
 представляет собой угол вокруг связи N-Cα, а пси (ψ) представляет собой угол вокруг Сα-C’ связи")

")
")









")




 в PDB")
 в PDB")
















")







")


")
")


 biology
biologySimilar presentations:




")





Структура белка. Лекция 6
1. Структура белка
Лекция 4Многие слайды и материалы используемые в презентации взяты из книги
Bioinformatics and Functional Genomics by Jonathan Pevsner Copyright ©
2015 by John Wiley & Sons, Inc. и соответствующего курса по
биоинформатики Johns Hopkins School of Medicine
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
1
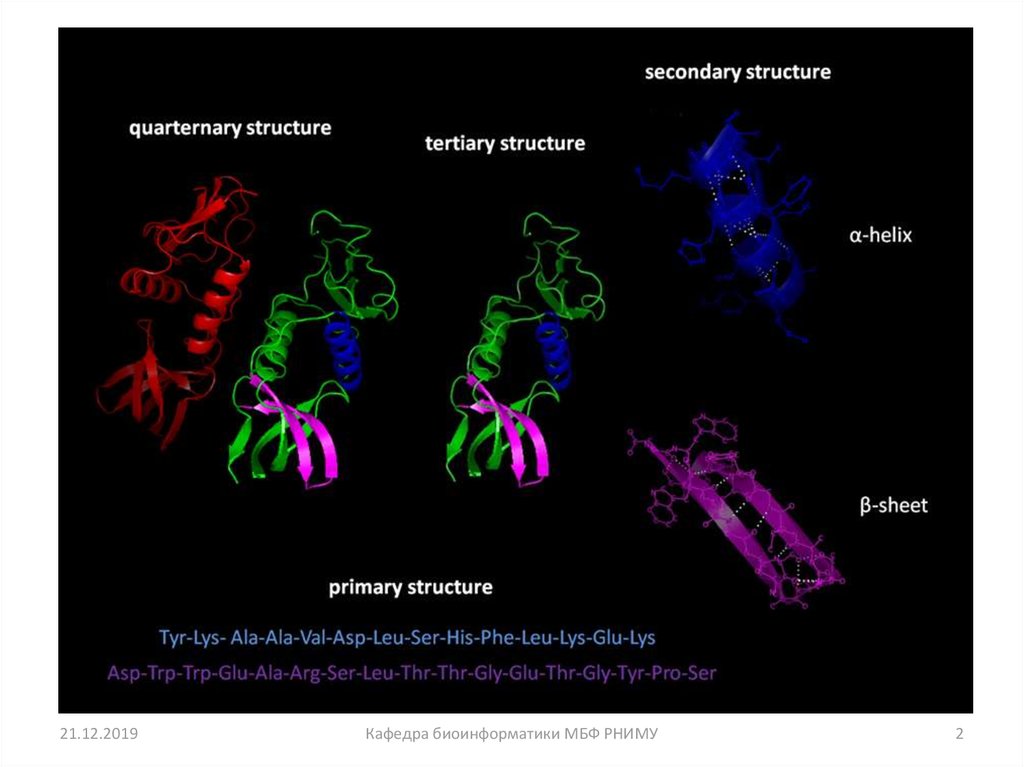
2.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
2
3. Фи (φ) представляет собой угол вокруг связи N-Cα, а пси (ψ) представляет собой угол вокруг Сα-C’ связи
21.12.2019Кафедра биоинформатики МБФ РНИМУ
3
4. Вторичная структура белка
Вторичные структуры белков определяютсяаминокислотными остатками боковых цепей
Миоглобин
Много альфа спиралей
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
Тиоредоксин
Много бета листов
4
5. Альфа спираль (α-helix)
Элемент вторичной структуры белков, который имеетформу правозакрученой винтовой линии и в котором
каждая аминогруппа (-NH2) в каркасе образует
водородную связь с карбонильной группой (-C=O)
аминокислоты, находящейся на 4 аминокислоты
раньше.
Функциональная роль:
1. Связывание с ДНК
21.12.2019
2. Пронизывание мембраны
Кафедра биоинформатики МБФ РНИМУ
3. Форма
5
6. β-лист (β-складчатый слой)
β-лист (β-складчатый слой)• β-листом (β-sheet) называют структуру из как минимум
двух β-цепочек, которые связаны между собой
водородными связями.
