


















































 chemistry
chemistrySimilar presentations:


")



")



Классификация электрохимических методов анализа
1.
КЛАССИФИКАЦИЯ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ АНАЛИЗАПо классификации Делахея, Лайтинена и Шарло все электрохимические
методы анализа можно разделить на три группы:
методы, основанные на протекании электродной реакции (группа А);
методы, не связанные с протеканием электродной реакции (группа В);
методы, связанные с изучением изменения структуры двойного слоя
(группа С).
Классификация по способу выполнения :
прямые методы, в которых концентрация компонента определяется
непосредственно из величины регистрируемого электрохимического параметра
(ионометрия, прямая амперометрия, потенциостатическая кулонометрия);
косвенные методы или титриметрия с электрохимическими способами
индикации конечной точки титрования (потенциометрическое,
амперометрическое, кулонометрическое титрование)
инверсионные методы, в которых с помощью электрохимической реакции
обычно на поверхности рабочего электрода проводят предварительное
концентрирование анализируемого вещества, а затем его определение путем
проведения той же реакции в обратном направлении (инверсионная
вольтамперометрия)
1
2.
2Классификация по количеству вещества, участвующего в
электрохимическом процессе:
методы, в которых все количество определяемого вещества участвует в
электрохимическом процессе (например, электрогравиметрия, прямая
кулонометрия)
методы, в которых лишь незначительная доля вещества подвергается
электропревращению (например, вольтамперометрия)
Классификационным признаком может также являться измеряемый
электрический параметр.
3.
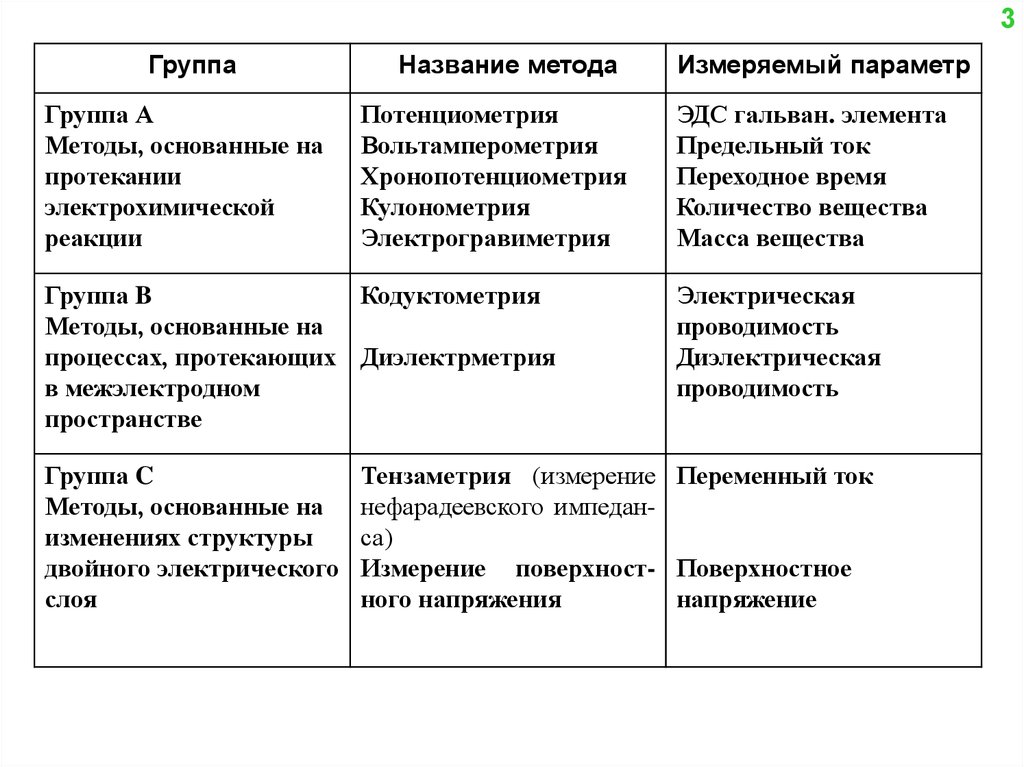
3Группа
Группа А
Методы, основанные на
протекании
электрохимической
реакции
Название метода
Потенциометрия
Вольтамперометрия
Хронопотенциометрия
Кулонометрия
Электрогравиметрия
Группа B
Кодуктометрия
Методы, основанные на
процессах, протекающих Диэлектрметрия
в межэлектродном
пространстве
Группа C
Методы, основанные на
изменениях структуры
двойного электрического
слоя
Измеряемый параметр
ЭДС гальван. элемента
Предельный ток
Переходное время
Количество вещества
Масса вещества
Электрическая
проводимость
Диэлектрическая
проводимость
Тензаметрия (измерение Переменный ток
нефарадеевского импеданса)
Измерение поверхност- Поверхностное
ного напряжения
напряжение
4.
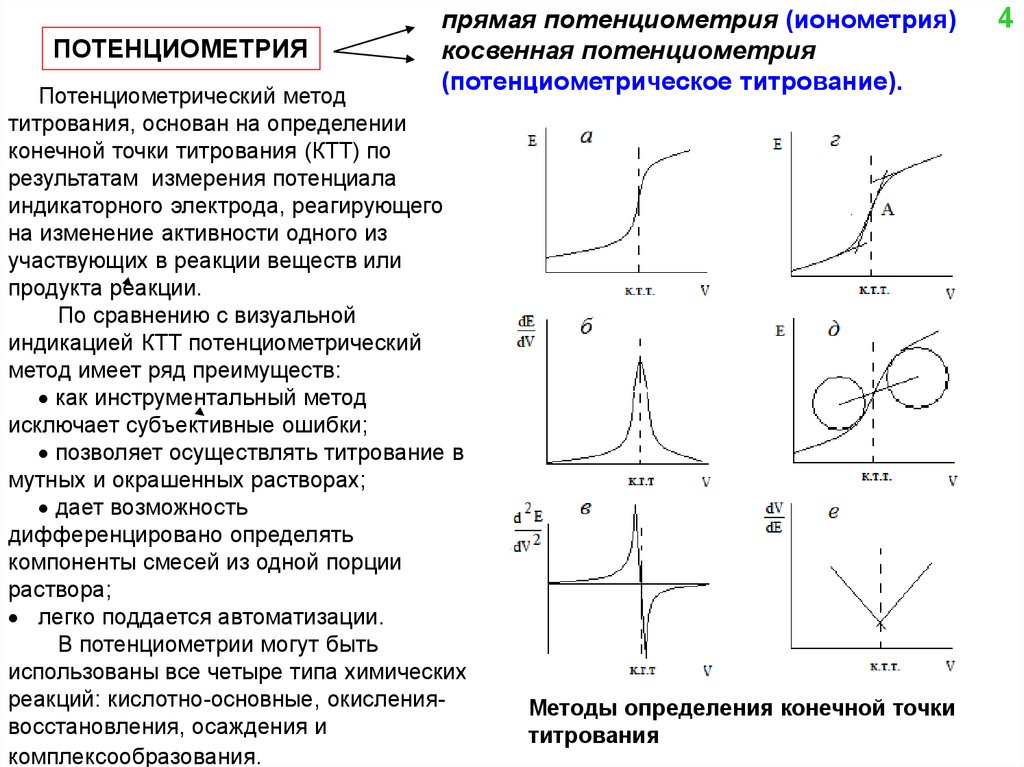
ПОТЕНЦИОМЕТРИЯпрямая потенциометрия (ионометрия)
косвенная потенциометрия
(потенциометрическое титрование).
Потенциометрический метод
титрования, основан на определении
конечной точки титрования (КТТ) по
результатам измерения потенциала
индикаторного электрода, реагирующего
на изменение активности одного из
участвующих в реакции веществ или
продукта реакции.
По сравнению с визуальной
индикацией КТТ потенциометрический
метод имеет ряд преимуществ:
как инструментальный метод
исключает субъективные ошибки;
позволяет осуществлять титрование в
мутных и окрашенных растворах;
дает возможность
дифференцировано определять
компоненты смесей из одной порции
раствора;
легко поддается автоматизации.
В потенциометрии могут быть
использованы все четыре типа химических
реакций: кислотно-основные, окислениявосстановления, осаждения и
комплексообразования.
Методы определения конечной точки
титрования
4
5.
5КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ. ПРОТОЛИТОМЕТРИЯ
Титрование растворами оснований называют алкалиметрия, а
растворами кислот - ацидиметрия
В кислотно-основном титровании независимо от природы определяемых
кислот или оснований необходимо зарегистрировать величину или изменение
концентрации ионов водорода, поэтому достаточно иметь любой индикаторный
электрод функционирующий как водородный (обратимый относительно ионов
H+).
К ним относятся газовый водородный, хингидронный, металл-оксидные
(например, сурьмяный) и стеклянный элетроды.
RT
E E
ln[H ] E 0 0,059 lg[H ] E 0 0,059 pH
F
0
Для каждого из выбранных водородных электродов эти уравнения будут
отличаться только величиной Eo, поэтому полученные с помощью разных
электродов кривые потенциометрического титрования будут идти симбатно.
(1)
6.
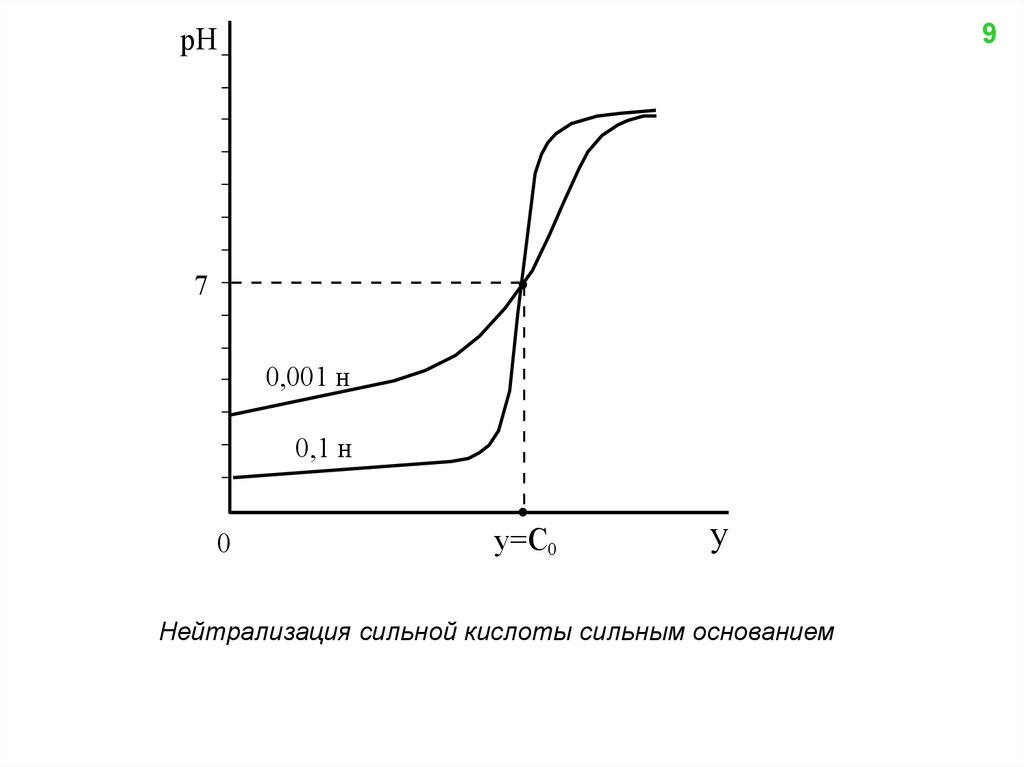
HA + BOH BA + H2OТитрование сильной кислоты в воде
Принцип электронейтральности раствора
[B+] + [H+] = [A-] + [OH- ] (2)
y – количество добавленной щелочи
y = сo − [H+] + Kw / [H+]
Ионное произведение
[OH-]= Kw / [H+]
с0 -
y = [B+] = [A-] − [H+] + Kw / [H+]
6
(3)
исходная концентрация кислоты
Для сильной кислоты
(4)
[A-]=с0
(5)
(6)
с y
с y
H
K
2
2
2
Решая данное квадратное уравнение,
получим:
Если y=с0 , то
H
y с0
K 10
W
0
7
(7)
0
(8)
или рН=7
w
7.
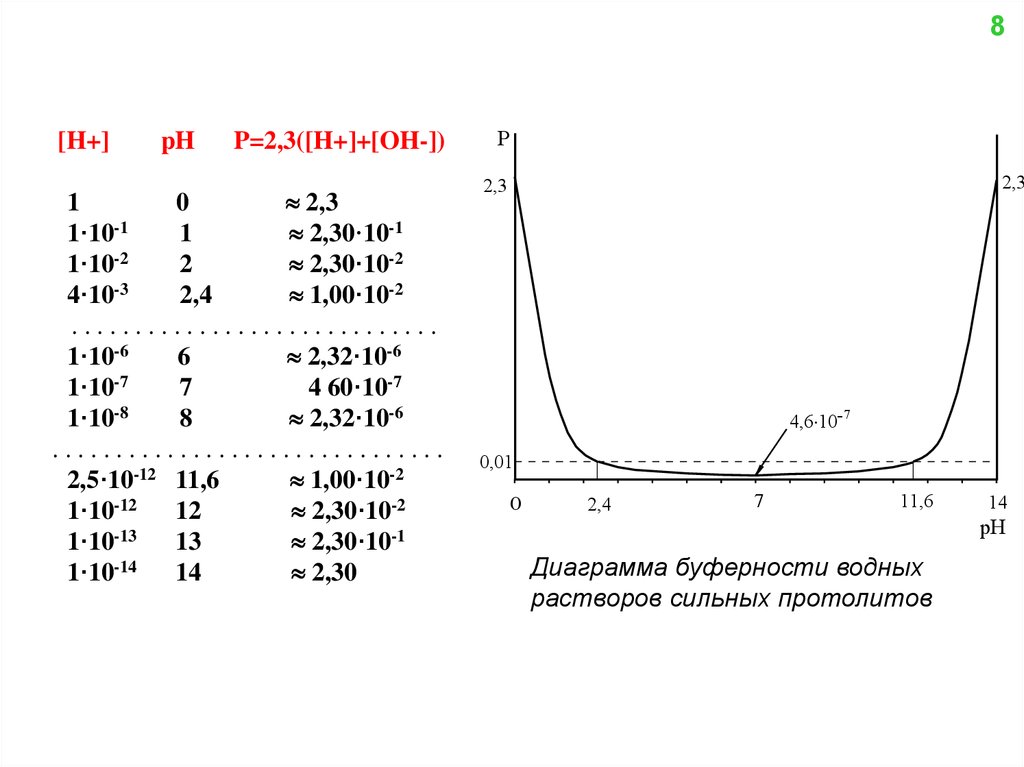
7Для оценки формы кривой титрования можно использовать “буферную емкость”
(или для краткости “буферность”) системы
Р = dy/dpH = d[B+] /dpH
(9)
Дифференциировать уравнение (4) по pH для нахождения буферности нельзя.
Учитывая, что
dpH = d(-lg[H+]) = -0,43(dln[H+]) = - d[H+] / 2,3[H+]
и умножая уравнение (4) на 2,3[H+] и дифференциируя по [H+] получим:
KW
dy
d [ B ] 2,3[ H ] dy
P
2
,
3
[
H
]
1
2
,
3
[
H
]
[
OH
]
2
dpH
dpH
d[H ]
[H ]
P = 2,3([H+] + [OH-]) = 2,3([H+] + Kw/[H+])
(10)
Используя это уравнение можно рассчитать зависимость Р водных растворов
сильных протолитов от рН
8.
8P=2,3([H+]+[OH-])
P
1
0
2,3
1·10-1
1
2,30·10-1
1·10-2
2
2,30·10-2
4·10-3
2,4
1,00·10-2
.............................
1·10-6
6
2,32·10-6
1·10-7
7
4 60·10-7
1·10-8
8
2,32·10-6
...............................
2,5·10-12 11,6
1,00·10-2
1·10-12
12
2,30·10-2
1·10-13
13
2,30·10-1
1·10-14
14
2,30
2,3
[H+]
pH
2,3
4,6 10-7
0,01
0
2,4
7
11,6
14
pH
Диаграмма буферности водных
растворов сильных протолитов
9.
pH9
7
0,001 н
0,1 н
0
y=C0
y
Нейтрализация сильной кислоты сильным основанием
10.
