



































































































































 medicine
medicineSimilar presentations:


")







Система качества, эффективности и безопасности лекарственных средств
1.
Лекция 5-7. Система качества,эффективности и безопасности
лекарственных средств
2.
Этапы становления службыконтроля качества лекарственных
средств в России
3.
I период - Царская Россия (до 1917года)
• I половина XYII в. - учреждение
Аптекарского приказа, одной из задач
которого был контроль качества ЛС
• 1789 г. – издание первого Аптекарского
Устава
4.
8 из 23 параграфов Аптекарского Уставабыли посвящены качеству ЛС
• «Аптекарь должен иметь добрые, свежие к употреблению
годные и расходу соразмерные запасы, и из таковых
приготовлять потребные сложные лекарства, в таковом только
количестве, чтобы оные, паче чаяния не причинили ни самому
убытка, ни вреда ближнему» (§ 3)
• «нигде, никто кроме аптек, лекарств в раздробь не продавал» (§
4);
• Уставом впервые в России был утвержден реестр разрешенных
к применению ЛС (ежегодно пересматривался Министерством
внутренних дел, которое ведало вопросами фармацевтического
законодательства)
5.
6.
• 1892 г – вопросы контроля качества вошли в Устав врачебный• При ввозе зарубежных ЛС допускалось освобождение их от
химического исследования, если представлялся «акт
химического анализа, произведенного таким русским или
иностранным учреждением, компетентность которого будет
признана Медицинским Советом»
• При изменении «дозы или оболочки» возбуждалось новое
ходатайство «с точным описанием состава и приложением
анализа, произведенного компетентной лабораторией».
7.
• В царской России все положения, касающиеся контролякачества ЛС имели законодательно закрепленную силу и
утверждались на самом высоком уровне
• Государственная система контроля качества ЛС была
ориентирована преимущественно на надзор за
соблюдением условий изготовления и реализации ЛС
8.
II период - Советский (1917-1992 гг.)• 1919 г. – на 1 Всероссийском съезде фармацевтических
подотделов Советов рабочих, крестьянских и
красноармейских депутатов подняты проблемы усиления
контроля качества ЛС
• 1923 г. при Аптечных управлениях крупных городов начали
открываться КАЛаборатории
• 1929 г. КАЛаб стали проводить выборочный контроль
качества ЛС аптечного изготовления
9.
• 1938 г. на совещании в НКЗ СССР былотмечен недопустимо высокий уровень брака ЛС:
- промышленный 7,1 %
- аптечный до
27,6 %
Основные причины:
- несоблюдение технологии;
- отсутствие «систематического наблюдения за постановкой контроля
качества на предприятиях» ;
- недоброкачественное сырье;
- крайне грязное содержание помещений и аппаратуры
НКЗ СССР пересмотрел задачи и методы контрольно-аналитической
службы
