


























 medicine
medicineSimilar presentations:
")
")








Генетика людини. Біосоціальна суть людини
1. Генетика людини
Біологічний вид Homo sapiens складає частина біосфери і продукт їїеволюції. Закономірності біологічних процесів, що відбуваються на
клітинному рівні і що мають універсальне значення в природі,
повною мірою прикладені до людини: організація еукаріотичної
клітини, її компартменталізация, основні шляхи метаболізму,
закономірності мітоза і мейоза і багато інших, розглянуті в
попередніх розділах. До людини застосовно більшість
фізіологічних закономірностей, характерних для царства тварин,
класу ссавців, загону приматів, сімейства гомінід, до яких він
відноситься. Все це підкреслює його зв'язок з світом живих істот.
До людини повною мірою прикладені закономірності спадковості і
мінливості.
2. Біосоціальна суть людини
Поза сумнівом, людина, як і всі інші організми, випробовує вплив мутаційного процесу - мутаційний тиск. Такі чинники, як міграція, абопотік генів, вибірковість спаровування, дрейф генів в ізольованих популяціях, зберігають своє значення і для людини. При цьому дія одних
чинників, наприклад міграції, посилюється, дія інших ослабляється, наприклад, все менше стає ізольованих популяцій з їх
близькоспорідненими браками і підвищеним коефіцієнтом інбрідінга і т.д. В той же час головний чинник еволюції - природний відбір - не
грає тієї ролі в людському суспільстві, яку він грає в популяціях всіх інших організмів.
Проте це не означає, що людина зовсім закінчила свою еволюцію. Еволюція людини перейшла переважно в сферу соціальну. Власне
соціальна сфера еволюції - це культура. Багато дослідників разом з генетичною спадковістю пропонують розглядати сигнальну спадковість
(М. Е. Лобашев), соціальну спадковість (Н. П. Дубінін), спадкоємність (З. Н. Давіденков), просто культуру (О. Солбріг і Д. Солбріг) і інші
поняття. Всі вони означають різні форми передачі досвіду між поколіннями.
Сигнальна спадковість є передачею навиків адаптивної поведінки від батьків нащадкам, а також в межах одного покоління і навіть від
нащадків батькам. Поза сумнівом, що сигнальна спадковість з'явилася вже у тварин і формувалася на основі різних способів комунікацій.
Сюди можна віднести явище імпринтінгу (відображення) поведінкових реакцій, що формуються на ранніх стадіях постнатального розвитку
хребетних, різні способи навчання і наслідування.
Серед ссавців сигнальна спадковість високого рівня досягла у приматів. О. Солбріг і Д. Солбріг наводять приклад розповсюдження
«культурної ознаки» в стаді макак (Масаса fuscata). «Це стадо приохотилося до нової їжі - бататів. Одна півторарічна самка, перш ніж з'їсти
батат, стала не просто відскрібати з нього пісок, а відмивати його в морі. Таку поведінку перейняли інші молоді особини. Матері навчилися
цьому у своїх підростаючих дитинчат і стали у свою чергу навчати такій поведінці молодших нащадків. Врешті-решт все стадо, за винятком
старих самців, які мало спілкуються з рештою стада, стало мити батати, перш ніж з'їсти їх».
Динаміка сигнальних, або культурних, ознак схожа з динамікою біологічних ознак, тобто ознак, контрольованих генами і хромосомами.
Популяції тварин можуть розрізнятися за сигнальними ознаками - поведінкою; ці ознаки, подібно до потоку генів, можуть передаватися
іншим популяціям. Можливий дрейф сигнальних ознак, і за цими ознаками, як і по біологічних, можливий відбір.
Разом з цими рисами схожості є щонайменше одна істотна відмінність - спадкоємство сигнальних ознак, придбаних в онтогенезі. При цьому
йдеться не про спадкове закріплення онтогенетичних змін, а про сигнальну спадковість.
Зачатки соціальної поведінки і явища сигнальної спадковості є вже у тварин, але найвищого розквіту сигнальна спадковість досягла в
людському суспільстві у формі культури. Соціальна еволюція людини склалася на фундаменті біологічної еволюції. В той же час
біосоціальна суть людини накладає відбиток на прояв біологічних, зокрема генетичних закономірностей, яким підкоряється його
індивідуальний і еволюційний розвиток.
3. Людина як об'єкт генетики
Людина як об'єкт генетикиГенетика людини сформувалася з урахуванням наступних особливостей, що
створюють труднощі при вивченні його спадковості і мінливості:
1) неможливості направлених схрещувань для генетичного аналізу;
2) неможливості експериментального отримання мутацій;
3) пізнього статевого дозрівання;
4) нечисленності потомства;
5) неможливості забезпечення однакових і строго контрольованих умов для
розвитку нащадків від різних браків;
6) недостатній точності реєстрації спадкових ознак і невеликих родоводів;
7) порівняно великого числа (2n=46) хромосом, що погано розрізняються.
Останнє утруднення, як, втім і всі інші, стосуються додатку так званих традиційних
методів генетичного і цитогенетичного аналізу. Останніми роками розвиток нових
методів в генетиці і застосування їх до людини дозволили усунути багато, але
далеко не всі труднощі роботи з людиною як з генетичним об'єктом.
4. Методи генетики людини
1. Генеалогічний метод дозволяє подолати складнощі, що виникають у зв'язку з неможливістю схрещування імалоплодністтю людини. Якщо є родоводи, то можна, використовуючи сумарні дані по декількох сім'ях,
визначити тип спадкоємства (домінантний, рецесивний, зчеплений зі статтю, аутосомний) ознаки, а також її
моногенність або полігенність.
Так, домінантна ознака «габсбургськая губа» (товста випнута нижня губа) простежується в династії Габсбургов,
починаючи з XV в. Аналогічне спадкоємство легко виявляється для ознаки брахідактилія, або короткопалість,
унаслідок недорозвинення (зрощення) кінцевих фаланг. За домінантним типом успадковується такий дефект, як
ахондроплазія - карликовість, пов'язана з різким укороченням кінцівок, і ін.
Використання генеалогічного методу дозволило встановити характер спадкоємства гемофілії А, або так званій
королівській гемофілії. Ця ознака - нездатність згущуватися крові унаслідок дефекту або відсутності в організмі
антігемофільного глобуліну (або чинника VIII) - виражається у кровотечах, що не зупиняються, які виникають
при щонайменших пораненнях, і приводить до загибелі хворих в ранньому віці. У хворих не утворюється фібрин
з фібріногена, а саме нитки фібрину грають важливу роль в реакції згортання крові.
Гемофілія А успадковується як рецесивна алель в Х-хромосомі, тобто виявляє зчеплення зі статтю. Відомо, що
носійкою (гетерозіготою) гемофілії А була англійська королева Вікторія. Один з її синів був гемофіліком і дві дочки
виявилися гетерозіготними носійками цієї хвороби. Надалі завдяки бракам між представниками царюючих
прізвищ Європи ця рецесивна, зчеплена зі статтю алель розповсюдилася серед правителів Німеччини, Росії і
Іспанії. Ознака виявляє типове крисс-крос спадкоємство. Чоловіки вражаються частіше, оскільки мають всього
одну Х-хромосому. Відомі випадки гемофілії у жінок. Лікування хворих засноване на введенні ним великих
кількостей антигемофільного глобуліну, що одержується з донорської крові.
Інший тип - гемофілія В - відкритий порівняно недавно. Він пов'язаний з дефектом іншого чинника згортання
крові - тромбопластину, або чинника IX. Це також рецесивна ознака, зчеплена зі статтю. Істотна трудність, з
якою зустрічається генеалогічний метод, - брак докладних родоводів.
