












")
 взаимодействия")







")








")







")






")

 chemistry
chemistrySimilar presentations:










Моделирование структуры биомакромолекул
1.
Моделирование структурыбиомакромолекул
Акберова Н.И., 2017
Лекция 1
2.
М о д е л и р о в а н и е стру к туры б и о м а к р о м о л е к у лДля чего?
3.
М о д е л и р о в а н и е стру к туры б и о м а к р о м о л е к у лС т ру к т ура б елков о п ределяет и х функ ц и ю
Структура
белка
Регуляция
Структура
Сигнальные
пути
Ф ункция
Д виж ение
К атализ
Транспорт
4.
Уровни белковой организации5. Первичная структура: последовательность
• Первичной структурой белка является его аминокислотная последовательностьМО Н О МЕР
П О Л И М ЕР
Аминокислота
П о липептид
Пептидная
сязь
>small ubiquitin-related modifier 3 precursor [Homo sapiens]
MSEEKPKEGVKTENDHINLKVAGQDGSVVQFKIKRHTPLSKLMKAYCERQG
LSMRQIRFRFDGQPINETDTPAQLEMEDEDTIDVFQQQTGGVPESSLAGHSF
6. Первичная структура: последовательность
• Д вадцать аминокислот,встречаю щихся в белках,имеют различные свойства.7. Первичная структура: последовательность
8. Вторичная структура: α-спирали, β-листы, петли
Вторичная структура: α -спирали, β-листы, петли• α -спирали и β-листы формируются путем образования водородных
между атомами кислорода и водорода главной цепи.
связей
9. Вторичная структура: α-спирали
Вторичная структура: α -спирали10. Вторичная структура: β-листы
Антипараллельный β-листПараллельный β-лист
β-лист
β-тяж
Смешанный β-лист
11. Структурные мотивы
12. Третичная структура: домены
13. Мозаичная структура белков
14.
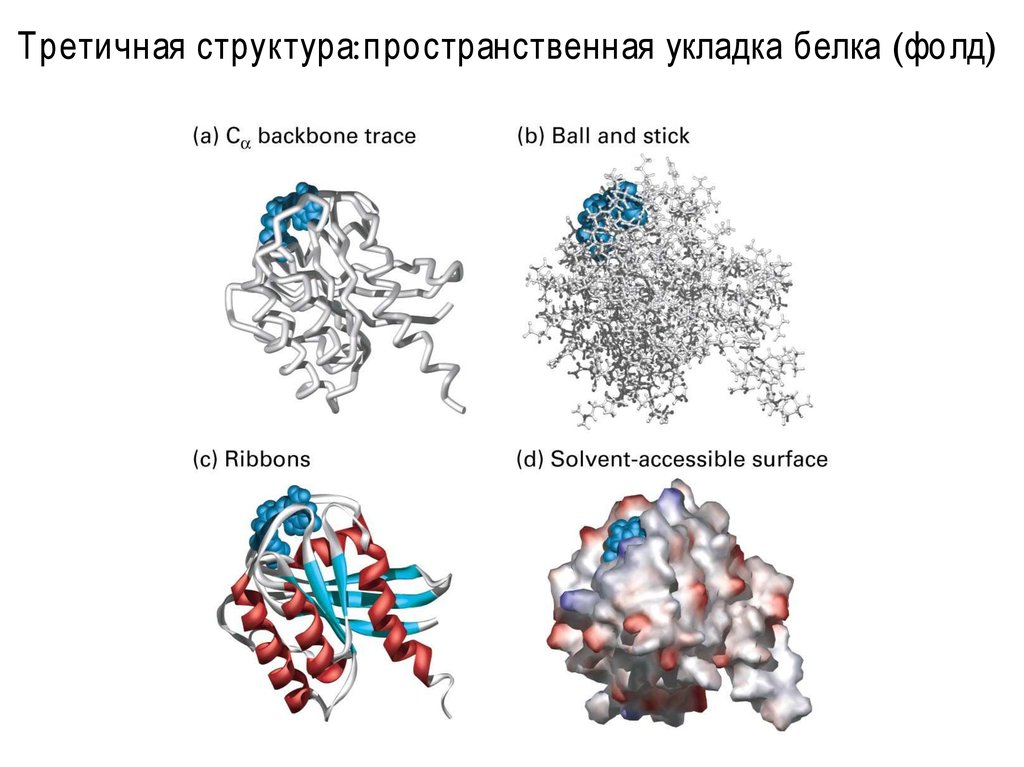
Третичная структура:пространственная укладка белка (фолд)15.
