




















 database
databaseSimilar presentations:





. Хранение, поиск и сортировка информации")




Базы данных для хранения биологической информации
1. Базы данных для хранения биологической информации
2.
Общие принципы организации баз данных для хранения биологической информации и функциональнаяспециализация.
Типы баз данных (последовательностей нуклеиновых кислот, последовательностей генов, аминокислотных
последовательностей белков, структуры белков и нуклеиновых кислот, кристаллические структуры малых
молекул, функции белков, данные по экспрессии генов и др.).
Оглавление базы данных и терминология поисковых систем.
Использование логических комбинаций и индексовых терминов.
Работа с контролируемыми словарями.
Примеры работы с базами данных.
3.
Важным звеном исследований молекулярной биологии является сравнение аминокислотных и нуклеотидныхпоследовательностей, которое позволяет идентифицировать семейства генов, относить к ним секвенированные
последовательности, устанавливать их структурные и функциональные взаимоотношения. Разработано большое
количество программ для сравнения последовательностей с последующим определением их сходства, но наиболее
часто используются программы серии BLAST (Basic Local Alignment Sequence Tool), предоставляемым сервером
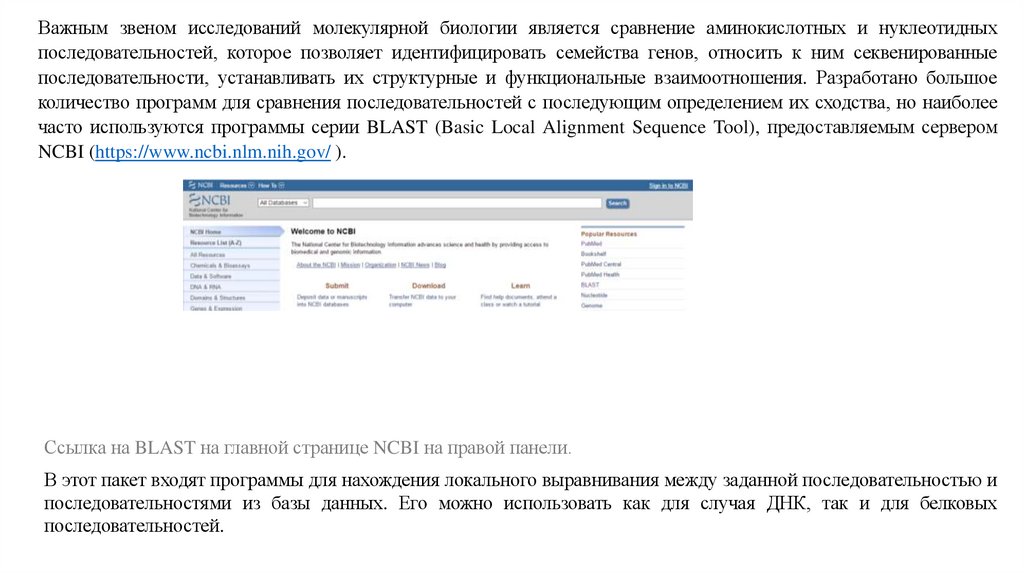
NCBI (https://www.ncbi.nlm.nih.gov/ ).
Ссылка на BLAST на главной странице NCBI на правой панели.
В этот пакет входят программы для нахождения локального выравнивания между заданной последовательностью и
последовательностями из базы данных. Его можно использовать как для случая ДНК, так и для белковых
последовательностей.
4.
Принцип работы BLASTСначала алгоритм BLAST создает таблицу всех «близких» слов (последовательностей нуклеотидов или
аминокислот) фиксированной длины - по умолчанию – длины 3 для белковых последовательностей и 11 для
нуклеотидных, - которые бы локально выравнивались с заданной последовательностью. Затем алгоритм сканирует
базу данных, и всякий раз, когда находит слово из списка, начинает процесс «расширения совпадения», чтобы
увеличить возможный участок выравнивания без разрывов, в обоих направлениях, до достижения максимального
веса. После этого вычисляется статистическая значимость найденных совпадений, и если она превышает
определенный порог, то выдается результат.
Для каждой обнаруженной последовательности необходимо определить значимость сходства с изучаемой
последовательностью.