• β-цепью (β-chain) или β-тяжем (β-strand) называют
участок полипептидной цепи длиной от 3 до 10
аминокислот, в вытянутой, практически линейной
форме
Антипараллельный b-лист
Параллельный b-лист
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
6
7. Диаграмма Рамачандрана отображает пси и фи углы для каждой аминокислоты белка (за исключением пролина, а в некоторых случаях
глицина)DeepView http://spdbv.vital-it.ch/
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
7
8.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
8
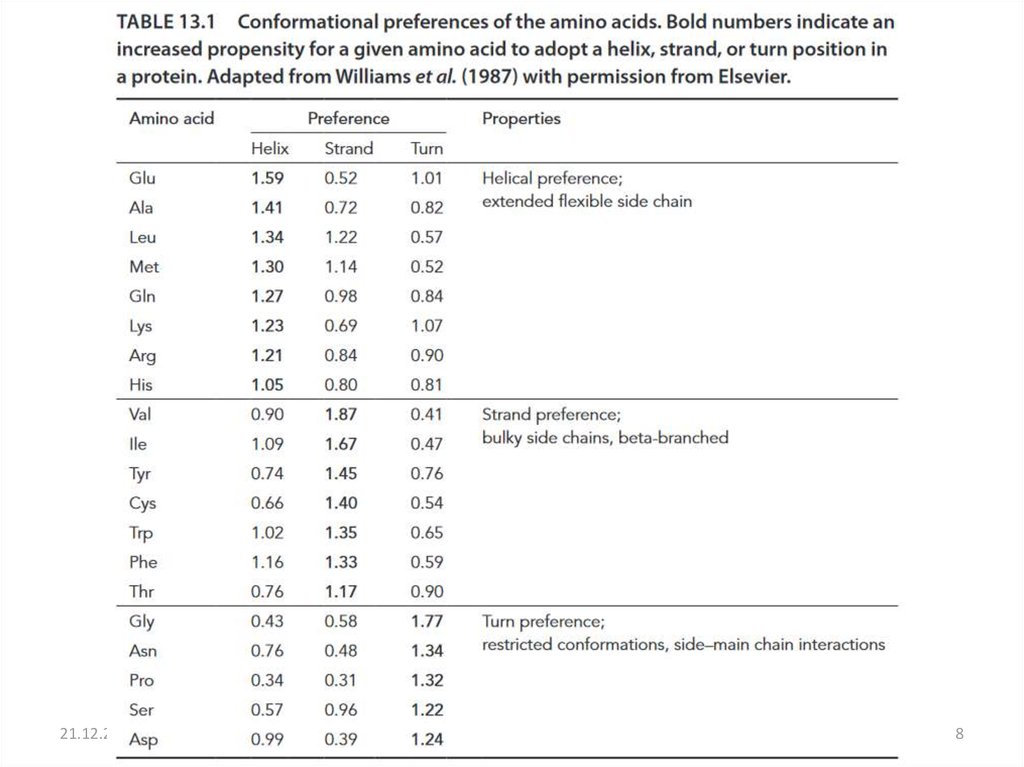
9. Предсказание вторичной структуры белка
Chou и Fasman (1974) разработали алгоритм,основанный на частотах аминокислот, обнаруженных
в α-спирали, β-листов, и повороты.
Proline: есть в поворотах, но не в α-спиралях.
GOR (Garnier, Osguthorpe, Robson): связанный
алгоритм
Современные алгоритмы: используют
множественное выравнивание последовательностей и
дают более высокий показатель успеха (около 70-75%)
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
9
10. On-line сервисы по прогнозу вторичной структуры доступные в интернете
21.12.2019Кафедра биоинформатики МБФ РНИМУ
10
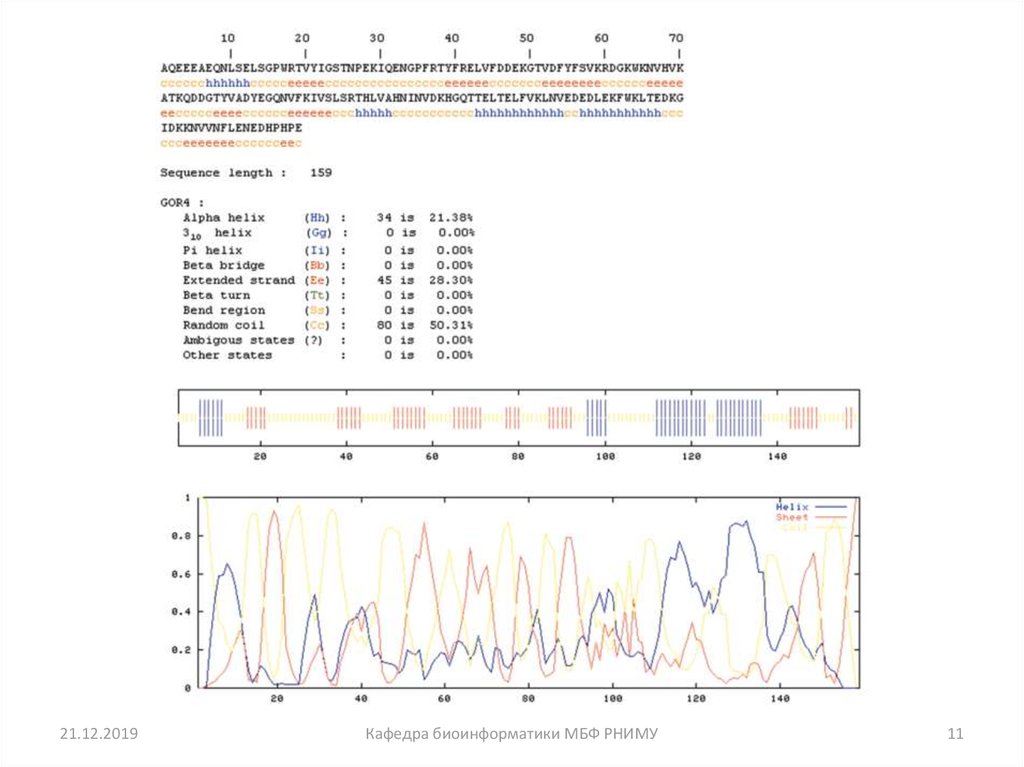
11.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
11
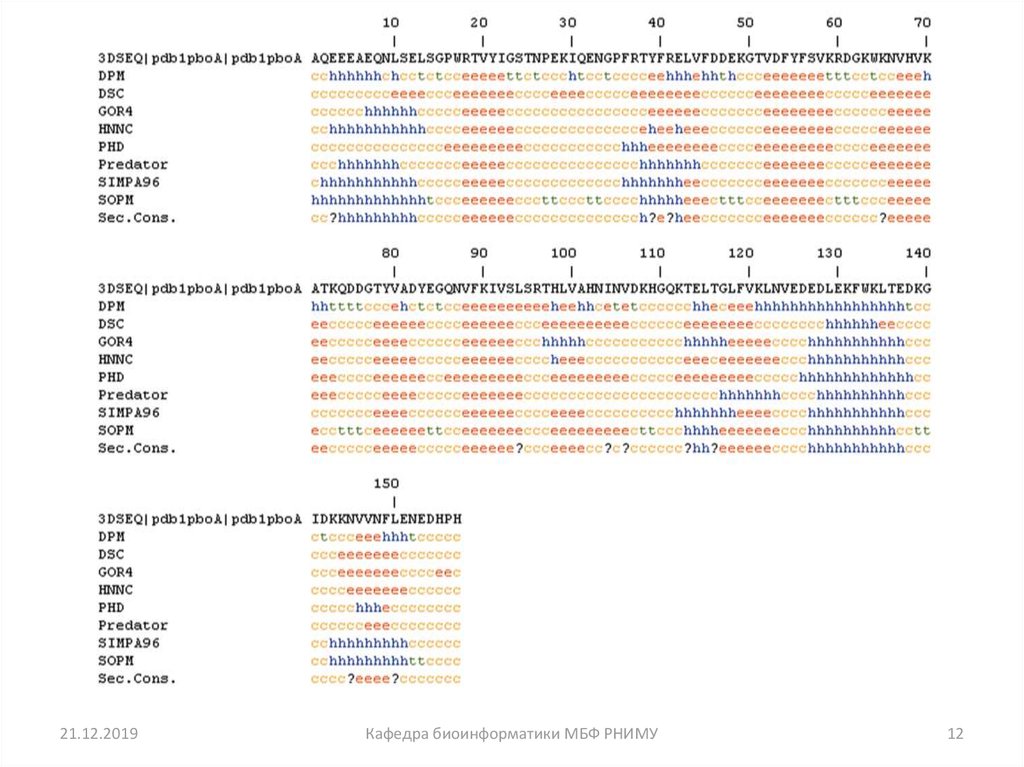
12.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
12
13.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
13
14. Третичная структура белка: сворачивание белка
• Основные подходы:– [1] Экспериментальное определение
(Рентгеновская кристаллография, ЯМР)
– [2] Прогнозирование
• сравнительное моделирование (на основе
гомологии)
• Threading
• Неэмпирический (De Novo) прогнозирование
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
14
15.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