Нейтрализация слабой кислоты сильным основаниемy = [B+] = [A-] − [H+] + Kw / [H+]
[H ][ A ]
KA
[HA]
y [B ]
[HA]=с0 -
(5)
Kс
[A ]
K [H ]
A
0
A
Kс
K
[H ]
K [H ]
[H ]
A
[A-] ≠ const
(4)
[A-]
10
0
W
(6)
Уравнение третьей степени, поэтому
задачу всегда упрощают
A
Kс
K
y=0: [ H ]
K [H ] [H ]
A
0
W
2
(7)
A
KA
KA
[H ]
KA C0
2
2
[ H ] K AC 0
(8)
C0 y
C0 y ; (9)
y C0: [ H ] K [ HA] (5)
;
A
[
H
]
K
pH
pK
lg
A
A
[A ]
y
y
Если y=1/2c0, то [H+]=KA
или
pH=pKA
(10)
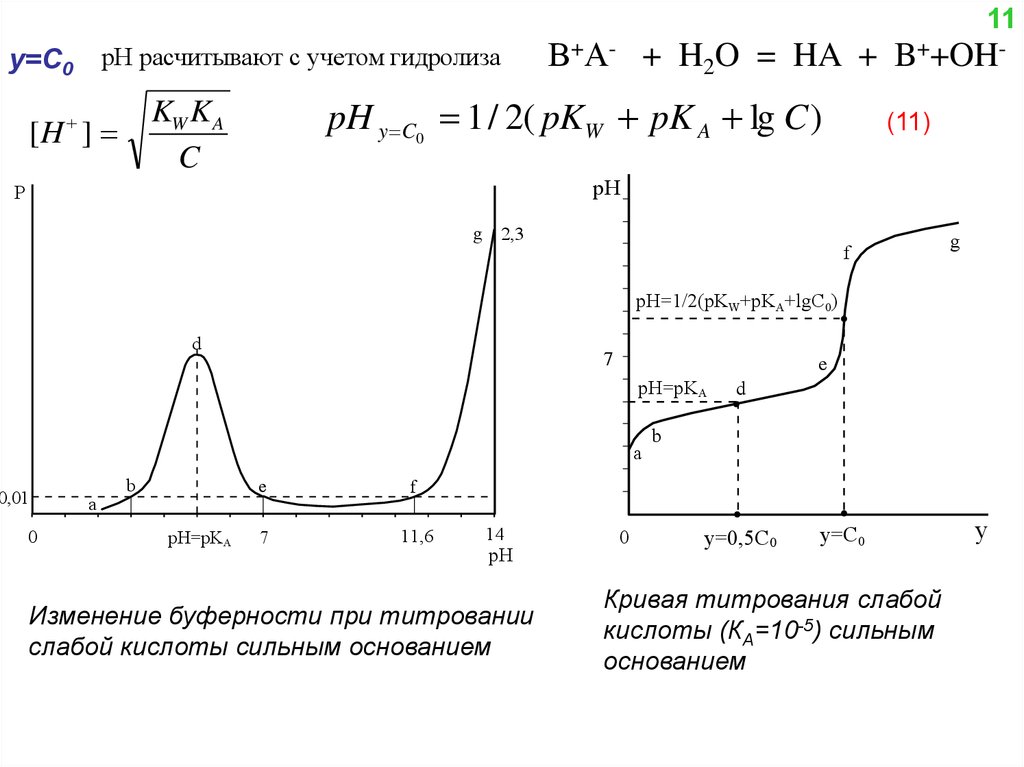
11.
11рН расчитывают с учетом гидролиза
y=C0
pH y C0 1 / 2( pKW pK A lg C )
KW KA
C
[H ]
В+А- + H2O = HA + B++OH(11)
pH
P
g 2,3
f
g
pH=1/2(pKW+pKA+lgC0)
d
7
e
pH=pKA
a
0,01
0
a
b
pH=pKA
e
f
7
11,6
14
pH
Изменение буферности при титровании
слабой кислоты сильным основанием
0
d
b
y=0,5C0
y=C0
Кривая титрования слабой
кислоты (КА=10-5) сильным
основанием
y
12.
12pH
pH pK A lg
0,1
pH pK A lg
pK A 3
99,9
10,42
7,85
7
pH pK A 3
8,88
7,90
3
ошибка 0,1 %
4,76
2
2,81
ошибка 0,01 %
y=0,5C0
pK A 8,6
C0 K A 10 8,6
1
0
99,9
pK A 3
0,1
y=C0
y
pK A 9,6
C0 K A 10 9,6
13.
13Титрование смесей протолитов
Допустим титруется смесь двух одноосновных кислот HA(1) и HA(2) различной силы,
причем рКА,1< рКА,2, а начальная концентрация этих кислот составляет соответственно
С0,1 и С0,2. Исходя из принципа электронейтральности раствора, будет справедливо
равенство
y = [B+] = [A(1)-]+[A(2)-] − [H+] + Kw / [H+]
y [B ]
K A,1C0,1
K A,1 [ H ]
K A, 2C0,2
K A, 2
KW
[H ]
[H ]
[H ]
K A,1C0,1 [ H ]
K A, 2 C 0 , 2 [ H ]
KW
dy
2,3[ H ] dy
P
2
,
3
[
H
]
2
2
dpH
d [H ]
( K A, 2 [ H ])
[H ]
( K A,1 [ H ])
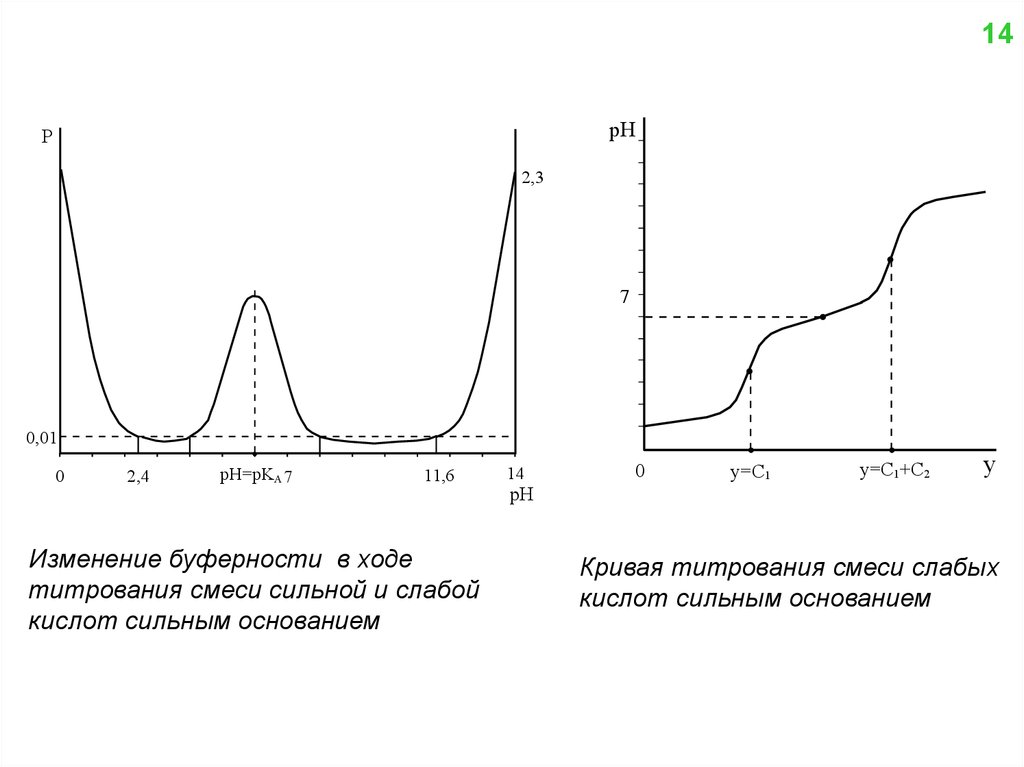
14.
14pH
P
2,3
7
0,01
0
2,4
pH=pKA 7
11,6
Изменение буферности в ходе
титрования смеси сильной и слабой
кислот сильным основанием
14
0
y=C1
y=C1+C2
y
pH
Кривая титрования смеси слабых
кислот сильным основанием
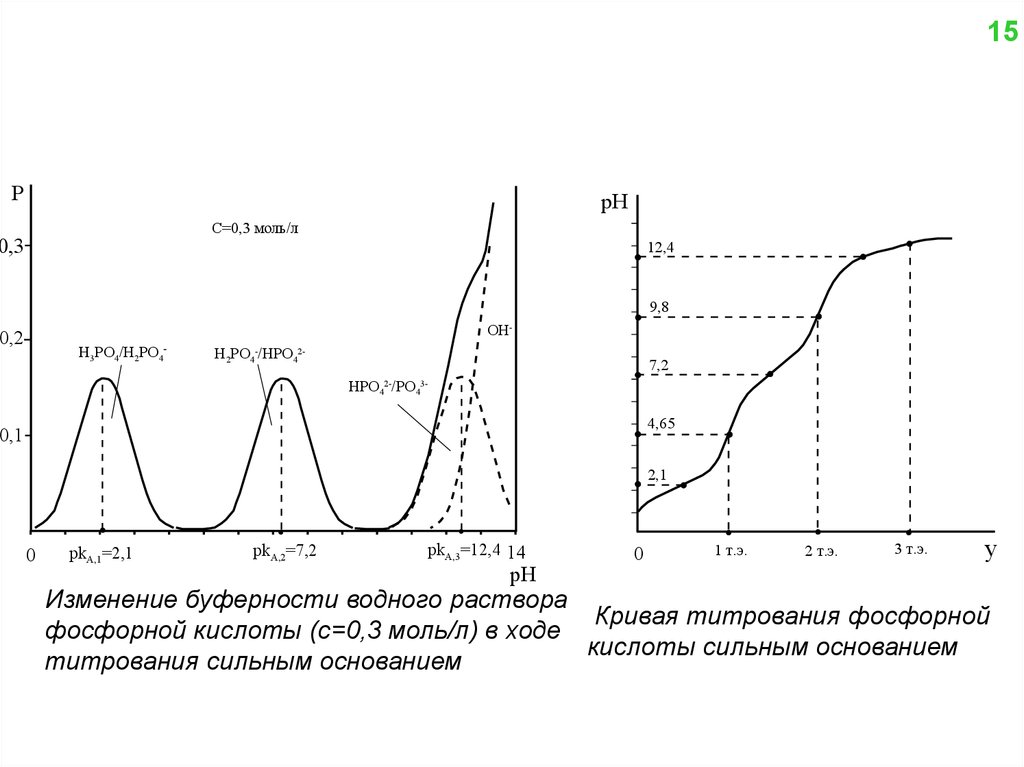
15.
15P
pH
С=0,3 моль/л
0,3
12,4
9,8
OH-
0,2
H3PO4/H2PO4-
H2PO4-/HPO42-
7,2
HPO42-/PO43-
4,65
0,1
2,1
0
pkA,1=2,1
pkA,2=7,2
pkA,3=12,4 14
pH
0
1 т.э.
2 т.э.
3 т.э.
y
Изменение буферности водного раствора
Кривая титрования фосфорной
фосфорной кислоты (с=0,3 моль/л) в ходе
кислоты сильным основанием
титрования сильным основанием
16.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ В НЕВОДНЫХ СРЕДАХ16
В подходящей неводной среде можно титровать смеси кислот,
константы, диссоциации которых различаются менее чем на четыре
порядка. Например, при потенциометрическом титровании в третбутанольном растворе смеси H2SO4 и НС1 раствором гидроксида
тетрабутиламмония в изопропаноле на кривой титрования
наблюдаются два скачка: первый соответствует титрованию НС1 и
половины количества H2S04, а второй ─ нейтрализации HSO4- до SO42Наибольший интерес представляют методы кислотно-основного
титрования неводных растворов органических веществ, анализ
которых в водных растворах невозможен.
Предел обнаружения F- при титриметрическом определении в смеси
предельный спирт–вода снижается на 2 - 3 порядка.
Уменьшение растворимости AgCl за счет введения в водный раствор 70%
метанола позволяет определять 2∙10-6 моль/л Сl- с хлорсеребрянымэлектродом.
17.
20Протолитическая теория кислот (Бренстед и Лоури)
А ↔ H+ + В
кислота
НС1 ↔
HNO3 ↔
СН3СООН ↔
Н2СO3 ↔
НСО3- ↔
Н3O+ ↔
H2O ↔
NH4+ ↔
Константа кислоты
КaA = (Н+)(В)/(А)
(i) – активността компонента i
основание
Н+ + С1H+ + NO3Н+ + СНзСООН+ + HCO3Н+ + СО32Н+ + Н2O
H+ + OHН+ + NH3
константа основания
КaB=(А)/[(Н+)(В)]
КaA = 1/КaB
(2)
(3)
Термодинамическая константа (Ka)
протолитического равновесия
Протолитическая реакция
А1 = H+ + В1
B2 + H+ = A2
--------------------A1 + B2 = B1 + A2
(1)
K Aa1
( B1 )( A2 )
K
a K Aa1 K Ba2
( A1 )( B2 ) K A2
a
(4)
18.
Органические растворители в соответствии с донорно-акцепторными21
свойствами по отношению к протону можно разделить на апротонные
(непротолитические), не участвующие в протолитических реакциях, и
протонные (протолитические).
Апротонные:(бензол, гексан, хлороформ, четыреххлористый углерод и т. п.)
Протонные растворители:
кислотные (протогенные), способные отщеплять протон (НСООН, СНзСООН,
С2Н5СООН и др.),
основные (протофильные), способные связывать протон (жидкий аммиак,
этилендиамин, пиридин и др.)
амфотерные (амфипротные), обладающие кислотной и основной группами
(этанол, пропанол, трет-бутанол, этиленгликоль и др.).
По способности изменять силу электролитов растворители делятся на
дифференцирующие и нивелирующие
Амфипротонный растворитель HSolv способен к автопротолизу (самоионизации),
HSolv + HSolv = H2Solv+ + SolvВ2
A1
A2
ион лиония
B1
лиат ион
19.
Константа автопротолиза(
H
Solv
)(
Solv
)
a
2
K HSolv
( HSolv ) 2
pKaHSolv = − lg KaHSolv;
pKaHSolv = pH + pSolv
22
Для чистого растворителя (HSolv) = 1
KaHSolv = (H2Solv+) (Solv-),
pH = − lg(H2Solv+);
ион лиония
pSolv = − lg(Solv-)
лиат ион
рKaH2O = рН + рОН
Нейтральная среда:
(H2Solv+) = (Solv-);
(H3O+) = (OH-);
рН = pSolv;
pH = pOH;
Кислая среда :
(H2Solv+) > (Solv-);
Щелочная среда :
(H2Solv+) < (Solv -); рН > pSolv ;
рН = 1/2 рKaHSolv ;
pH = 1/2pKaH2O
рН < pSolv;
Нормальная шкала кислотности: от pH=0 до pH=pKaHsolv
(5)
20.
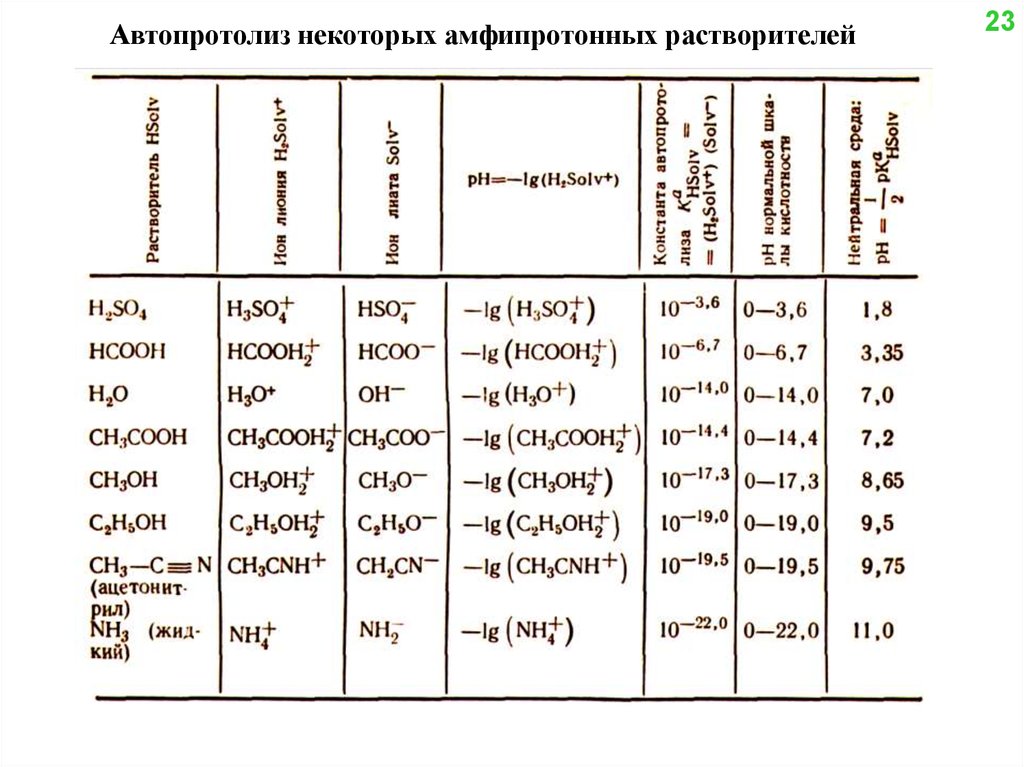
Автопротолиз некоторых амфипротонных растворителей23
21.