Для этого программа вычисляет вес (score) выравнивания и величину E (expected value, E–value). E–value – это
ожидаемое количество последовательностей с весом выравнивания равным или большим веса для анализируемой
последовательности, которые, вероятно, будут обнаружены при поиске в базе данных. Чем выше вес, тем больше
сходство двух последовательностей. Чем меньше величина Е, тем достовернее выравнивание. При этом следует
учитывать, что гомология ниже 50 % при больших значениях E–value, как правило, несущественна.
5.
Ориентировочно значения E-value можно интерпретировать следующим образом:Е < 0.02 – вероятно, последовательности являются гомологами;
0,02 < E < 1 – гомология не очевидна;
Е > 1 – следует ожидать, что это случайное совпадение.
Следует отметить, что статистические оценки полезны и необходимы, но
существует также и множество эмпирических правил интерпретации процента
идентичных аминокислотных остатков в оптимальном выравнивании белковых
последовательностей.
6.
1. Если два белка содержат более 45% идентичных остатков в их оптимальномвыравнивании, то есть все основания предполагать, что эти белки имеют
подобные структуры и, скорее всего, общую или сходную функцию.
2. Если они содержат более 25% идентичных осттков, они, вероятно, имеют
подобный фолдинг. С другой стороны, низкая степень сходства
последовательностей не может исключить возможность гомологии.
3. Область
18-25%
была
определена
как
область
двусмысленности
(неоднозначности), для которой предположение о гомологии можно высказывать
только в качестве гипотезы. Парные выравнивания, которые находятся ниже этой
области, малоинформативны. При этом отсутствие значимого сходства
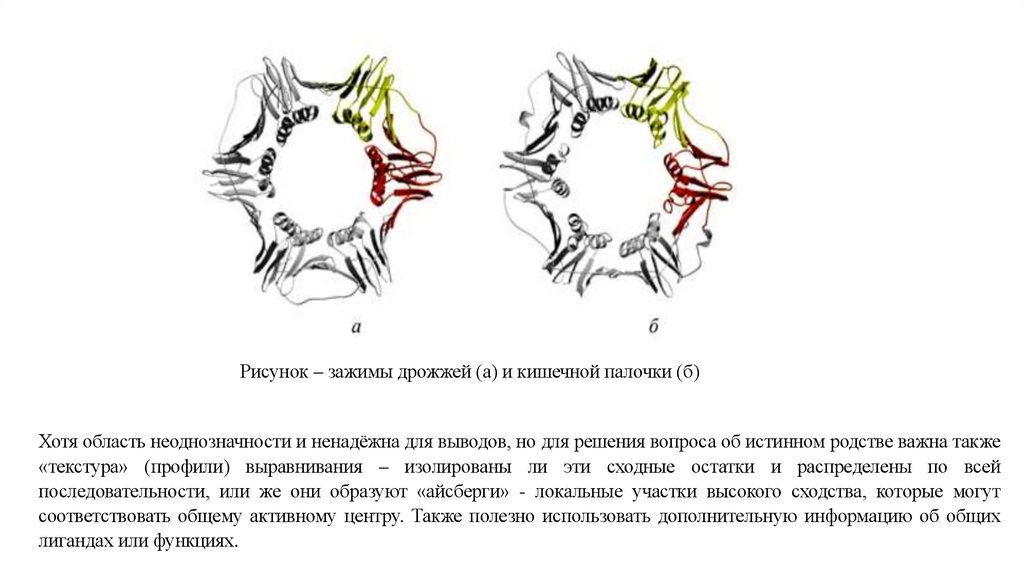
последовательностей совсем не означает отсутствие сходства структур. Например,
аминокислотные последовательности гомологичных белковых зажимов в ДНКполимеразе дрожжей (белок 1p1q) и E. coli (белок 2pol) подобны только на 12%,
но они практически одинаковы по структуре и функциям (см. рис.)
7.
Рисунок – зажимы дрожжей (а) и кишечной палочки (б)Хотя область неоднозначности и ненадёжна для выводов, но для решения вопроса об истинном родстве важна также
«текстура» (профили) выравнивания – изолированы ли эти сходные остатки и распределены по всей
последовательности, или же они образуют «айсберги» - локальные участки высокого сходства, которые могут
соответствовать общему активному центру. Также полезно использовать дополнительную информацию об общих
лигандах или функциях.
8.
Состав пакетаВообще сам поиск в BLAST состоит из 4 компонентов:
запроса (последовательности или её кода),
базы данных,
программы (алгоритм, по которому будет осуществляться поиск)
цели поиска (схожие последовательности).
Базы по типу хранящейся в них информации делятся:
на базы данных белковых и нуклеотидных последовательностей.
