





 medicine
medicineSimilar presentations:










Наблюдения из практики Case reports
1.
Наблюдения из практикиCase reports
Журнал неврологии и психиатрии им. С.С. Корсакова
2021, т. 121, №9, с. 104-110
https://doi.org/10.17116/jnevro2021121091104
S.S. Korsakov Journal of Neurology and Psychiatry
2021, vol. 121, no. 9, pp. 104-110
https://doi.org/10.17116/jnevro2021121091104
Случай нейроакантоцитоза, связанный с мутацией VPS13A
© Н.А. СКРИПКИНА, В.К. ДАТИЕВА, О.С. ЛЕВИН
ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава России, Москва, Россия
Резюме
Нейроакантоцитоз — группа нейродегенеративных заболеваний, проявляющихся сочетанием неврологической симптоматики (чаще всего хореический гиперкинез) и наличием повышенного количества акантоцитов в периферической крови.
В эту группу входят хорея-акантоцитоз, синдром МакЛеода, пантотенаткиназа-ассоциированная нейродегенерация, гентингтоноподобное заболевание 2-го типа и некоторые другие очень редкие заболевания. В данной статье представлен генетически подтвержденный клинический случай хореи-акантоцитоза, связанный с компаунд-мутацией в гене VPS13A, подробно обсуждаются этапы диагностического поиска, представлен алгоритм обследования пациентов с хореей.
Ключевые слова: нейроакантоцитоз, хорея-акантоцитоз, хорея, мутация VPS13A.
Информация об авторах:
Скрипкина Н.А. — https://orcid.org/0000-0003-1331-0764
Датиева В.К. — https://orcid.org/0000-0002-8446-064X
Левин О.С. — https://orcid.org/0000-0003-3872-5923
Автор, ответственный за переписку: Левин О.С. — e-mail: neurolev@mail.ru
Как цитировать:
Скрипкина Н.А., Датиева В.К., Левин О.С. Случай нейроакантоцитоза, связанный с мутацией VPS13A. Журнал неврологии
и психиатрии им. С.С. Корсакова. 2021;121(9):104–110. https://doi.org/10.17116/jnevro2021121091104
Case-report of neuroacanthocytosis associated with a compound mutation in the VPS13A gene
© N.A. SKRIPKINA, V.K. DATIEVA, O.S. LEVIN
Russian Medical Academy of Continuous Professional Education, Moscow, Russia
Abstract
Neuroacanthocytosis is a group of neurodegenerative diseases manifested by a combition of neurological symptoms (most often
choreic hyperkinesis) and the presence of an increased number of acanthocytes (erythrocytes with horns) in the peripheral blood.
This group includes chorea-acanthocytosis, MacLeod’s syndrome, pantothenate kinase-associated neurodegeneration, Huntington-like disease type 2, and some other very rare diseases. This article presents a genetically confirmed clinical case of chorea-acanthocytosis associated with a compound mutation in the VPS13A gene, discusses in detail the stages of a diagnostic search, presents an algorithm for examining patients with chorea.
Keywords: neuroacanthocytosis, chorea-acanthocytosis, chorea, mutation VPS13A.
Information about the authors:
Skripkina N.A. — https://orcid.org/0000-0003-1331-0764
Datieva V.K. — https://orcid.org/0000-0002-8446-064X
Levin O.S. — https://orcid.org/0000-0003-3872-5923
Corresponding author: Levin O.S. — e-mail: neurolev@mail.ru
To cite this article:
Skripkina NA, Datieva VK, Levin OS. Case-report of neuroacanthocytosis associated with a compound mutation in the VPS13A gene.
S.S. Korsakov Journal of Neurology and Psychiatry = Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova. 2021;121(9):104–110. (In Russ.).
https://doi.org/10.17116/jnevro2021121091104
Феномен хореи известен на протяжении уже многих веков. Его патогенез обусловлен органическими или
психогенными нарушениями. Термин хорея берет начало от древнегреческого слова χορει′α, которое описывает
разновидность традиционного кругового танца и предпо-
104
лагает быстрые отрывистые, непредсказуемые движения.
Впервые подобные непроизвольные движения были описаны в XVI веке врачом и алхимиком Парацельсом, использовавшим термин «пляска Святого Вита» — отсылающего к своеобразной эпидемии «маниакального танца»,
Журнал неврологии и психиатрии им. С.С. Корсакова, 2021, т. 121, №9
2.
Наблюдения из практикиCase reports
Рис. 1. Алгоритм обследования пациента с хореей.
БГ — болезнь Гентингтона; АС-О — антистрептолизин-О; АВМ — артериовенозные мальформации; АТ — антитела; СКВ — системная красная волчанка.
Fig. 1. Algorithm for examining a patient with chorea.