Если растворенный протолит по отношению к молекулам растворителя проявляет 24кислые свойства, устанавливается следующее равновесие:
А + HSolv = H2SoIv+ + В
(6)
А + Н 2O = Н 3O+ + В
CH3COOH + H2O = H3O+ + CH3COO−
NH4+ + H2О = H3O+ + NH3
Константу этого равновесия называют константой кислоты протолитической
пары «А, В» в растворителе HSolv.
a
(
H
Solv
)(
B
)
(
H
Solv
)
K
(
H
)(
B
)
a
2
2
K Aa HSolv
a A
K Aa K HSolv
, H 2 Solv (7)
( A)( HSolv )
( A) ( HSolv )( H ) K H Solv ,HSolv
2
KaHSolv,H2Solv+ –константа основания протолитической пары «H2Solv+, HSolv »,
константа основности растворителя: HSolv + Н+ = H2Solv+
В водных растворах ускусная кислота принадлежит к слабым кислотам и равновесие
СН3СООН + Н2О = СН3СОО- + Н3O+ смещено влево.
В жидком аммиаке уксусная кислота сильная кислота и равновесие
СН3СООН + Н2О = СН3СОО- + Н3O+ смещено вправо.
22.
Если растворенный протолит по отношению к молекулам растворителяпроявляет основные свойства, устанавливается равновесие:
В + HSolv = A + Solv-
25
(8)
Константу этого равновесия называют константой основания
протолитической пары «А, В» в растворителе HSolv.
a
K
(
A
)(
Solv
)
(
H
)(
A
)
(
Solv
)
a
K Ba HSolv
a B
K Ba K HSolv
, Solv
( B)( HSolv )
( B) ( HSolv )( H ) K Solv ,HSolv
(9)
где KaHSolv,Solv- − константа кислоты растворителя, т.е. константа
равновесия процесса HSolv = Solv - + H+,
Между константами КaA{HSolv} и KaB{HSolv} одной и той же протолитической
пары в одном и том же растворителе существует простая зависимость.
Если перемножить уравнения (7) и (9), получим
a
K Aa{HSolv} K Ba{Solv} ( H 2 Solv )( Solv ) K HSolv
pКaA{HSolv} + pKaB{HSolv} = pKaHSolv
(10)
(11)
23.
26pKaA
Кислота А
Основание В
pKa
B
В водных растворах
~−9
НСlO4 + Н2O ↔ Н3O+ + СlO4-
~23
~−8
HI + Н2O ↔ Н3O+ + I-
~22
~−7
НС1 + Н2O ↔ Н3O+ + Сl-
~21
~−3
H2SO4 + H2O ↔ H3O+ + HSO4-
~17
− 1,4
HNO3 + H2O ↔ H3O+ + NO3-
15,4
0,0
Н3O+ + Н 2O ↔ Н2O + Н3O+
14,0
2,1
Н3РO4 + Н2O ↔ Н3O+ + Н2РО4-
11,9
О+
3,2
HF + Н2O ↔ Н3
3,8
HCOOH + H2O ↔ Н3O+ + HCOO-
4,8
CH3COOH+ H2O ↔ Н3O+ + CH3COO-
+
F-
3,6
10,2
9,2
9,8
9,2
B безводной уксусной кислоте
7,6
7,0
H2S + Н2O ↔ Н3O+ + HS-
7,0
7,2
H2PO4- + Н2O ↔ Н3O+ + HPO42-
6,8
9,2
NH4+ + Н2O ↔ Н3O+ + NH3
4,8
9,3
HCN + Н2O ↔ Н3O+ + CN-
4,7
+ С6Н5
НСООН + С2Н5ОН ↔ С2Н6ОН2+ + НСООСН3СООН + С2Н5ОН ↔ C2H5OH2+ +
СН3СОО -
Н2СO3 + Н2O ↔ Н3O+ + НСО3-
9,9
С6Н5ОН + Н2O ↔ Н3
10,3
НСО3- + Н2O ↔ Н3O+ + СО32-
3,7
12,4
HPO42- + Н2O ↔ Н3O+ + PO43-
1,6
14,0
Н2O + Н2O ↔ Н3O+ + ОН-
0,0
23
NH3 + Н2O ↔ Н3O+ + NH2-
−9
24
OH- + Н2O ↔ Н3O+ + O2-
− 10
O-
HNО3 + С2Н5ОН ↔ С2Н5ОН2+ + NO315,4
10,8
6,4
O+
В этанольных растворах
4,1
10,3
8,7
2,9
НС1О4 + СН3СООН ↔ CH3COOH2+ + СlO4-
11,5
4,1
H2SО4 + СН3СООН ↔ СН3СООН2+ + HSO4-
10,3
5,0
НС1 + СН3СООН ↔ CH3COOH2+ + Cl-
9,4
9,4
HNО3 + CH3COOH ↔ CH3COOH2+ + NO3-
5,0
24.
Сильными называют протолиты, при растворении которыхравновесия (6) или (8) смещены вправо.
Для кислот pKaA < О (KaA > 1), для оснований pKaB < 0; ( КaB > 1).
27
Протолиз протекает сильнее в растворителях, имеющих высокие
значения диэлектрической постоянной ε.
1. Титрование очень слабой кислоты следует проводить в полярном
растворителе с высокой основностью. Наоборот, слабых оснований ─
в кислых растворителях, также по возможности с высокой
диэлектрической постоянной.
2. Если замена растворителя невозможна, то при анализе очень
слабого протолита в среде выбранного растворителя можно
применять метод обратного титрования.
Например, в воде фенол очень слабая кислота (КА{H2O}≈10-10) и не может
быть определен титрованием сильным основанием. Сопряженное фенолу
основание − фенолят-ион (C6H5O-), будет достаточно сильным основанием в
воде.
25.
283. При анализе смесей протолитов важно, чтобы растворитель был
высоко дифференцирующим. Дифференцирующее действие растворителей
зависит от кислотно-основных свойств, диэлектрической проницаемости,
способности к образованию водородных связей и сольватирующей
способности.
В протогенных растворителях происходит дифференцирование силы
кислот, т. е. большое количество веществ, которые в воде проявляют
кислотные свойства, в кислых растворителях уже не являются кислотами. В
среде муравьиной и уксусной кислот проявляют кислые свойства только
минеральные кислоты, в то время как карбоновые кислоты таких свойств не
проявляют.
По отношению к основаниям увеличение кислых свойств растворителя
приводит к нивелированию силы оснований. В среде муравьиной кислоты почти
все основания нивелированы по силе, а в среде уксусной кислоты основания,
рКВ которых в воде более 10, также нивелированы.
Основные растворители, наоборот, нивелируют силу кислот и
дифференцируют силу оснований. Органические соединения, проявляющие
в некоторых растворителях основные свойства, не проявляют основных свойств
в основных растворителях, сильные же в воде основания оказываются в них
дифференцированными по силе.
26.
294. Растворители амфипротонного характера обладают более высоким
дифференцирующим действием как в отношении кислот, так оснований.
Чем меньше константа автопротолиза, т.е. чем больше нормальная
шкала рН растворителя, тем больше наблюдаемые при титровании в
среде такого растворителя скачки титрования и тем больше возможность
дифференцированного титрования смесей электролитов.
Однако константы автопротолиза определены для немногих
растворителей, в число которых не вошли некоторые растворители,
получившие наибольшее практическое применение в аналитической химии
(например, кетоны, нитропроизводные углеводородов, смешанные
растворители). Поэтому практический интерес представляет
определение относительной шкалы кислотности органических
растворителей путем титрования в их среде наиболее сильных кислот и
оснований, например хлорной кислоты и гидроокиси тетраариламмония.
Указанные электролиты обычно используются в качестве наиболее сильных
кислых или основных титрантов при определении оснований и кислот в
неводных растворах.
27.
Относительную шкалу кислотности данного растворителявыражают числом милливольт, получаемым путем
вычитания потенциала полунейтрализации гидроокиси
тетраэтиламмония из потенциала полунейтрализации
хлорной кислоты как электролитов, которые занимают
крайние положения на шкале кислотности:
ESolv=E1/2(A) – E1/2(B)
где ESolv − относительная шкала кислотности растворителя, мB;
E1/2(A) − потенциал полунейтрализации НС1O4;
E1/2(B) − потенциал полунейтрализации (C2H5)4NOH.
30
28.
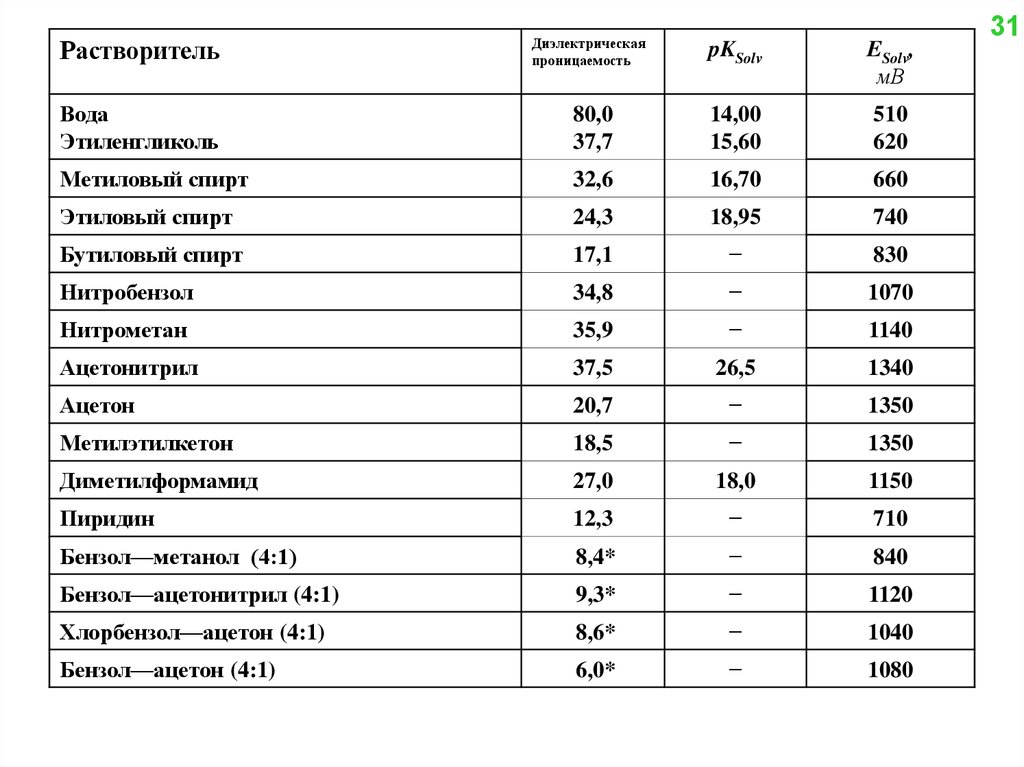
РастворительДиэлектрическая
проницаемость
pKSolv
ESolv,
мВ
Вода
Этиленгликоль
80,0
37,7
14,00
15,60
510
620
Метиловый спирт
32,6
16,70
660
Этиловый спирт
24,3
18,95
740
Бутиловый спирт
17,1
−
830
Нитробензол
34,8
−
1070
Нитрометан
35,9
−
1140
Ацетонитрил
37,5
26,5
1340
Ацетон
20,7
−
1350
Метилэтилкетон
18,5
−
1350
Диметилформамид
27,0
18,0
1150
Пиридин
12,3
−
710
Бензол—метанол (4:1)
8,4*
−
840
Бензол—ацетонитрил (4:1)
9,3*
−
1120
Хлорбензол—ацетон (4:1)
8,6*
−
1040
Бензол—ацетон (4:1)
6,0*
−
1080
31
29.
325. Величина диэлектрической проницаемости растворителей также
оказывает влияние на дифференцирующее действие растворителей.
Дифференциирующая способность уксусной кислоты (ε = 6) значительно
выше, чем у муравьиной кислоты (ε = 57)
6. Дифференцирующее действие растворителей зависит от
сольватирующей способности и способности молекул
растворителя образовывать водородные связи с молекулами
электролита.
Диссоциация электролита в растворе часто идет по схеме:
HA + nSolv ↔HA(Solv)n ↔ HSolv+cольв. + A- cольв.
30.
Удобным методом оценки влияния растворителей на силу кислот и оснований, 33также как и для оценки относительной шкалы кислотности растворителей, является
метод определения относительной кислотности электролитов по потенциалам
полунейтрализации. В момент, когда нейтрализовано 50% определяемой слабой
кислоты или слабого основания, рН = рК. Следовательно, величина потенциала
полунейтрализации определяется величиной константы ионизации титруемого
электролита и может характеризовать его относительную силу в неводных растворах.
В качестве стандарта при определении потенциалов полунейтрализации кислот
обычно используют бензойную кислоту, а при определении потенциалов
полунейтрализации оснований — дифенилгуанидин. Разность Е(1/2) исследуемой
кислоты (или основания) и стандартного вещества дает представление о возможности
дифференцированного титрования смеси кислот (оснований), т.е. величина ΔE(1/2)
может служить критерием их силы в неводных растворах
ΔЕ(1/2) = Е(1/2),x – E(1/2),ст. ,
(13)
где ΔЕ(1/2),x, и ΔE(1/2),ст. − потенциалы полунейтрализации исследуемого и стандартного
вещества. При определении ΔЕ(1/2) существенную роль играют сила растворенных
кислот и оснований, а также природа растворителя и растворенного вещества. Чем
больше величина ΔE(1/2), тем выше сила протолита и дифференциирующая способность
растворителя. Различие потенциалов полунейтрализации в 200 - 300 мВ в большинстве
случаев оказывается достаточным для осуществления избирательного титрования.
31.
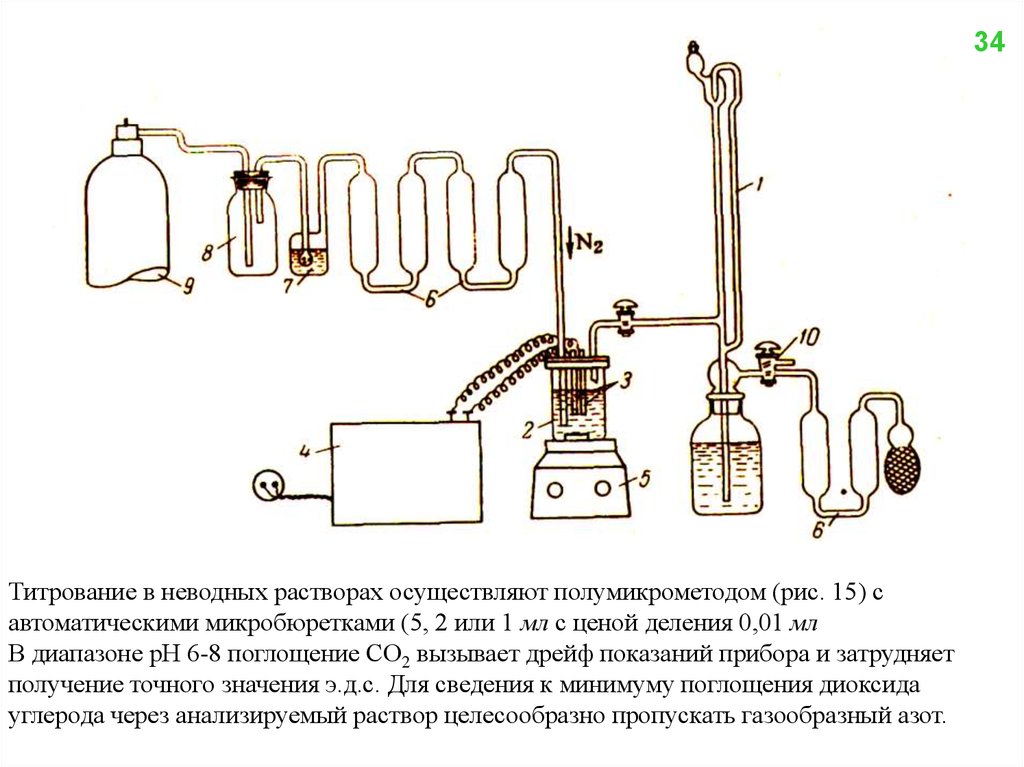
34Титрование в неводных растворах осуществляют полумикрометодом (рис. 15) с
автоматическими микробюретками (5, 2 или 1 мл с ценой деления 0,01 мл
В диапазоне рН 6-8 поглощение СО2 вызывает дрейф показаний прибора и затрудняет
получение точного значения э.д.с. Для сведения к минимуму поглощения диоксида
углерода через анализируемый раствор целесообразно пропускать газообразный азот.
