сравниваете удаленные гомологи, то лучше всего
довольно высокий штраф за внесение делеции и низкий
за удлинение
Близкие гомологи – штрафы ближе друг к другу
30.
31.
32.
33.
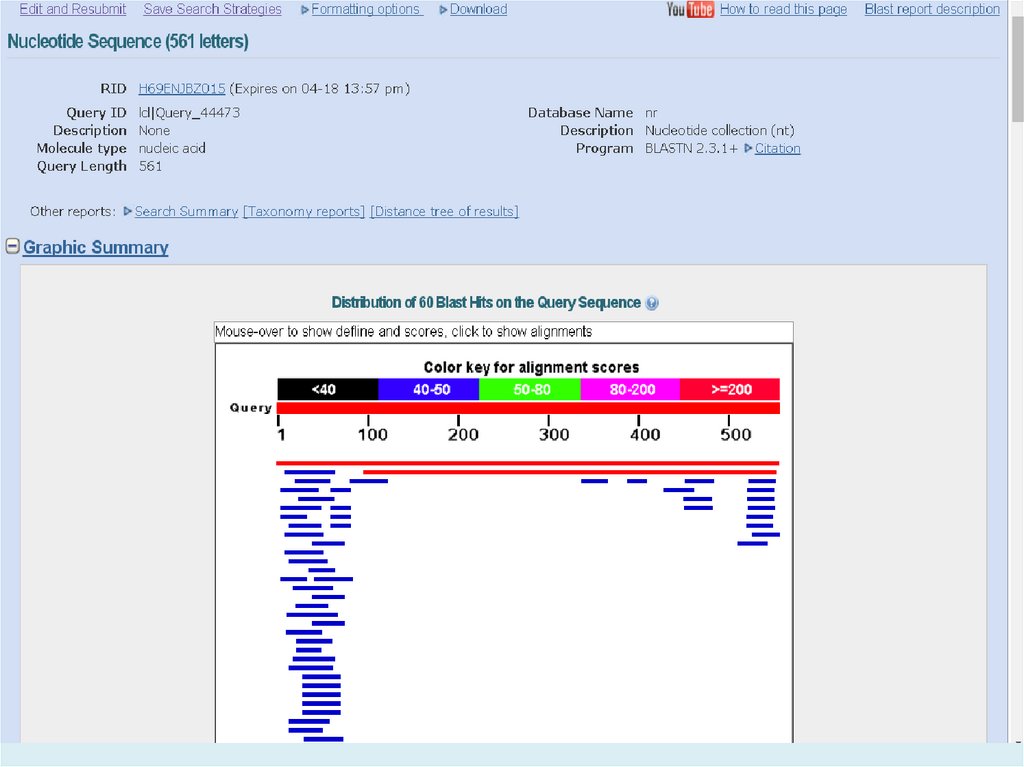
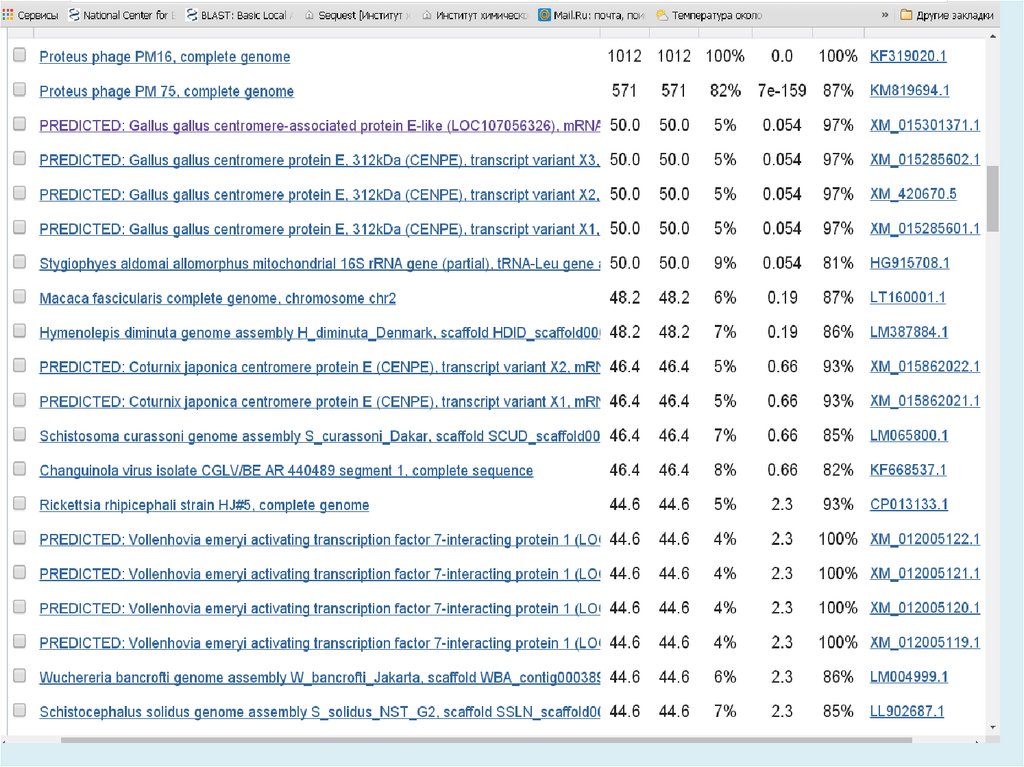
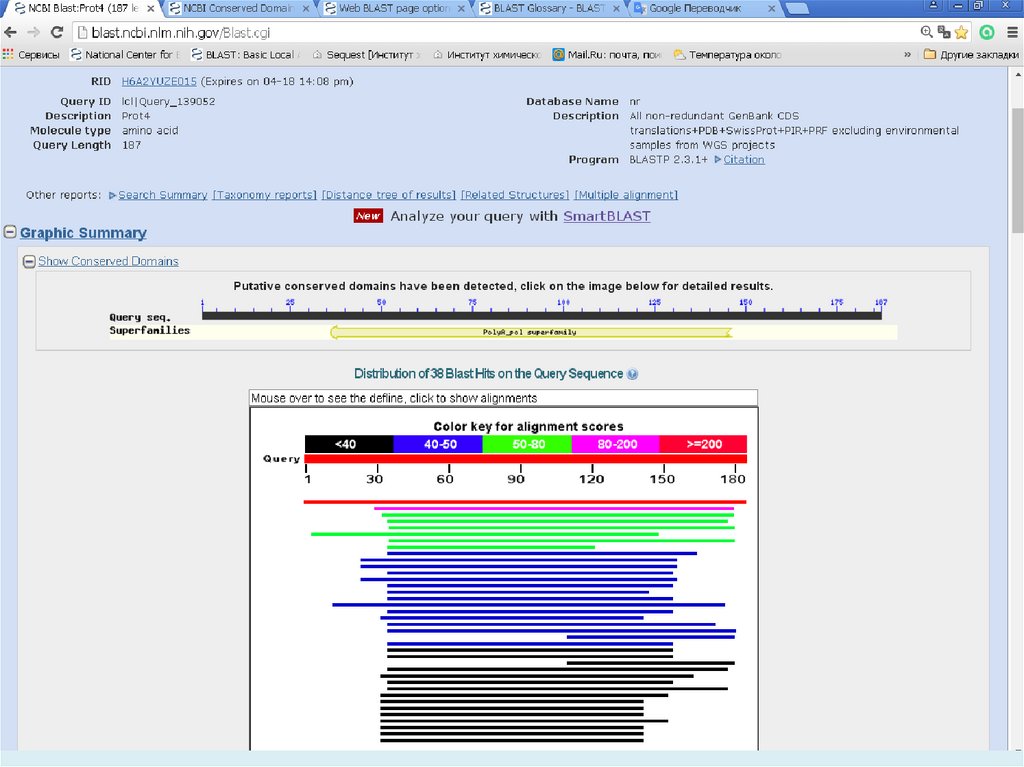
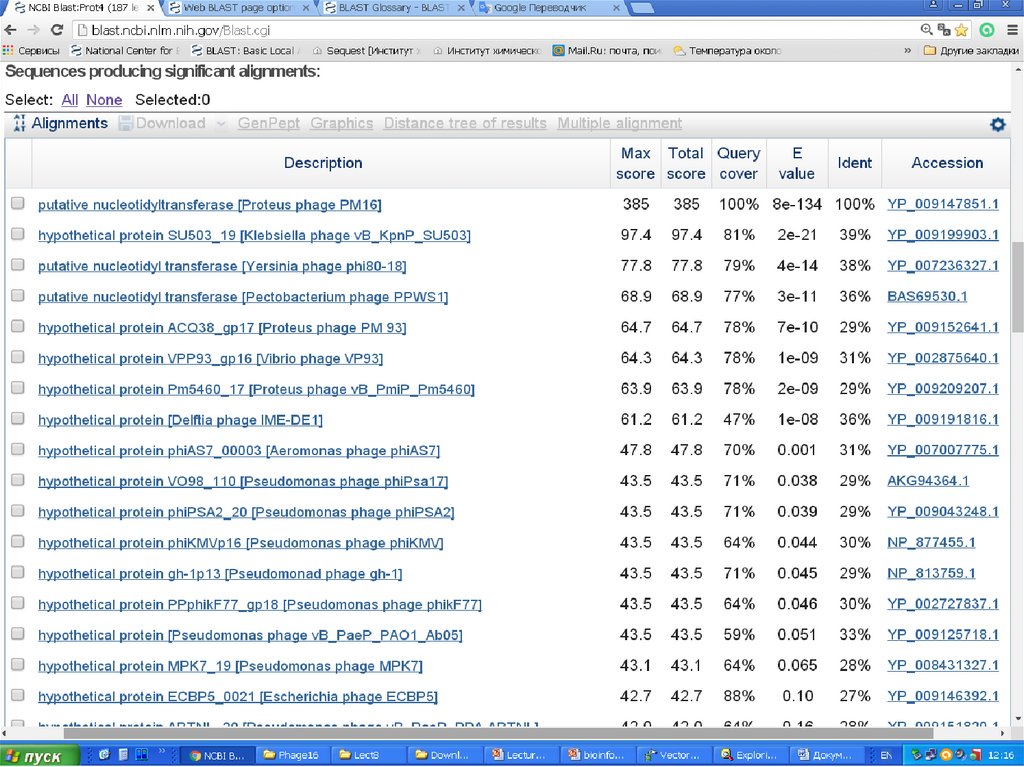
E-value (the expectation value) – представляет числоразличных элайментов с баллами эквивалентными
или лучшими, чем S, которое, как ожидается, будет
найдено при поиске базе данных случайно. Чем ниже
значение Е, тем больше значимость выравнивания. (0
- e-6 – хорошо, > 0.001 - 0.01 – плохо). Этот параметр
определяет статистический порог значимости
выравнивания. Для низких значений, равна р,
вероятности случайного выравнивания.
Как правило, BLAST недооценивает e-value!
Bit Score – мера статистической значимости (вес – сумма
стоимостей всех точечных замен) выравнивания,
(меньше 50 – плохо). Bit Score могут быть
использованы для сравнения выравниваний из
различных поисков.
34.
35.
36.
37.
38.
39.