BG — Huntington’s disease; AC-O — antistreptolysin-O; AVM — arteriovenous malformations; AT — antibodies; SLE — systemic lupus erythematosus.
появившегося в средневековой Европе. Ссылка на святого Вита могла появиться в связи с тем, что больные исцелялись после танца под куполом капеллы, посвященной
святому Виту, известному как покровитель танцоров, однако в литературе встречаются и другие объяснения. Безусловно, подобные эпидемии носили психогенный характер. Некоторые ученые связывают их появление с явлениями эрготизма или отравлением ртутью, хотя хорея
не является типичным симптомом ртутной интоксикации.
В XVII веке английский врач Т. Сиденгам отметил развитие хореи у детей. Он объяснил появление этого синдрома локальной эпидемией «танцевальной мании». Это двигательное
расстройство у детей в дальнейшем получило известность как
«малая хорея» в отличие от «большой хореи» — больших эпидемий танцевальных маний у взрослых. Связь «малой хореи»
с ревматической лихорадкой была отмечена французским неврологом Ж.М. Шарко. Им же было предложено использование эпонимического термина «хорея Сиденгама» в 1887 г.
Наследственный характер хореи у пациентов с дебютом в более старшем возрасте в Нью-Йорке впервые был
описан Дж. Гентингтоном в 1872 г. Подобное предположение было встречено критически Ж.М. Шарко. Подроб-
ное описание наследственных и приобретенных форм хореи было проведено В. Ослером в 1894 г.
Установление причины развития хореи у пациента требует проведения достаточного широкого диагностического
поиска. Направление исследования зависит от нескольких
факторов: возраста пациента, времени развертывания гиперкинеза, лекарственного анамнеза, наличия подобных случаев в семье пациента, сопутствующих расстройств [1—5].
Алгоритм исследования пациента с хореей представлен на рис. 1.
При этом необходимо помнить про более редкие причины развития хореи, которые относятся к группе нейродегенеративных заболеваний, для которых характерной особенностью является развитие нейроакантоцитоза.
Нейроакантоцитоз — это синдром, развивающийся
при различных заболеваниях, генетически отличающихся
друг от друга. Для него характерно появление в периферической крови акантоцитов — эритроцитов с многочисленными отростками (акантами) и прогрессирующей дегенерацией базальных ганглиев [6]. Синдром нейроакантоцитоза развивается исключительно редко, частота встречаемости
1—5 на 1 000 000 для каждой нозологии. Основными заболе-
S.S. Korsakov Journal of Neurology and Psychiatry, 2021, vol. 121, no. 9
105
3.
Наблюдения из практикиCase reports
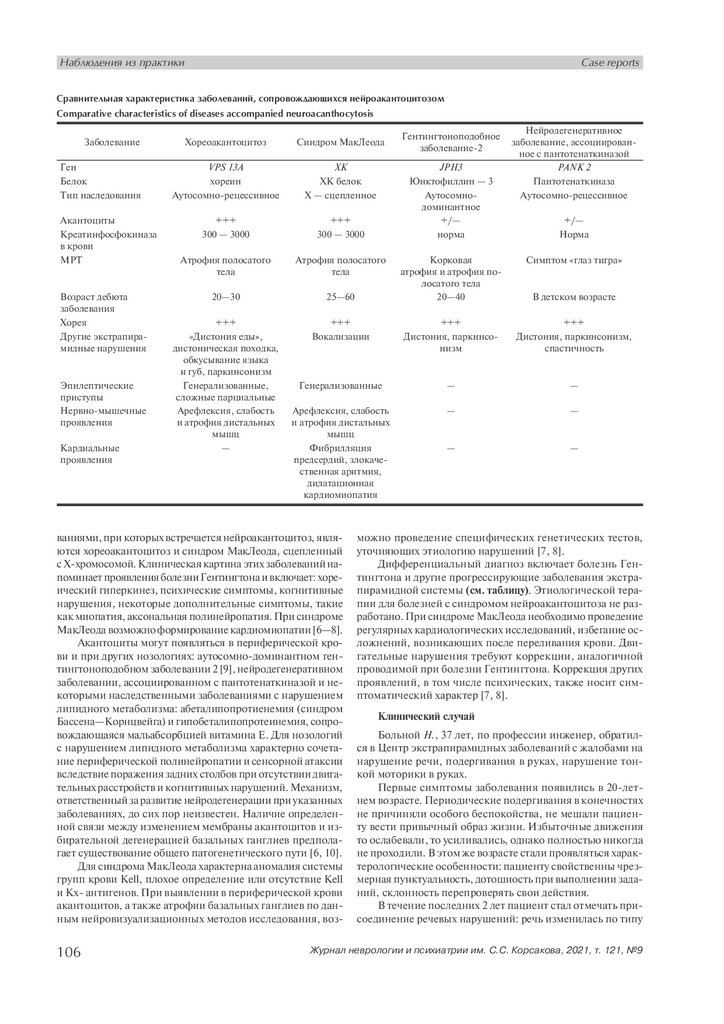
Сравнительная характеристика заболеваний, сопровождающихся нейроакантоцитозом
Comparative characteristics of diseases accompanied neuroacanthocytosis
Заболевание
Ген
Белок
Тип наследования
Акантоциты
Креатинфосфокиназа
в крови
МРТ
Возраст дебюта
заболевания
Хорея
Другие экстрапирамидные нарушения
Эпилептические
приступы
Нервно-мышечные
проявления
Кардиальные
проявления
Хореоакантоцитоз
Синдром МакЛеода
VPS 13A
хореин
Аутосомно-рецессивное
XK
XK белок
X — сцепленное
+++
300 — 3000
+++
300 — 3000
Атрофия полосатого
тела
Атрофия полосатого
тела
20—30
25—60
+++