32.
В качестве титрантов используют в основном неводные растворыхлорной кислоты. Хлорная кислота наиболее сильная кислота в среде неводных
35
растворителей, что и обуславливает ее широкое применение. В качестве титрантов
применяются также алкил- или арилсульфоновые кислоты. В уксуснокислой среде
фторсульфоновая кислота является более сильной по сравнению с хлорной кислотой, а
стабильность ее растворов и простота приготовления позволяют считать ее одним из
лучших титрантов. Иногда используются также растворы салициловой, пикриновой,
трихлоруксусной, иодной, азотной и серной кислот
Из неорганических оснований наиболее широко применяются метаноловые,
этаноловые, изопропаноловые и смешанные стандартные растворы едкого кали и
едкого натра. С успехом используются уксуснокислые растворы ацетатов
щелочных металлов, которые по силе не уступают спиртовым растворам щелочей.
Широко применяются спиртовые растворы алкоголятов щелочных, металлов:
метилаты, этилаты, изопропилаты натрия, калия, лития в среде спиртов и
аминоэтилат натрия в среде этилендиамина.
Самыми сильными основными титрантами в среде органических растворителей
являются растворы четвертичных аммониевых оснований: гидроокиси тетраметил-,
тетраэтил- и тетрабутиламмония, а также некоторые их производные, например,
трибутилметил-, триметилбензил-, гексадецилтриметил-, тетрапропил-,
триметилфениламмония и другие.
33.
Расчет кривой протолитического титрования в неводных средахКαA = (Н+)(В)/(А)
КαВ =(А)/[(Н+)(В)]
А ↔ H+ + В
протолитическая пара
константа кислоты
константа основания
Автопротолиз неводного растворителя
HSolv + HSolv = H2Solv+ + SolvCH3OH + CH3OH = CH3OH2+ + CH3OИон лиония
a
K HSolv
( H 2 Solv )( Solv )
( HSolv ) 2
H2O + H2О = Н3O+ + ОН−
Автопротолиз воды
лиат ион
стандартные
условия
(HSolv)=1:
KaHSolv =(H2Solv+) (Solv -)
KcHSolv =[H2Solv+] [Solv -]
Титрование раствора сильного протолита раствором сильного протолита
H2Solv+ + Solv- = 2HSolv;
Solv- + H2Solv+ = 2HSolv
Н3O+ + ОН− = 2H2О
(алкалиметрия)
ОН− + Н3O+ = 2H2О
(ацидиметрия)
Константа равновесия в данном случае
Kc=1/KcHSolv
35
34.
Титрование сильной кислоты сильным основанием (алкалиметрия)36
0 < 1;
pH = - lgc0 - lg(1- )
1;
pH = -lgc0+ ½lg Kc c02 = ½pKcHSolv
>1;
pH = -lgc0+ lg Kc c02 + lg( -1) = lgc0 + pKcHSolv + lg( -1)
Титрование сильного основания сильной кислотой (ацидиметрия)
0< <1
pH = pKcHSolv + lgc0 - lg(1- )
= 1;
pH = ½pKcHSolv
>1;
pH = - lgc0 - lg( -1)
35.
37Расчет рН кривой титрования сильной кислоты сильным основанием
36.
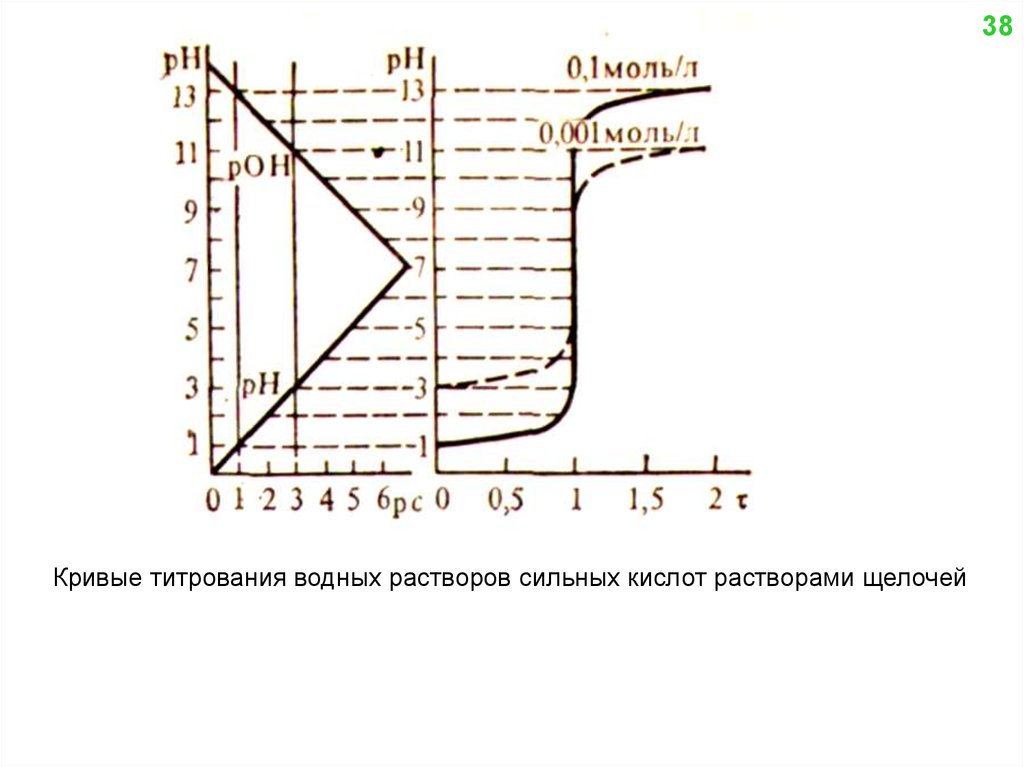
38Кривые титрования водных растворов сильных кислот растворами щелочей
37.
Расчет кривой титрования слабой кислоты сильным основанием39
A + Solv- = B + HSolv
- степень оттитрованности
pH = ½pKсA- ½ lgc0
= 0;
0 < < 1; pH = pKсA+ lg[c0 /c0(1- )] = pKсA+ lg[ /(1- )]
= 1;
>1;
pH = ½pKcHSolv + ½lg KcA + ½lg c0
pH = ½pKсHSolv + lg c0( -1)
Расчет кривой титрования слабого основания сильной кислотой
B +H2Solv+ = A + HSolv
= 0;
0 < < 1;
pH = ½pKcHSolv + ½lg KcA + ½lg c0
pH = pKсA+ lg[(1- ) / ]
= 1;
pH = ½pKcA - ½lg c0
>1;
pH = - ½lg c0( -1)
38.
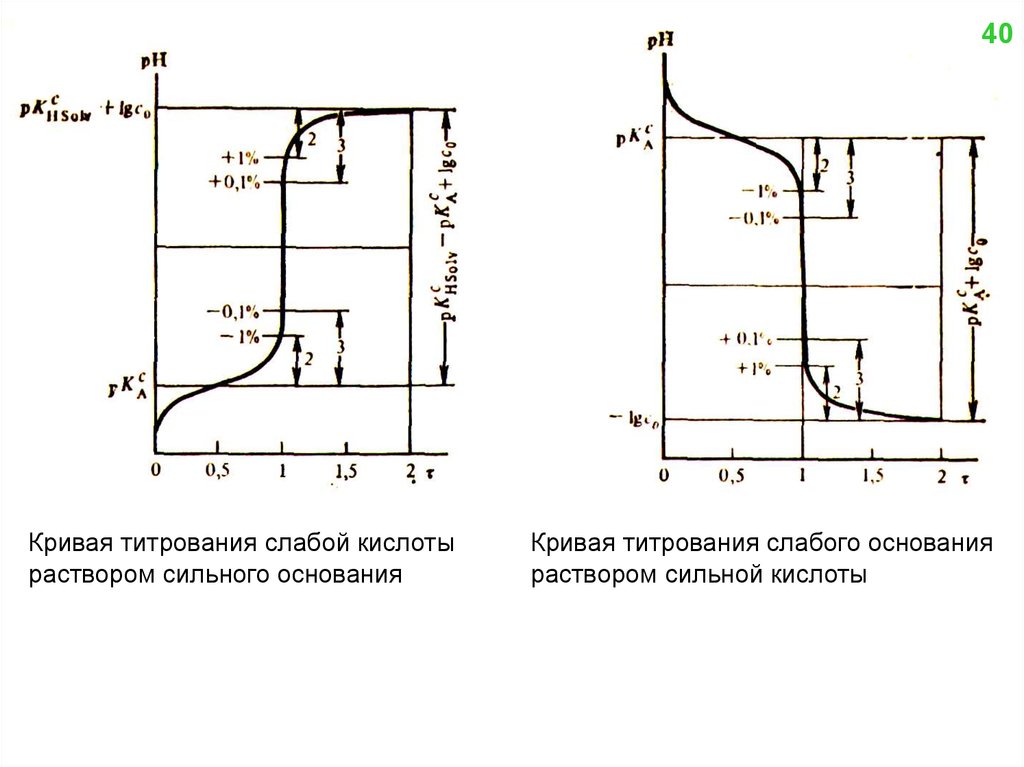
40Кривая титрования слабой кислоты
раствором сильного основания
Кривая титрования слабого основания
раствором сильной кислоты
39.
Окислительно-восстановительное потенциометрическое титрование41
Ox + ne = Red,
EOx,Re d E
MnO4─ + 5e + 8H+ = Mn2+ + 4H2O;
0
Ox , Re d
E MnO ,H ,Mn2 E MnO ,H ,Mn2
4
aRed1 + bOx2 = aOx1 + bRed2
RT
[Ox1 ]
E1 E
ln
n1F
[Re d1 ]
0
1
[ Ox1 ]a [Re d2 ]b
K
[Re d1 ]a [ Ox2 ]b
RT
[Ox ]
ln
nF
[Re d ]
(3)
(5)
4
(1)
[ MnO4 ][ H ]8
RT
ln
5F
[ Mn 2 ]
2Fe3+ + 2I- = 2Fe2+ + I2.
(2)
RT
[Ox2 ]
E2 E
ln
n2 F
[Re d2 ]
0
2
(4)
Константу равновесия
окислительно-восстановительного
процесса легко рассчитать, зная
стандартные потенциалы E01 и E02
40.
В равновесии Е1=Е2, поэтому приравнивая правые части уравнений 3 и 4,получим
a
b
RT
[
Ox
]
RT
[
Ox
]
0
1
2
E10
ln
E
ln
2
an1F
[Re d1 ]a
bn2 F
[Re d2 ]b
Учитывая, что
n1α = n2b = q, где q − общее число электронов, получим
E
lg K
RT
ln K E20 E10
qF
0
2
E10 q
(6)
0,059
Для реакции, схема которой соответствует уравнению 4, в момент
эквивалентности
[Ox1 ] [Re d2 ]
[Re d1 ] [Ox2 ]
[Ox1 ]
K
[Re d1 ]
a b
[Re d2 ]
[Ox2 ]
a b
1
[ Ox1 ]
K a b (8)
(7)
[Re d1 ]
Отношение [Ox1] / [Red1] характеризует полноту протекания реакции, а
обратная величина − ошибку титрования, т.е. долю исходного вещества не
вступившего в реакцию.
41.
Расчет кривой титрования и анализ ее формыКогда y<C0 удобно использовать уравнение 3, если y>C0 − уравнение 4
В точке эквивалентности, когда y=C0, общий потенциал смеси для можно
рассчитать, используя уравнения 3, 8 и 6:
EY C0
0,059
[Ox1 ]
0,059
0
E1 E
lg
E1
lg K
n1
[Re d1 ]
n1 (a b)
0
1
0,059 ( E20 E10 ) q
E
n1 (a b) 0,059
0
1
EY C0
EY C0
bE10 aE20
a b
E10 E20
2
(9)
(10)
(11)
Учитывая, что q = n1a, уравнение
9 можно переписать в виде
В частном случае, когда
а в 1
Начальную точку кривой титрования, когда
титрант еще не добавлен и y=0, рассчитать
невозможно. Этот момент формально
соответствует полностью восстановленной
системе, при этом Е1 = - ∞.
42.
Расчет кривой титрования и анализ ее формыКогда y<C0 удобно использовать уравнение 2, если y>C0 − уравнение 3
В точке эквивалентности, когда y=C0, общий потенциал смеси для можно
рассчитать, используя уравнения 2, 8 и 6:
EY C0
0,059
[Ox1 ]
0,059
0
E1 E
lg
E1
lg K
n1
[Re d1 ]
n1 (a b)
0
1
0,059 ( E20 E10 ) q
E
n1 (a b) 0,059
0
1
EY C0
bE10 aE20
a b
(9)
Учитывая, что q = n1a, уравнение 9
можно переписать в виде
В частном случае, когда
(10)
EY C0
E10 E20
2
а в 1
(11)
Начальную точку кривой титрования, когда титрант еще не добавлен и y=0,
рассчитать невозможно. Этот момент формально соответствует полностью
восстановленной системе, при этом Е1 = - ∞.
43.
Все остальные точки кривой титрования можно рассчитать, зная исходную концентрациюС0 определяемого вещества и концентрацию y добавленного титранта.
Red1 + Ох2 = Ox1 + Red2,
С0
(С0–y+x)
Cu+ + Fe3+ = Cu2+ + Fe2+
y
x
y-x.
в равновесии
y-x
х – концентрация исходного вещества, образовавшегося за счет обратимости реакции.
RT
y x
E1 E
ln
n1 F
C0 y x
0
1
(12)
Практически всю кривую титрования,
принебрегая обратимостью реакции, можно
рассчитать по уравнению (14)
( y x)2
K
(C 0 y x ) x
RT
y
E1 E
ln
n1 F
C0 y
0
1
Если ввести степенью окисленности (оттитрованности) системы
C=[Ox]+[Red], α =([Ox]/C)∙100
[Ox]=αC/100
[Red]=(100-α)C/100
(13)
EOx,Re d E
0
Ox , Re d
(14)
α (% или доля)
RT
ln
nF
(100 )
(15)
44.
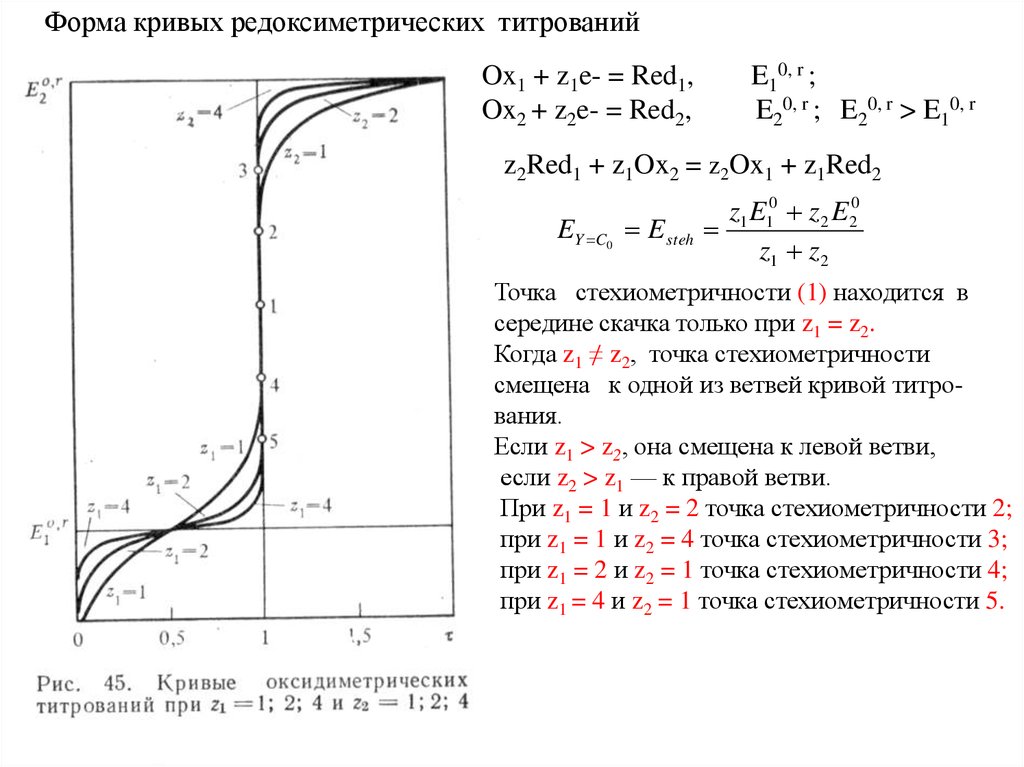
Форма кривых редоксиметрических титрованийOx1 + z1e- = Red1,
Ox2 + z2e- = Red2,
E10, r ;
E20, r ; E20, r > E10, r
z2Red1 + z1Ox2 = z2Ox1 + z1Red2
EY C0 E steh
z1 E10 z 2 E 20
z1 z 2
Точка стехиометричности (1) находится в
середине скачка только при z1 = z2.