5.
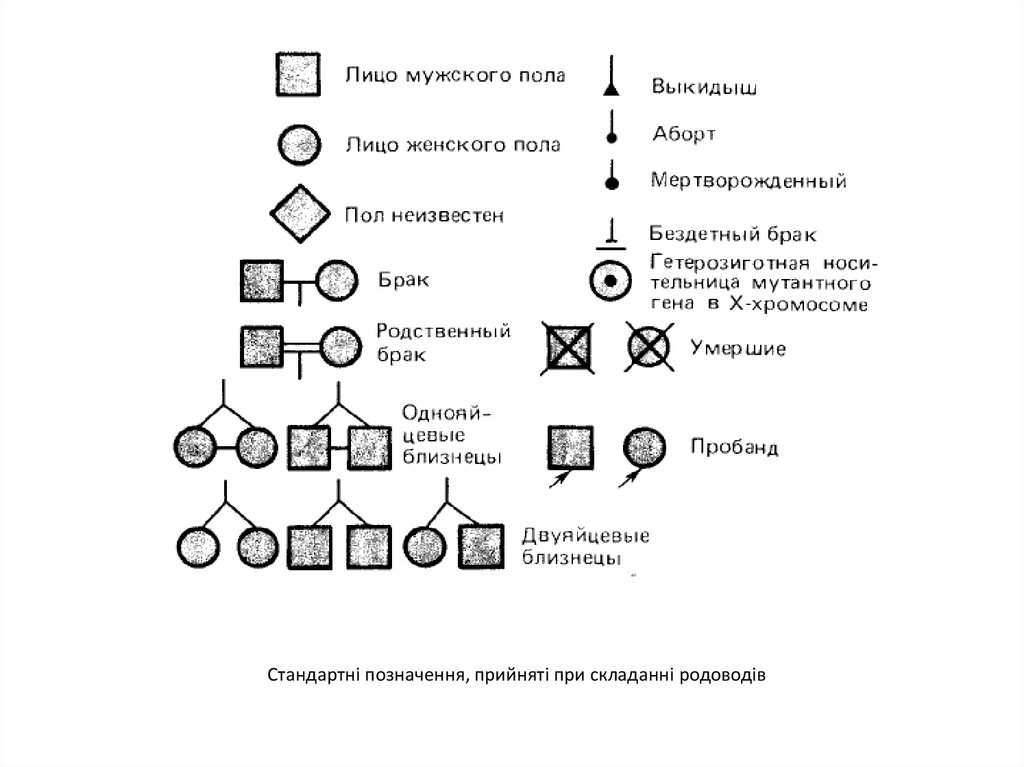
Стандартні позначення, прийняті при складанні родоводів6.
Родовід, що ілюструє спадкоємство гемофілії А. Червоні значки - хворі індивідууми; I - IV покоління7.
Приклад успадкування за домінантним типом – успадкування ахондроплазії.8.
2. Близнюковий метод використовується для з'ясування ступеня спадковоїобумовленості досліджуваних ознак. Явище поліембріонії відоме у деяких
тварин. Воно характеризується появою декілька ідентичних, або однояйцевих
близнят (ОБ) - монозіготних близнят. Разом з такими Про існують різнояєчні
близнята (РБ), що народжуються при заплідненні двох одночасно дозріваючих
яйцеклітин. Якщо Про як результат клонового розмноження однієї заплідненої
яйцеклітини завжди ідентичні по підлозі і дуже схожі, часто практично невиразні,
то РБ можуть мати як однакову, так і різну підлогу. Зустрічаються РБ, що сильно
розрізняються за зовнішніми ознаками, як розрізняються особини, що виникли в
результаті самостійних випадків запліднення. В цьому випадку РБ представляють
результат розщеплювання при схрещуванні.
Основоположником близнюкового методу в генетиці людини був Ф. Гальтон, (XIX
в.), який звернув увагу на те, що є близнята схожі і розрізняються і запропонував
порівнювати їх між собою для з'ясування впливу спадковості і «живлення», тобто
впливи середовища на прояв ознак. Не дивлячись на те що Ф. Гальтон у той час не
міг точно вказати, які близнята є ОБ, а які РБ, він вперше застосував підхід,
широко використовуваний в генетиці людини в даний час. Близнюковий метод
заснований на трьох положеннях:
ОБ мають ідентичні генотипи, а РБ - різні генотипи.
Середовище, в якому розвиваються близнята і під дією якої з'являються
відмінності ознак у ОБ, може бути однаковим і неоднаковим для однієї і
тієї ж пари ОБ.
Всі властивості організму визначаються взаємодією тільки двох чинників:
генотипу і середовища.
9.
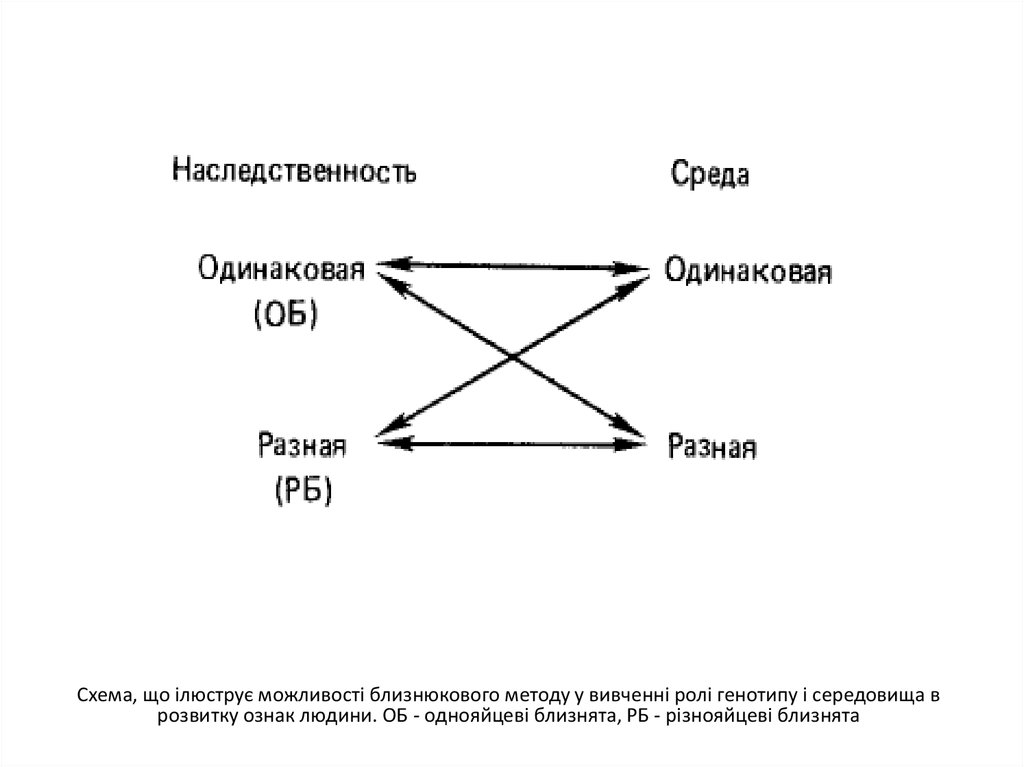
Схема, що ілюструє можливості близнюкового методу у вивченні ролі генотипу і середовища врозвитку ознак людини. ОБ - однояйцеві близнята, РБ - різнояйцеві близнята
10.