«Дистония еды»,
дистоническая походка,
обкусывание языка
и губ, паркинсонизм
Генерализованные,
сложные парциальные
Арефлексия, слабость
и атрофия дистальных
мышц
—
JPH3
Юнктофиллин — 3
Аутосомнодоминантное
+/—
норма
Нейродегенеративное
заболевание, ассоциированное с пантотенаткиназой
PANK 2
Пантотенаткиназа
Аутосомно-рецессивное
+/—
Норма
Корковая
атрофия и атрофия полосатого тела
20—40
Симптом «глаз тигра»
+++
Вокализации
+++
Дистония, паркинсонизм
+++
Дистония, паркинсонизм,
спастичность
Генерализованные
—
—
Арефлексия, слабость
и атрофия дистальных
мышц
Фибрилляция
предсердий, злокачественная аритмия,
дилатационная
кардиомиопатия
—
—
—
—
ваниями, при которых встречается нейроакантоцитоз, являются хореоакантоцитоз и синдром МакЛеода, сцепленный
с X-хромосомой. Клиническая картина этих заболеваний напоминает проявления болезни Гентингтона и включает: хореический гиперкинез, психические симптомы, когнитивные
нарушения, некоторые дополнительные симптомы, такие
как миопатия, аксональная полинейропатия. При синдроме
МакЛеода возможно формирование кардиомиопатии [6—8].
Акантоциты могут появляться в периферической крови и при других нозологиях: аутосомно-доминантном гентингтоноподобном заболевании 2 [9], нейродегенеративном
заболевании, ассоциированном с пантотенаткиназой и некоторыми наследственными заболеваниями с нарушением
липидного метаболизма: абеталипопротиенемия (синдром
Бассена—Корнцвейга) и гипобеталипопротеинемия, сопровождающаяся мальабсорбцией витамина E. Для нозологий
с нарушением липидного метаболизма характерно сочетание периферической полинейропатии и сенсорной атаксии
вследствие поражения задних столбов при отсутствии двигательных расстройств и когнитивных нарушений. Механизм,
ответственный за развитие нейродегенерации при указанных
заболеваниях, до сих пор неизвестен. Наличие определенной связи между изменением мембраны акантоцитов и избирательной дегенерацией базальных ганглиев предполагает существование общего патогенетического пути [6, 10].
Для синдрома МакЛеода характерна аномалия системы
групп крови Kell, плохое определение или отсутствие Kell
и Kx- антигенов. При выявлении в периферической крови
акантоцитов, а также атрофии базальных ганглиев по данным нейровизуализационных методов исследования, воз-
106
Гентингтоноподобное
заболевание-2
В детском возрасте
можно проведение специфических генетических тестов,
уточняющих этиологию нарушений [7, 8].
Дифференциальный диагноз включает болезнь Гентингтона и другие прогрессирующие заболевания экстрапирамидной системы (см. таблицу). Этиологической терапии для болезней с синдромом нейроакантоцитоза не разработано. При синдроме МакЛеода необходимо проведение
регулярных кардиологических исследований, избегание осложнений, возникающих после переливания крови. Двигательные нарушения требуют коррекции, аналогичной
проводимой при болезни Гентингтона. Коррекция других
проявлений, в том числе психических, также носит симптоматический характер [7, 8].
Клинический случай
Больной Н., 37 лет, по профессии инженер, обратился в Центр экстрапирамидных заболеваний с жалобами на
нарушение речи, подергивания в руках, нарушение тонкой моторики в руках.