Когда z1 ≠ z2, точка стехиометричности
смещена к одной из ветвей кривой титрования.
Если z1 > z2, она смещена к левой ветви,
если z2 > z1 — к правой ветви.
При z1 = 1 и z2 = 2 точка стехиометричности 2;
при z1 = 1 и z2 = 4 точка стехиометричности 3;
при z1 = 2 и z2 = 1 точка стехиометричности 4;
при z1 = 4 и z2 = 1 точка стехиометричности 5.
45.
Вычислить скачок можно задаваясь допустимой погрешностью анализа. Допустим,устраивает погрешность 0,1%, тогда в конце титрования восстановителя
(начало скачка) редокси-потенциал:
Конец скачка:
Величина скачка:
E E10,r
0,059
0,999
0,177
lg
E10,r
z1
1 0,999
z1
E E20,r
0,059
0,001
0,177
lg
E20,r
z2
1 0,001
z2
E 0 , r
0,177 0,177
z1
z2
Как должны различаться стандартные потенциалы реагирующих систем, чтобы
можно было с заданной точностью зарегистрировать конец титрования.
Допустим устраивает погрешность 0,1%.
Если (z1=z2=1),
то минимальная разность
∆Е0=(Е02 – Е01) = 0,354 В
Если z1=1 и z2=2, то ∆Е0=0,265 В; при z1=1 и z2=3 ∆Е0 = 0,236 В.
Концентрация титруемого раствора не влияет на кривые редоксиметрических
титрований, если только не меняется число частиц; например, из одной частицы
окисленной формы возникают несколько частиц восстановленной формы той же пары
или наоборот.
46.
Титрование смесей восстановителей (окислителей)Рассмотрим оксидиметрическое титрование смеси двух восстановителей Red1 и Red2.
Титрантом при этом служит раствор окислителя Ох. Редоксипереходы:
Редоксипереходы:
Ox1 + z1e- = Red1, E10, r
Ox2 + z2e- = Red2, E20, r
титрант
E0, r
Ox + ze- = Red,
У первой точки стехиометричности имеется
скачок, если выполнены условия:
погрешность до ±0,1%
E 2O ,r E1O ,r
0,177 0,177
z1
z2
вторая точка стехиометричности
E O ,r E2O ,r
0,177 0,177
z1
z2
47.
ОСАДИТЕЛЬНОЕ ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕHal- + Ag+ = AgHal↓
Индикаторный электрод - серебянный
До точки эквивалентности (y<c0):
E E
1
0,059 lg[ Ag ] E
0
Ag / Ag
0
Ag / Ag
0,059 lg ПР
0,059 lg[ Hal ]
AgHal
E2 E 0 Ag / Ag 0,059 lg[ Ag ]
После точки эквивалентности (y>c0):
(1)
(2)
Если проводить титрование 0,1 моль/л раствора галогенида 0,1 моль/л раствором
нитрата серебра, то при степени оттитрованности 99,9% (погрешность 0,1%)
E1 E 0 Ag / Ag 0,059 lg ПРAgHal 0,059 lg 10 4
(3)
При появлении избытка Ag+ в таком количестве чтобы [Ag+]=10-4 моль/л
E2 E 0 Ag / Ag 0,059 lg 10 4
Величина скачка
(4)
E E2 E1 0,059 lg 10 8 0,059 lg ПРAgHal
Потециал в точке эквивалентности
y=c0
E
y c0
E
0
Ag / Ag
0,059 lg ПР
AgHal
(5)
(6)
48.
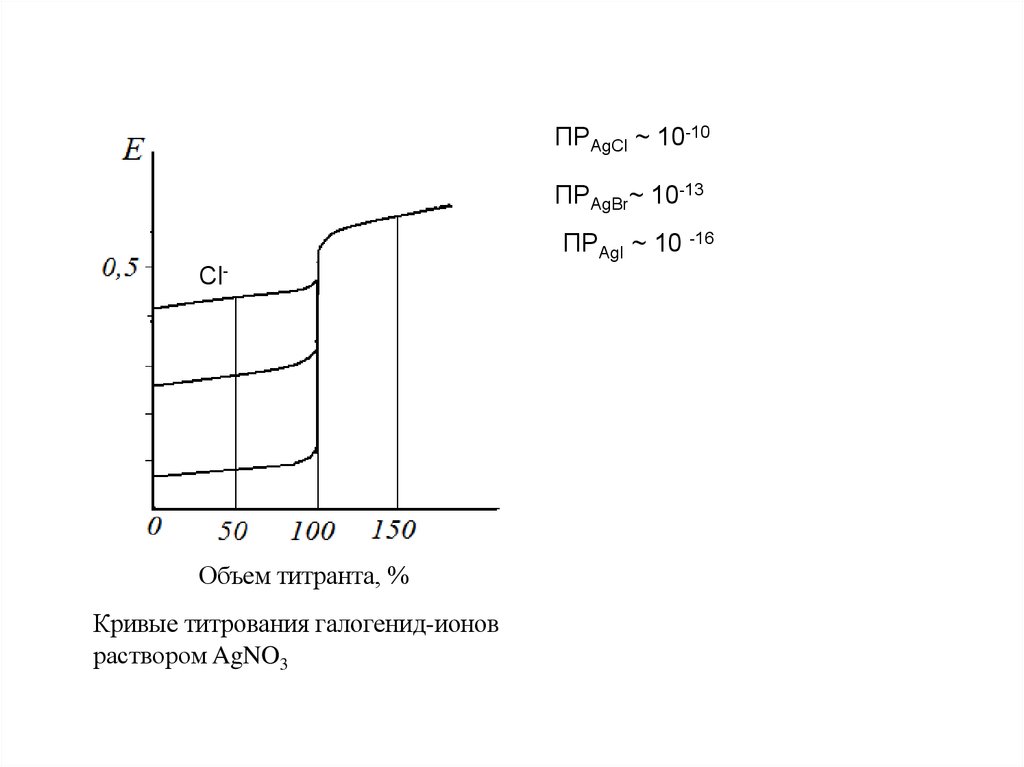
ПРAgCl ~ 10-10ПРAgBr~ 10-13
ПРAgI ~ 10 -16
Cl-
Объем титранта, %
Кривые титрования галогенид-ионов
раствором AgNO3
49.
АI- + B+ = BAI↓ и AII2- + 2B+ = B2AII↓,[B ]
ПРВАI
[ AI ]
ПРВ2 АII
2
[ AII ]
[ AI ]
2
[ AII ]
ПРВАI
ПРВ2 АII
Например, ионом серебра титруется смесь анионов Cl- и CrO42-. Произведения
растворимости хлорида и хромата серебра приблизительно равны, соответственно,
10-10 и 10-12. Первым осаждается хлорид-ион, хромат-ион начнет осаждаться, когда
[Cl ]
2
[CrO4 ]
[Cl ] 10
4
10 10
В момент начала осаждения хромата серебра концентрация
недотитрованного хлорид-иона составит
10 12
2
[CrO4 ]
Эта величина и определяет абсолютную ошибку
титрования.
50.
КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕBn+ + pA z- = BAp(n-pz)
[ BA p( n pz ) ]
n
z p
[ B ][ A ]
(1)
Потенциал индикаторного электрода 1-го рода до точки эквивалентности
E1 E
o
B n / B
0 / 059
lg[ B n ]
n
(2)
а после точки эквивалентности:
o
E2 E BA
p
o
E BA
/ B , Az
Z
P / B , A
( n pz )
]
0,059 [ BA p
lg
n
[ Az ] p
E Bo n / B
0,059
lg
n
(3)
(4)
, где
51.
осложненияОбщие констант прочности
Ступенчатые константы прочности
Bz+ + A↔BAz+
[ BA z ]
K1 z
[ B ][ A]
Bz+ + A↔BAz+
z
z
BAz+ + A↔BA2z+
K2
[ BA z ]
1 z
[ B ][ A]
[ BA2 ]
[ BАz ][ A]
Bz+ + 2A↔BA2z+
[ BA ]
2 z 2 2
[ B ][ A]
………………………………………………………………………………………………………
BAi-1z+ + A↔BAiz+
z
z
Ki
[ BAi ]
z
[ BАi 1 ][ A]
Bz+ + iA↔BAiz+
[ BA ]
i z i i
[ B ][ A]
……………………………………………………………………………………………
z
z
BAn-1z+ + A↔BAnz+
Kn
[ BAn ]
z
[ BАn 1 ][ A]
Из сравнения K и β видно, что
β1= K1; β2= K1 K2; βi= K1 K2··· Ki;
Bz+ + nA↔Banz+
βn= K1 K2···Ki···Kn.
[ BA ]
n z n n
[ B ][ A]
52.
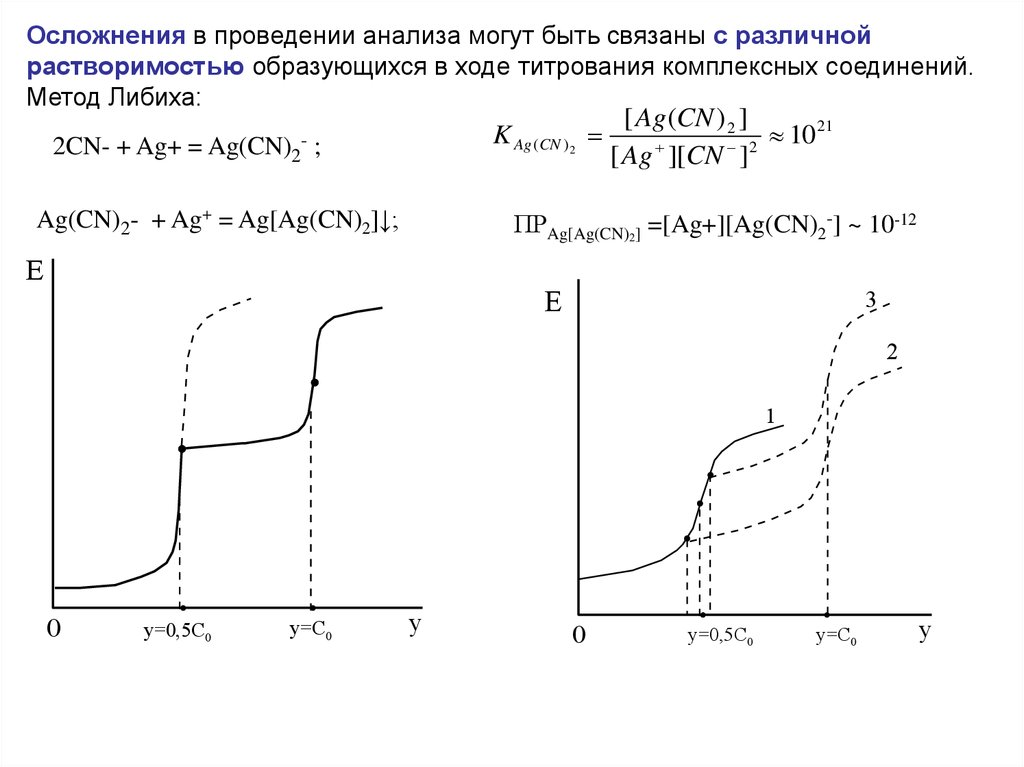
Осложнения в проведении анализа могут быть связаны с различнойрастворимостью образующихся в ходе титрования комплексных соединений.
Метод Либиха:
K Ag ( CN )2
-
2CN- + Ag+ = Ag(CN)2 ;
Ag(CN)2- + Ag+ = Ag[Ag(CN)2]↓;
[ Ag (CN ) 2 ]
10 21
2
[ Ag ][CN ]
ПРAg[Ag(CN)2] =[Ag+][Ag(CN)2-] ~ 10-12
E
E
3
2
1
0
y=0,5C0
y=C0
y
0
y=0,5C0
y=C0
y
53.
Хелатометрия (комплексонометрия)2
2
g
2
g
2
2
K равн.
[ g 2 ][ ]2
[ g 2 ][ 2 2 ]
(5)
При использовании ртутного электрода как индикаторного на его поверхности
протекает реакция
0,059
g
2
2e g
E1 E o g 2 / g
2
lg[ g 2 ]
(6)
При добавлении в раствор ЭДТА образуется устойчивый комплексонат ртути HgY2(lgβHgY2-=21,8), который диссоциирует в очень небольшой степени
HgY2- = Hg2+ + Y4+ , что и определяет величину потенциала ртутного электрода:
E Hg 2 / Hg E
o
Hg 2 / Hg
0,059
[ g 2 ]
lg 4
2
[ ] HgY 2
(7)
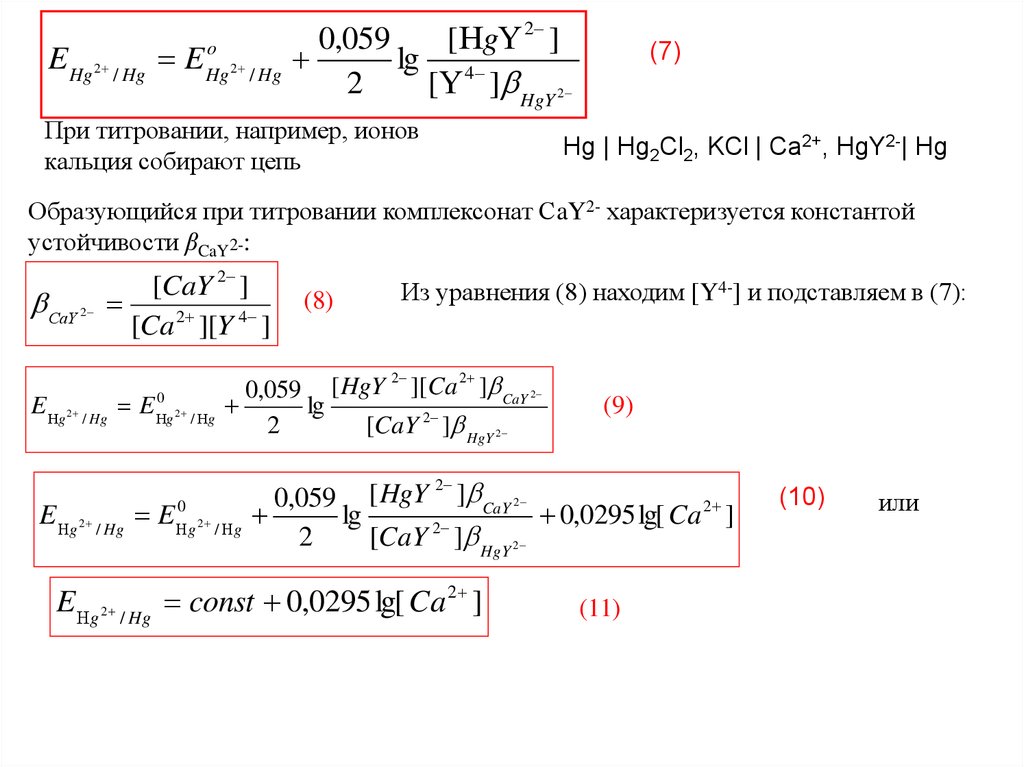
54.
E Hg 2 / Hg Eo
Hg 2 / Hg
0,059
[ g 2 ]
lg 4
2
[ ] HgY 2
При титровании, например, ионов
кальция собирают цепь
(7)
Hg | Hg2Cl2, KCl | Ca2+, HgY2-| Hg
Образующийся при титровании комплексонат СaY2- характеризуется константой
устойчивости βCaY2-:
СaY
2
[CaY 2 ]
[Ca 2 ][Y 4 ]
E g 2 / Hg E
0
g 2 / g
E g 2 / Hg E
(8)
Из уравнения (8) находим [Y4-] и подставляем в (7):
2
2
0,059 [ HgY ][Ca ] CaY 2
lg
2
[CaY 2 ] HgY 2
0
g 2 / g
(9)
2
0,059 [ HgY ] CaY 2
2
lg
0
,
0295
lg[
Ca
]
2
[CaY 2 ] HgY 2
E g 2 / Hg const 0,0295 lg[ Ca 2 ]
(11)
(10)
или
55.