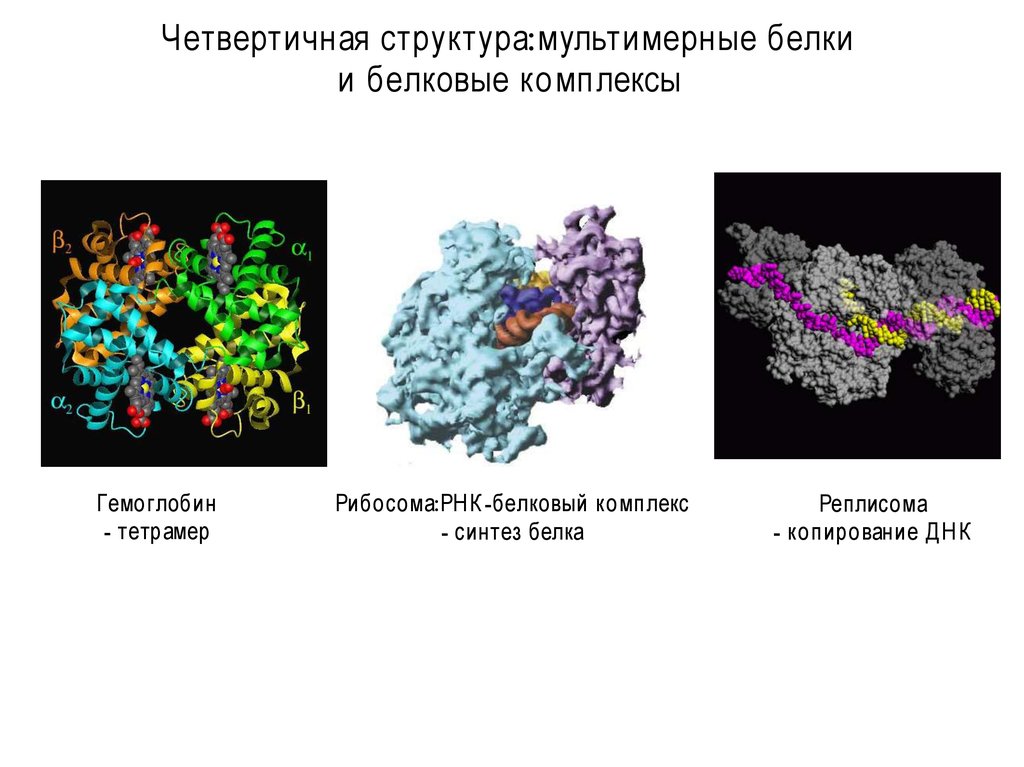
Четвертичная структура:мультимерные белкии белковые комплексы
Гемоглобин
- тетрамер
Рибосома:РН К -белковый комплекс
- синтез белка
Реплисома
- копирование Д Н К
16. Сворачивание белка (фолдинг)
• Сворачивание белка - процесс укладки полипептидной цепи вкомпактную пространственную структуру.
• Аминокислотная последовательность белка одназначно
определяет его пространственную структуру [Anfinsen et al.,
1960s].
• Пространственная структура белка определяет его функцию .