Первые симптомы заболевания появились в 20-летнем возрасте. Периодические подергивания в конечностях
не причиняли особого беспокойства, не мешали пациенту вести привычный образ жизни. Избыточные движения
то ослабевали, то усиливались, однако полностью никогда
не проходили. В этом же возрасте стали проявляться характерологические особенности: пациенту свойственны чрезмерная пунктуальность, дотошность при выполнении заданий, склонность перепроверять свои действия.
В течение последних 2 лет пациент стал отмечать присоединение речевых нарушений: речь изменилась по типу
Журнал неврологии и психиатрии им. С.С. Корсакова, 2021, т. 121, №9
4.
Наблюдения из практикиCase reports
Рис. 2. МРТ головного мозга.
Fig. 2. MRI of the brain.
«каши во рту». Его стали часто переспрашивать на работе,
дома. Пациент обратил внимание, что участились эпизоды прикусывания языка, губ, щек. Избыточные движения
в конечностях стали более продолжительными, затрудняли выполнение привычной работы.
У деда пациента по отцовской линии отмечалась болезнь Паркинсона с дебютом в возрасте 80 лет. О какойлибо значимой патологии у других ближайших родственников пациент не упоминает.
При осмотре обращают внимание грубая орофациальная дистония, сопровождающаяся причмокиванием, а также
насильственные движения в конечностях, шее, туловище.
Движения глазных яблок, как медленные следящие,
так и саккадические, в полном объеме. Лицо симметричное. Речь дизартрофонична. Голос монотонный. При высовывании языка по команде он располагается по средней
линии, следов прикуса на языке нет.
Пробы на выявление гипокинезии отрицательны, пациент удовлетворительно справляется со всеми заданиями. При выполнении повторяющихся двигательных актов
рисунок движений прерывается избыточной активностью
(подергиванием рук, ног), которым пациент старается придать вид произвольных, включить их в актуальную двигательную программу. Тонус в конечностях несколько снижен. Феномена рекурвации не отмечается. При вытягивании рук вперед отмечается дистоническая установка правой
кисти в виде ее легкого сгибания с разведением большого
пальца и переразгибанием I и IV — V пальцев.
Сила мышц не снижена. Сухожильные рефлексы
с верхних конечностей — средней живости, без четкой разницы сторон, с нижних — коленные сохранны, ахилловы
рефлексы отсутствуют с двух сторон. Патологических рефлексов нет. Дисметрии при выполнении координаторных
проб нет. При выполнении пальценосовой пробы интенционное или терминальное усиление тремора отсутствует
как в правой, так и левой руке. Поверхностная и глубокая
чувствительность снижены по типу «носков».
Походка изменена по типу «танцующей». Длина шага
неравномерна. Скорость ходьбы достаточна. Шарканья, застываний не отмечается. При ходьбе отмечаются эпизоды
с угрозой утраты равновесия. Туловище выпрямлено, несколько отклонено назад. Ахейрокинеза нет. В пробе Ромберга устойчив. В пробе Тевенара на постуральную устой-
чивость при подталкивании кзади пациент совершает 5 шагов, удерживая при этом равновесие. Вставание со стула,
присаживание не затруднены. Тандемная ходьба нарушена.
При нейропсихологическом исследовании выявлены
нейродинамические нарушения в виде флуктуаций внимания, быстрой истощаемости. При выполнении заданий нацелен на достижение результата. Активен преимущественно
в начале задания, где достигается максимальная продуктивность. Умеренные нарушения слухоречевой памяти, однако
опосредованные приемы при воспроизведении оказываются эффективными. Снижение объема вербальных ассоциаций при поиске слов, относящихся к заданному признаку. Фонетическая речевая активность нарушена в большей
степени в сравнении с семантической (13 и 23 слова соответственно). При выполнении серийных счетных операций наблюдалось увеличение латентного периода умственных действий при понимании условий задания. В тестах
на мышление (арифметический тест, поиск обобщающего слова, интерпретация пословиц) выявлено некоторое
уплощение мышления. Общая оценка когнитивных функций согласно Краткому исследованию психического статуса (MMSE) составила 30 баллов. При выполнении заданий из лобной батареи тестов отмечалась склонность пациента к импульсивности. Таким образом, констатирована
умеренная дисфункция лобной коры при заинтересованности глубинных отделов мозга и относительной сохранности
мнестических и интеллектуальных процессов.
В психологическом статусе отмечалось снижение критики к своему состоянию. Повышенная тревожность, обсессивно-компульсивный синдром, выражающийся в желании постоянно перепроверять свои действия, прикасаться к окружающим предметам.
В биохимическом анализе крови обращает на себя
внимание повышение содержания креатинфосфокиназы
до 2554 Ед/л (норма — 0—190), незначительное увеличение печеночных ферментов аланинаминотрансферазы —
54 Ед/л (норма — 0—40), аспартатаминотрансферазы —
67 Ед/л (норма — 0—40). Другие показатели (мочевина,
креатинин, мочевая кислота, общий билирубин, холестерин, триглицериды, глюкоза) в пределах нормы.