ОБ і РБ зазвичай порівнюють по ряду показників на великому матеріалі. На основіодержаних даних обчислюють показники конкордантності (частоти схожості) і
дискордантності (частоти відмінностей). Деякі приклади порівняння ОБ і РБ,
надані в таблиці, показують, що у ОБ конкордантність значно вище, ніж у РБ,
проте ступінь схожості для різних ознак варіює. Це дозволяє оцінити роль
генотипу і середовища в їх прояві.
Запропоновано декілька способів кількісного виразу частки спадковості в
розвитку ознаки. Простий з них:
де Н - частка спадковості, М - конкордантность у ОБ, D - конкордантность у РБ.
Близнюковий метод дає цінні результати при вивченні морфологічних і
фізіологічних ознак. Його можливості не слід переоцінювати, особливо в
застосуванні до показників соціальної поведінки людини. Багато авторів
намагалися аналізувати схильність до злочинів, використовуючи близнюковий
метод.
Конкордантність деяких ознак людини у однояйцевих і різнояйцевих близнят.
11. Близнюковий метод дає цінні результати при вивченні морфологічних і фізіологічних ознак. Його можливості не слід переоцінювати, особливо
Близнюковий метод дає цінні результати при вивченні морфологічних і фізіологічних ознак. Його можливості не слідпереоцінювати, особливо в застосуванні до показників соціальної поведінки людини. Багато авторів намагалися
аналізувати схильність до злочинів, використовуючи близнюковий метод
Конкордантність деяких ознак людини у однояйцевих і різнояйцевих близнят
Конкордантність, %
Признаки
ОБ
РБ
100
64
Нормальні ознаки
Групи крові системи АВ0
Форма брів
100
51
Колір очей
99,5
28
Колір волосся
97
23
Папілярні лінії кистей рук
92
40
Клишоногість
23
2
Грижа спинного мозку
77
33
Синдром Дауна
89
7
Рахіт
88
22
Паралітинчний поліомієліт
36
6
Кір
95
87
Скарлатина
84
47
Дифтерит
50
38
Рак
16
14
Епілепсія
67
3
Слабоумство
91
53
Шизофренія
80
Маніакально-депресивний психоз
77
13
19
Патологічні стани
З цієї таблиці виходить, що якщо один з пари близнят скоює злочин, то вірогідність того, що і другий близнюк
злочинець, значно вище для ОБ, чим для РБ.
12.
Полярні координати, що ілюструють обмін речовин в організмі людиниА - гіпотетичний середній індивідуум; Б, В - фактичні приклади неспоріднених дорослих
індивідуумів; Г, Д -однояйцевые близнята.
Як показники (довжина променя) використані значення інтенсивності обміну таких з'єднань, як: 1
- креатинін, 2 - сахароза, 3 - KCl, 4 - NaCl, 5 - HCl, 6 - сечова кислота, 7 - глюкоза, 8 - лейцин, 9 валін, 10 - цитруллін, 11 - аланін, 12 - лізин, 13 - таурін, 14 - гліцин, 15 - серіy, 16 - глутамінова
кислота, 17 - аспарагинова кислота, 18 - солі лимонної кислоти і т.д.
13.
Висновки, що далеко йдуть, про роль спадковості в асоціальній поведінці людейбули б поспішними, оскільки в приведених дослідженнях не враховувалися ряд
важливих соціальних показників, характерних для близнят. Перш за все те, що
ОБ, як правило, дружніші РБ. Для ОБ характерні більш схожий соціальний досвід і
замкнутість. У більшості досліджень не вказано, які саме злочини скоїли
близнята, чи були їх вчинки схожими або різними. Необхідний також аналіз
формування особи близнят, їх спілкування, середовища і т.д. Схожість поведінки
Про може бути результатом їх однакового соціального досвіду, їх поведінка може
бути обумовлена поєднанням однакового генотипу і однаковою або дуже
схожого соціального середовища.
Для застосування близнюкового методу потрібне критичне відношення до його
можливостей. Приклад послідовного застосування близнюкового методу
представляє дослідження ролі генотипу і модифікацій у формуванні обміну
речовин людини. На попередньому малюнку були графічно умовно представлені
полярні координати, що відображають обмін різних з'єднань у двох однояйцевих
близнят і двох неспоріднених індивідуумів порівняно з якимсь гіпотетичним
середнім варіантом. За деякими показниками різко розрізняються навіть
однояйцеві близнята - результат клонового розмноження організмів. Такі
відмінності можуть бути пов'язані як з випадковими змінами середовища, що
відбувалися в онтогенезі близнят, так і з природними (внутрішніми) причинами
модифікаційної мінливості.
Таким чином, застосування близнюкового методу показує, що такі генетично
детерміновані ознаки, як показники обміну речовин, можуть сильно піддаватися
модифікаційній мінливості. Це слід враховувати, зокрема, при описі симптомів
спадкових захворювань, що дуже важливе в медичній генетиці.
14. Порівняння злочинної поведінки однояйцевих і різнояйцевих близнят
Автор, рікдослідження
ОБ
РБ
Страна
Кількіс Другий близнюк Кількіс Другий близнюк
ть пар
ть пар
Теж
Не
Теж
Не
злочине злочине
злочине злочине
ць
ць
ць
ць
Ланге, 1929
Легра, 1932
Кранц, 1936
Штумпфль, 1939
Боргстрем, 1939
Розанов и др., 1941
Иосимасу, 1957
Хайяси, 1967
Христиансен, 1968
Суммарные данные
Германия
Голландия
Германия
»
Финляндия
США
Япония
»
Дания
13
4
31
18
4
45
28
15
91
10
4
20
11
3
35
14
11
48
3
0
11
7
1
10
14
4
43
17
2
15
5
0
5
43
23
20
19
7
12
5
2
3
27
6
21
26
0
26
Данные недостаточны
122
29
93
249
156
62,6 %
93
264
69
25,4%
195
15. Цитогенетичний метод
Як уже згадувалося, досить велике число важко відмітних один від одного (в межах груп) хромосом створювали труднощів застосуванні цитологичеського методу і в розвитку цитогенетіки людини. Розробка методів діференційнного
забарвлення спростила проблему ідентифікації всіх хромосом людини. Завдяки культивуванню клітин людини in vitro
можна одержувати чималий матеріал для опису цитологічних особливостей досліджуваного індивідуума. Для цього
зазвичай використовують короткочасну культуру лейкоцитів периферичної крові.
Цитологичний контроль застосовують при діагностиці ряду спадкових захворювань, які пов'язані з явищами анеуплоїдії і
різною хромосомною аберацією. Разом з вивченням мітотичних хромосом корисну інформацію одержують і при
спостереженні інтерфазних клітин. Зокрема, чоловіків і жінок розрізняють по наявності в інтерфазному ядрі так званого
тельця Барра, або статевого хроматіна. Він є у жінок і відсутній у чоловіків. Статевий хроматін є результатом
гетерохроматинізациі одній з двох Х-хромосом, що інактивується у жінок. Ідентифікація статевого хроматину в клітинах
зіскрібку слизистої оболонки рота широко застосовується для визначення генетичної підлоги пацієнтів в практиці медичної
генетики, а також в спортивній медицині.
Шляхом цитологичеськіх досліджень людей, що мають різне число Х- і Y-хромосом унаслідок нерозбіжності, виявили, що
число тілець статевого хроматіна відповідає формулі
В = X — Р/2,
де В - число тілець Барра, X - число Х-хромосом, Р - плоїдность. Для диплоїдних клітин це означає, що число грудок
статевого хроматіна рівне числу Х-хромосом мінус одиниця.
Цитологичний метод придбав велике значення у зв'язку з можливостями, які відкрила гібридизація соматичних клітин.
Як було показано в гл. 11, отримання гібридів між соматичними клітинами людини і миші дозволяє в значній мірі
подолати проблеми, пов'язані з неможливістю схрещувань, і картировать багато генів, контролюючих метаболізм клітини.