––
–
–
PSI – BLAST
Алгоритм:
Несколько раундов поиска
Первый раунд – просто blastp (BLOSUM62)
Построение PSSM на основе полученных хитов
Следующий раунд на основе этой PSSM
Методов итераций, пока множество хитов не перестанет
меняться
PHI - BLAST
– Query – белок + паттерн, которому этот белок удовлетворяет
– Пример: >P28332|ADH6_HUMAN Alcohol dehydrogenase 6 - Homo sapiens
(Human)
MSTTGQVIRCKAAILWKPGAPFSIEEVEVAPPKAKEVRIKVVATGLCGTEMKVLGSKHLDLLYPTILGHEGAGI
VESIGEGVSTVKPGDKVITLFLPQCGECTSCLNSEGNFCIQFKQSKTQLMSDGTSRFTCKGKSIYHFGNT
STFCEYTVIKEISVAKIDAVAPLEKVCLISCGFSTGFGAAINTAKVTPGSTCAVFGLGGVGLSVVMGCKAA
GAARIIGVDVNKEKFKKAQELGATECLNPQDLKKPIQEVLFDMTDAGIDFCFEAIGNLDVLAAALASCNES
YGVCVVVGVLPASVQLKISGQLFFSGRSLKGSVFGGWKSRQHIPKLVADYMAEKLNLDPLITHTLNLDKI
NEAVELMKTGKW
– G - H - E - x - {EL} - G - {AP} - x(4) - [GA] - x(2) - [IVSAC]
40.
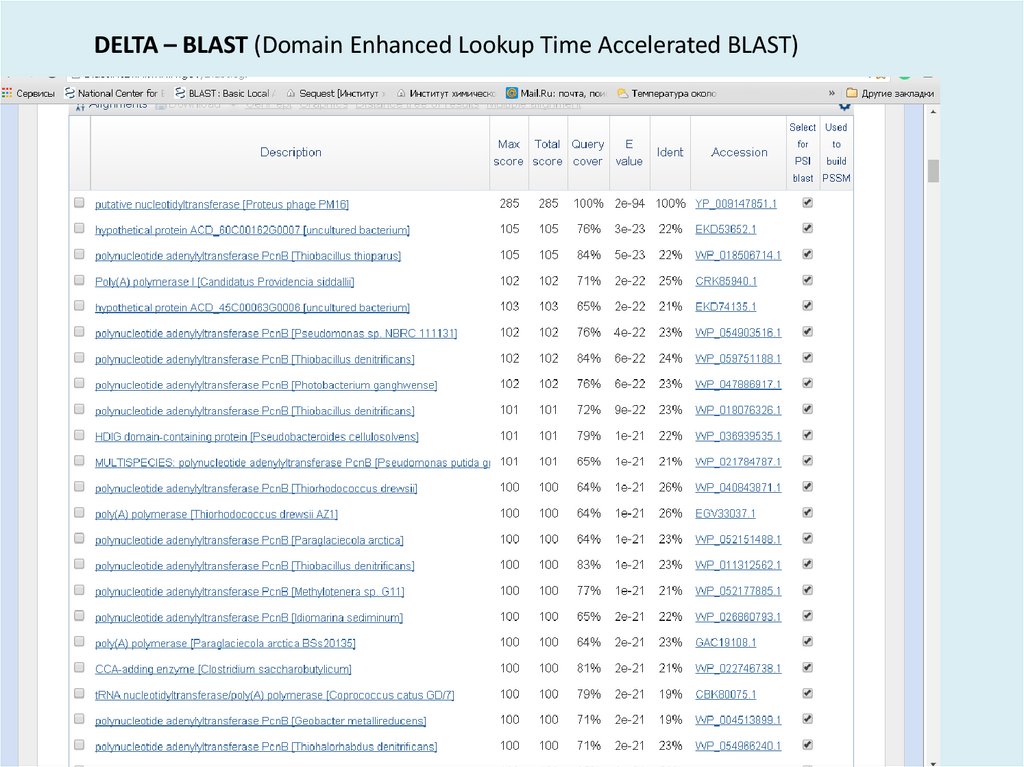
41.
DELTA – BLAST (Domain Enhanced Lookup Time Accelerated BLAST)42.
