


















")

")




































 biology
biologySimilar presentations:




")

")



Биоинформатика. Поиск гомологов в базах данных. (Тема 5)
1.
2. Поиск гомологов в базах даных
BLASTFASTA
3.
>EC_Tr : MQNRLTIKDIARLSGVGKSTVSRVLNNEYR>EC_Fr : MKLDEIARLAGVSRTTASYVINGKAKQYR
При аналізі первинних структур процедура
вирівнювання
виявляє
сходство
між
послідовностями (sequence similarity), яке
може свідчити про гомологію (homology),
тобто еволюційну спорідненість макромолекул.
Геп – пропуск в
послідовності
>EC_Tr : MQNRLTIKDIARLSGVGKSTVSRVLNNE---YR
>EC_Fr : ----MKLDEIARLAGVSRTTASYVINGKAKQYR
4.
Гомологичные последовательности –последовательности, имеющие общее
происхождение (общего предка).
Признаки гомологичности белков
сходная 3D-структура
в той или иной степени похожая
аминокислотная последовательность
разные другие соображения…
5. Что изображено?
Названиепоследовательнос
ти
Консервативный
остаток
Номер столбца
выравнивания
Функционально
консервативная
позиция
Номер последнего в строке остатка
ИЗ ЭТОЙ ПОСЛЕДОВАТЕЛЬНОСТИ
6.
«Идеальное» выравнивание – записьпоследовательностей одна под другой так, чтобы
гомологичные фрагменты оказались друг под
другом.
домовой
скупидом
водомерка
лесовоз
ледоход
?
?
Гэп – пропуск в
последовательности
---лесо---воз
лед---оход---
7.
Попарное выравнивание:*
20
XYLR_ECOLI : GYPSLQYFYSVFKKAYDTTPKEYR : 24
XYLR_HAEIN : GYPSIQYFYSVFKKEFEMTPKEFR : 24
Множественное выравнивание:
*
20
APPY_ECOLI : GYNSTSYFICAFKDYYGVTPSHYF
CELD_ECOLI : GYSSPSLFIKTFKKLTSFTPKSYR
CFAD_ECOLI : GISSASYFIRVFNKHYGVTPKQFF
ENVY_ECOLI : GYSSTSYFISVFKAFYGLTPLNYL
FAPR_ECOLI : GYTSVSYFIKTFKEYYGVTPKKFE
MELR_ECOLI : GFRSSSRFYSTFGKYVGMSPQQYR
RHAS_ECOLI : GFSDSNHFSTLFRREFNWSPRDIR
ROB_ECOLI : RFDSQQTFTRAFKKQFAQTPALYR
TETD_ECOLI : QFDSQQSFTRRFKYIFKVTPSYYR
XYLR_ECOLI : GYPSLQYFYSVFKKAYDTTPKEYR
XYLR_HAEIN : GYPSIQYFYSVFKKEFEMTPKEFR
g s
F
Fk
tP
:
:
:
:
:
:
:
:
:
:
:
24
24
24
24
24
24
24
24
24
24
24
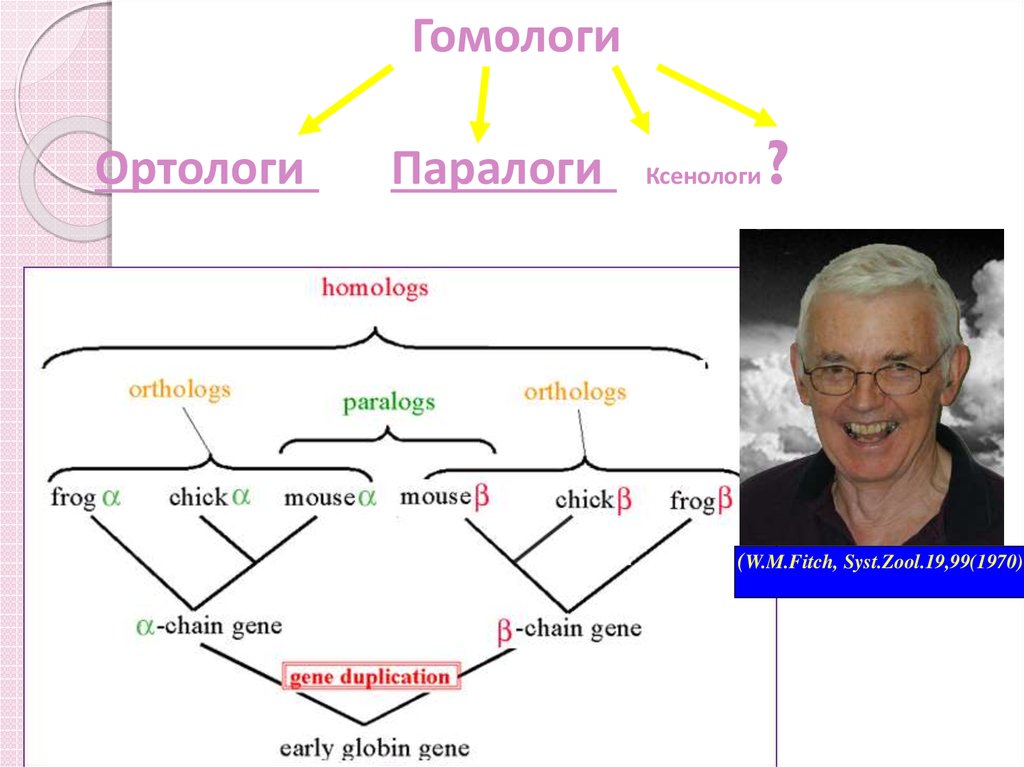
8. Ортологи и паралоги
Ортологи – гени з різних організмів, щорозійшлися при видоутворенні.
◦ Мається на увазі, що ортологи мають
спільного «предка» і однакову функцію (якщо
тиск відбора слабкий, то функція может
«плисти»).
Паралоги – гени, що розійшлися при дуплікації
(«копіюванні»).
◦ Копії гена не зазнавали тиска відбора, а
значить, могли змінити функцію.
9. BLAST
Что такое выравниваниеВыравнивание 2х последовательностей