Клонування ряду генів людини дозволило виявити поліморфізм по цих генах на основі використання рестрикційного
аналізу хромосомною ДНК. Фрагменти, що розрізняються, ДНК, відповідна даному гену або його ділянкам, виявляється за
допомогою електрофорезу рестриктів і подальшій гібридизації їх з радіоактивним зондом - клонованою ділянкою ДНК, по
якій вивчають поліморфізм. Такі зонди використовують також для локалізації генів безпосередньо на препаратах
хромосом. Об'єднання цього підходу з методом диференціального забарвлення дає можливість «прив'язувати» конкретні
гени до ділянок конкретних хромосом. Об'єднання генеалогічного методу з цитогенетичним, а також з новітніми
методами генної інженерії значно прискорило процедуру картування генів у людини.
16. Популяційний метод
Популяційний метод, або методи генетики популяцій, про які мовилося в гл. 18, широко застосовуються вдослідженнях людини. Він дає інформацію про ступінь гетерозиготності і поліморфізму людських популяцій,
виявляє відмінності частот алелей між різними популяціями. Так, добре вивчено розповсюдження алелей
системи груп крові АВ0. Різну концентрацію конкретних алелей локуса І пов'язують з відомими даними про
чутливість різних генотипів до інфекційних хвороб Це допомагає поня1ь напрям еволюції і відбору, що діяв в
різних регіонах, в історії людства.
Передбачається, що зниження концентрації аллелі І0 у ряді популяцій було пов'язано з розповсюдженням чуми,
оскільки збудник чуми Pasteurella pestis володіє властивістю антигена 0. Зниження концентрації аллелі ІА
пов'язують з епідеміями віспи, які переважно винищували носіїв антигена А.
Метод популяції дозволяє визначити адаптивну цінність конкретних генотипів. Багато ознак і відповідно
обумовлюючі їх гени адаптивний нейтральні і виявляються як природний поліморфізм людських популяцій
(наприклад, багато морфологічних ознак: колір очей, волос, форма вух і т. д.). Інші ознаки виникли як адаптивне
по відношенню до певних умов існування; наприклад, темна пігментація шкіри негрів оберігає від дії сонячної
радіації. Відомі приклади умовно адаптивних алелей. До їх числа відноситься така генетична аномалія, як
серповидноклітинна анемія. Рецесивна алель, така, що викликає в гомозиготному стані це спадкове
захворювання, виражається в заміні всього одного амінокислотного залишку у в-цепі молекули гемоглобіну
(глутамінова кислота або валін). Хворі серповидноклітинною анемією гинуть в ранньому віці. Ця хвороба
поширена в популяціях тропічної Африки і тропічної Азії. Порівняно висока частота летальною алелі у вказаних
районах спантеличувала дослідників, поки А. Аллісон не виявив, що гетерозіготи по серповидноклітинній анемії
набагато стійкіше до малярії, чим гомозіготи по нормальній алелі. Таким чином, в природних умовах
розповсюдження малярії в місцевих популяціях відбір йшов у бік підтримки в гетерозігі алелі, очевидно,
шкідливою в гомозиготному стані.
У популяціях людини так само, як і в популяціях інших організмів, в гетерозіготному стані міститься значний
генетичний тягар, тобто рецесивні алелі, що приводять до розвитку різних спадкових хвороб. Підвищення
ступеня інбридінгу в популяціях повинне приводити до підвищення частоти гомозіготізаціїрецесивних алелей. Ця
закономірність повинна застерігати від укладення близькоспоріднених шлюбів. Порівняння частот спадкових
аномалій у нащадків від неспоріднених браків і від браків між кузенами (двоюрідними братами і сестрами)
показує, що в другому випадку частота аномалій зазвичай значно вище.
17.
Джерелом генетичного тягаря служать мутації, що виникають в популяціях людини спонтанно абопід дією чинників навколишнього середовища, серед яких все більшої питомої ваги набувають так
звані антропогенні чинники. Не дивлячись на насущну необхідність знання частот спонтанної
мутації у людини, загальна оцінка цього показника поки вельми приблизна. Значно простіше
оцінити частоту домінантних, ніж рецесивних, мутацій. Наприклад, за даними данських генетиків,
частота домінантної мутації ахондроплазії складає 0,00004 на локус за покоління, тобто 4 гамети з
100 000 несуть знов виниклі мутації. Дані, що стосуються інших мутацій, коливаються між
частотами близько 1 на 10 000 і 1 на 100 000 на локус.
Якщо прийняти, що число структурних генів в гаплоїдному наборі людини від 100 000 до 1 000 000
(за оцінками різних дослідників), то сумарна частота генних мутацій складе від 1 до 10 на
гаплоїдний геном.
При вивченні летальних ефектів в близькоспоріднених браках показано, що близько 8 % всіх
людей несуть знов виниклу мутацію. Таким чином, приблизні оцінки, що одержуються різними
методами, співпадають.
Частота хромосомної аберації і геномних змін значно перевищує частоту генних мутацій.
Наприклад, частота нерозбіжності 21-ої пари хромосом близько 1 %. Якщо прийняти цей показник
як середню частоту нерозбіжності, то сумарна частота нерозбіжності складе близько 20 %. Мабуть,
високий рівень загибелі зігот на ранніх стадіях розвитку у людини пояснюється високою сумарною
частотою мутацій різного типу.
Як і будь-яка інша дисципліна, сучасна генетика людини використовує багато методів суміжних
наук: біохімії, фізіології, молекулярній біології, генній інженерії і т.д. Велика питома вага у
вирішенні проблем генетики людини і медичної генетики має онтогенетичний метод, згідно
якому розвиток нормальних і патологічних ознак розглядається в ході індивідуального розвитку.
Деякі спадкові хвороби виявляються тільки в певному віці. Прикладом може служити хорея
Хантінгтона, що успадковується як аутосомний домінантна ознака. Це захворювання
виражається в розладі психіки у хворих у віці 25-45 років. До цієї ж категорії відноситься хвороба
Альцгеймера (старече недоумство) і ін.
18. Медична генетика
Генетичний тягар в людських популяціях виявляється у великому числі спадковихзахворювань. Їх відомо близько 2000. Частоту спадкових захворювань можна
оцінити на прикладі дослідження, проведеного А. Стівенсоном в Північній
Ірландії. Він виявив, що близько 4 % новонароджених несуть серйозні генетичні
дефекти. Ця цифра не включає викиднів і мертвонародження, частота яких
складає близько 14 % від зареєстрованих вагітностей. Частина з них, поза
сумнівом, відбувається з причин генетичних аномалій. Ця цифра (4 %) не включає
такі поширені хвороби, як діабет і шизофренія, у виникненні яких істотну роль
також грає спадковість компоненту.
А. Стівенсон встановив, що близько 26 % лікарняних ліжок в Північній Ірландії
займають пацієнти, страждаючі спадковими хворобами. Крім того, 6-8 %
пацієнтів, що консультуються з лікарями, теж відносяться до цієї категорії.
Згадувана вже шизофренія зустрічається в середньому з частотою 1 %, а в деяких
країнах і частіше. Наприклад, на півночі Швеції частота цього захворювання
коливається між 2 і 3 %.
Вивчення і можливе запобігання наслідкам генетичних дефектів людини предмет медичної генетики. Умовно спадкові хвороби можна підрозділити на три
великі групи: хвороби обміну речовин, молекулярні хвороби, які зазвичай
викликаються генними мутаціями, і хромосомні хвороби.
19.