17. Нековалентные (“слабые”) взаимодействия
Водородныесвязи
И о нные связи
Гидрофобные
взаимодействия
Ван-дер-Ваальсовы
взаимодействия
18. Гидрофобность аминокислот
• Гидрофобные эффекты играю т важ ную роль в сворачивании белкаЭкспериментально измеренные уровни гидрофобности аминокислот
19. Определение пространственной структуры белка
О пределение пространственной структуры белка• Экспериментальный подход
- Рентгеноструктурный анализ
- Метод многомерного ЯМР
- Метод криоэлектронной микроскопии
• Вычислительный подход
- Предсказание пространственной структуры белка на основе
информации о его последовательности
Molecular Conceptor v. 2.11, Synergix ltd., USA
20. Преимущества метода рентгеноструктурного анализа.
•принципиально достижимовысокое разрешение. Разрешение
выше 1Å позволяет определять
степень протонирования а/к
остатков в белках
•возможность разрешать структуры
объектов большого размера
(вирусная капсида, рибосома,
фотосинтетический реакционный
центр, т.д.), состоящих из
нескольких десятков тысяч атомов.
Molecular Conceptor v. 2.11, Synergix ltd., USA
21.
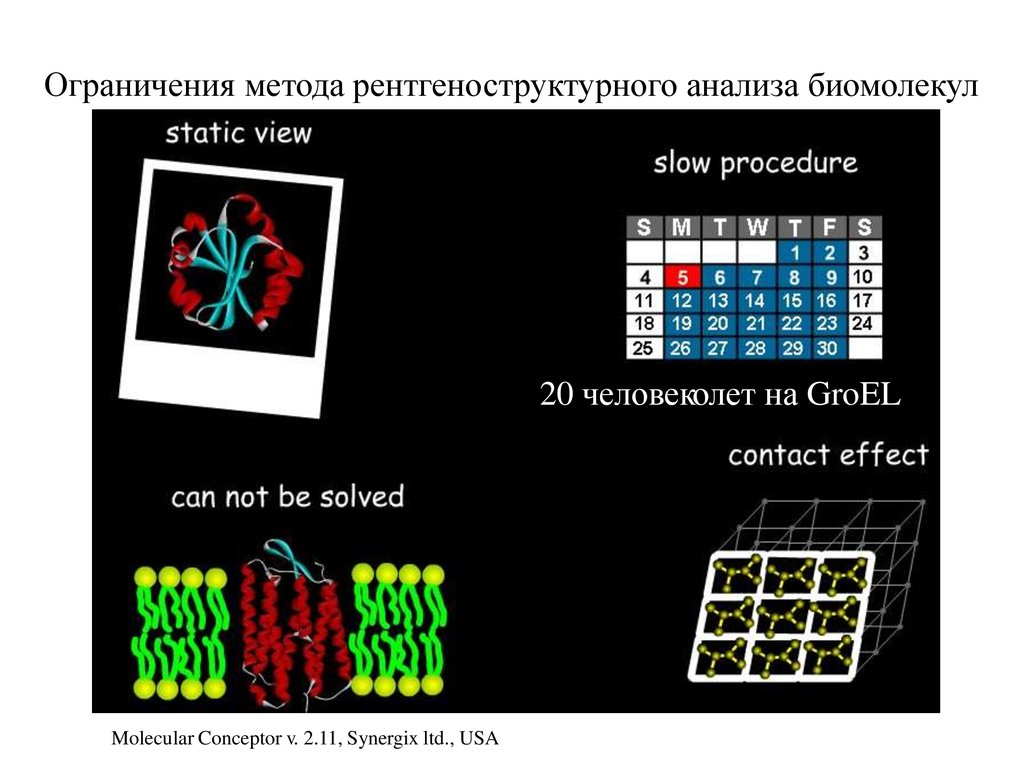
Ограничения метода рентгеноструктурного анализа биомолекул20 человеколет на GroEL
Molecular Conceptor v. 2.11, Synergix ltd., USA
22. Схема рентгеноструктурного исследования
23. Наработка и очистка белка
Выращивание кристалл(а/ов)Molecular Conceptor v. 2.11, Synergix ltd., USA
24. Снятие рентгенограмм кристаллов
РегулярныйКристалл
Размером
От 0,3 мм
Molecular Conceptor v. 2.11, Synergix ltd., USA
25. Определение координат тяжелых атомов биомолекулы
Molecular Conceptor v. 2.11, Synergix ltd., USA26. Protein Structure Initiative (NIGMS, NIH, USA, 2001-2010, 2011-2015)
Выбор объекта$750M
Экспрессия белка
С высоким выходом
Новая структура каждые 2
GroEL за 2 месяца.