BLAST на NCBI:
◦
◦
◦
◦
◦
Что это такое
Как выбрать правильную программу
Как выбрать правильную базу данных
Как запустить
Как интерпретировать результаты
10. Почему нас интересует локальное сходство последовательностей?
Мы верим, что:1. функцию, структуру и многие другие свойства
белка/ДНК определяет последовательность;
2. родственные белки имеют похожие свойства
молекулы, похожие по последовательности,
похожи и по свойствам
Т.о. свойства можно предсказать, анализируя
изученные последовательности, похожие на
данную
11.
ГомологиОртологи
Паралоги
Ксенологи
?
(W.M.Fitch, Syst.Zool.19,99(1970)
12. Схожие 3D структуры
Вставка в «синей»последовательности
13. Матрицы замен
Матрица 20*20 на пересечении 2х aa их уровеньсходства (?):
◦ Похожесть по свойствам (объем, гидрофильность, заряд
и т.д.)
◦ Эволюционное родство – частота замен 1ой aa на другую
в изученных белках
2 сорта последних:
РАМ (Point Accepted Mutations) – на выравниваниях очень
близких белков (РАМ20 = РАМ^20)
BLOSUM (BLOck Scoring Matrix) – на блоках выравниваний
далеких белков (без делеций) (BLOSUM62 – на белках со
средним уровнем сходства 62% попарно)
14. Делеции/инсерции
Общий штрафЗначительно чаще 1 длинная делеция, чем
много коротких => штраф за внесение
делеции + штраф за удлинение делеции
15. Типы выравнивания
Локальное – поиск фрагментов наиболее похожихдруг на друга
домовой
домовой
скупидом
водомерка
домовой
водомерка
Глобальное – сравнение последовательностей
целиком: каждый нуклеотид (аминокислота) находит
себе пару
лесовоз
ледоход
?
---лесо---воз
лед---оход---
16. Критерии качества выравнивания
Количество идентичных (похожих)аминокислот/нуклеотидов
◦ Для белков – более 25% id при длине > 100 aa
◦ Для ДНК – более 70% id при длине > 100 nt
Длина выравнивания
Вероятность наблюдать такое сходство
случайным образом
◦ Зависит от базы данных
Score
– общая мера сходства:
◦ Зависит от программы
17. Поиск гомологов в базах даных
FASTA (Pearson and Lipman, 1988)BLAST (Altschul et al., 1990)
18. FASTA
1.A lookup table is generated consisting of short stretches of amino acids ornucleotides from a database. The size of these stretches is determined
from the ktup parameter. If ktup ј 3 for a protein search, then the query
sequence is examined in blocks of three amino acids against matches of
three amino acids found in the lookup table. The FASTA program
identifies the 10 highest scoring segments that align for a given ktup.