Спадкові хвороби, причинами яких є блокування ферментативних реакцій перетворення фенілаланінуТовсті стрілки - метаболічний шлях, розриви в стрілках - переривання реакцій, тонкі стрілки указують на
спадкові захворювання
20. Молекулярні хвороби
Межа між хворобами обміну речовин і молекулярними хворобами умовна і скоріше відображає ступінь обізнаності про причини спадковоїаномалії. Детально вивчено значне число так званих гемоглобінопатій, тобто хвороб, що виникають із-за спадкового порушення структури
гемоглобіну. Їх відомо більше 100 для α і β-ланцюгів глобіну. Гемоглобінопатії розрізняють залежно від молекулярних причин виникнення:
заміни амінокислот (вже наводився приклад серповидноклітинної анемії - HBS) і таласемії. Останній тип пов'язаний з порушенням синтезу
α або β-ланцюгів, наприклад з уповільненням або припиненням їх синтезу.
Переважна більшість мутацій, що зачіпають амінокислотні залишки, відповідальні за формування третинної і чверткової структури,
порушують функції гемоглобіну і внаслідок цього приводять до захворювань людей - носіїв відповідних аномалій. Більшість замін (58 з 66)
амінокислот в тих частинах α - і β-ланцюгів, які розташовуються зовні молекули гемоглобіну в результаті складання поліпептидних ланцюгів,
не робить якого-небудь істотного впливу на функціонування всієї молекули. У той же час заміни у внутрішній - гидрофобній частині майже
завжди супроводжуються порушеннями функцій.
Причинами згаданих вище таласемій можуть бути делеції, наприклад, одного або більш структурних генів Нbа. Для β-ланцюгів є в нормі
чотири гени. Тяжкість захворювання корелює з числом втрачених копій гена.
Талассемии можуть бути пов'язані як з делеціями, так і з точковими мутаціями структурного гена, що пригнічують або повністю припиняють
синтез поліпептиду. До цієї ж категорії відносять і складніші перебудови. Наприклад, відомі форми гемоглобіну з множинними замінами.
Походження одного з типів гемоглобіну - Лепоре пояснюється результатом нерівного кросинговера між розташованими поряд генами для
α- і β-ланцюгів, що мають високий рівень гомології первинної структури. В результаті вийшов поліпептидний ланцюг з N-термінальною
частиною α- і С-термінальной частиною β-глобіну. Виявлений і реципрокний продукт такого обміну - гемоглобін анті-Лепоре.
Мутації - вставки і випадання поблизу термінаторного кодону в межах генів для α- і β-ланцюгу - приводять до появи довших, ніж в нормі,
відповідно α- і β--ланцюгів глобіну, оскільки природні кодони-термінатори (у обох випадках UAA) опиняються «не у фазі» і трансляція іРНК
продовжується далі, поки рібосома не зустрічає новий кодон-нонсенс. Це, зокрема, гемоглобін Констант Спрінг (HBCS), що містить 31
зайвий амінокислотний залишок в α-цепи, і деякі інші форми.
До молекулярних хвороб відносяться аномалії генетичних процесів, наприклад, порушення реплікації і репарації ДНК. Найдетальніше
вивчені різні форми пігментної ксеродерми - рецесивний аутосомний дефект ексцизійної репарації і самої репаративної ДНК-полімерази.
На основі гібридизації соматичних клітин встановлено не менші шість груп комплементації, мабуть, відповідних самостійним генам,
контролюючим репарацію. Хворі пігментною ксеро-дермою проявляють підвищену чутливість до дії сонячного світла, у них розвивається
рак шкіри.
Дефекти систем репарації виявлені і при інших спадкових захворюваннях. При анемії Фанконі дефектний етап вирізування пошкодженої
ділянки з молекули ДНК (порушений синтез екзонуклеази). У разі атаксії - телеангіоектазії (синдром Луї Бар) також пошкоджені системи
репарації, що виражається в підвищенні чутливості клітин хворих до дії випромінювань і хімічних мутагенів, що різко збільшують в них
частоту хромосомної аберації, що має високий спонтанний рівень (близько 7,5% для лімфоцитів периферичної крові). При радіотерапії таких
хворих спостерігаються ускладнення іноді із смертельним результатом. Механізми репарації порушені і у разі прогерії, або передчасного
старіння, і синдрому Блюма.
21.
Загальна схема лікування молекулярних хвороб22. Хромосомні хвороби
Цей тип спадкових захворювань пов'язаний із змінами числа або структури хромосом. Характерною відмінністю більшостіхромосомних хвороб від хвороб, причинами яких виявляються генні мутації, є їх повторне виникнення, а не спадкоємство
від попередніх поколінь. Хромосомні і геномниє мутації утворюються як в гаметогенезі батьків, так і безпосередньо у
зиготі або на ранніх стадіях дроблення. У останньому випадку форма спадкового захворювання буде мозаїчною.
У людини відомі всі типи хромосомних і геномних мутацій, включаючи поліплоїдію. Описані рідкісні триплоїди і
тетраплоїди в основному серед спонтанно абортованих ембріонів або плодів і серед мертвонароджених. Новонароджені з
такими порушеннями живуть декілька днів. Рідкісні серед живонароджених і моносомії по аутосомах. Описані
моносомики по 21-ій і 22-ій хромосомах. Звичайно це мозаїчні організми із значною часткою нормальних клітин. Будь-які
хромосомні перебудови приводять до розвитку патологічного стану.
Моносомія всього організму описана для Х-хромосоми. Це синдром Шерешевського-Тернера, перший клінічний опис
якого в 1925 р. дав радянський учений Н. А. Шерешевській, а потім в 1938 р. - Дж. Тернер. Оскільки у людини Y-хромосома
грає визначальну підлогу роль, індивідууми ХО - жінки, проте з порушеннями в розвитку первинних і вторинних статевих
ознак. Друга Х-хромосома необхідна для нормального диференціювання гонад за жіночим типом. У хворих не
виявляється статевий хроматін.
Женщини-трісоміки з каріотипом 47, XXX нормальні в розумових і фізичних відносинах, плодовиті і не виявляють
відхилень в статевому розвитку. Із збільшенням числа Х-хромосом збільшується частота відхилення від норми: розумова
неповноцінність, аномалії зубів, порушення форми черепа, зміни інших частин скелета, порушення системи статевих
органів.
Різні комбінації Х- і Y-хромосом при полісомії по статевих хромосомах, окрім XYY, об'єднують під загальною назвою
синдрому Клайнфельтера. Y-хромосома визначає чоловічу стать, і хлопчики до періоду статевого дозрівання мало
відрізняються від людей з нормальним каріотипом 46, XY. Надалі спостерігається недорозвинення чоловічих статевих
ознак, перш за все гонад, що спричиняє за собою недорозвинення чоловічих вторинних статевих ознак. Хворі зазвичай
високорослі, але з жіночим типом скелета і з проявом деяких жіночих вторинних статевих ознак (характер волосяного
покриву, гинекомастія) (мал. 20.12). У клітинах хворих з синдромом Клайнфельтера ідентифікується статевий хроматін.
З числа аутосомних хвороб найдетальніше вивчена трісомія по 21-ій хромосомі, або синдром Дауна. Його хромосомна
природа була встановлена Ж. Леженом і ін. в 1959 р. Типові ознаки хворих з трісомієй 21-широке перенісся, широка
відстань між ніздрями, косоокі очі з характерною складкою століття - епікантом (мал. 20.13). Хворі виявляють розумову
відсталість. Близько половини хворих має порок серця і крупних судин. Найнебезпечніша риса хвороби Дауна - її висока
частота. Неодноразово відмічено, що частота новонароджених з синдромом Дауна збільшується з віком матері. 22-40 %
дітей з синдромом Дауна народжують матері старше 40 років, складають лише 2-3 % жінок дітородного віку.