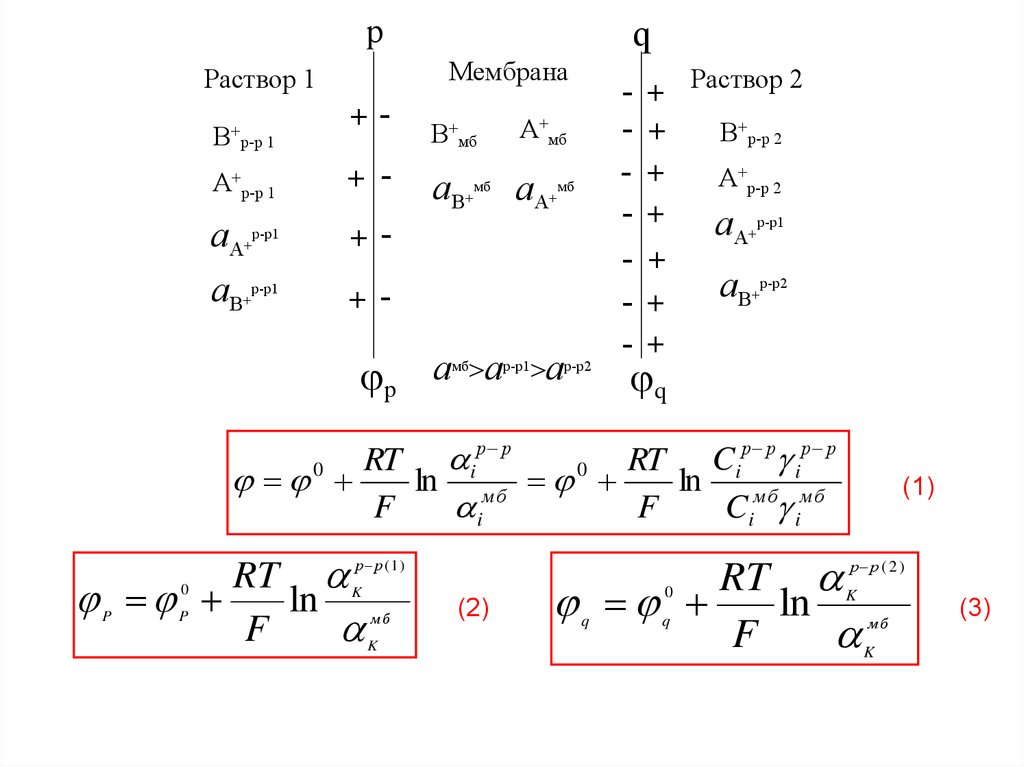
pМембрана
Раствор 1
B+р-р 1
+ -
А+р-р 1
+ -
aA
aВ
+ -
р-р1
+
р-р1
+
q
B+мб
А+мб
a
a
мб
В+
мб
А+
+ -
p a >a
мб
>aр-р2
р-р1
-
+
+
+
+
+
+
+
Раствор 2
B+р-р 2
А+р-р 2
aA
р-р1
+
aВ
р-р2
+
q
р р
р р р р
C
i
RT
RT
0
0
i
i
ln м б
ln
F
i
F
Ciм б iм б
RT
ln
F
р р (1)
0
P
P
K
мб
K
(1)
RT
ln
F
р р ( 2 )
(2)
0
q
q
K
мб
K
(3)
56.
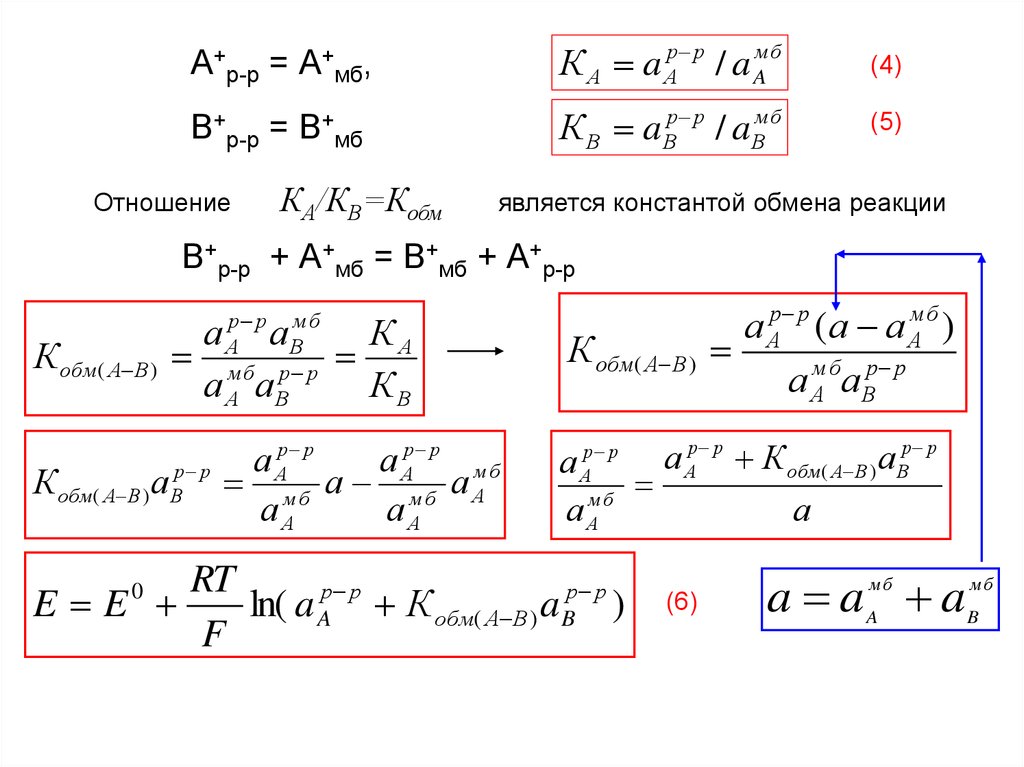
A+р-р = А+мб,К А a Ар р / a Aмб
(4)
В+р-р = В+мб
К В a Вр р / a Вмб
(5)
Отношение
КА/КВ=Кобм
является константой обмена реакции
В+р-р + А+мб = В+мб + А+р-р
К обм( А В )
р р м б
А
В
м б р р
А
В
а а
а а
К обм( А В ) а Вр р
КА
КВ
а Ар р
а Ар р м б
мб а мб аА
аА
аА
К обм( А В )
р р
А
мб
А
а
а
RT
E E
ln( a Aр р К обм( А В ) a Bр р )
F
0
а Ар р ( а а Амб )
мб р р
а А аВ
а Ар р К обм( А В ) а Вр р
а
(6)
a a a
мб
мб
A
B
57.
Уравнение (6) не учитывает диффузионный потенциал внутри мембраны,возникающий из-за различий в активностях потенциалопределяющих ионов в
поверхностных слоях мембраны, прилегающих к внутреннему и внешнему
раствору. Его величина зависит от чисел переноса t ионов в мембране;
u Aa A
tA
u Aa A uB a B
(7)
E E0
u
RT
ln( a Aр р B К обм( А В ) a Bр р )
F
uA
Величина (иB/иА)Кобм(А-В) в этом выражении называется коэффициентом
(8)
селективности (КA/B) электрода по отношению к ионам А+ и является
основным параметром, характеризующим селективность мембранного
электрода. Селективность электрода зависит также от соотношения активностей
определяемых и мешающих ионов (аА/аB). Чем меньше (КA/B), тем более
селективен электрод по отношению к определяемому иону.
Коэффициент селективности можно определить экспериментально,
58.
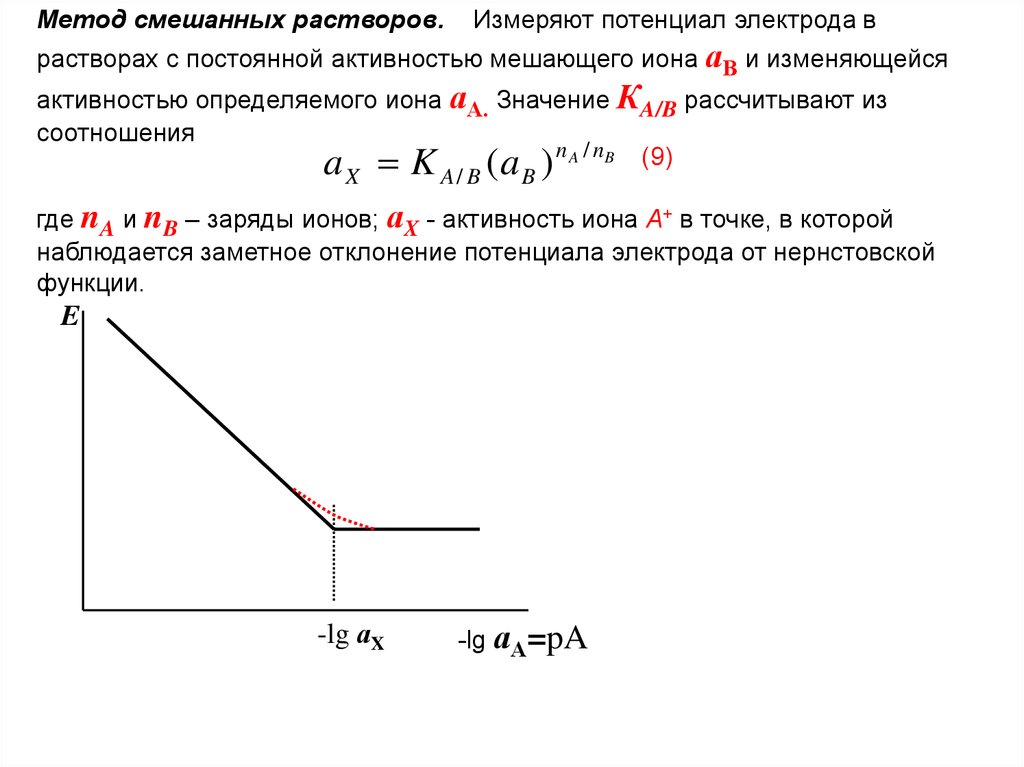
Метод смешанных растворов.Измеряют потенциал электрода в
растворах с постоянной активностью мешающего иона аВ и изменяющейся
активностью определяемого иона аА. Значение КA/B рассчитывают из
соотношения
a K ( a ) n A / nB (9)
X
A/ B
B
где nA и nB – заряды ионов; аX - активность иона А+ в точке, в которой
наблюдается заметное отклонение потенциала электрода от нернстовской
функции.
E
-lg аX
-lg аА=pA
59.
Метод отдельных растворов. Измеряют потенциал электрода в раствореопределяемого иона в отсутствие мешающего иона (EА) , а затем в растворе
мешающего иона в отсутствие определяемого иона (EB ), причем активности
ионов в этих растворах должны быть одинаковыми aA = aB .
Если соответствующие значения потенциалов для обоих ионов описываются
уравнением Нернста, то lg КA/B можно рассчитать из зависимости:
lg K A / B
n
EB E A
1 A lg a A
2,303RT / n A F nB
(10)
В общем случае влияние мешающих ионов на потенциал мембранного
электрода можно выразить с помощью уравнения Никольского:
E E0
RT
n /n
ln a A K A / B a B A B
nAF
(11)
где знак «+» для катионов, а «–» – для анионов; nA и nB – заряды определяемых и
мешающих ионов; aА и aB – соответствующие активности ионов в растворе;
КA/B - коэффициент селективности.
60.
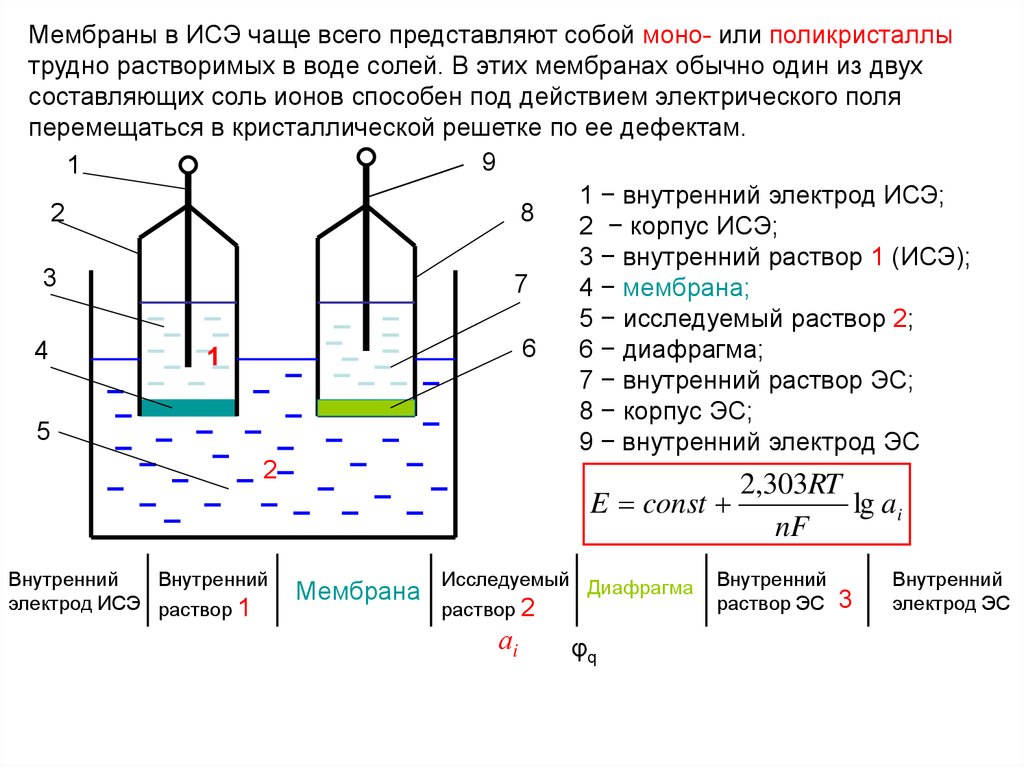
Мембраны в ИСЭ чаще всего представляют собой моно- или поликристаллытрудно растворимых в воде солей. В этих мембранах обычно один из двух
составляющих соль ионов способен под действием электрического поля
перемещаться в кристаллической решетке по ее дефектам.
9
1
1 − внутренний электрод ИСЭ;
2
8
2 − корпус ИСЭ;
3 − внутренний раствор 1 (ИСЭ);
3
7
4 − мембрана;
5 − исследуемый раствор 2;
6
6 − диафрагма;
4
1
7 − внутренний раствор ЭС;
8 − корпус ЭС;
5
9 − внутренний электрод ЭС
2
2,303RT
E const
lg ai
nF
Внутренний
Внутренний
электрод ИСЭ раствор 1
Мембрана
Исследуемый Диафрагма
раствор 2
аi
φq
Внутренний
раствор ЭС
3
Внутренний
электрод ЭС
61.
Электроды с кристаллической мембранойЭлектрод
Мембрана
Определяемый
Мешающие
рН
_______________________________ ион________________ ионы___________________
1. ЛантанLaF3
FAl3+, Fe3+, Ce4+, Li+, Th4+
фторидный
100–10-6 М
ОН≈5
2. Сульфидcеребряные
электроды
Ag2S
AgCl-Ag2S
Ионоселект.
CN- и SCN- - электроды
Электроды
на основе
MeS+Ag2S
Гетероген.
мембраны
Пунгора
AgBr-Ag2S
AgI-Ag2S
AgCN-Ag2S
AgSCN-Ag2S
CuS-Ag2S
PbS-Ag2S
CdS-Ag2S
Ag+; S2100 –10-7 М
10-2 –10-7 М
Ag+; Hal[Cl-] >5∙10-5
[Br-] >5∙10-6
[I-] >5∙10-8
[CN-] >10-6
[SCN-] >5∙10-6
Hg2+; CN-
сильные восстановители
10-1>[Me2+]>10-7
F-, Сl-, Вr-, I-, S2-,
Ag+, Ва2+,Са2+,
SO42-, PO43-,
62.
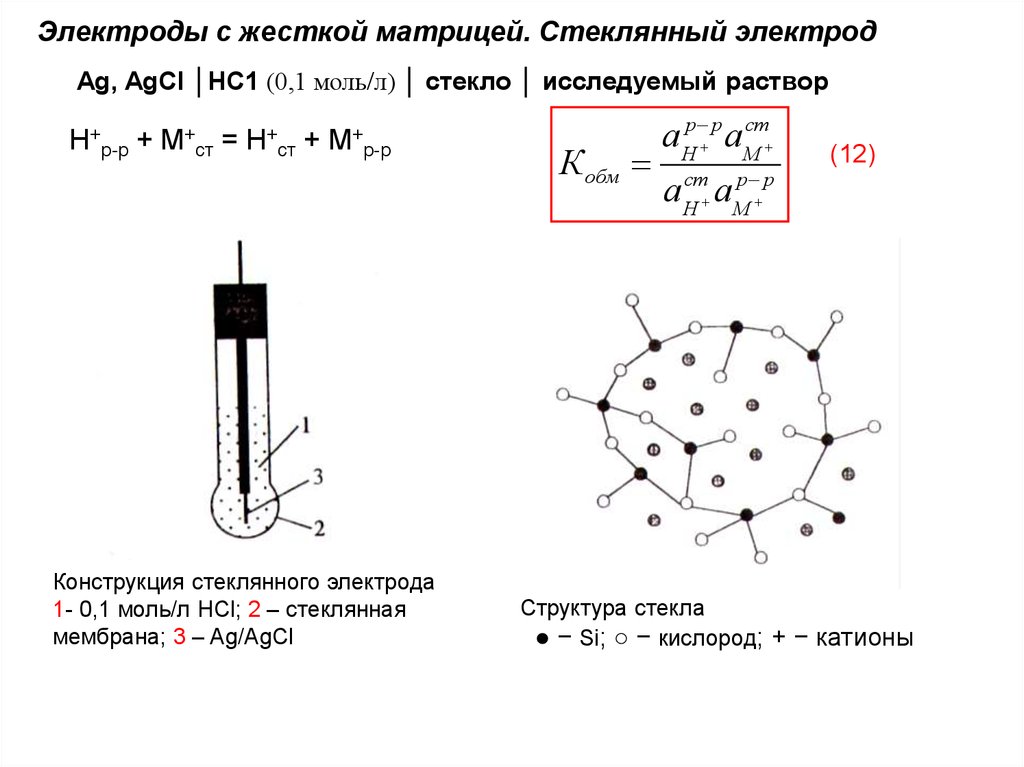
Электроды с жесткой матрицей. Стеклянный электродAg, AgCl │HC1 (0,1 моль/л) │ стекло │ исследуемый раствор
Н+р-р + М+ст = Н+ст + М+р-р
Конструкция стеклянного электрода
1- 0,1 моль/л HCl; 2 – стеклянная
мембрана; 3 – Ag/AgCl
К обм
а Нр р а Мст
а Нст а Мр р
(12)
Структура стекла
● − Si; ○ − кислород; + − катионы
63.