Кроме таких общих баз BLAST использует и специальные базы, такие как база данных векторного скрининга,
база данных разнообразия геномов различных организмов и др. Примеры баз, в которых BLAST осуществляет
поиск, приведены на таблице.
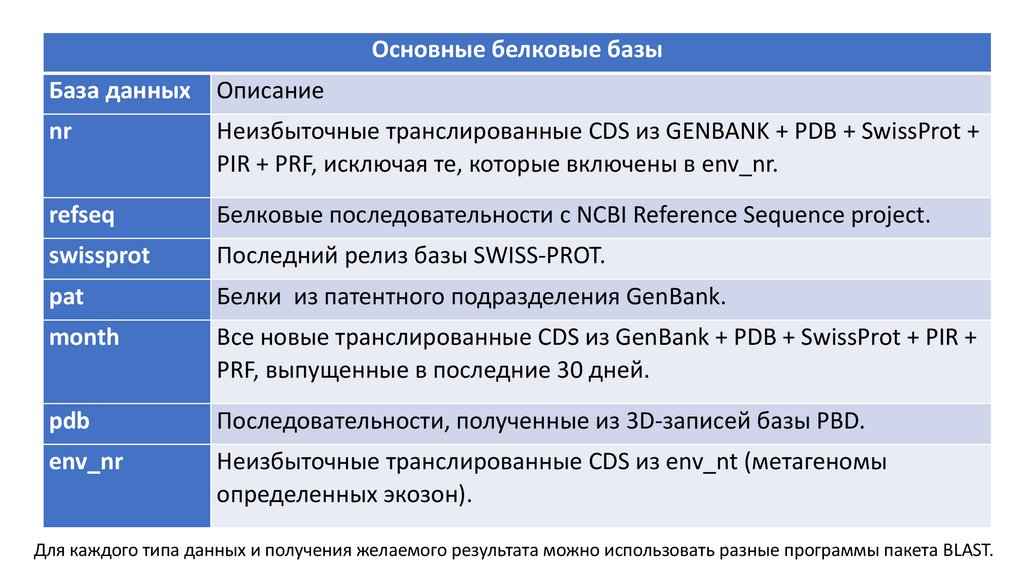
9.
Основные белковые базыБаза данных Описание
nr
Неизбыточные транслированные CDS из GENBANK + PDB + SwissProt +
PIR + PRF, исключая те, которые включены в env_nr.
refseq
Белковые последовательности с NCBI Reference Sequence project.
swissprot
Последний релиз базы SWISS-PROT.
pat
Белки из патентного подразделения GenBank.
month
Все новые транслированные CDS из GenBank + PDB + SwissProt + PIR +
PRF, выпущенные в последние 30 дней.
pdb
Последовательности, полученные из 3D-записей базы PBD.
env_nr
Неизбыточные транслированные CDS из env_nt (метагеномы
определенных экозон).
Для каждого типа данных и получения желаемого результата можно использовать разные программы пакета BLAST.
10.
nrrefseq_mrna
refseq_genomic
est
est_human
est_mouse
est_others
gss
pat
alu_repeats
dbsts
chromosome
wgs
env_nt
Основные нуклеотидные базы
Все последовательности GenBank + EMBL + DDBJ + PDB (но не EST, STS, GSS
последовательности).
mRNA-последовательности из NCBI Reference Sequence Project.
Геномные последовательности из NCBI Reference Sequence Project.
Последовательности баз GenBank + EMBL + DDBJ из подразделения EST.
Набор человеческих est.
Набор мышиных est.
Набор est других организмов.
Изученные последовательности генома, включающие определенные границы
экзонов и Alu PCR последовательности.
Нуклеотиды из патентного подразделения GenBank.
Выбранные из REPBASE Alu-повторы, которые подходят для маскировки их в
последовательности, по которой осуществляется поиск.
Database of Sequence Tag Sites из подразделения STS GenBank + EMBL + DDBJ.
Полные геномы и хромосомы. Перекрываются с refseq_genomic.
Сборки последовательностей, полученных с помощью Whole Genome Shotgun.
Последовательности из образцов окружающей среды, таких как метагеном
бактерий, выделенных из почвы или из морской воды. Самый большой ресурс –
11.
Алгоритмы анализа белковСтандартный blastp используется как для идентификации последовательности
аминокислот, так и для поиска похожих последовательностей в базах данных белков.
Для более точных результатов поиска похожих последовательностей можно снять
галочку с пункта «low complexity filter».