23.
Мікрофотографія клітини букального епітелію. Знизу – клітини чоловіка (тільцяБарра не видно). Зверху – жінки (тільце Барра позначено стрілкою)
24. Приклади анеуплоїдії у людини
ХромосомиСтатеві хромосоми (♀)
Х0 моносомия
Синдром
Частота при
народженні
Шерешевського-Тернера
1 :5000
XXX трисомія
ХХХХ тетрасомія
ХХХХХ пентасомія
Мета-жінки
1 :700
Статеві хромосоми (♂)
XYY трисомия
XXY трисомия
Нормальные
1 : 1000
XXYY тетрасомия
XXXY тетрасомия
XXXXXY гексасомия
Клайнфельтера
1:500
Дауна
Патау
Эдвардса
1 :700
1 : 5 000
1 : 10 000
Аутосоми
Трисомія 21
Трисомія 13
Трисомія 18
25.
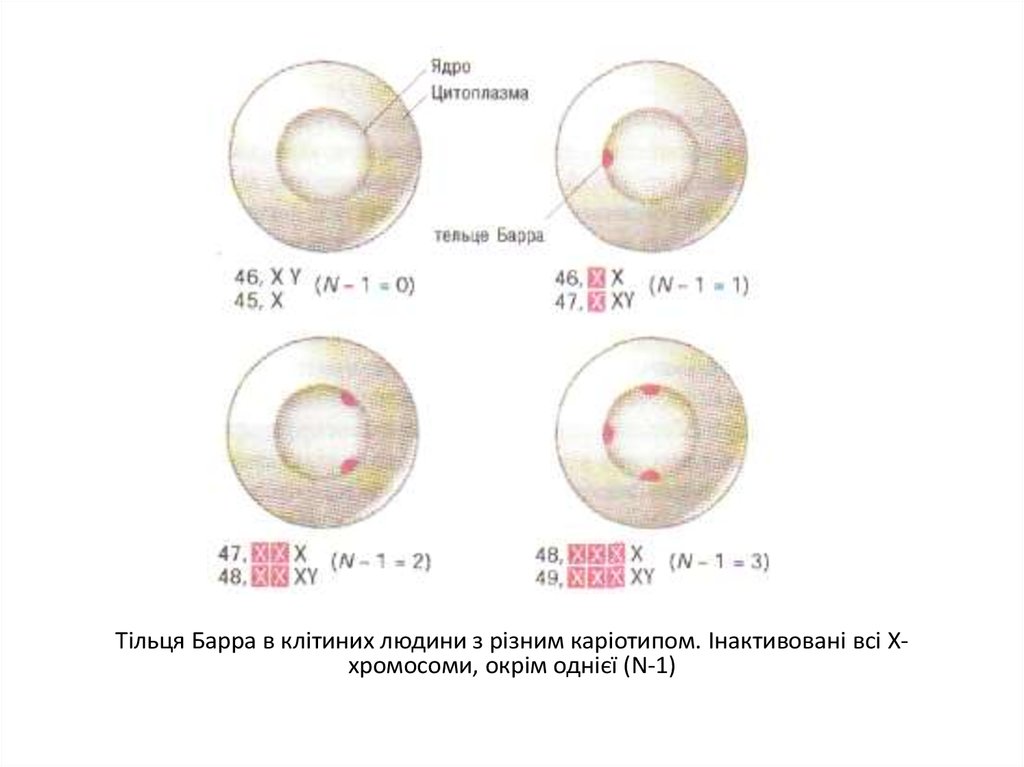
Тільця Барра в клітиних людини з різним каріотипом. Інактивовані всі Ххромосоми, окрім однієї (N-1)26.
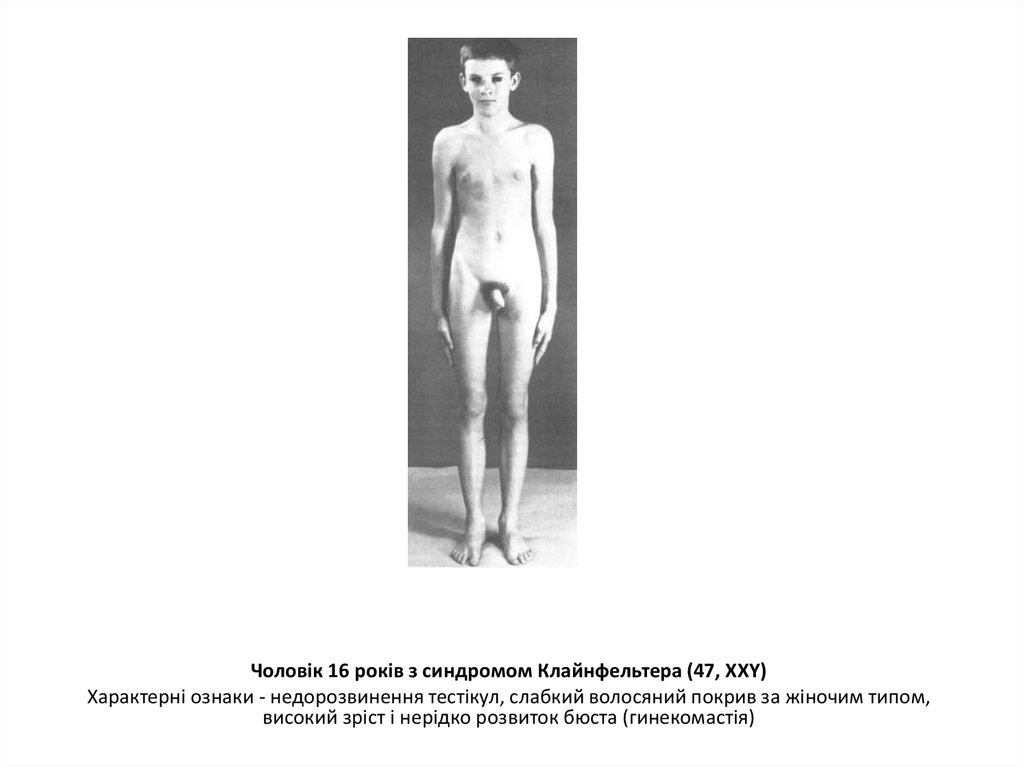
Чоловік 16 років з синдромом Клайнфельтера (47, XXY)Характерні ознаки - недорозвинення тестікул, слабкий волосяний покрив за жіночим типом,
високий зріст і нерідко розвиток бюста (гинекомастія)
27.
Синдром Шерешевського-Тернера. Шийні крилоподібні складки, широко розташованінедорозвинені соски молочних залоз
28.