15
16. Шаги получения структуры белка
21.12.2019Кафедра биоинформатики МБФ РНИМУ
16
17. Основные этапы определения структуры из TargetDB, регистрационной базы данных мишеней
21.12.2019Кафедра биоинформатики МБФ РНИМУ
17
18. Приоритеты для выбора мишени для определения ее структуры
• Исторические - небольшие, растворимые,распространенные белки (гемоглобин, цитохромы с,
инсулин).
• Современные критерии:
– Представляют все ветви жизни
– Представляют, ранее неохарактеризованные семейства
– Медицинская польза мишеней
– Некоторые пытаются получить все структуры внутри
отдельного организма (Methanococcus jannaschii,
микобактерии туберкулеза)
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
18
19. The Protein Data Bank (PDB)
• PDB является основным хранилищем длябелковых структур
• Основан в 1971 году
• Доступ в http://www.rcsb.org/pdb или
просто http://www.pdb.org
• В настоящее время содержит более 155000
структурных объектов
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
19
20.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
20
21. Рост количества структур в PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
21
22. Распределение данных PDB по качеству разрешения
21.12.2019Кафедра биоинформатики МБФ РНИМУ
22
23. Статистика PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
23
24. Поиск гемоглобина
21.12.2019Кафедра биоинформатики МБФ РНИМУ
24
25. Запись гемоглобина (accession 2H35) в PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
25
26. Визуализация структуры гемоглобина (accession 2H35) в PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
26
27. Инструменты для интерактивной визуализация белковых структур
21.12.2019Кафедра биоинформатики МБФ РНИМУ
27
28. Загрузка файла гемоглобина
21.12.2019Кафедра биоинформатики МБФ РНИМУ
28
29.