2. These 10 aligned regions are rescored, allowing for conservative
replacements, using a scoring matrix such as PAM250.
3. High-scoring regions are joined together if they are part of the same
proteins.
4. FASTA then performs a global (Needleman–Wunsch) or local (Smith–
Waterman) alignment on the highest scoring sequences, thus optimizing
the alignments of the query sequence with the best database matches.
Thus, dynamic programming is applied to the database search in a limited
fashion, allowing FASTA to return its results very rapidly because it
evaluates only a portion of the potential alignments.
19. FASTA
http://www.ebi.ac.uk/Tools/sss/fasta/20. BLAST – Basic Local Alignment and Search Tool
Локальное выравниваниеГлавная задача – поиск похожих
последовательностей в базах данных (=> главное
достоинство – скорость)
Неточно восстанавливает сходство
Основная программа поиска по БД
Для специализированных БД часто предлагается
на сайте БД
Для поиска среди известных
последовательностей есть специальные сервера
21. Родной BLAST – NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)
22.
Алгоритм BLASTBasic Local Alignment Search Tool
Также, как FASTA, требует параметр k (длина
слова).
◦ Белки k= 3 letter words
◦ ДНК k= 11 letter words.
23. Алгоритм BLAST (шаг 1)
1. Поиск идентичных\похожих участков2. Попытка «удлинить» эти участки насколько
возможно (т.е. пока score растёт)
В результате: High-scoring Segment Pairs (HSPs)
THEFIRSTLINIHAVEADREAMESIRPATRICKREAD
INVIEIAMDEADMEATTNAMHEWASNINETEEN
24.
Алгоритм BLAST (шаг 2)Попытка соединить соседние HSPs путем
выравнивания последовательностей
между ними:
THEFIRSTLINIHAVEADREA____M_ESIRPATRICKREAD
INVIEIAMDEADMEATTNAMHEW___ASNINETEEN
25. Blast
Blast – это семейство программ: BlastN, BlastP, BlastX,tBlastN
Query:
ДНК
Белок
Database:
ДНК
Белок
BlastN - ДНК vs ДНК
BlastP – белок vs белок
BlastX - translated ДНК vs белок
tBlastN - белок vs translated ДНК
26. Blast
27. Одною з розповсюджених прикладних задач є пошук гомологів відомих білків у повністю розшифрованих геномах.
пряме співставлення амінокислотноїпослідовності проти нуклеотидної є
неможливим
дві можливі стратегії аналізу
28. Стратегія 1
Миперетворюємо
цільову
амінокислотну
послідовність в набір нуклеотидних послідовностей,
згідно стандартного генетичного коду.
На виході отримуємо величезну купу нуклеотидних
послідовностей, кількість яких не може бути
спрогнозовано,
внаслідок
виродженності
генетичного коду. Як наслідок, задачу співставлення
цього масиву послідовностей з вмістом нуклеотидної
бази даних, взагалі не може бути вирішено за
розумний проміжок часу
29. Стратегія 2
Ми перетворюємо (транслюємо) вміст нуклеотидноїбази даних в амінокислотні послідовності.
На виході отримуємо 6 варіантів на кожну
нуклеотидну послідовність, відповідно до кількості
можливих рамок зчитування. Таким чином, кількість
порівняльних процедур системи замість з неосяжної
кількості зменшується до шести, і може бути
проведено за розумний проміжок часу, що і виконує
програма TBLASTN
30. Поиск гомологов
По ДНК или по белку?Какой поиск предпочтительней?
31. ДНК или белок?
Какая последовательность болеепостоянна в эволюционном плане?
UCAUAC
Or
Serine -Tyrosine
32.
Поиск гомологовГенетический код избыточен – почти все
аминокислоты кодируются более, чем 1 кодоном
(тройка нуклеотидов)
Последовательность ДНК может меняться, в то
время, как последовательность белка остается
постоянной.
Ser-Tyr….
UCAUAC
UCUUAC
UCGUAC
U……
33.
Поиск гомологовНуклеотиды – 4-х буквенный алфавит.
Аминокислоты – 20-и буквенный алфавит
Две случайные последовательности ДНК будут
идентичны ~ 25%.