Пациенту было выполнено МРТ головного мозга
(рис. 2). На представленных изображениях аксиальных срезов в T2, FLAIR и DWI-режимах прослеживается атрофия
S.S. Korsakov Journal of Neurology and Psychiatry, 2021, vol. 121, no. 9
107
5.
Наблюдения из практикиCase reports
головки хвостатого ядра, отложение железа в области базальных ганглиев симметрично с обеих сторон.
На электроэнцефалограмме были выявлены умеренные
общемозговые изменения биоэлектрической активности
головного мозга с признаками дисфункции в срединных
структурах. Эпилептиформной активности зарегистрировано не было. Наличия локальных нарушений биопотенциалов обнаружено не было.
При выполнении ЭКГ установлено: ритм синусовый,
частота сердечных сокращений 74 в мин, нормальное положение электрической оси сердца, нарушение внутрижелудочковой проводимости, изменения миокарда в заднебоковой стенке левого желудочка и в высоких боковых
отделах. На ЭХО-кардиограмме обнаружено, что размеры полостей сердца не изменены, глобальная систолическая функция левого желудочка сохранена, диастолическая
функция левого желудочка не нарушена. Умеренное пролабирование миксоматозноизмененных створок митрального клапана с митральной регургитацией 2 ст, трикуспидальной регургитацией 1 ст.
Таким образом, диагностический поиск в случае выявления у пациента хореического гиперкинеза, сопровождающегося когнитивными нарушениями, требует исключения болезни Гентингтона. Отсутствие мутации при проведении генетического исследования в рассматриваемом
примере потребовало продолжения поиска причины указанных расстройств. После исключения симптоматических
форм хореического гиперкинеза на фоне аутоиммунных
нарушений, воспалительного процесса, сосудистых, дисгормональных, лекарственных, метаболических изменений
диагностический поиск был направлен на выявление более редких причин. При выполнении рутинных клинических исследований внимание привлекло повышение уровня креатинфосфокиназы более чем в 13 раз.
Учитывая особенности клинической картины, в том
числе отсутствие ахилловых рефлексов, пациенту было
предложено проведение исследование мазка крови на выявление акантоцитов. Наличие акантацитов в периферической крови, молодой возраст, отсутствие семейного анализа и кардиальной патологии потребовало проведения
анализов, подтверждающих диагноз хореоакантацитоза:
методом прямого автоматического секвенирования было
выполнено исследование образца ДНК пациента с целью
поиска мутаций в гене VPS13A, изменения в котором приводят к развитию хореоакантоцитоза. В результате анализа выявлены мутация c.85_86delCT в экзоне 1 в гетерозиготном состоянии и мутация c.799C>T(p. rg267Term) в экзоне 11 в гетерозиготном состоянии. Таким образом, диагноз
хореоакантоцитоза у данного пациента подтвержден молекулярно-генетическим методом.
Пациенту была подобрана терапия, направленная на
коррекцию двигательных нарушений: препараты амантадина, типичные нейролептики. Однако существенного эффекта на фоне приема этих препаратов достичь, к сожалению, не удалось. В дальнейшем пациенту была рекомендована терапия тетрабеназином.
Хореоакантоцитоз — аутосомно-рецессивное нейродегенеративное заболевание с относительно поздним дебютом клинической картины [8, 11, 12].
Как правило, симптомы заболевания проявляются после 20 лет. По мере прогрессирования заболевания у большинства пациентов формируется характерный фенотип болезни, включающий хорею, а также своеобразную ороман-
108
дибулярную дистонию с протрузией языка — «дистония еды»
(во время еды язык выталкивает пищу, помещенную в полость рта), непроизвольные вокализации, дизартрию, обкусывание губ и языка [11—14]. Походка пациентов с хореоакантоцитозом изменяется по типу гуттаперчивой с формированием туловищной неустойчивости, резкими грубыми
туловищными мышечными спазмами [15]. У большинства
больных отмечается генерализованная хорея, реже встречается паркинсонизм [16]. Помимо орофациальной дистонии
нередко развивается дистония конечностей. У 1/3 пациентов первыми проявлениями заболевания являются генерализованные приступы. Часто развиваются нарушение памяти и регуляторных функций, однако подобные нарушения
не являются облигатным проявлением заболевания [16, 17].
При патологоанатомических исследованиях обнаруживаются глиоз и гибель нейронов в указанных областях. Никаких дополнительных включений или других нейропатологических изменений не регистрируется [18].
Как правило, появление симптомов заболевания происходит в том возрасте, когда перед родителями ребенка
с хореоакантоцитозом вопроса о рождении других детей
нет. Тем не менее риск развития болезни у любого сиблинга составляет 25%. Все дети пациента с хореоакантоцитозом унаследуют один мутантный аллель, клиническая картина заболевания у них не проявится [19].