Структура PDB файла21.12.2019
«TITLE» – в этом разделе
указывается название модели и
краткий
перечень
молекул,
входящих в состав pdb-файла
«COMPND» – немного более
подробный
список
молекул,
входящих в состав файла
«SOURCE» – определяет организмисточник содержащейся в pdbфайле модели молекулы белка
«EXPDTA» – в данном разделе
указывается
метод,
который
использовался
для получения
данной модели. Для получения
моделей используются методы
рентгеноструктурной
кристаллографии (X-RAY) и ядерномагнитного резонанса (NMR).
Кафедра биоинформатики МБФ РНИМУ
29
30.
21.12.2019«SEQRES» – аминокислотная
последовательность
содержащегося в pdb-файле
белка.
«HETNAM»
–
названия
веществ, находящихся в файле
помимо белка (активаторов,
ингибиторов и растворителя,
использованного
при
получении модели).
«HELIX» – указание того, какие
участки белка свернуты в
α-спираль.
«SHEET» – указание того,
какие участки белка уложены
в β-складки.
Кафедра биоинформатики МБФ РНИМУ
30
31.
«SITE» - описывает остатки, содержащие
каталитические, взаимодействующие с
кофактором, анти-кодона, регуляторные и
другие необходимые сайты присутствующие
в структуре в окружающем пространстве
лигандов.
«ATOM» - является самым большим
разделом и содержит информацию обо всех
атомах, входящие в молекулу белка. Для
каждого атома указаны: его порядковый
номер в молекуле белка; химический
элемент
атома;
в
состав
какой
аминокислоты белка он входит; к какой
субъединице белка принадлежит эта
аминокислота; порядковый номер этой
аминокислоты; декартовы координаты (x, y,
z) атома.
«TER» – обозначает конец раздела «ATOM».
«HETATM» – та же информация, что и в
разделе «ATOM», но для веществ,
находящихся в файле помимо белка:
активаторов, ингибиторов и растворителя.
http://www.wwpdb.org/documentation/file-format-content/format33/v3.3.html
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
31
32.
Пути доступа к PDB файламSwiss-Prot, NCBI, EMBL
Protein Data Bank
CATH, Dali, SCOP, FSSP
базы данных интерпретирующие PDB файлы
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
32
33. CATH: http://www.cathdb.info/
21.12.2019Кафедра биоинформатики МБФ РНИМУ
33
34. CATH иерархия
CATH организуетбелковые структуры в
кластеры на четырех
основных уровнях: Класс
(C), архитектура (А),
топологии (T), и
гомологичных
сверхсемейств (H)
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
34
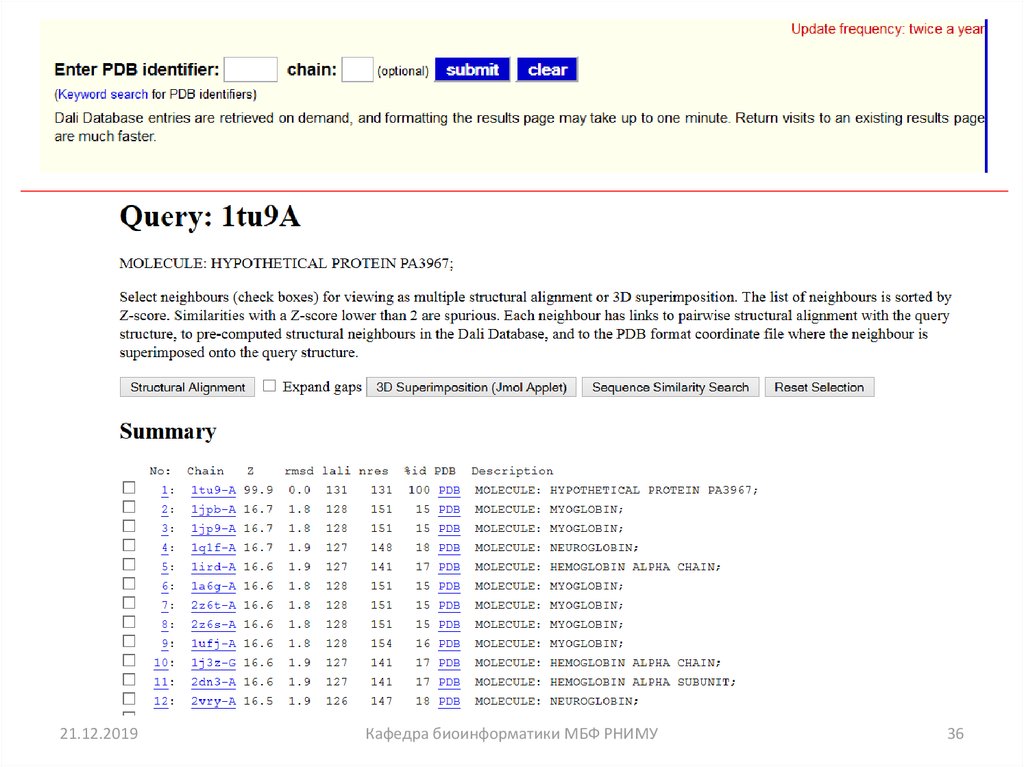
35. Dali
• Обеспечивает классификацию всех структур из PDB иописание семейств белковых последовательностей,
связанных с представительными белков с известной
структурой.