Две случайные белковые последовательности будут
идентичны ~ 5%.
34.
Поиск гомологовМатрицы для сравнения белков более
чувствительны, чем матрицы для ДНК.
Базы данных ДНК намного больше
белковых → будут случайные совпадения.
35.
Поиск гомологовИспользование белковых
последовательностей более
предпочтительно при поиске гомологов
36. Специализированные инструменты
ДНК:◦ megaBLAST – другой алгоритм для сравнения ДНК.
Оптимизирован для длинных похожих
последовательностей. Оптимален для поиска хитов в
родном геноме или очень близких видах
◦ Discontiguous megaBLAST – аналогично, параметры
подобраны для более далеких видов
Белок:
◦ PSI-BLAST (Position-Specific Iterated -BLAST) поиск
удаленных белковых гомологов с использованием PSSM
(position-specific scoring matrix)
◦ PHI-BLAST (Pattern-Hit Initiated -BLAST) ищет
гомологичные белки, удовлетворяющие заданному
паттерну
37. Специализированные инструменты
ДНК:◦ megaBLAST – другой алгоритм для сравнения ДНК.
Оптимизирован для длинных похожих
последовательностей. Оптимален для поиска хитов в
родном геноме или очень близких видах
◦ Discontiguous megaBLAST – аналогично, параметры
подобраны для более далеких видов
Белок:
◦ PSI-BLAST (Position-Specific Iterated -BLAST) поиск
удаленных белковых гомологов с использованием PSSM
(position-specific scoring matrix)
◦ PHI-BLAST (Pattern-Hit Initiated -BLAST) ищет
гомологичные белки, удовлетворяющие заданному
паттерну
38. Какую программу выбрать?
БелокДНК
Локализацию в
родном геноме
Изученного
гомолога
Свой ген или гомолога в
определенном геноме
Гомолога
с заданной
Возможно,
весьма
функцией
удаленного
гомолога
Ортологичный локус
в близком виде
BLAST
Похожую, возможно,
некодирующую, ДНК
Белок-кодирующий ген в
последовательности
39. Стандартный input
40. Промежуточная страница - СD
41. Output - I
42. Output - II
43. Output - III
44. Output IV
45. E-value, bit score
E-value(математическое ожидание, the
expectation value) – оценка числа раз
наблюдать хит такого же качества при таком
размере базы данных (0 - e-6 – хорошо, > 0.001
- 0.01 – плохо)
Как правило, BLAST недооценивает e-value!
46.
47. E-value, bit score
BitScore – мера статистической значимости
(вес – сумма стоимостей всех точечных замен)
выравнивания - меньше 50 – плохо)
48. Параметры выравнивания
Матрица:BLOSUM для локального выравнивания обычно лучше,чем PAM
◦ Чем выше номер BLOSUM – тем строже выравнивание (BLOSUM80 вместо
BLOSUM45 – более короткие выравнивания)
◦ РАМ – чем ниже, тем строже
Штрафы за делеции:
◦ Чем больше штраф за внесение, тем короче выравнивания
◦ Меняете матрицу – надо менять и штраф
◦ Чем ниже номер BLOSUM (выше РАМ), тем меньше штраф за внесение
делеции
◦ Штраф за удлинение ~10 раз ниже, чем за внесение
Если сравниваете удаленных гомологов, то лучше всего
довольно высокий штраф за внесение делеции и низкий за
удлинение
Близкие гомологи – штрафы ближе друг к другу
49. Сообщение о параметрах
В конце файла текстовая информация обиспользованный параметрах:
◦
◦
◦
◦
◦
◦
◦
Использованная матрица замен
Штрафы за внесение и продление делеции
Дата
Использованная БД
Размер БД
Количество полученных хитов
…
50. Выбор параметров
Меняйте параметры только, если поумолчанию не работает (параметры по
умолчанию подобраны хорошо для
большинства ситуаций)
Для того, чтобы выбрать более
подходящие параметры надо очень
ТОЧНО сформулировать задачу
51. Какие параметры менять? Фильтрация
Low-complexity region – другой aa-составФильтрация: если Ваш белок содержит
большой регион низкой сложности –
попробуйте использовать BLAST без
соответствующей фильтрации
Если Ваш белок содержит очень часто
встречающиеся домены, их тоже можно
отфильтровать – в ручную
ДНК – геном-специфичные повторы!