43.
44.
45.
46.
47.
48.
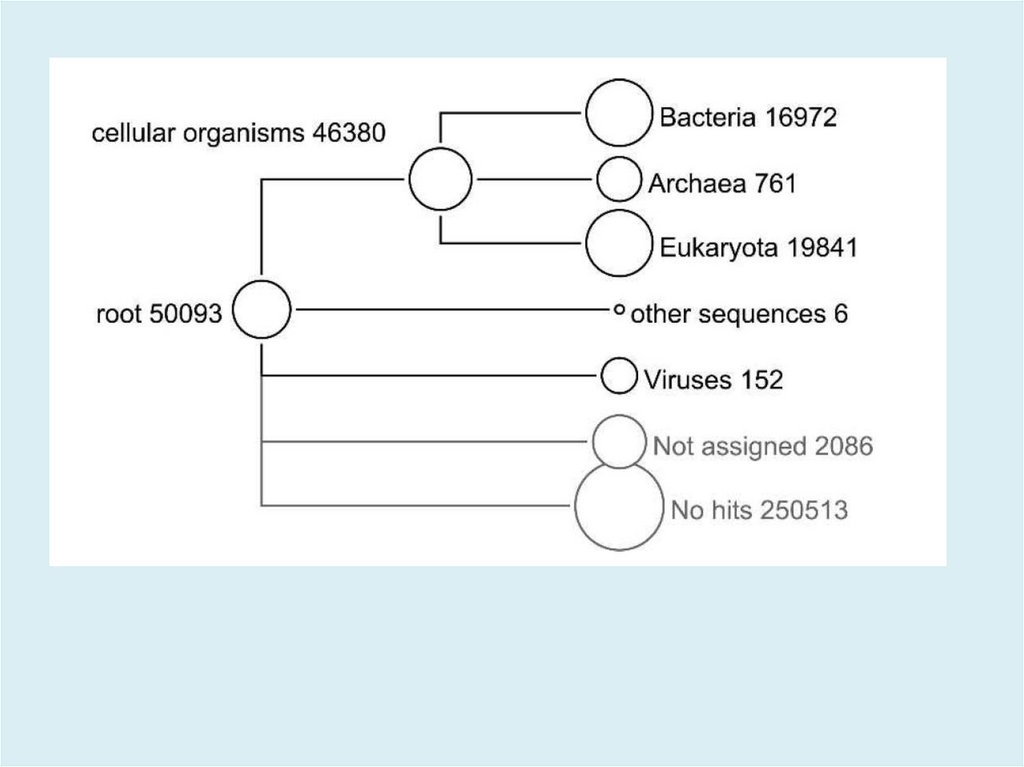
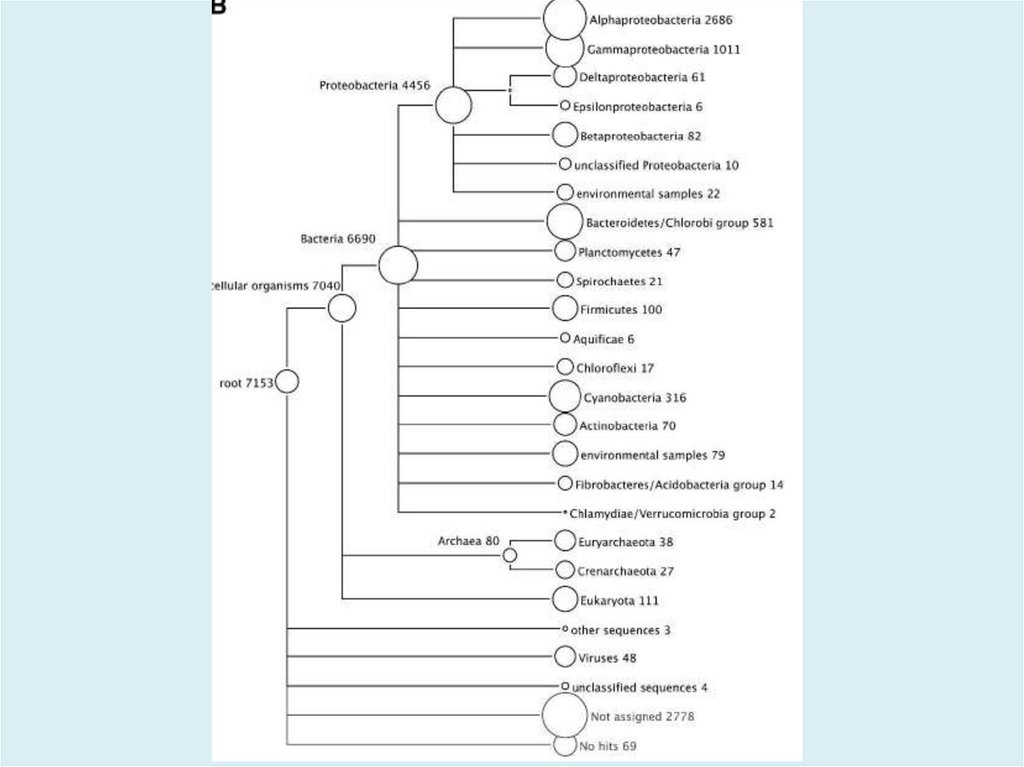
MEGAN V5.11.3http://ab.inf.uni-tuebingen.de/software/megan/
Основное применение программы для разбора и анализа
результата BLAST сравнения набора ридов против одной
или нескольких референсных баз данных, как правило, с
использованием BLASTN, BLASTX или BLASTP для
сравнения с NCBI-NT, NCBI-NR или конкретной геномной
базы данных. Результатом такого анализа является оценка
таксономического содержания (профиль вида) образца, из
которого риды были получены. Программа использует
несколько различных алгоритмов чтобы поместить риды в
систематику, присваивая каждый риду к таксону на
определенном уровне в иерархии NCBI, на основе
обращений к известным последовательностям, как
записаным в файле BLAST.
49.
MEGAN V5.11.3http://ab.inf.uni-tuebingen.de/software/megan/
The aim of MEGAN is to provide a tool for studying the taxonomic
content of a set of DNA reads, typically collected in a metagenomics
project. In a preprocessing step, a sequence alignment of all reads
against a suitable database of reference DNA or protein sequences
must be performed to produce an input file for the program.
MEGAN is suitable for DNA reads (metagenome data), RNA reads
(metatranscriptome data), peptide sequences (metaproteomics
data) and, using a suitable synonyms file that maps SILVA ids to
taxon ids, for 16S rRNA data (amplicon sequencing). At start-up,