• Для попарного выравнивания, Дали использует матрицу
расстояний, которая содержит все попарные оценки
расстояния между Cα атомами в двух структурах. Эти
оценки
структурных
группировок
являются
производными в виде взвешенной суммы сходства
внутримолекулярных расстояниях.
• Dali основана на сравнения 3D белковых структур в PDB
(все против всех).
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
35
36.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
36
37. Доступ к PDB через NCBI
• Перейти на сайт БД Structure, на домашнейстранице NCBI
• Через Entrez
• Выполнить поиск BLAST, ограничивая
результаты данными из PDB
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
37
38. БД Structure, на домашней странице NCBI
21.12.2019Кафедра биоинформатики МБФ РНИМУ
38
39.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
39
40.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
40
41.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
41
42. Поиск BLAST по данными из PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
42
43. Поиск BLAST по данными из PDB
21.12.2019Кафедра биоинформатики МБФ РНИМУ
43
44.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
44
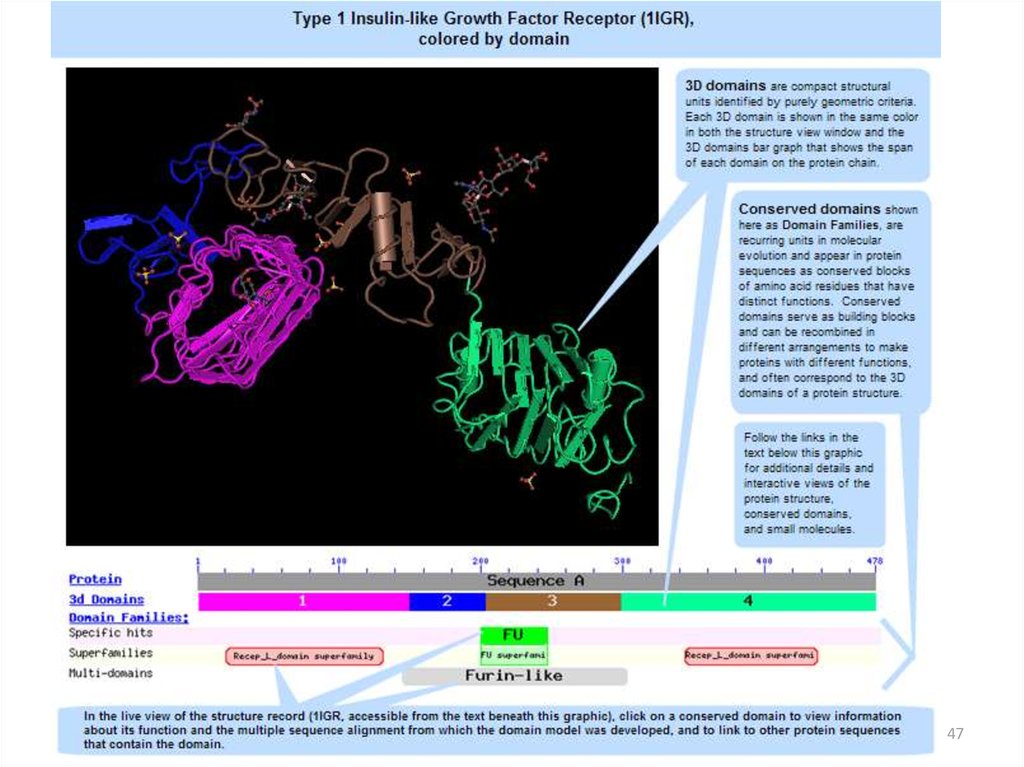
45. Conserved Domain Database (CDD)
21.12.2019Кафедра биоинформатики МБФ РНИМУ
45
46.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
46
47.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
47
48.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
48
49.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
49
50.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
50
51.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