12.
13.
Position-Specific Iterated (PSI)-BLAST – наиболее чувствительный алгоритм BLAST,который позволяет находить сильно непохожие на входную последовательность
белки. Этот инструмент можно использовать, когда стандартный blastp не дал
результатов, или выдал гипотетические белки.
Первый раунд PSI-BLAST – обычный blastp. Программа строит PSS-матрицу, или
профиль, из множественного выравнивания последовательностей с низкими Evalue. Далее эта матрица будет использоваться для построения выравнивания этих
белков (с низким E-value) с белками с более высоким, неподходящим E-value, при
следующей итерации, за счет чего белки с низким E-value, то есть те, которые очень
слабо похожи на входную последовательность, но соответствуют составленному
программой профилю, будут перемещаться в зону высокого E-value. После первого,
обычного выравнивания можно выбрать те последовательности с низким E-value,
по которым программа будет строить матрицу, а не использовать все. Кроме того,
полученную PSS-матрицу можно сохранить и использовать при поиске в других
базах.
14.
Pattern-Hit Initiated (PHI)-BLAST разработан для поиска белков, содержащихспецифические паттерны, определяемые пользователем, и фланкированные
последовательностями, схожими с таковыми во входной последовательности. Это
двойное «требование» к алгоритму предназначено для уменьшения времени поиска
за счет ограничения по базам, однако оно скорей всего не отразится на степени
гомологии целевых последовательностей со входной. Чтобы выполнить PHI-BLAST,
необходимо ввести свою последовательность и PHI-паттерн (на основе синтаксиса
PROSITE) в опциях. Пример:
Последовательность
>gi|4758958|ref|NP_004148.1| Human cAMP-dependent protein kinase
MSHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARAPASVLPAATPRQSLGHPPPEPGPDRVADAKGDSESEEDE
DLEVPVPSRFNRRVSVCAETYNPDEEEEDTDPRVIHPKTDEQRCRLQEACKDILLFKNLDQEQLSQVLDAMFERIVKADEHVID
QGDDGDNFYVIERGTYDILVTKDNQTRSVGQYDNRGSFGELALMYNTPRAATIVATSEGSLWGLDRVTFRRIIVKNNAKKRK
MFESFIESVPLLKSLEVSERMKIVDVIGEKIYKDGERIITQGEKADSFYIIESGEVSILIRSRTKSNKDGGNQEVEIARCHKGQYFGE
LALV
Паттерн
[LIVMF]-G-E-x-[GAS]-[LIVM]-x(5,11)-R-[STAQ]-A-x-[LIVMA]-x-[STACV].
15.
Алгоритмы анализа нуклеиновых кислотMEGABLAST
– алгоритм, спроектированный для эффективного поиска и выравнивания длинных
очень похожих последовательностей, поэтому он хорошо подходит для идентификации
неизвестных последовательностей.
Discontiguous MEGABLAST
больше подходит для поиска похожих, а не идентичных последовательностей.
Алгоритмы пакета BLAST разбивают последовательность на «слова», ищут по ним и
строят по ним выравнивания. Длина «слов» - один из самых важных параметров,
регулирующих чувствительность алгоритма. MEGABLAST использует слова длиной в 11
«буков», что меньше, чем в blastn. Длину «слов» можно изменять вручную до
минимальных семи.
16.
Более чувствительный поиск может производиться в discontiguous MEGABLAST.Вместо того, чтобы требовать точного совпадения «слов», являющихся «затравками»
для расширяющихся выравниваний, этот алгоритм расширяет «окно» матричной
последовательности, которое выравнивается на последовательность из базы.
Эта программа также учитывает третью wobble-позицию каждого кодона, игнорируя
несоответствия в ней при выравнивании.
Поиск через discontiguous MEGABLAST более чувствителен и эффективен, чем
стандартный blastn с той же длиной «слова», поэтому сейчас чаще рекомендуют
использовать именно его.
Параметры для discontiguous MEGABLAST (пункт discontiguous word options):
Размер «слова» (word size): 11-12
Длина матрицы: 16, 18 или 21.
Тип матрицы: кодирующая, максимальная, две.
17.
Рисунок – Пункт выбора алгоритма поиска и выравнивания нуклеотидных последовательностей18.
Смежные алгоритмыК смежным алгоритмам относят blastx, tblastn и tblastx.