Для хореоакантоцитоза характерно медленное развитие
в течение 15—30 лет. Однако возможна внезапная смерть
вследствие поражения вегетативной нервной системы.
Диагностика нейроакантоцитоза
Диагноз нейроакантоцитоза может быть выставлен с высокой степени вероятности исходя лишь из особенностей
клинической картины. На сегодняшний день критериев для
диагноза или обязательных исследований, подтверждающих
диагноз, не разработано. Учитывая, что не всегда происходит
формирование классической клинической картины, в некоторых случаях требуется проведение генетического анализа или определение специфического белка — хореина [20].
Двигательные нарушения представлены преимущественно хореей, однако, у некоторых пациентов развивается
паркинсонизм. Нередко развивается дистония, она преимущественно поражает оральную область, затрагивает язык.
Это приводит к выраженной дизартрии и дисфагии. Указанные расстройства сопровождаются потерей веса. Двигательные расстройства нарастают со временем [11, 14, 16].
Когнитивные и поведенческие нарушения напоминают
лобный синдром, представляют собой расторможенность и нарушение регуляторных функций. В некоторых случаях именно они являются первыми проявлениями болезни [6, 12, 16].
Психические симптомы представляют собой шизофреноподобный психоз или обсессивно-компульсивное расстройство. При ретроспективном анализе было установлено, что в части случаев психические симптомы могут
опережать неврологические. Назначение нейролептиков
может затруднять оценку клинической картины. Развивающиеся двигательные нарушения могут быть ошибочно расценены как осложнения терапии нейролептиками,
а не симптомы нейродегенеративного заболевания [6, 13].
Эпилептические приступы развиваются примерно у половины пациентов. Они могут быть начальным проявлением заболевания. Их происхождение, как правило, височное. В некоторых случаях у больных отмечаются семейные
случаи височной эпилепсии [13, 16].
Журнал неврологии и психиатрии им. С.С. Корсакова, 2021, т. 121, №9
6.
Наблюдения из практикиДля миопатии характерно прогрессирующее течение,
вовлечение преимущественно дистальных групп мышц,
сопровождающееся их похудением и слабостью. Однако
в большей части случаев она может оставаться на субклиническом уровне. Угнетение глубоких сухожильных рефлексов и вибрационной чувствительности происходит из-за
аксональной полинейропатии. Это может усугублять клинические проявления миопатии [10, 13, 16].
При тонком исследовании глазодвигательных функций могут отмечаться ограничение взора вверх, замедление саккадических движений. Патологических изменений
на сетчатке не происходит [13, 17].
При нейровизуализационных методах исследования регистрируется прогрессирующая атрофия полосатого тела,
особенно характерно вовлечение головки хвостатого ядра
с расширением передних рогов боковых желудочков. Степень атрофии базальных ганглиев оптимально регистрируется на срезах во фронтальной плоскости. При МРТ на
Т2 взвешенных изображениях отмечается усиление сигнала в области хвостатого ядра и скорлупы. При проведении
исследований с радиофармпрепаратами отмечается снижение метаболизма глюкозы в стриатуме, аналогичное развивающемуся при болезни Гентингтона. Морфометрия при
МРТ головного мозга демонстрирует специфическое вовлечение головки хвостатого ядра. В отличие от болезни
Гентингтона выраженной корковой атрофии не происходит. Патологические изменения наблюдаются в таламусе
и черном веществе [13, 21].
У большинства пациентов с нейроакантоцитозом отмечается повышенный уровень креатинфосфокиназы. Реже отмечается повышение в крови содержания печеночных
ферментов: лактатдегидрогеназы, аспартатаминотрансферазы, аланинаминотрансферазы [10, 12, 13].
Электрофизиологические методы исследования выявляют аксональную полинейропатию с нормальной скоростью проведения. В отличие от синдрома МакЛеода миопатия и аксональная полинейропатия выражены умеренно
или незначительно. Мышечная биопсия и электромиография как правило демонстрирует нейропатические изменения, редко миопатические [13, 16, 17].
Лабораторные исследования, необходимые для установления диагноза, включают:
— выявление акантоцитов в периферической крови. Содержание акантоцитов в периферической крови варьирует от 5 до 50%. В части случаев они могут выявляться не сразу, а появляться в ходе дальнейшего течения
болезни [10, 13, 16].
— для анализа крови на выявление акантоцитов разработан специальный алгоритм исследования. Используется кровь, разведенная физиологическим раствором
в соотношении 1:1, с добавлением 10 ед/мл гепарина. Затем мазок крови подвергают фазовоконтрастной
микроскопии. В норме регистрируется меньше 6,3%
акантоцитов. Высушенные мазки крови, как правило, неинформативны.