Высокоэффективная
очистка
Кристаллизация
дня!
4 крупных и 6 малых центров разрешили за 7 лет более
3000 белковых структур (40% новых структур)
Полуавтоматическая
Валидация стр-ры,
публикация
Помещение в PDB
Service R.B., Science 319, 1610 (2008)
Отбор кристаллов,
Сбор данных
Полуавтоматическое
Разрешение структуры
27. Многомерная ЯМР спектроскопия
Преимущества:•молекулы в растворе
(тяжѐлая вода), не нужно
выращивать кристалл
•положения атомов
водорода м.б. определены
• информация о динамике
атомов м.б. определена
В белках 103 – 104 протонов
Метильные и метиленовые
группы 0,8-3,5 ppm,
ароматические, индольные и
иммидазольные кольца 6,5-8 ppm
В ДНК/РНК Н-2/Н-8 пуринов 8,49 ppm, Н-5 пиримидинов 6,36,6 ppm, Н-6 8,0-8,5 ppm,
метильная группа тимидина
2,3-2,4 ppm.
28.
Bruker Biospin AVANCE 1000The World’s First 1 Gigahertz NMR Spectrometer
World’s First 1 Gigahertz
NMR Spectrometer based on
unique
23.5 Tesla Standard-Bore,
Persistent Superconducting
Magnet (12-tonne, 4.5-metretall machine)
€11.7-million
Dr. Lyndon Emsley,
European Centre for High
Field NMR (CRMN) in
Lyon, France.
Nature 463,605(2010).
(US$16.3-million)
http://www.bruker-biospin.com/av1000-dir.html
29. Ограничение метода многомерного ЯМР
Molecular Conceptor v. 2.11, Synergix ltd., USA•Структура белков < 300 а/к остатков м.б. Определена этим методом, что
составляет менее половины известных белковых последовательностей.
Известны примеры разрешения структур белков из 700 а/к.
• Дороговизна получения образцов с изменѐнным изотопным составом
(13С, 19F, 31P)
• Невысокая точность разрешения структуры
30. Физические принципы метода ядерного магнитного резонанса
Характерные спектрыхимических групп и
соединений
Molecular Conceptor v. 2.11, Synergix ltd., USA
31. Сбор данных
Анализ,Соотнесение
(assignment)
Molecular Conceptor v. 2.11, Synergix ltd., USA
32.
Sequential NOEs ("NOESY walks") in aromaticH1'/H5 region of TWJ-TC acquired in D20 at 30'Cwith a 300 ms mixing time. (A) Strand 1
connectivities. (B) Strand 2 connectivities.
Leontis, N. et al., Biophysical Journal, 68, 251
(1995).
33. Определение координат атомов молекулы
Molecular Conceptor v. 2.11, Synergix ltd., USAДля структур разрешѐнных методом многомерного
ЯМР представлено 10-20 структур. Усреднѐнная
структура имеет наибольшую достоверность.
34.
Электронная микроскопияОпределяется форма крупных межмолекулярных
комплексов методом диффракции электронных пучков.
Типичное разрешение этого метода 3-5 Å не позволяет
определять координаты атомов.