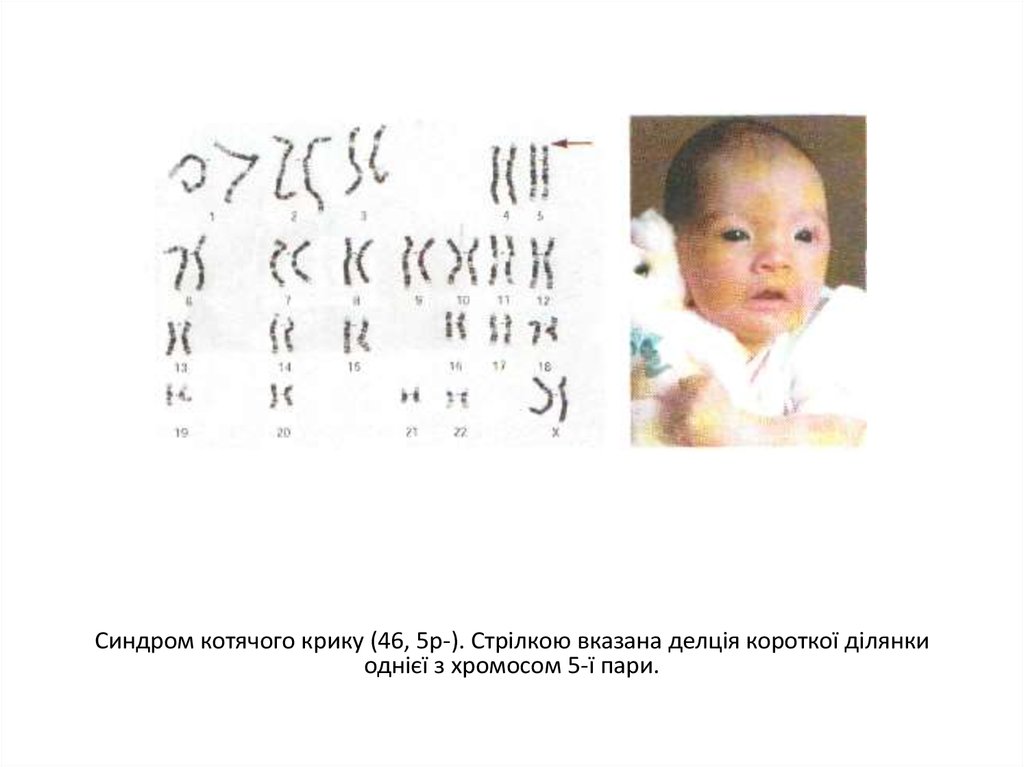
Синдром котячого крику (46, 5р-). Стрілкою вказана делція короткої ділянкиоднієї з хромосом 5-ї пари.
29.
Вік матеріЧастота синдрому Дауна
у новонарожденных, %
15-19
20—24
25—29
30—34
35—39
40 и більше
0,03—0,04
0,02—0,04
0,04—0,08
0,11—0,13
0,33—0,42
0,80—1,88
Хворий з синдромом Дауна (трісомія 21). Типові для синдрому Дауна риси
обличчя - широка голова, епікант, широкий ніс
30.
Унаслідок зниженого імунітету хворі з синдромом Дауна рано вмирають, тому вони практично незустрічаються серед дорослих людей. Застосування сульфаніламідних препаратів і антибіотиків
підвищило середню тривалість життя цих хворих з 9 до 15 років. Хворі зазвичай безплідні, але
відомо 11 випадків, коли жінки з хворобою Дауна мали дітей. З 11 дітей п'ятеро успадкували цей
синдром. Отже, зайва 21-а хромосома передається з вірогідністю 50 % відповідно до теоретично
очікуваним.
Тут розглянуті лише деякі приклади генних (метаболічних і молекулярних) і хромосомних хвороб
людини. Приведені приклади до деякої міри дають уявлення про тяжкість генетичного тягаря
людства, складності і крихкості генетичної організації людини.
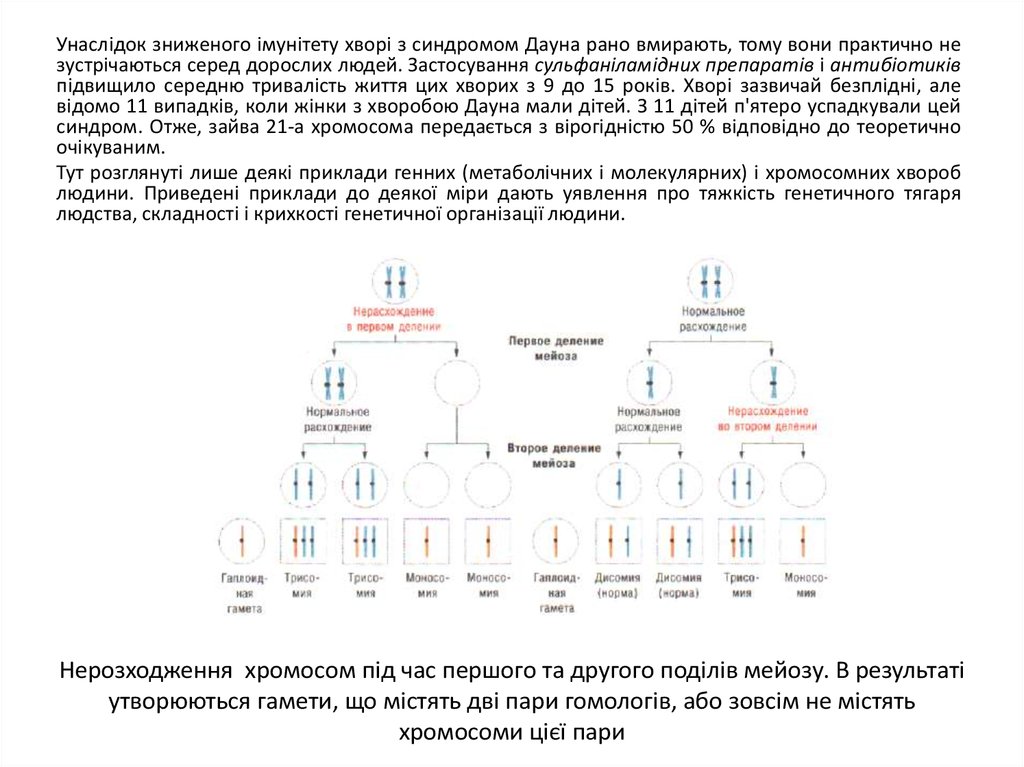
Нерозходження хромосом під час першого та другого поділів мейозу. В результаті
утворюються гамети, що містять дві пари гомологів, або зовсім не містять
хромосоми цієї пари
31. Значення діагностики і лікування від спадкових хвороб
У міру розвитку медицини можливість виявлення спадкових захворювань збільшується. Цей чинник указує назначення медичної генетики і генетики людини, що росте. Заходи, прийняті при ранньому виявленні спадкових
хвороб, можуть запобігти їх розвитку. Дієтологічні заходи дозволяють уникнути патологічних наслідків,
наприклад при галактоземії, фенілкетонурії і інших спадкових хворобах обміну.
При діагностові спадкових захворювань Н. П. Бочков із співробітниками рекомендує керуватися наступним:
Застосовувати клинико-генеалогічний метод, який дозволяє виявляти «сімейні» хвороби.
Часто до спадкових відносяться захворювання, що повторюються хронічно і тривало непіддатливі
лікуванню, особливо в дитячому віці.
На можливу спадкову форму захворювання указують специфічні симптоми, що рідко зустрічаються.
То ж відноситься до патологічних змін багатьох органів і систем.
Дійсно, плейотропія, специфічні прояви мутантних ознак характерні для спадкової мінливості. І навіть в тих
порівняно небагатьох випадках, коли втручання медицини допомагає хворим з генетичними дефектами,
говорити про лікування не можна, оскільки причина хвороби - зміна генетичного матеріалу. Проте успіхи біології
і медицини в цьому напрямі вселяють великі надії.