В простейшем случае электродный процесс сводитсяк обмену ионами водорода между раствором и
стеклом и отвечает перемещению единичного
заряда:
Если предположить, что сумма активностей
ионов водорода и щелочного металла в
стекле постоянна и выразить отношение
активностей ионов в (13) через константу
обмена, то получим выражение
RT
E
ln( a Н К обмa М )
F
0
(14)
RT
2,303RT
0
E
ln a Н E
lg a H E 0 0,059 pH
F
F
Если
(13)
aH+>>Kобма М+, то:
Если
E ст
E ст
Ecт
р р
RT
0
E
ln Hст
F
H
0
aH+ <<Kобм аМ+ , то
E ст
RT
E
ln a М
F
0
(15)
(15)
64.
рН<2: в кислой среде с высокой ионнойсилой активность воды по обеим сторонам
стеклянной мембраны не остается
одинаковой, поэтому в воде возникает
концентрационная ячейка, потенциал
которой также входит в измеряемую
разность потенциалов.
-Е
рН>9: щелочная ошибка возникает,
когда aH+ ≈ Kобма М+,
2
11
рН
Натриевое стекло, состоящее из
72% SiО2, 6% СаО и 22% Na2О
Литиевое стекло:
72% SiО2, 6% СаО и 22% Li2О.
Стекло натрий-чувствительных электродов: 11% Na2О, 18% А12О3 и
71% SiО2; K Na/K =10-3–10-5
Электроды на основе халькогенидных стекол (28% Ge, 60% Se, 12% Sb),
легированных Fe (≈2%) имеют нернстовский отклик к ионам Fe (III) и Сu (II). Еще
один электрод из халькогенидного стекла состава Cu6As4S9 чувствителен к
ионам Сu (II),
65.
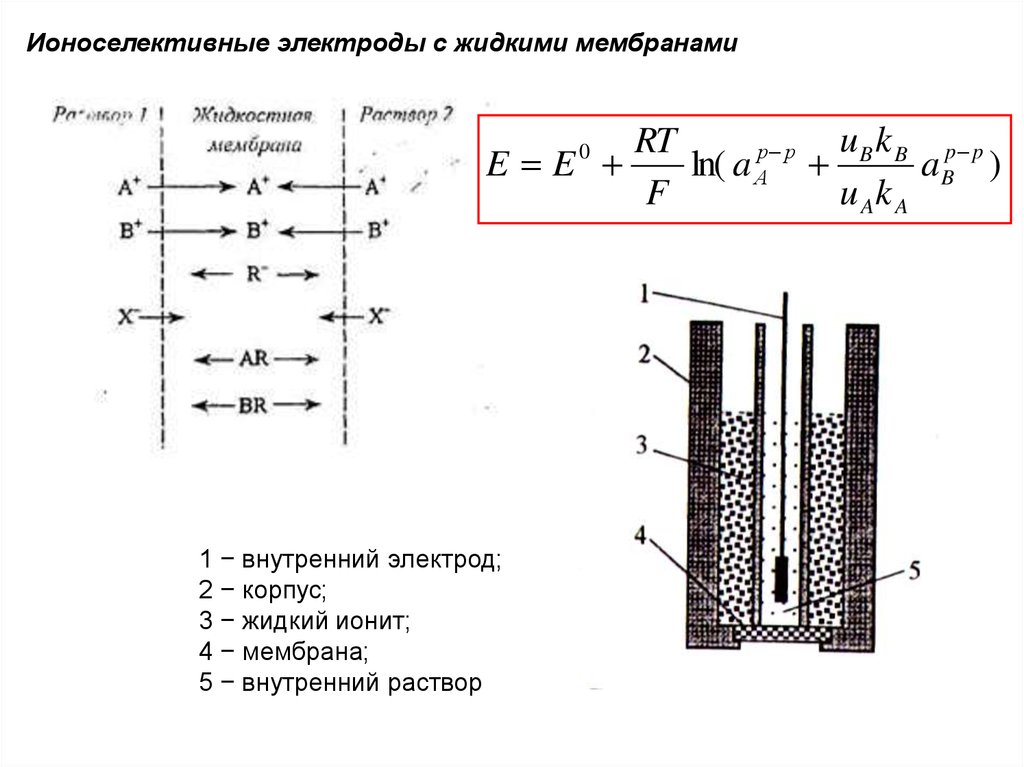
Ионоселективные электроды с жидкими мембранамиuB k B p p
RT
р р
E E
ln( a А
aB )
F
u Ak A
0
1 − внутренний электрод;
2 − корпус;
3 − жидкий ионит;
4 − мембрана;
5 − внутренний раствор
66.
Кальциевый электродПервым ИСЭ с жидкой мембраной был кальций-селективный электрод
на основе кальциевой соли додецилфосфорной кислоты, растворенной в
диоктилфенилфосфате. В выпускаемых в настоящее время электродах для
определения кальция в качестве ионофоров применяют эфиры фосфорной
кислоты с двумя алифатическими радикалами, содержащими от 8 до 16
углеродных атомов, или нейтральные переносчики. В случае эфиров
фосфорной кислоты на поверхности мембраны устанавливается равновесие
[(RO)2POO]2Ca = 2(RO)2POО- + Са2+
[Ca2+]: 10-1 ─ 10-5 M. Определению кальция мешают ионы стронция, магния, бария
Высокой селективностью по отношению к ионам тяжелых металлов обладают
иониты с активными группами, содержащими серу (замещенные тиогликолевой
кислоты и др.). Использование таких электродов ограничено из-за склонности
серосодержащих соединений к окислению и взаимодействию с ионами
водорода.
67.
Электроды с анионной функциейК электродам с анионной функцией относятся мембраны, которые в качестве
ионитов содержат комплексы положительно заряженных переходных
металлов с нейтральными органическими лигандами, например с о-фенантролином. Комплексные соли типа ML3(NO3)2, где L - лиганд, функционируют
как анионообменники. На основе комплексов с Ni разработаны электроды,
селективные к ионам СlO4-, NO3-, BF4-, Сl-.
RT
E const
ln a NO
3
F
Фенантролиновые комплексы Со
используются для определения ионов
СlO4-, Вr- и I-.
Для определения нитрат-ионов используются
также четвертичные аммониевые и
фосфониевые соли. Электроды
характеризуются крутизной электродной
функции, близкой к теоретической, в
диапазоне концентраций от 10-1 до 10-5 моль/л.
Коэффициенты селективности по отношению к
ионам Сl-, NO2-, SO42- не превышают 10-2.
68.
Ионообменники на основе солей тетраалкиламмония применяют дляизготовления хлоридных электродов. В качестве органического катиона в них
используется диметилдистеариламмоний. Электрды можно применять для
измерения активности ионов хлора в присутствии сульфид-ионов,
Комплексные соединения макроциклических лигандов, в том числе
циклических антибиотиков, с катионами щелочных и щелочноземельных
металлов. Прочность комплексов, состав которых обычно отвечает
соотношению 1:1, определяется тем, что в них катион металла, попадая во
внутреннюю полость, удерживается в ней электростатическим притяжением
атомов кислорода, электронные пары которых ориентированы внутрь цикла.
Основное условие для образования таких комплексов - соответствие
внутримолекулярной полости лиганда размерам иона металла.
Благодаря хорошей растворимости макроциклов в неводных средах они
способны переводить катионы щелочных и щелочноземельных металлов из
водной фазы в органическую. При этом макроциклические соединения можно
уподобить челноку, снующему через границу раздела мембрана/анализируемый раствор. Подобные соединения называют еще мембрано-активными
комплексонами (МАК) или нейтральными переносчиками.
69.
Структурные формулывалиномицина (II) и некоторых
соединений с открытой цепью
(III, IV), являющихся
нейтральными переносчиками
Раствор валиномицина в
дифениловом эфире является
основой мембраны калийселективного электрода. При
определении калия в
присутствии натрия коэффициент селективности < 10-4.
Электрод позволяет
определять калий в диапазоне
концентраций от 10-1 до 10-5
моль/л. Единственным
мешающим ионом, является
ион аммония.
70.
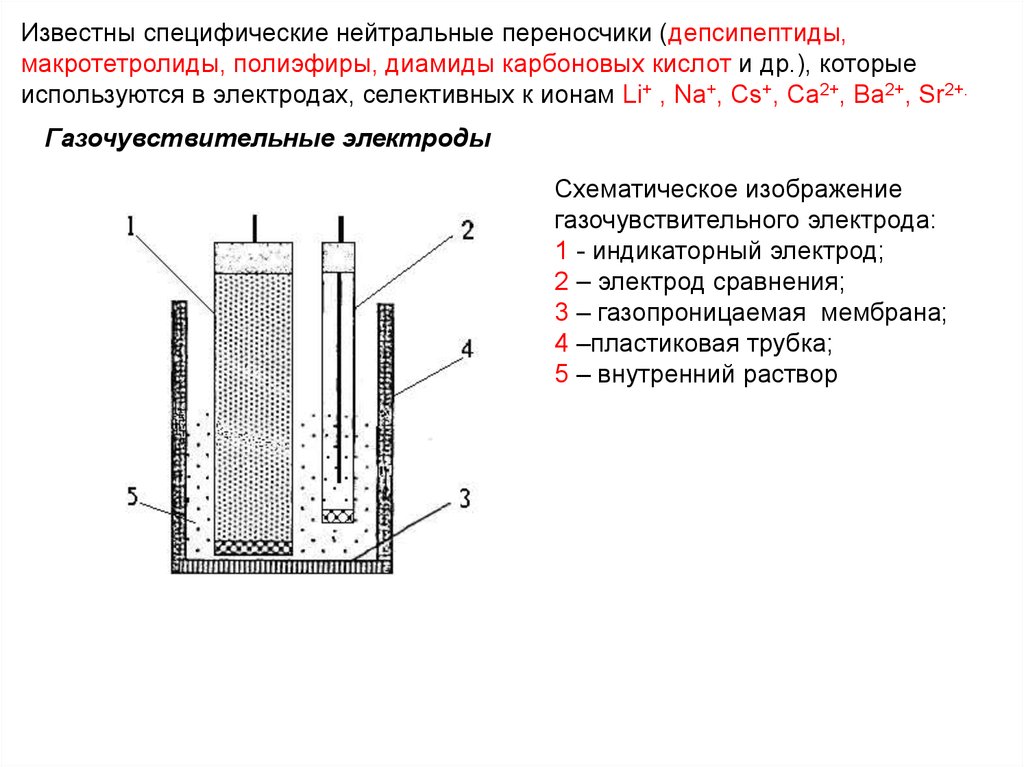
Известны специфические нейтральные переносчики (депсипептиды,макротетролиды, полиэфиры, диамиды карбоновых кислот и др.), которые
используются в электродах, селективных к ионам Li+ , Na+, Cs+, Са2+, Ba2+, Sr2+.
Газочувствительные электроды
Схематическое изображение
газочувствительного электрода:
1 - индикаторный электрод;
2 – электрод сравнения;
3 – газопроницаемая мембрана;
4 –пластиковая трубка;
5 – внутренний раствор
71.
Типы газочувствительных электродовОпределяемое
вещество
NH3
SO2
NO2
H2 S
HCN
HF
CO2
Cl2,Br2,I2
Равновесие во внутреннем
растворе
NH3+H2O=NH4++OHxNH3+Mn+=M(NH3)xn+
SO2+ H2O= H++HSO32 NO2+ H2O=NO3-+ NO2-+2H+
H2S=S2-+2H+
2HCN+Ag+=Ag(CN)2-+ 2H+
HF= H++FCO2+H2O=HCO3-+H+
X2+ H2O= 2H++XO-+X-
Ион, к которому
чувствителен
электрод
H+
Ag+, Cd2+, Cu2+
H+
H+, NO3S2Ag+
FH+
H+, X-
72.
Потенциометрические биосенсорыЕсли индикаторная реакция катализируется ферментами, то
электрохимические системы называют ферментными электродами.
В ферментных электродах в качестве электрохимических датчиков применяют
платиновые, серебряные, графитовые, различные ионоселективные и
газочувствительные электроды. При контакте фермента с исследуемым
веществом в приэлектродном слое происходит ферментативная реакция.
Если продукт этой реакции электрохимически активен, то по изменению
потенциала (или тока) электрода можно судить о количестве определяемого
вещества.
Основные типы реакций, приводящих к изменению потенциала
ферментного электрода:
реакции с образованием Н2О2;
реакции, сопровождающиеся изменением рН среды;
реакции с выделением СО2 или NH3;
реакции с образованием NH4+, CN- или других ионов;
реакции с образованием обратимой редокс-пары, например, I2/I или хинон/гидрохинон.
73.
Определяемоевещество
Фермент
Мочевина
Уреаза
Глюкоза
Глюкозоксидаза
Креатинин
Креатининаза
L-Аминокислоты L-Аминокислотная
оксидаза
L – Тирозин
L-Тирозиндекарбоксилаза
Пенициллин
Пенициллиназа
Амигдалин
Β-Глюкозидаза
Нитрит
Нитритредуктаза
Специфичность
датчика
Диапазон
концентраций, моль/л
NH4+
NH3
CO2
H+
ICO2
NH4+
10-2-10-5
10-2-10-4
10-2-10-4
10-1-10-3
10-3-10-4
10-2-10-4
10-2-10-4
CO2
10-1-10-4
H+
CNNH3
10-2-10-4
10-2-10-5
10-2-10-4
74.
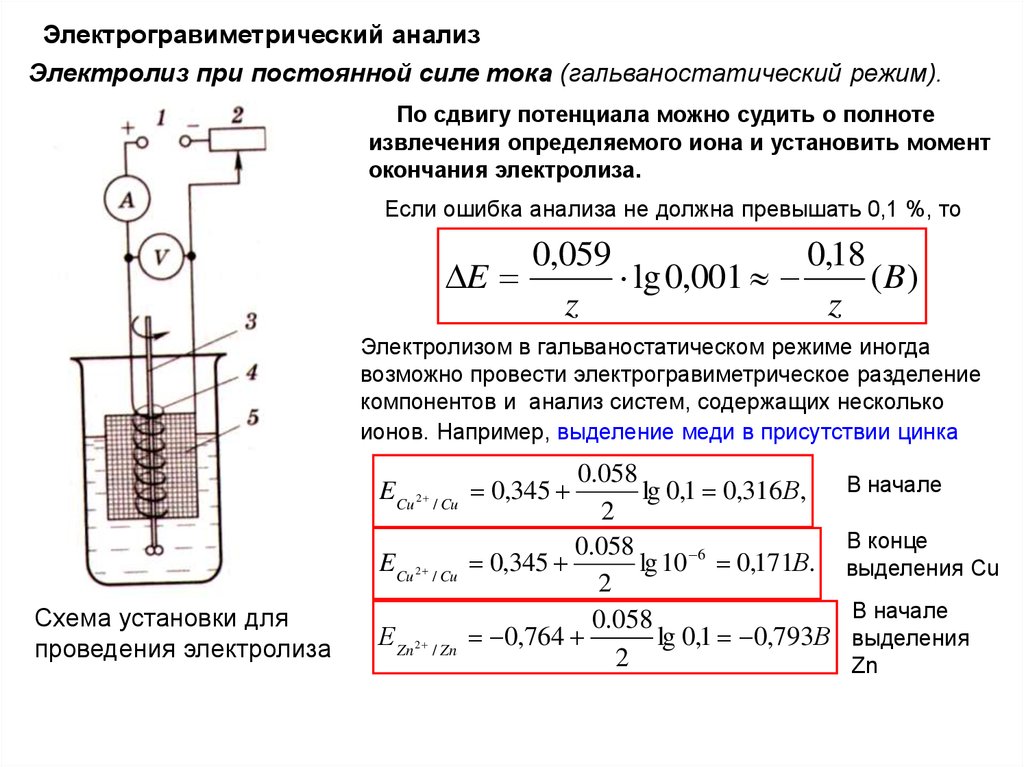
Электрогравиметрический анализЭлектролиз при постоянной силе тока (гальваностатический режим).