Образцы в замороженном состоянии, что предотвращает
радиационные повреждения и удерживает их в нативном
состоянии.
Molecular Conceptor v. 2.11, Synergix ltd., USA
35. Банк белковых структур. Protein Data Bank (PDB)
http://www.rcsb.org/pdbResearch Collaboratory for Structural Bioinformatics
Каждая структура имеет свой идентификатор (4 символа) и ей соответствует
файл, в котором приведены координаты тяжѐлых атомов.
Структура PDB файла. (Brookhaven Protein Data Bank)
HEADER
COMPND
COMPND
SOURCE
AUTHOR
………
HELIX
HELIX
………
ATOM
ATOM
ATOM
ATOM
……
END
OXIDOREDUCTASE(ALDEHYDE(D),NAD+(A))
2NAD
2
NAD-DEPENDENT FORMATE DEHYDROGENASE (E.C.1.2.1.2)
2 (HOLO FORM) COMPLEXED WITH NAD AND AZIDE
(METHYLOTROPHIC BACTERIUM PSEUDOMONAS SP. 101)
V.S.LAMZIN,Z.DAUTER,V.O.POPOV,E.H.HARUTYUNYAN,K.S.WILSON
2NAD
2NAD
2NAD
2NAD
3
4
5
6
1 H1A GLY A 55
LEU A
59
5 LEFT-HANDED
2 H1 LEU A 59 GLY A 67 1 3/10 FOR RES 63 - 67
2NAD
2NAD
194
195
2NAD
2NAD
2NAD
2NAD
584
585
586
587
281
282
283
284
CB LEU A
CG LEU A
CD1 LEU A
CD2 LEU A
36
36
36
36
65.524 35.308
64.771 34.257
64.749 34.636
63.336 34.097
06-JUL-94 2NAD
0.941
1.735
3.209
1.458
1.00
1.00
1.00
1.00
14.15
17.41
28.05
22.50
36.
https://www.dnastar.com/blog/structural-biology/why-structure-prediction-matters/37. Предсказание пространственной структуры белка
• Ab initio - моделирование укладки “из первых принципов” - безиспользования дополнительной информации о структурах
схож их белков.
• Предсказание на основе гомологии (homology modeling) моделирование на основе известных структур схожих белков.
• Тридинг (Threading) - моделирование на основе слабой
гомологии.
38. Предсказание структуры ab initio
• Ф ункция потенциальной энергии- Модель водного раствора
- О ценка попарного взаимодействия между аминокислотами
• П о иск в пространстве всевозможных конформаций
-
Модель на основе “решетки”
Молекулярная динамика
Использование библиотек известных 3D фрагментов
• Предсказание вторичной структуры
39. Предсказание структуры с использованием решетки
• HP-модель (Hydrophobic-Polar) - рассматривает гидрофобныевзаимодействия как наиболее важные.
-
Не существует эффективных алгоритмов
Плохо отраж ает реальность
40. Предсказание структуры с использованием решетки
41. ROSETTA
Используются структурно консервативные фрагменты
длиной 4-10 аминокислот
П о иск в пространстве конформаций осуществляется
методом Монте К арло
П о лученные структуры кластеризуются и в качестве
результата выдаю тся наилучшие структуры для каждого
кластера
42. Предсказание структуры на основе гомологии
Выравнивание рассматриваемой последовательности с
последовательностями белков с известной 3D структурой
(обычно >30% сходства)
Наложение моделируемой последовательности на
известную структуру согласно выравниванию
Л о кальное улучшение полученной пространственной
структуры
-
Число уникальных укладок (фолдов),наблюдаю щихся в белках,ограничено
(несколько тысяч)
90% помещаемых в PDB структур имеют уже известные укладки (фолды)
43. Примеры укладок (фолдов)
44. Предсказание структуры на основе гомологии
Raw modelLoop modeling
Side chain placement
Refinement
45.