— у пациентов с хореоакантоцитозом отмечается отсутствие белка хореина при проведении вестерн-блоттеста в эритроцитах [20].
— единственным геном, ответственным за развитие НА,
известным на сегодняшний день, является VPS13A. Патологический ген VPS13A локализуется на 9 хромосоме. Он ответственен за синтез белка хореина [18, 20].
Данный белок вовлечен в процесс внутриклеточного
Case reports
белкового метаболизма, однако его точная физиологическая роль не установлена. Хореин широко представлен в головном мозге и различных внутренних органах.
Все мутации в гене, ответственном за синтез хореина,
приводят к его исчезновению, при этом развивается полная клиническая картина, нет промежуточных
форм, например, при гетероносительстве [20]. Анализ
ДНК, подтверждающий мутацию гена VPS13A, сложен
из-за большого размера и гетерогенности мест мутаций. На сегодняшний день он доступен лишь в единственной коммерческой лаборатории (http://www.mgzmuenchen.de).
Таким образом, в случаях, когда клиническая картина напоминает проявления нейроакантоцитоза, для подтверждения/установления диагноза необходимо определение уровня ферментов печени и креатинфосфокиназы.
Если результаты анализов демонстрируют повышенные
значения ферментов, следующим шагом должно стать выявление акантоцитов в мазке свежей крови. При количестве акантоцитов более 6% следует провести анализ крови
на выявление Kell-антигена на эритроцитах для исключения синдрома МакЛеода. При обнаружении указанных антигенов следует определить содержание хореина с помощью вестерн блот-теста (для исключения хореоакантоцитоза). Если хореин в периферической крови определяется,
но клиническая картина соответствует хореоакантоцитозу, следует рассмотреть вопрос о выполнении молекулярно-генетического исследования.
В описанном случае особенно интересным представляется достаточно быстрое прогрессирование когнитивных
и поведенческих нарушений. По-видимому, в данном случае в происхождении подобных расстройств участвуют подкорковые структуры, в частности хвостатое ядро и скорлупа. В проекции обоих образовании на МРТ изображениях
в T2, DWI-режиме отмечается гиперинтенсивность сигнала, что может указывать на атрофию указанных структур,
а также отложение железа, которое в подобных ситуациях
свидетельствует о нейродегенеративном процессе. Кроме того, расширение передних рогов боковых желудочков
указывает на атрофию базальных ганглиев, что также может сопровождаться развитием поведенческих и когнитивных расстройств, поскольку корковая атрофия не достигает выраженных значений.
В лечении симптомов хореоакантоцитоза целесообразно достичь оптимального контроля над двигательными нарушениями, которые позволят пациенту оставаться максимально длительное время независимым в быту. При отсутствии депрессии для коррекции двигательных расстройств
препаратом выбора является тетрабеназин. Использование
нейролептиков для коррекции хореического гиперкинеза
сопряжено с риском нарастания паркинсонизма, а также
когнитивных расстройств [11].
В литературе описаны случаи коррекции гиперкинеза
проведением стимуляции внутреннего сегмента бледного
шара. Однако в рассматриваемом нами случае превалирование атрофии в области базальных ганглиев снижает вероятность положительного эффекта после проведения подобного вмешательства. Кроме того, наличие выраженных
когнитивных нарушений повышает риск негативного результата [22, 23].
S.S. Korsakov Journal of Neurology and Psychiatry, 2021, vol. 121, no. 9
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
109
7.
Наблюдения из практикиCase reports
ЛИТЕРАТУРА/REFERENCES
1.
Гринберг Д.А., Аминофф М.Дж., Саймон Р.П. Клиническая неврология. Пер. с англ. Под ред. Левина О.С. М.: 2004.
Grinberg DA, Aminoff MJ, Simon RP. Clinical neurology. Per. from English. Ed. Levina O.S. M.: 2004. (In Russ.).
2.
Мументалер М., Бассетти К., Дэтвайлер К. Дифференциальный диагноз
в неврологии. М. 2010.
Mumentaler M, Bassetti K, Detweiler K. Differential diagnosis in neurogii.
M. 2010. (In Russ.).
3.
Шток В.Н., Левин О.С. Справочник по формулированию клинического
диагноза болезней нервной системы. М. 2006.
Shtok VN, Levin OS. Guide to the formulation of the clinical diagnosis of diseases of the nervous system. M. 2006. (In Russ.).
4.
Экстрапирамидные расстройства. Руководство по диагностике и лечению. Под ред. Штока В.Н. М. 2002.
Extrapyramidal Disorders. A Guide to Diagnosis and Treatment. Ed.
Stock V.N. M. 2002. (In Russ.).