52. Параметры output-формата
Количество хитовВыбор базы данных (организм)
Выбор порога - Expect (если хитов
мало, то можно смотреть на более
подозрительные)
Entrez query – ключевые слова
(например, “protease AND human”)
53. PSI - BLAST
Алгоритм:◦ Несколько раундов поиска
◦ Первый раунд – просто blastp (BLOSUM62)
◦ Построение PSSM на основе полученных хитов
(можете выбрать те, что надо)
◦ Следующий раунд на основе этой PSSM
◦ Методов итераций, пока множество хитов не
перестанет меняться
54. PSSM
Portion of a PSSM from a PSI-BLAST search using RBP4 (NP_006735) as a query. The 199 aminoacid residues of the query are represented in rows; the 20 amino acids are in columns. Note that for
a given residue such as alanine the score can vary (compare A14, A15, and A16, which receive scores
of 3, 2, and 4). The tryptophan in position 40 is invariant in several hundred lipocalins. Compare the
score of W40, W3, or W5 (each receives ю12) with W13 (ю7); in the W3, W5, and W40 positions a
match is rewarded more highly, and the penalties for mismatches are substantially greater. A PSSM
such as this one allows PSI-BLAST to perform with far greater sensitivity than standard blastp
searches
55. PHI - BLAST
Query – белок + паттерн, которому этот белокудовлетворяет
Пример:
>P28332|ADH6_HUMAN Alcohol dehydrogenase 6 - Homo sapiens
(Human)
MSTTGQVIRCKAAILWKPGAPFSIEEVEVAPPKAKEVRIKVVATGLCGTEMKVLGSKHLD
LLYPTILGHEGAGIVESIGEGVSTVKPGDKVITLFLPQCGECTSCLNSEGNFCIQFKQSK
TQLMSDGTSRFTCKGKSIYHFGNTSTFCEYTVIKEISVAKIDAVAPLEKVCLISCGFSTG
FGAAINTAKVTPGSTCAVFGLGGVGLSVVMGCKAAGAARIIGVDVNKEKFKKAQELGATE
CLNPQDLKKPIQEVLFDMTDAGIDFCFEAIGNLDVLAAALASCNESYGVCVVVGVLPASV
QLKISGQLFFSGRSLKGSVFGGWKSRQHIPKLVADYMAEKLNLDPLITHTLNLDKINEAV
ELMKTGKW
G - H - E - x - {EL} - G - {AP} - x(4) - [GA] - x(2) - [IVSAC]
56. НММ-профиль
Hidden Markov models describealignments based on the probability of
amino acids occurring in an aligned column.
This is conceptually related to the positionspecific scoring matrix used by PSI-BLAST.
(a) An alignment of five globins is shown.
The five proteins are a nonsymbiotic plant
hemoglobin from rice (Oryza sativa)
(1D8U), human neuroglobin (1OJ6A),
human beta globin (2hhbB), leghemoglobin
from the soybean Glycine max(1FSL), and
human myoglobin (2MM1). (b) The
probability of each residue occurring in
each aligned column of residues is
calculated. (c) From these probabilities, a
score is derived for any query such as
HARTV. Note that the actual score will also
account for gaps and other parameters.
Also note that this is a position-specific
scoring scheme; for example, there is a
different probability of the amino acid
residue lysine occurring in position 3
versus 4. (d) The probabilities associated
with each position of the alignment can be
displayed in boxes representing states.
57. Пример простого мотива
Алкогольдегидрогеназа 6(человек)
68 - 82:
GHEgAGIvesiGegV
Алкогольдегидрогеназа
класса 3 (рис)
70 - 84:
GHEaAGIvesvGegV
Алкогольдегидрогеназа,
специфичная к пропанолу
(кишечная палочка)
57 - 71:
GHEgIGVvaevGpgV
Распознающее правило типа «паттерн»:
G - H - E - x - {EL} - G - {AP} - x(4) - [GA] - x(2) - [IVSAC]
Паттерн – регулярное выражение UNIX’a:
Например, выражение [AC]-x-V-x(4)-{ED} читается как
Ala или Cys- х-Val- х- х- х - х- (любой остаток, но не Glu и не Asp)
58.
Другие программы поиска по БД:◦ FASTA (www.ebi.ac.uk/fasta33/)
◦ Ssearch (алгоритм Smith-Waterman)
(www.ch.embnet.org)
◦ BLAT (genome.ucsc.edu)