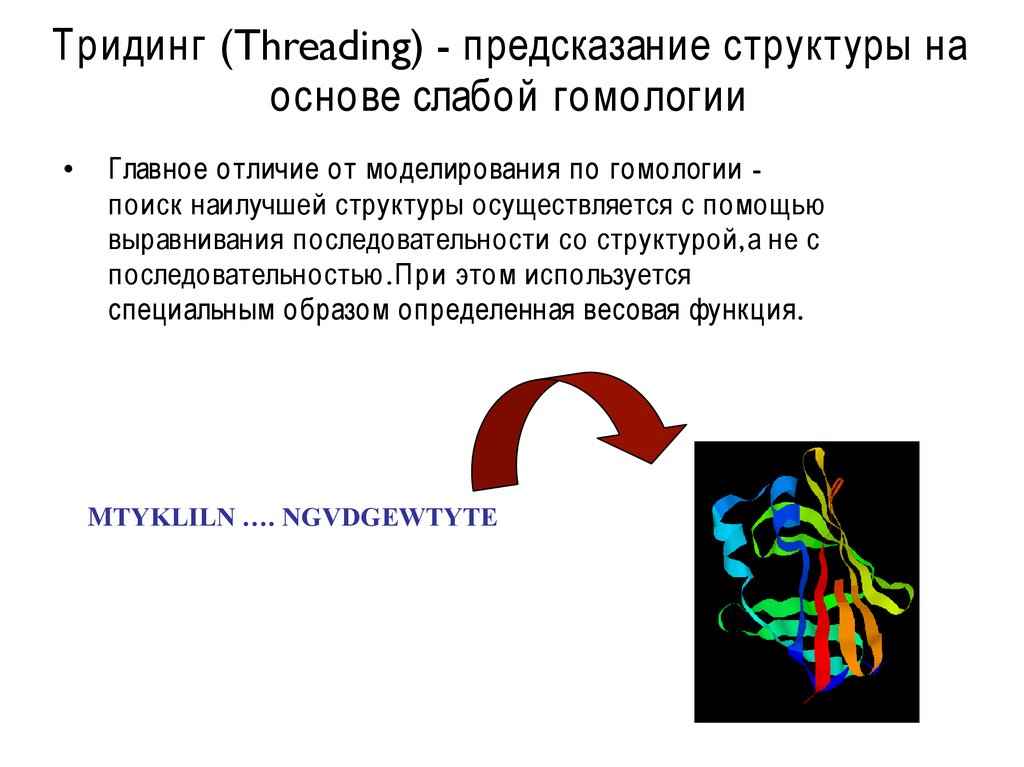
Тридинг (Threading) - предсказание структуры наоснове слабой гомологии
Главное отличие от моделирования по гомологии поиск наилучшей структуры осуществляется с помощью
выравнивания последовательности со структурой,а не с
последовательностью .При этом используется
специальным образом определенная весовая функция.
MTYKLILN …. NGVDGEWTYTE
46. Основные компоненты тридинга
О сновные компоненты тридингабиблиотека уникальных укладок (фолдов)
функция,определяю щая вес выравнивания
последовательности со структурой
алгоритм нахождения наилучшего выравнивания
47. CASP - конкурс методов предсказания структуры белков
Critical Assessment of protein Structure Prediction, CASPFoldIt
48. Гомологическое моделирование третичной структуры белка на основе первичной структуры
Стратегия построения пространственной структуры белков методоммоделирования по гомологиям:
Определения круга гомологичных белков;
Нахождение структурно-консервативных элементов в структуре гомологов (SCRs);
Выравнивание последовательности модельного белка с
последовательностями гомологов, с учѐтом наличия SCR;
Присвоение координат атомов остатков, входящих в SCR, соответствующим
атомам модельного белка согласно выравниванию;
Предсказание конформации петель, соединяющих SCR, а также N- и Сконцов пептидной цепи белка;
Поиск оптимальной конформации боковых остатков аминокислот модельного
белка, отличающихся от остатков опорного белка;
Использование методов регуляризации структуры (энергетическая минимизация и
молекулярная динамика) для уточнения молекулярной структуры с целью
устранения стерических напряжений созданных при построении моделей.