MEGAN first reads in the current NCBI taxonomy (consisting of over
one million taxa). A first application of the program is that it
facilitates interactive exploration of the NCBI taxonomy.
50.
MEGAN V5.11.3http://ab.inf.uni-tuebingen.de/software/megan/
However, the main application of the program is to parse and
analyze the result of an alignment of a set of reads against one or
more reference databases, typically using BLASTN, BLASTX or
BLASTP or similar tools including DIAMOND to compare against
NCBI-NT, NCBI-NR or genome specific databases. The result of a such
an analysis is an estimation of the taxonomical content (“species
profile”) of the sample from which the reads were collected. The
program uses a number of different algorithms to “place” reads into
the taxonomy by assigning each read to a taxon at some level in the
NCBI hierarchy, based on their hits to known sequences, as recorded
in the alignment file. Alternatively, MEGAN can also parse files
generated by the RDP website or the Silva website
51.
MEGAN V5.11.3http://ab.inf.uni-tuebingen.de/software/megan/
New input to the program is usually provided as a BLAST file
obtained from a BLAST alignment of the given set of reads against a
reference database such as NCBI-nr or NCBI-nt. MEGAN supports
BLASTN, BLASTX and BLASTP standard text-format, and BLAST XML
format as well as multiple output formats provided by tools similar
to BLAST, like RapSearch2 and DIAMOND.