12.
Lossos A, Dobson-Stone C, Monaco AP. Early clinical heterogeneity in
choreoacanthocytosis. Arch Neurol. 2005;62:611-614.
https://doi.org/10.1001/archneur.62.4.611
13.
Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: clinical, radiological, and neurophysiological findings in an Italian family. Neurol Sci. 2003;24:188-189.
https://doi.org/10.1007/s10072-003-0123-1
14.
Bader B, Walker RH, Vogel M. Tongue protrusion and feeding dystonia: a
hallmark of chorea-acanthocytosis. Mov Disord. 2010;25:127-129.
https://doi.org/10.1002/mds.22863
15.
Schneider SA, Lang AE, Moro E. Characteristic head drops and axial extension in advanced chorea-acanthocytosis. Mov Disord. 2010;25:1487-1491.
https://doi.org/10.1002/mds.23052
16.
Walker RH, Jung HH, Dobson-Stone C. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68:92-98.
https://doi.org/10.1212/01.wnl.0000250356.78092.cc
5.
Штульман Д.Р., Левин О.С. Неврология. Справочник практического
врача. 8-е изд. М.: МЕДпресс-информ; 2012.
Shtulman DR, Levin OS. Neurology. Reference practitioner. 8th ed. M.
2012. (In Russ.).
17.
Walker RH, Liu Q, Ichiba M. Self-mutilation in chorea-acanthocytosis:
Manifestation of movement disorder or psychopathology? Mov Disord.
2006;21:2268-2269.
https://doi.org/10.1002/mds.21156
6.
Danek A, Jung HH, Melone MA. Neuroacanthocytosis: new developments in
a neglected group of dementing disorders. J Neurol Sci. 2005;229-230:171-186.
https://doi.org/10.1016/j.jns.2004.11.024
18.
7.
Danek A, Rubio JP, Rampoldi L. McLeod neuroacanthocytosis: genotype
and phenotype. Ann Neurol. 2001;50:755-764.
https://doi.org/10.1002/ana.10035
Ishida C, Makifuchi T, Saiki S. A neuropathological study of autosomal-dominant chorea-acanthocytosis with a mutation of VPS13A. Acta
Neuropathol. 2009;117:85-94.
https://doi.org/10.1007/s00401-008-0403-1
19.
Walker RH, Danek A, Dobson-Stone C. Developments in neuroacanthocytosis: Expanding the spectrum of choreatic syndromes. Mov Disord.
2006;21:1794-1805.
https://doi.org/10.1002/mds.21108
20.
Dobson-Stone C, Velayos-Baeza A, Filippone LA. Chorein detection for
the diagnosis of chorea-acanthocytosis. Ann Neurol. 2004;56:299-302.
https://doi.org/10.1002/ana.20200
21.
Gautam G, Hashmi MA, Pandey A. Neuroacanthocytosis: A rare movement disorder with magnetic resonance imaging. J Neurosci Rural Pract.
2011;2:111-112.
https://doi.org/10.4103/0976-3147.80078
22.
Burbaud P, Rougier A, Ferrer X. Improvement of severe trunk spasms by bilateral high-frequency stimulation of the motor thalamus in a patient with
chorea-acanthocytosis. Mov Disord. 2002;17:204-207.
https://doi.org/10.1002/mds.1260
23.
Ruiz PJ, Ayerbe J, Bader B. Deep brain stimulation in chorea-acanthocytosis. Mov Disord. 2009;24:1546-1547.
https://doi.org/10.1002/mds.22592
8.
9.
Mohiddin SA, Fananapazir L. Cardiac involvement in the neuroacanthocytosis syndromes. In: Danek A., ed. Neuroacanthocytosis Syndromes. Dordrecht, The Netherlands: Springer; 2004;139-152.
https://doi.org/10.1007/1-4020-2898-9_16
Walker RH, Rasmussen A, Rudnicki D. Huntington’s disease-like 2 can
present as chorea-acanthocytosis. Neurology. 2003;61:1002-1004.
https://doi.org/10.1212/01.WNL.0000085866.68470.6D
10.
Rampoldi L, Danek A, Monaco AP. Clinical features and molecular bases
of neuroacanthocytosis. J MolMed. 2002;80:475-491.
https://doi.org/10.1007/s00109-002-0349-z
11.
Schneider SA, Aggarwal A, Bhatt M. Severe tongue protrusion dystonia: clinical syndromes and possible treatment. Neurology. 2006;67:940-943.
https://doi.org/10.1212/01.wnl.0000237446.06971.72
Поступила 10.02.2021
Received 10.02.2021
Принята к печати 11.02.2021
Accepted 11.02.2021
110
Журнал неврологии и психиатрии им. С.С. Корсакова, 2021, т. 121, №9