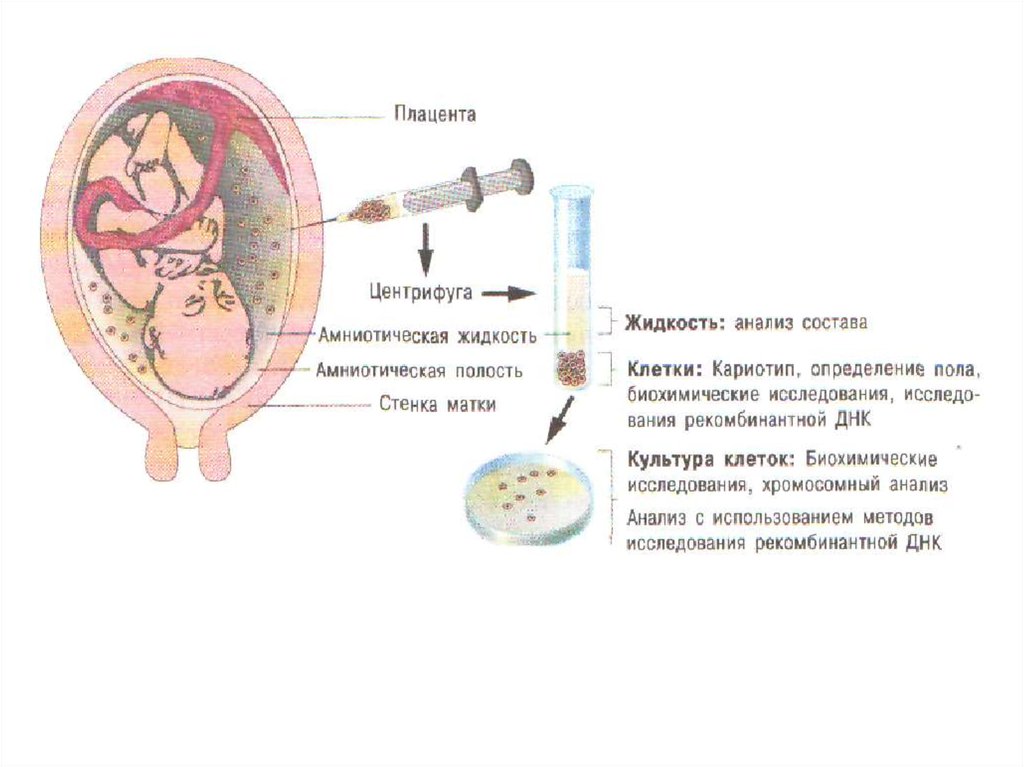
Для багатьох спадкових захворювань стала можливою так звана преначальная (тобто до народження)
діагностика. Це метод амніоцентеза, який полягає в отриманні за допомогою шприца 10-15 мл амніотічної
рідини, в якій знаходяться клітини плоду. Такий зразок центрифугуванням розділяють на клітини, які культивують
на штучному середовищі, і надосадкову рідину, в якій визначають співвідношення метаболітів, що відображають
нормальний або патологічний стан плоду. Культивовані ембріональні клітини використовують для визначення
числа хромосом і виявлення можливих хромосомних аномалій.
Цим методом можна визначити більше 100 хромосомних і генних аномалій вже в перші тижні вагітності, що
дозволяє при несприятливому діагнозі ухвалити рішення про продовження або переривання вагітності.
З'ясування метаболічних і молекулярних причин того або іншого спадкового захворювання дає можливість
компенсувати дефект у порядку модифікаційних змін - фенокопіювати норму.
32.
33. Амніоцентез — метод пренатального діагностування спадкових хвороб
Цим методом можна визначити більше 100 хромосомних і генних аномалій вже в перші тижні вагітності, щодозволяє при несприятливому діагнозі ухвалити рішення про продовження або переривання вагітності.
Вже згадувалася гемофілія Л, при лікуванні якої потрібне введення великої кількості чинника VIII. Оскільки
препарат одержують з донорської крові, вартість його дуже висока. Застосування методів генної інженерії
дозволило і в цьому випадку наблизитися до вирішення проблеми. На основі знання частини первинної
структури чинника VIII була синтезована ДНК - зонд розміром в 36 п. н. Її використовували для пошуків гена, що
кодує чинник VIII, в банці генів людини, одержаному за допомогою фага λ. Виділений врешті-решт ген виявився
рекордно великим, завдовжки 186 000 п. н. Він містить 26 екзонів і 25 інтронов. Первинну структуру чинника VIII
складають 2332 амінокислотних залишку, які залишаються після того, як від його попередника відрізується 19
амінокислот так званого лідерного пептиду, необхідного для секреції. Із-за великих розмірів гена для чинника
VIII жодна фагова частинка в банці генів не містила його цілком.
Виділений по частинах і зшитий з шматків ген ввели в культивовані клітини китайського хом'яка, які стали
виробляти чинник VIII і виділяти його в культуральне середовище. Так був одержаний ще один препарат для
медичних цілей методами генної інженерії.
На прикладі дослідження гена, що кодує чинник VIII, можна показати, як в даний час досліджується молекулярна
природа спадкових змін. Маючи в розпорядженні радіоактивну ДНК гена і суміш рестрікційних фрагментів
геномної ДНК людини, страждаючої гемофілією, можна провести електрофорез цієї геномної ДНК і потім
гібридизувати фрагменти з радіоактивним зондом. Якщо мутація торкнулася послідовності нуклеотидів якогонебудь рестрикційного сайту, то картина розподілу радіоактивних смуг, що виявляються при електрофорезе,
відрізнятиметься від тієї, яка виходить для геномної ДНК здорової людини. Застосовуючи цей метод, Р. Лон і Г.
Віхар показали, що у різних хворих-гемофіліков спостерігаються як точкових мутації - заміни пар нуклеотидів, так
і делеції різної довжини.
Виділення і клонування нормальних генів людини все більше дозволяє сподіватися на те, що виникне нова
галузь медицини - генотерапія, передбачена ще у середині 70-х років Дж. Меррілом і ін. Генотерапія дозволить
замінювати дефектні гени нормальними і тим самим усувати причину хвороби, а не її симптоми.
34. Медико-генетичне консультування
Велике значення для медичної генетики має визначення гетерозиготного носійства, тобто виявлення впопуляції індивідуумів, які самі не страждають від спадкового захворювання, але гетерозиготні по рецесивній
мутації, яка здатна його викликати. Якщо такими гетерозиготами поодинці і тому ж гену опиниться подружжя,
яке вирішить завести дитину, то вони легко можуть обчислити вірогідність появи у них нащадка - гомозиготного
по рецесивній алелі і, отже, страждаючого від спадкової хвороби.
Вся біда полягає в тому, що в більшості випадків молоді люди, одружуючись, навіть не підозрюють про таку
можливість і вже звичайно не «проходили» генетику. Для того, щоб допомогти зорієнтуватися тим, хто хоче
дізнатися, яка вірогідність появи в їх сім'ї тієї або іншої спадкової аномалії, існує медико-генетичне
консультування. Звичайно, завдання цієї служби значно складніші, ніж прогноз простих варіантів
розщеплювання. Знання генетики людини, детальне знайомство з родоводами людей, що звертаються за
консультацією, дозволяють лікареві-генетикові оцінювати ступінь ризику у кожному конкретному випадку.
Ухвалення рішення - мати або не мати дітей, перервати або не перервати вагітність - завжди залишається за
подружжям.
Таким чином, сучасні тенденції генетики людини - це активна протидія несприятливим чинникам, що викликають
спадкові аномалії на основі знання генетичних закономірностей. Проте ця позиція має мало загального з
наївними і жорстокими ідеями поліпшення людської породи шляхом селекції, які висували прихильники колись
модної євгеніки на зорі існування генетики.
Людство гетерогенне за своїми морфологічними, фізіологічними ознаками, за здібностями до різного роду
діяльності. Ця гетерогенність - різноманітність здібностей - застава процвітання людства, гарантія його
подальшого прогресу. Спроби проповідувати ідеї нерівності між якими б то не було етнічними групами або
расами людей на основі їх генетичних відмінностей не мають нічого спільного ні з біологічними законами, ні із
законами соціальної справедливості. Подальший прогрес соціалістичного суспільства багато в чому залежить від
правильного розуміння законів генетики в застосуванні до людини, перш за все в створенні рівних можливостей
для забезпечення максимального розкриття людської особи.