52.
53.
54.
DNA Masterhttp://cobamide2.bio.pitt.edu/
DNA Master это Multiple Document Interface (MDI)
программа для создания, модификации и анализа ДНК,
РНК и белковых последовательностей.
Why do you call it the world's greatest sequence explorer?
Try it; see what it does. Did you find what you wanted? How easy was that? Need
something different? Let us know.
55.
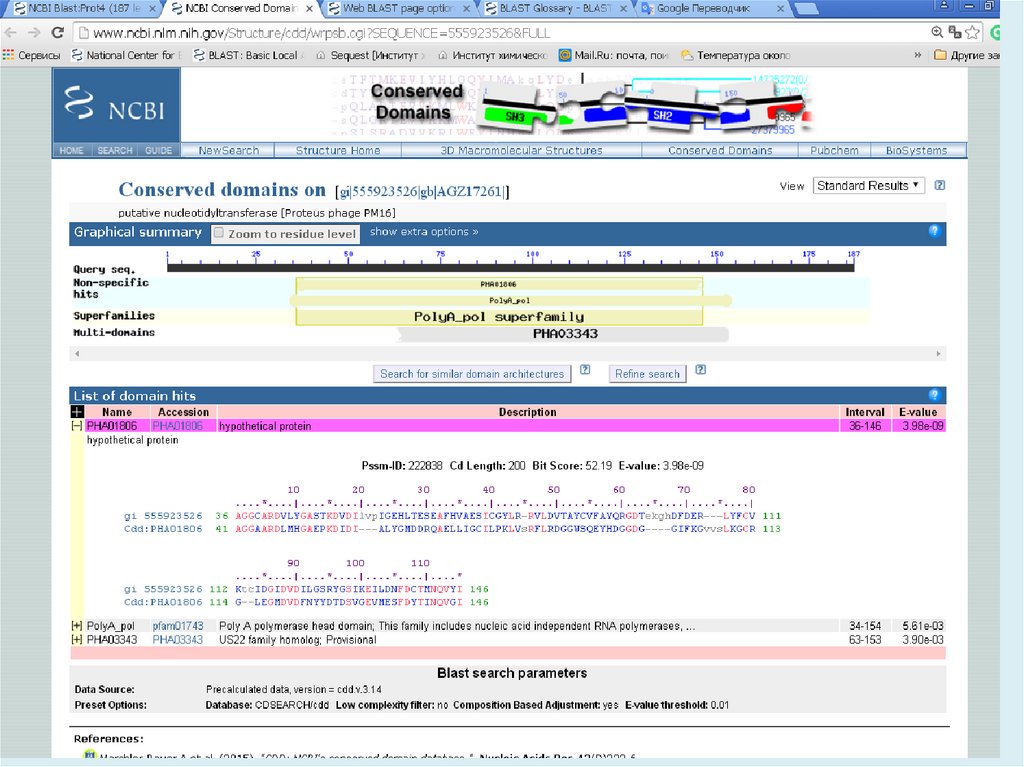
domainA discrete portion of a protein assumed to fold independently of the rest of the
protein and possessing its own function.
E value
The Expectation value or Expect value represents the number of different alignments
with scores equivalent to or better than S that is expected to occur in a database
search by chance. The lower the E value, the more significant the score and the
alignment.
filtering
Filtering, also known as masking, removes regions of (nucleic acid or amino acid)
sequence having characteristics that may lead to spurious high scores.
gap
homology
Similarity attributed to descent from a common ancestor. Homologous biological
components (genes, proteins, structures) are called homologs. See also orthologs and
paralogs.
motif
A short conserved region in a protein sequence. Motifs are frequently highly
conserved parts of domains. PSSM
A Position-Specific Scoring Matrix (PSSM) is a profile that gives the log-odds score for
finding a particular matching amino acid in a target sequence.
query
The input sequence (or other type of search term) to which all of the entries in a
database are to be compared.