Blastx осуществляет трансляцию входной нуклеотидной последовательности и сравнение её с белковой
базой. Алгоритм транслирует последовательность в 6-ти рамках считывания и статистически оценивает
каждую из них, поэтому его хорошо применять при анализе последовательности с неизвестной рамкой
или при риске фреймшифта. Так, этот алгоритм всегда используют на первом этапе анализа неизвестной
нуклеотидной последовательности. Этот поиск более чувствителен, чем blastn, так как сравнение
производится на уровне белков.
Tblastn применим для поиска гомологов белков из неаннотированных нуклеотидных
последовательностей. Этот алгоритм позволяет сравнивать входную белковую последовательность с
шестью способами транслированными нуклеотидными последовательностями нуклеотидных баз
данных.
Tblastx – это алгоритм, осуществляющий трансляцию входной нуклеотидной последовательности и
сравнивающий её с такими же транслированными нуклеотидными последовательностями баз данных.
Этот поиск более чувствителен, чем blastp, который можно осуществить после ручной трансляции
нуклеотидной последовательности. Tblastx обходит вероятные фреймшифты, которые могут
препятствовать поиску открытых рамок считывания. Это очень удобно при идентификации
потенциальных белков, кодируемых однократно прочитанными EST; кроме того, с помощью этого
инструмента можно легко идентифицировать новые гены. Этот инструмент следует использовать только
в крайнем случае, так как он очень ресурсозатратный.
19.
Специализированные инструментыПанель специализированных инструментов находится внизу главной страницы BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
20.
CD-search – поиск консервативных доменов, который использует reverse positionspecific BLAST (RPS-BLAST). Он более чувствительный, чем стандартный поиск.
Принцип работы заключается в том, что алгоритм строит PSS-матрицу на основе
выравнивания входной последовательности с наиболее похожими, и сравнивает
полученную матрицу с базой данных матриц выравнивания доменов из баз Smart,
Pfam, COG и др.
Алгоритм CDART (protein homology by domain architecture) позволяет искать
последовательности со схожей доменной организацией. Первым этапом является
поиск консервативных доменов через RPS-BLAST, вторым – поиск белков, содержащих
как можно большее количество доменов, содержащихся во входном белке.
Результаты сортируются от самых схожих к самым отличающимся.
Программа VecScreen определяет присутствие во входной последовательности
участков векторного происхождения.
Более полное описание инструментов пакета BLAST можно найти на главной странице
(ссылка приведена выше) или в мануале.
21.
Как выбирать алгоритм?22.
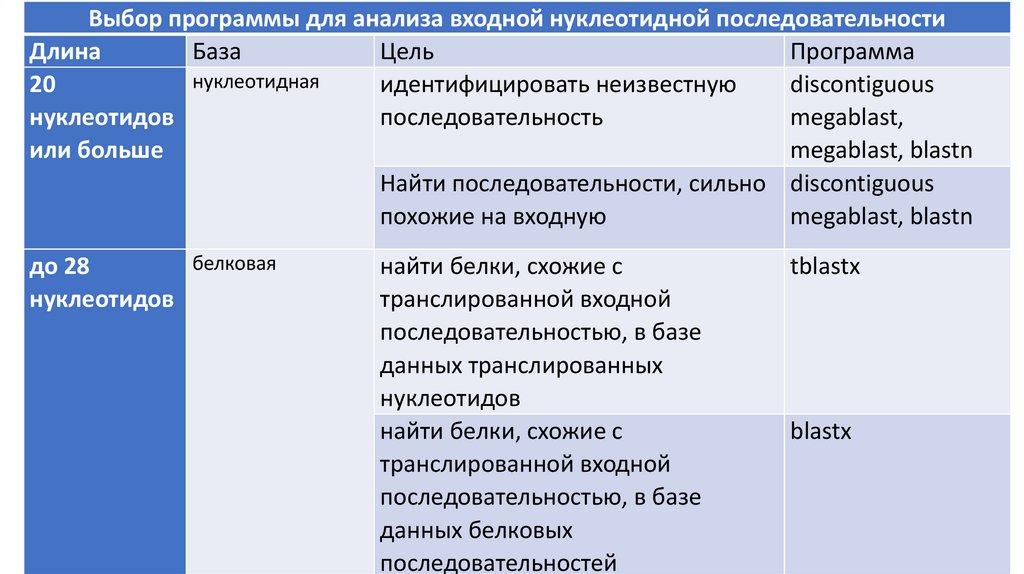
Выбор программы для анализа входной нуклеотидной последовательностиДлина
База
Цель
Программа
нуклеотидная
20
идентифицировать неизвестную
discontiguous
нуклеотидов
последовательность
megablast,
или больше
megablast, blastn
Найти последовательности, сильно discontiguous
похожие на входную
megablast, blastn
белковая
до 28
нуклеотидов
найти белки, схожие с
транслированной входной
последовательностью, в базе
данных транслированных
нуклеотидов
найти белки, схожие с
транслированной входной
последовательностью, в базе
данных белковых
последовательностей
tblastx
blastx
23.