51
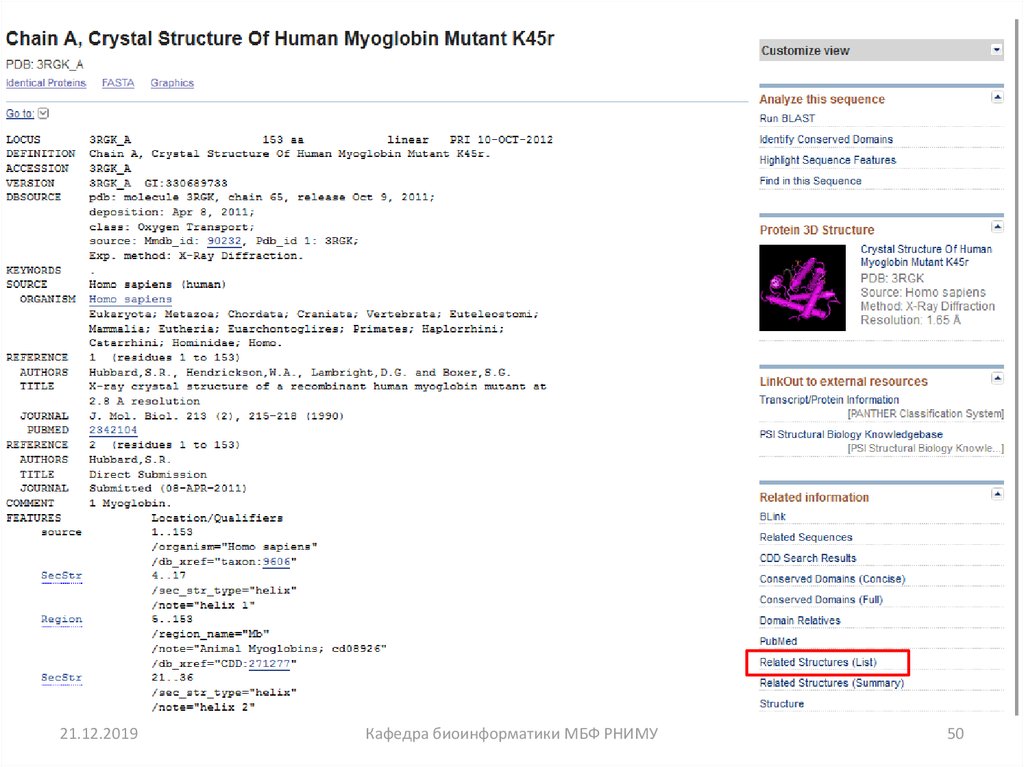
52. БД Structure, на домашней странице NCBI
21.12.2019Кафедра биоинформатики МБФ РНИМУ
52
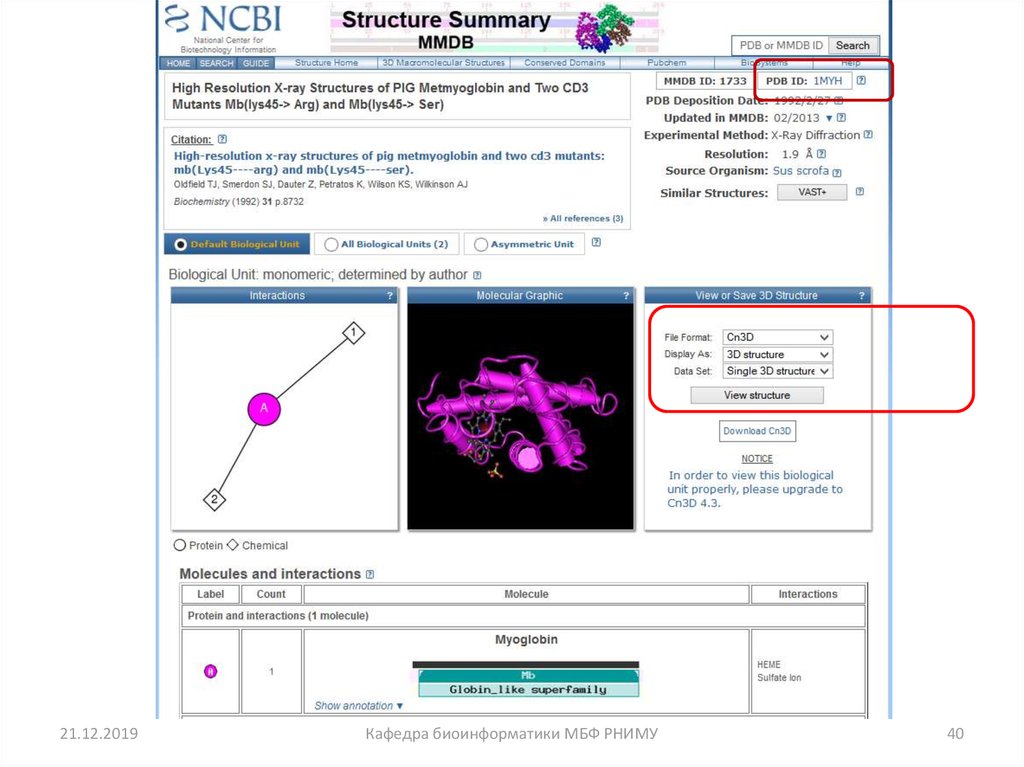
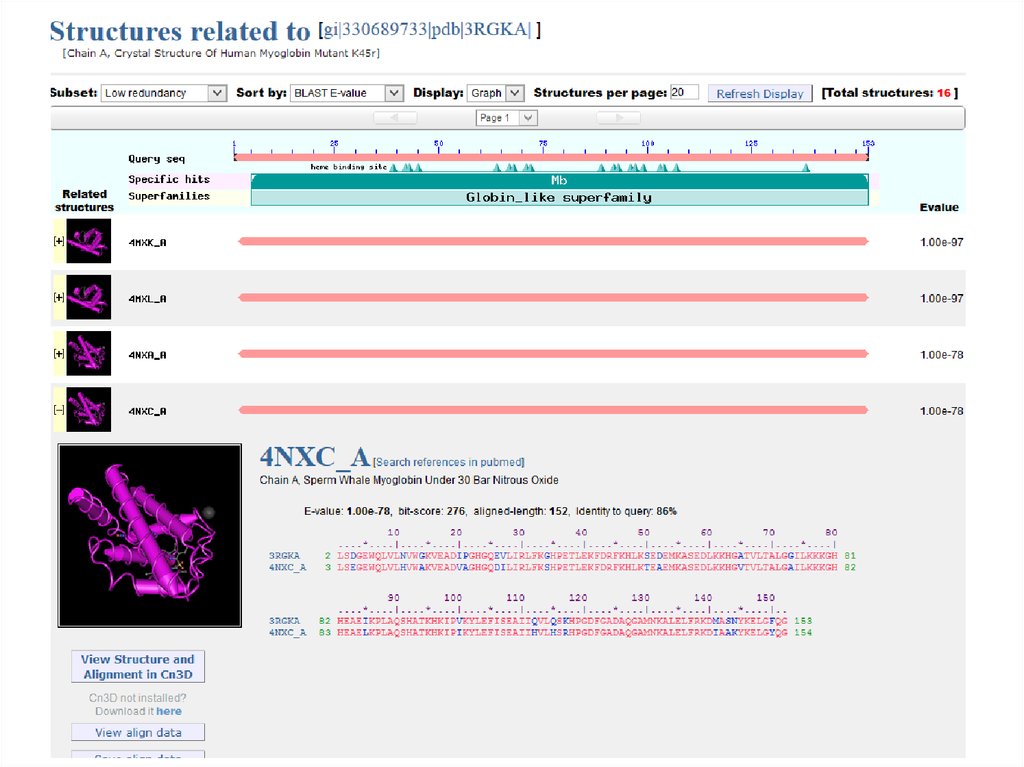
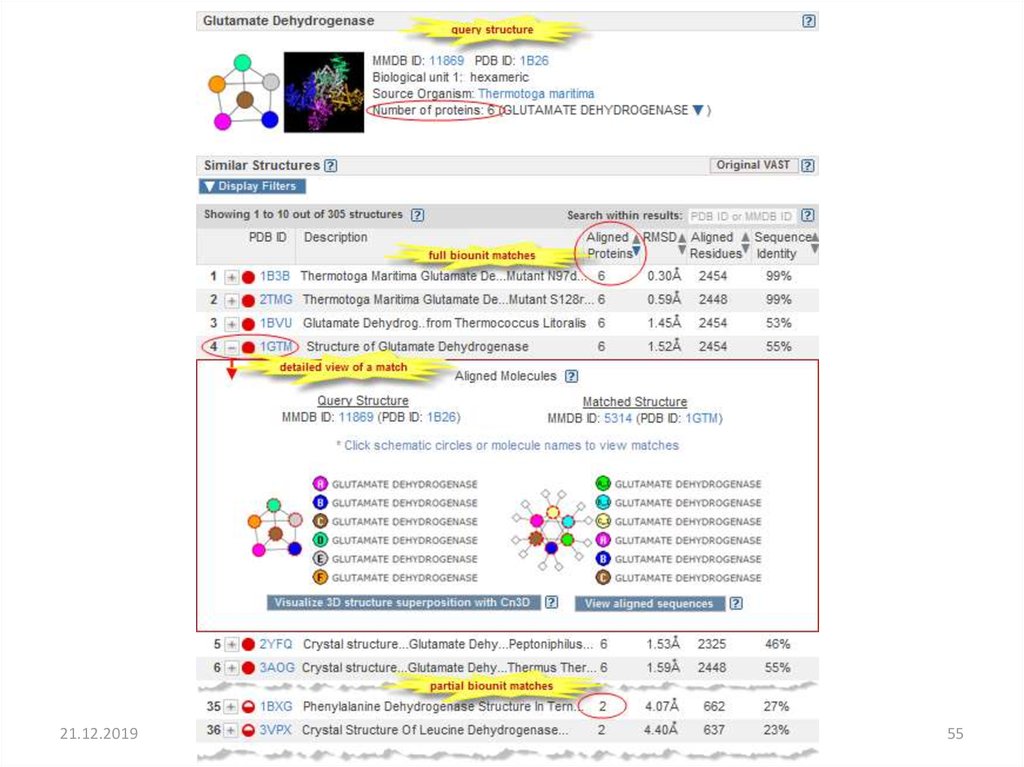
53. VAST
Vector Alignment Search Tool являетсякомпьютерным
алгоритмом, разработанным в
NCBI
и
используется
для
выявления сходных 3-мерных
структур белков по чисто
геометрическим критериям, а
также
для
определения
отдаленных гомологов, которые
не могут быть распознаны при
сравнении последовательностей.
Суперпозиции сходных структур,
и
их
соответствующие
выравнивания,
можно
посмотреть в интерактивном
режиме в программе просмотра
структура NCBI Cn3D.
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
53
54.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
54
55.