56.
RAST (Rapid Annotation using Subsystem Technology) is afully-automated service for
annotating bacterial and archaeal genomes. It provides
high quality genome annotations for these genomes across
the whole phylogenetic tree.
57.
58.
LOCUSDEFINITION
ACCESSION
VERSION
KEYWORDS
SOURCE
ORGANISM
FJ795028
701 bp
mRNA
linear
PRI 06-APR-2009
Homo sapiens tumor necrosis factor alpha (TNF) mRNA, partial cds.
FJ795028
FJ795028.1 GI:226201420
.
Homo sapiens (human)
Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE
1 (bases 1 to 701)
AUTHORS
Guan,W.J., Ma,Y.H., Yu,L.L., Na,R.S. and Liu,S.
TITLE
Direct Submission
JOURNAL
Submitted (28-FEB-2009) Academy of Agricultural Sciences, Institute
of Animal Science, Quanmingyuan West, Beijing 100193, People's
Republic of China
FEATURES
Location/Qualifiers
source
1..701
/organism="Homo sapiens"
/mol_type="mRNA"
/db_xref="taxon:9606"
/chromosome="6"
/map="6p21.3"
/sex="male"
/tissue_type="placenta"
/country="China"
/collection_date="2008"
gene
<1..701
/gene="TNF"
59.
CDS<1..701
/gene="TNF"
/note="APC1 protein"
/codon_start=3
/product="tumor necrosis factor alpha"
/protein_id="ACO37640.1"
/db_xref="GI:226201421"
/translation="STESMIRDVELAEEALPKKTGGPQGSRRCLFLSLFSFLIVAGAT
TLFCLLHFGVIGPQREEFPRDLSLISPLAQAVRSSSRTPSDKPVAHVVANPQAEGQLQ
WLNRRANALLANGVELRDNQLVVPSEGLYLIYSQVLFKGQGCPSTHVLLTHTISRIAV
SYQTKVNLLSAIKSPCQRETPEGAEAKPWYEPIYLGGVFQLEKGDRLSAEINRPDYLD
FAESGQVYFGIIAL«
ORIGIN
1 tgagcactga aagcatgatc cgggacgtgg agctggccga ggaggcgctc
61 caggggggcc ccagggctcc aggcggtgct tgttcctcag cctcttctcc
121 tggcaggcgc caccacgctc ttctgcctgc tgcactttgg agtgatcggc
181 aagagttccc cagggacctc tctctaatca gccctctggc ccaggcagtc
241 ctcgaacccc gagtgacaag cctgtagccc atgttgtagc aaaccctcaa
301 agctccagtg gctgaaccgc cgggccaatg ccctcctggc caatggcgtg
361 ataaccagct ggtggtgcca tcagagggcc tgtacctcat ctactcccag
421 agggccaagg ctgcccctcc acccatgtgc tcctcaccca caccatcagc
481 tctcctacca gaccaaggtc aacctcctct ctgccatcaa gagcccctgc
541 ccccagaggg ggctgaggcc aagccctggt atgagcccat ctatctggga
601 agctggagaa gggtgaccga ctcagcgctg agatcaatcg gcccgactat
661 ccgagtctgg gcaggtctac tttgggatca ttgccctgtg a
//
cccaagaaga
ttcctgatcg
ccccagaggg
agatcatctt
gctgaggggc
gagctgagag
gtcctcttca
cgcatcgccg
cagagggaga
ggggtcttcc
ctcgactttg
60. Базы данных
• GenBank US http://www.ncbi.nlm.nih.gov/• EMBL European Molecular Biology
Laboratory http://www.embl.org/
• DDBJ DNA Data Bank of Japan —
http://www.ddbj.nig.ac.jp/
INSDC (International Nucleotide Sequence
Database Collaboration)