Выбор программы для анализа входной белковой последовательностиДлина
База
Цель
Программа
Идентифицировать неизвестную
blastp
аминокислотную последовательность или
найти похожие белки
Найти членов белкового семейства или
PSI-BLAST
белковая построить PSS-матрицу
Найти белки, похожие на входной и
PHI-BLAST
содержащий определенный пользователем
паттерн
Найти консервативные домены во входной CD-search (RPSпоследовательности
BLAST)
Найти консервативные домены во входной CDART
последовательности и найти белки со
15
схожей доменной архитектурой (составом)
аминокислот
По входной аминокислотной
tblastn
или больше
нуклеотид последовательности найти схожие в базе
данных транслированных нуклеотидных
ная
последовательностей
24.
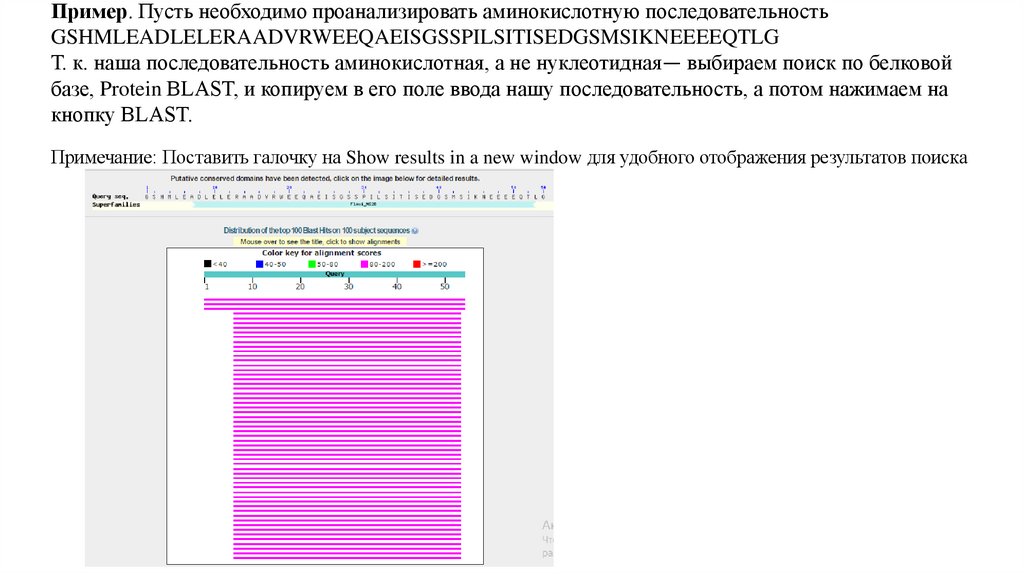
Пример. Пусть необходимо проанализировать аминокислотную последовательностьGSHMLEADLELERAADVRWEEQAEISGSSPILSITISEDGSMSIKNEEEEQTLG
Т. к. наша последовательность аминокислотная, а не нуклеотидная— выбираем поиск по белковой
базе, Protein BLAST, и копируем в его поле ввода нашу последовательность, а потом нажимаем на
кнопку BLAST.
Примечание: Поставить галочку на Show results in a new window для удобного отображения результатов поиска
25.
После нескольких секунд поиска база данных выдаст нам результат — наша последовательность больше всегопохожа на последовательности 2FOM Chain A и Dengue Virus Ns2bNS3 PROTEASE.
Тут каждая строка содержит совпадение с нашей последовательностью. Цвета обозначают «вес» (score) –
количественный критерий качества совпадения нашей последовательности и обнаруженных. Интуитивно понятно,
что чем больше сходство между двумя последовательностями, тем они ближе по конформации и функциям. Однако
бывает, что совпадение между двумя последовательностями есть по всей длине молекулы, но через очень
небольшие интервалы есть несовпадающие звенья. В таком случае полоски будут равны по длине, но не красного
или розового, а зеленого, синего или черного цвета.
Для изменения опций можно использовать Formatting options Reformat.