21.12.2019Кафедра биоинформатики МБФ РНИМУ
55
56. Предсказание структуры белка
21.12.2019Кафедра биоинформатики МБФ РНИМУ
56
57. Моделирование по гомологии (Comparative Modeling)
21.12.2019Кафедра биоинформатики МБФ РНИМУ
57
58. Есть несколько основных типов ошибок, которые происходят при моделировании по гомологии
• Ошибки в упаковке боковой цепи• Искажения в пределах выровненных участков
• Ошибки в участках мишени, которые не имеют совпадают
с шаблоном
• Ошибки в выравнивании последовательностей
• Использование неправильных шаблонов
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
58
59. Веб-сайты для предсказания структур гомологичным моделированием, а также для оценки качества полученных моделей
21.12.2019Кафедра биоинформатики МБФ РНИМУ
59
60. Распознавание фолдинга (Threading)
Хотя в настоящее время в PDB более 100 000 записей, может быть лишьнесколько тысяч различных белковых типов сворачивания белков в природе.
Распознавания фолдинга, также называемый Threading, полезно, когда для
интересующей нас последовательности мишени не хватает возможности
идентифицировать совпадающую последовательность с известной
структурой. Входная последовательность разбивается на субфрагменты и
сопоставляется (продевается) со структурами известных белков. Скоринг
функции позволяют оценить, на сколько наша последовательность
совместима с последовательностью с известными структурами.
3D-PSSM (http://www.sbg.bio.ic.ac.uk/~3dpssm/index2.html)
PHYRE (http://www.sbg.bio.ic.ac.uk/~phyre/)
FUGUE (http://tardis.nibio.go.jp/fugue/prfsearch.html )
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
60
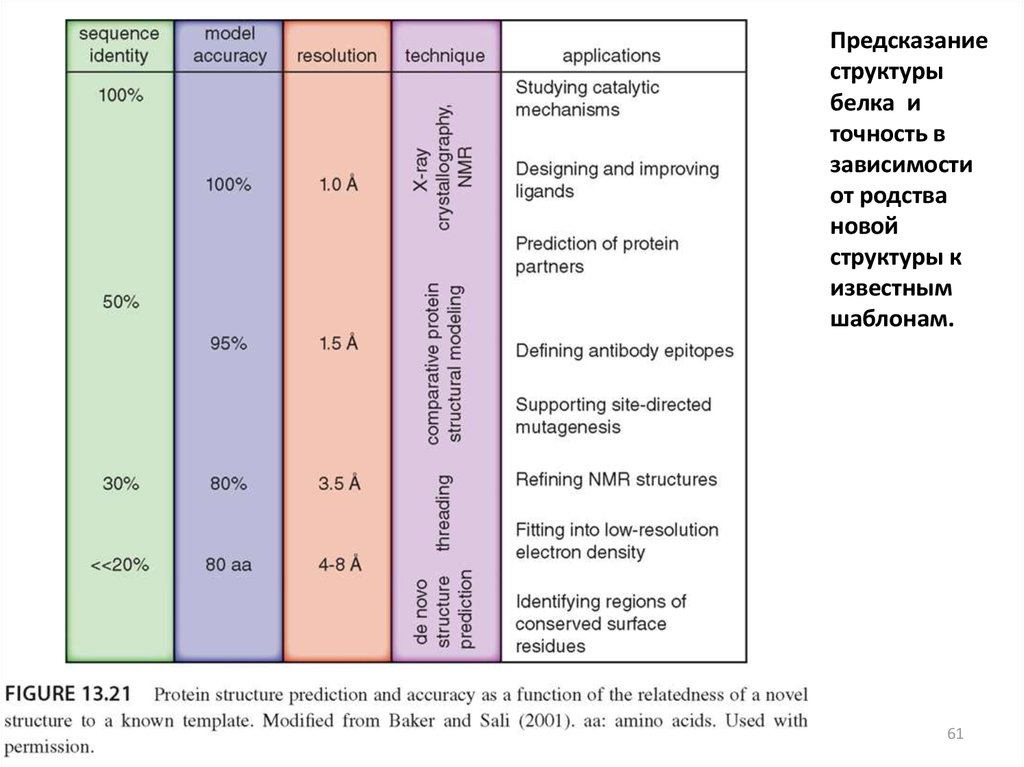
61.
Предсказаниеструктуры
белка и
точность в
зависимости
от родства
новой
структуры к
известным
шаблонам.
21.12.2019
Кафедра биоинформатики МБФ РНИМУ
61
62. Соревнование по предсказанию структуры белка (CASP13 - 2018)
http://predictioncenter.org/casp8/index.cgi21.12.2019
Кафедра биоинформатики МБФ РНИМУ
62
63.
Расстояние экспер vs п2
1
3
Большинство
групп сделали
правильное
предсказание (1)
за исключением
(2)
Большинство
групп сделали
неправильное
предсказание,
но (3) лучшее
Половина групп с
4
плохим
предсказанием (4),
половина с
хорошим (5);
5
ключевое
21.12.2019
63
Кафедра биоинформатики МБФ РНИМУ
Процент а.к. остатков
(Ca)различие
64. Структура белка и болезни человека
БолезньМуковисцидоз
Серповидноклеточная анемия
"Коровье бешенство"
Болезнь Альцгеймера
21.12.2019
Белок
CFTR
гемоглобин бета
прионный белок
белок-предшественник амилоида
Кафедра биоинформатики МБФ РНИМУ
64