49. Присвоение координат атомов
В первую очередь присваиваются координаты атомам полипептидной цепи.Затем присваиваются координаты атомам боковых цепей. Благоприятный случай,
когда аминокислота модельного белка совпадает с соответствующей кислотой белкагомолога. В этом случае конформация боковой цепи остаѐтся неизменной. Если
боковая цепь аминокислоты модельного белка короче, чем соответствующая цепь
аминокислоты гомолога, более короткая цепь повторяет насколько это возможно
более длинную (торсионные углы
одинаковы). Если же аминокислота
модельного белка более длинная, то начальный ход повторяет ход боковой цепи в
белке-гомологе, а последующие атомы цепи помещаются в развѐрнутую (extended)
конформацию,
вероятно вызывая сильные напряжения в структуре модельного белка.
50.
Поиск конформации соединяющих петельПосле того, как присвоены координаты
атомам, составляющим петли, мы имеем
модельную структуру, которая нуждается в
приведении еѐ в соответствие со
следующими требованиями:
Геометрия пептидной цепи модельной
структуры должна быть регулярной (трансконформация пептидных групп, близкие к
равновесным значения валентных углов и
дли связей);
Атомы не должны перекрываться, т.е.
расстояния между несвязанными атомами
не должны быть существенно меньше, чем
сумма их ван-дер-ваальсовских радиусов;
Боковые цепи аминокислот должны
находиться в равновесной конфигурации;
Если в молекуле имеются дисульфидные
мостики (Cys-Cys связи), то расстояния
между соответствующими атомами серы
должны быть приведены в соответствие с
геометрией;
В структуру должны быть помещены
необходимые простетические группы.
51. Построение пространственной структуры D-amino-acid oxidase из Trigonopsis variabilis (Yeast)
В качестве опорного белка была использована пространственная структура DAmino Acid Oxidase из Rhodotorula gracilis (PDB идентификатор 1C0L)52.
53. Типичная процедура регуляризации модельной структуры белка
1.2.
3.
4.
5.
6.
7.
Энергетическая минимизация участков сочленения SCR и петель с упором на
восстановление нормальной пептидных связей;
Энергетическая минимизация пептидной цепи и боковых остатков петель;
Энергетическая минимизация боковых цепей аминокислот, принадлежащих SCR,
подвергшихся замене при присваивании координат;
Энергетическая минимизация всех боковых остатков белка;
Энергетическая минимизация (500-1000 шагов) всей структуры модельного белка;
Молекулярная динамика модельного белка в вакууме на протяжении 20-50 пикосекунд;
Финальная энергетическая минимизация структуры белка (200-500 шагов).
Результатом этой процедуры будет белковая структура с правильной стереохимией (длины
валентных связей и значения валентных углов не будут существенно отличаться от равновесных
значений), с отрицательной энергией несвязанных взаимодействий (свидетельство того, что не
наблюдается перекрытие ван-дер-ваальсовских радиусов атомов), с отрицательной энергией
электростатических взаимодействий (произошло сближение противоположно заряженных атомов)
и с ненулевой энергией водородных связей (в молекуле установились водородные связи).
Дальнейшая регуляризация структуры приведѐт к еѐ улучшению с точки зрения стереохимии, но
при этом возрастут искажения структуры активного центра (центра связывания) вашей структуры.
Модельная структура построена и отрелаксирована. Она обладает участками структурноконсервативных областей, унаследованных от белков гомологов, правильной стереохимией
(результат регуляризации). Дальнейшие манипуляции с этой структурой (подгонка геометрии
активного центра, точечные мутации) зависят от цели исследований. Полученную структуру
надо рассматривать как средство иллюстрации результатов вашей работы
(объяснения экспериментальных фактов, гипотезы).