По сдвигу потенциала можно судить о полноте
извлечения определяемого иона и установить момент
окончания электролиза.
Если ошибка анализа не должна превышать 0,1 %, то
0,059
0,18
E
lg 0,001
(B)
z
z
Электролизом в гальваностатическом режиме иногда
возможно провести электрогравиметрическое разделение
компонентов и анализ систем, содержащих несколько
ионов. Например, выделение меди в присутствии цинка
0.058
lg 0,1 0,316 В,
2
0.058
0,345
lg 10 6 0,171В.
2
0.058
0,764
lg 0,1 0,793В
2
ECu 2 / Cu 0,345
ECu 2 / Cu
Схема установки для
проведения электролиза
Е Zn 2 / Zn
В начале
В конце
выделения Cu
В начале
выделения
Zn
75.
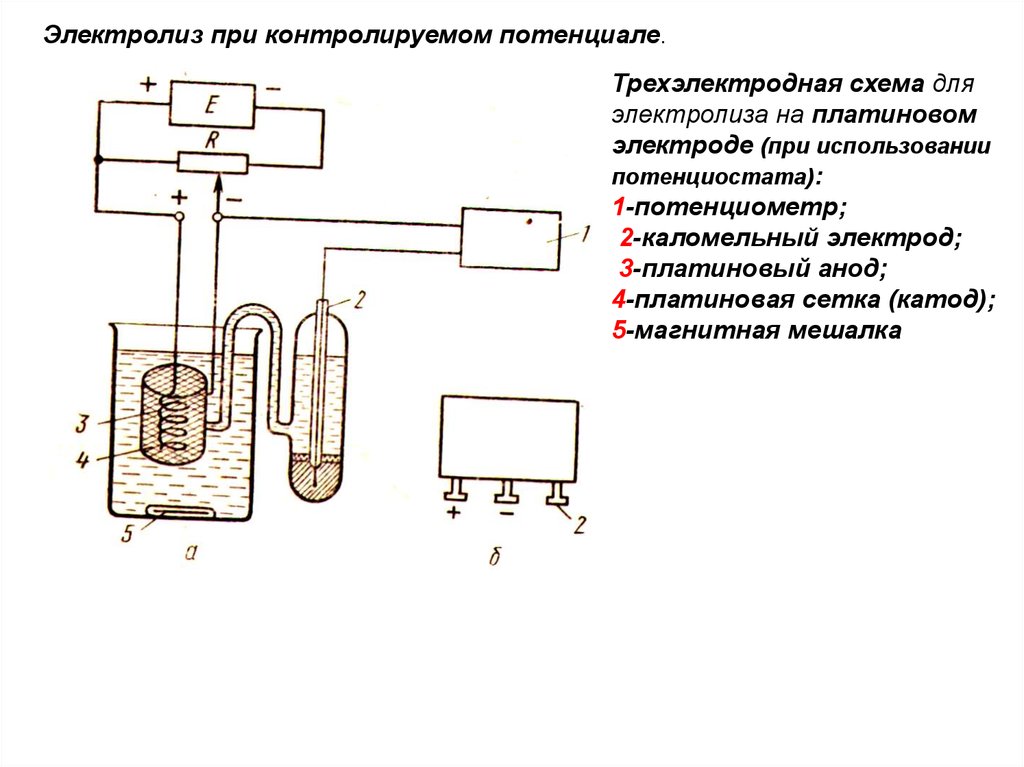
Электролиз при контролируемом потенциале.Трехэлектродная схема для
электролиза на платиновом
электроде (при использовании
потенциостата):
1-потенциометр;
2-каломельный электрод;
3-платиновый анод;
4-платиновая сетка (катод);
5-магнитная мешалка
76.
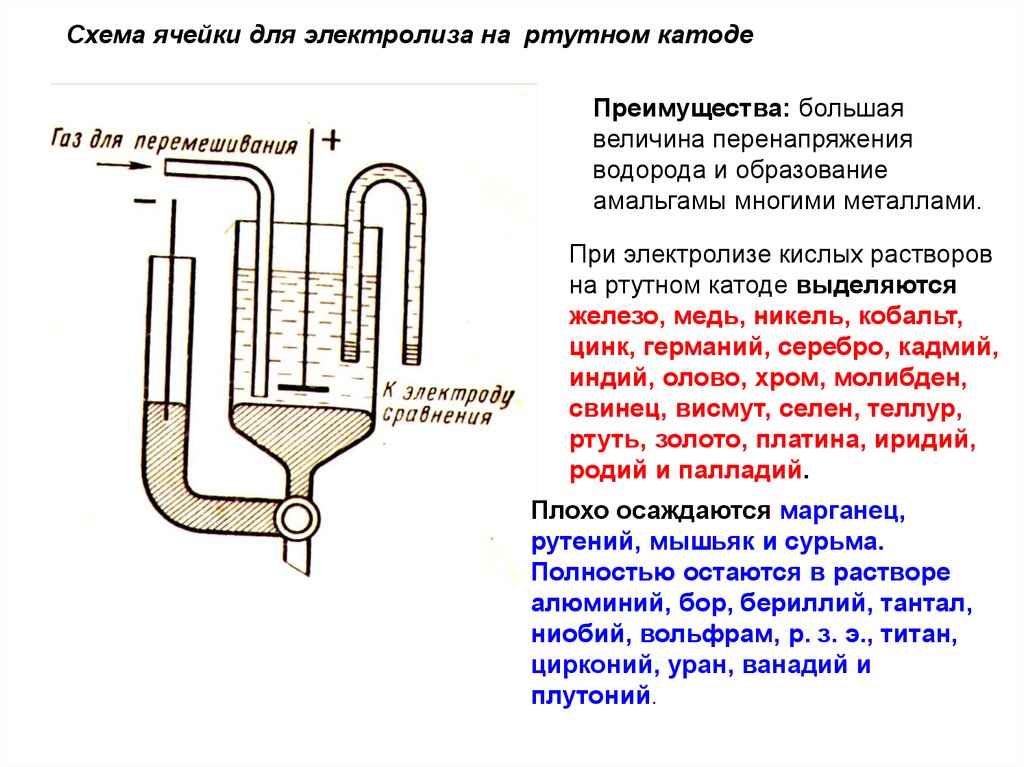
Схема ячейки для электролиза на ртутном катодеПреимущества: большая
величина перенапряжения
водорода и образование
амальгамы многими металлами.
При электролизе кислых растворов
на ртутном катоде выделяются
железо, медь, никель, кобальт,
цинк, германий, серебро, кадмий,
индий, олово, хром, молибден,
свинец, висмут, селен, теллур,
ртуть, золото, платина, иридий,
родий и палладий.
Плохо осаждаются марганец,
рутений, мышьяк и сурьма.
Полностью остаются в растворе
алюминий, бор, бериллий, тантал,
ниобий, вольфрам, р. з. э., титан,
цирконий, уран, ванадий и
плутоний.
77.
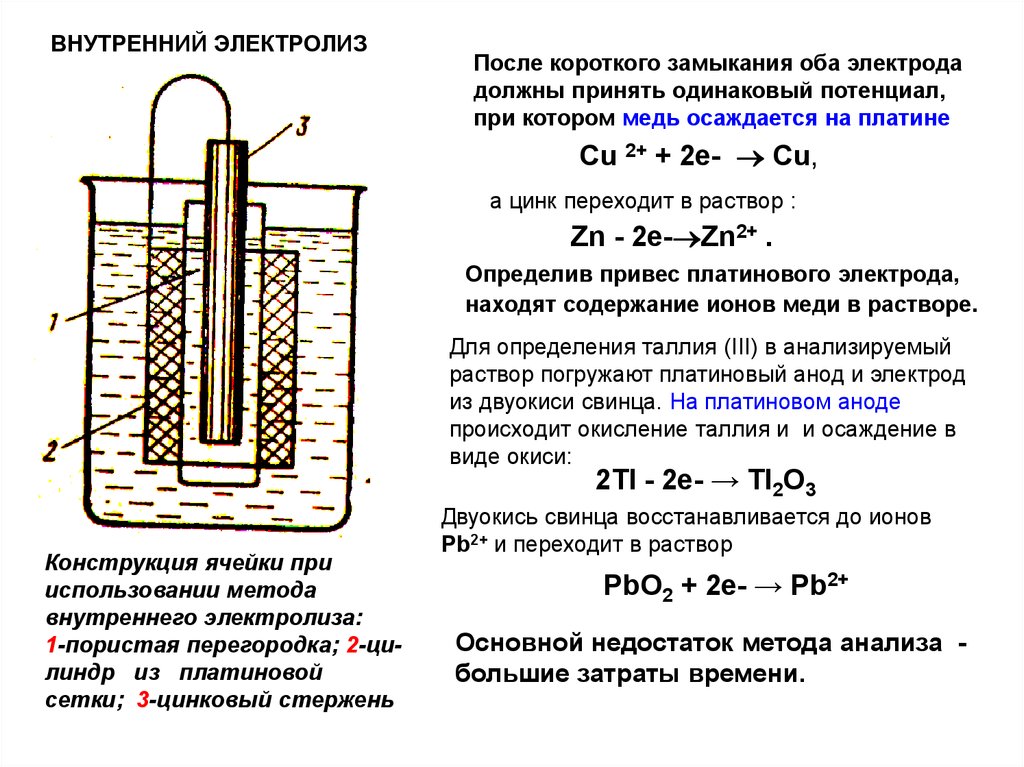
ВНУТРЕННИЙ ЭЛЕКТРОЛИЗПосле короткого замыкания оба электрода
должны принять одинаковый потенциал,
при котором медь осаждается на платине
Cu 2+ + 2e- Cu,
а цинк переходит в раствор :
Zn - 2e- Zn2+ .
Определив привес платинового электрода,
находят содержание ионов меди в растворе.
Для определения таллия (III) в анализируемый
раствор погружают платиновый анод и электрод
из двуокиси свинца. На платиновом аноде
происходит окисление таллия и и осаждение в
виде окиси:
2Тl - 2е- → Тl2О3
Конструкция ячейки при
использовании метода
внутреннего электролиза:
1-пористая перегородка; 2-цилиндр из платиновой
сетки; 3-цинковый стержень
Двуокись свинца восстанавливается до ионов
Рb2+ и переходит в раствор
PbO2 + 2e- → Pb2+
Основной недостаток метода анализа большие затраты времени.
78.
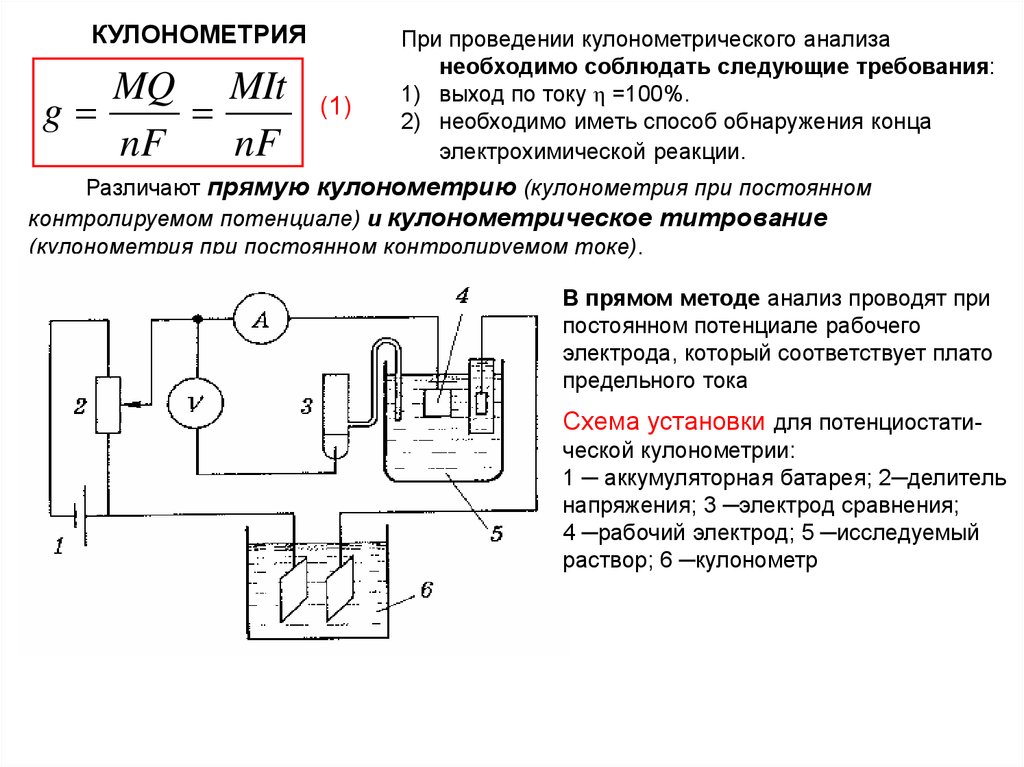
КУЛОНОМЕТРИЯMQ MIt
g
nF
nF
(1)
При проведении кулонометрического анализа
необходимо соблюдать следующие требования:
1) выход по току η =100%.
2) необходимо иметь способ обнаружения конца
электрохимической реакции.
Различают прямую кулонометрию (кулонометрия при постоянном
контролируемом потенциале) и кулонометрическое титрование
(кулонометрия при постоянном контролируемом токе).
В прямом методе анализ проводят при
постоянном потенциале рабочего
электрода, который соответствует плато
предельного тока
Схема установки для потенциостатической кулонометрии:
1 ─ аккумуляторная батарея; 2─делитель
напряжения; 3 ─электрод сравнения;
4 ─рабочий электрод; 5 ─исследуемый
раствор; 6 ─кулонометр
79.
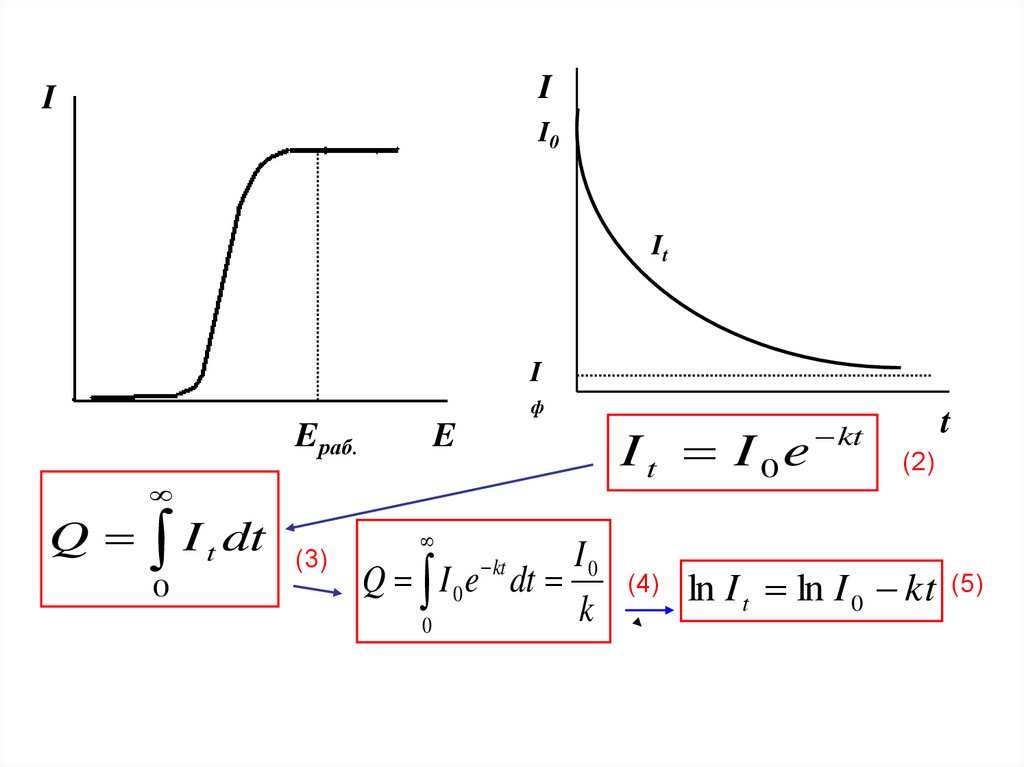
II
I0
It
I
ф
Eраб.
I t I 0e
E
Q
I
0
t
dt
(3)
I0
Q I 0 e dt
k
0
kt
(4)
t
kt
(2)
ln I t ln I 0 kt
(5)