Было организовано 73 КАЛаб (во всех областных центрах)
Утверждена Инструкция о внутриаптечном контроле
(Уровень брака в 1940 г. снизился до 1 %)
10.
• 1951 г. - ГАПУ МЗ СССР обязало КАЛаб контролировать качество всехЛС, поступающих на склады, независимо от наличия результатов
заводского анализа
• 1963 г. – дифференцированный порядок контроля качества ЛС:
• обязательный посерийный контроль всех новых ЛС в течение 2 лет
с начала промышленного выпуска;
• ядовитых ЛС;
• растворов для инъекций
Создана Государственная инспекция по контролю за качеством ЛС и
медтехники МЗ СССР
Все химико-фармацевтические препараты, антибиотики, витамины,
препараты из животного сырья, выпускаемые предприятиями
различных министерств подлежали госконтролю в ГНИИСКЛС,
который осуществлял:
- предварительный;
- выборочный последующий;
- арбитражный контроль
11.
III период (с 1992 года - по настоящее время)• 1998 г. – приняты основополагающие документы,
формирующие государственную систему контроля качества
ЛС в Российской Федерации:
• 86-ФЗ «О лекарственных средствах»
• «Система сертификации ЛС Системы сертификации ГОСР
Р»
• «Об обращении лекарственных средств» (от 12.04.2010 №
61);
12.
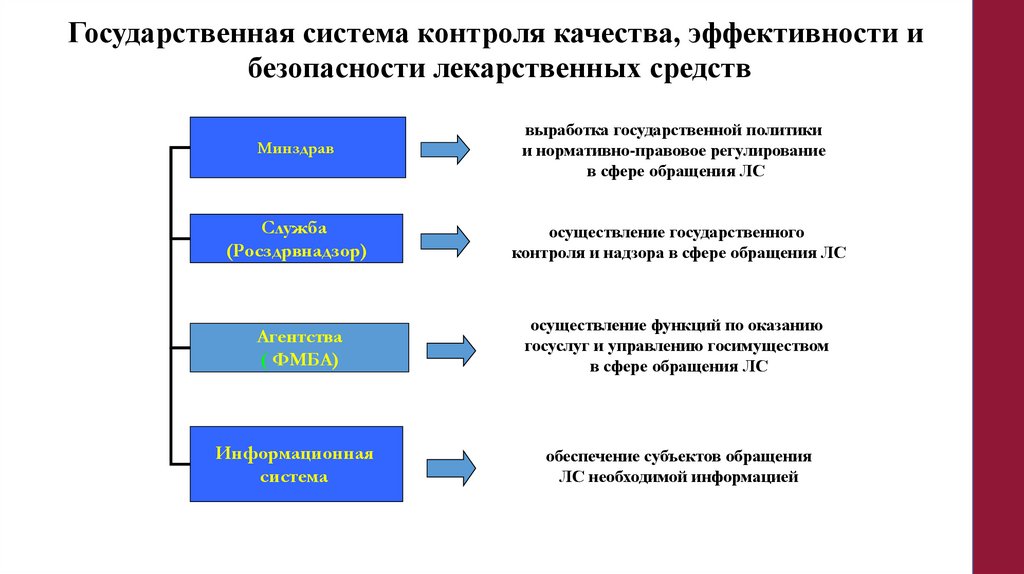
Государственная система контроля качества, эффективности ибезопасности лекарственных средств
Минздрав
выработка государственной политики
и нормативно-правовое регулирование
в сфере обращения ЛС
Служба
(Росздрвнадзор)
осуществление государственного
контроля и надзора в сфере обращения ЛС
Агентства
( ФМБА)
осуществление функций по оказанию
госуслуг и управлению госимуществом
в сфере обращения ЛС
Информационная
система
обеспечение субъектов обращения
ЛС необходимой информацией
13.
Система качества лекарственных средствСистема качества лекарственных средств – совокупность организационной
структуры, ресурсов и комплекса мер по обеспечению эффективности и
безопасности ЛС и их соответствия установленным требованиям по показателям
качества
- Законодательство в сфере обращения ЛС;
- Система контроля и надзора в сфере обращения ЛС (доклинические,
клинические испытания, регистрация, производство, качество, ввоз,
изготовление, реклама, оптовая и розничная торговля, применение,
уничтожение);
- Стандартизация в области качества ЛС и методов контроля;
- Система независимых лабораторий, осуществляющих экспертизу качества
ЛС;
- Система научно-исследовательских учреждений
14.
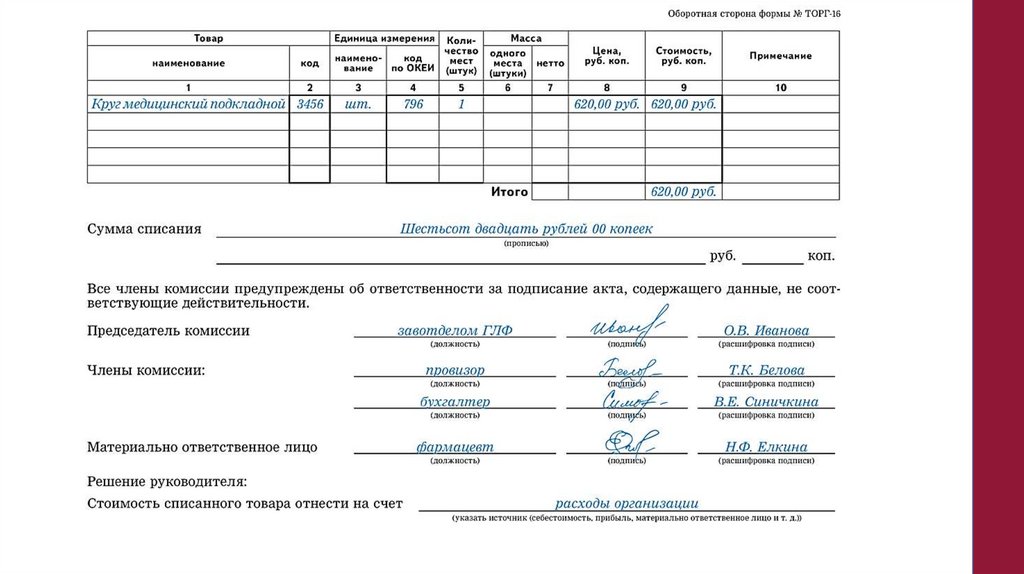
Главная задача системы качества,эффективности и безопасности ЛС
защита потребителей от негативных последствий
применения лекарственных средств, что может быть
связано с:
недостаточной изученностью ЛС на этапе разрешения и
внедрения в практику ЛС
выпуском внутри страны или ввозом в страну
субстандартных ЛС
нарушениями условий хранения лекарственных средств
на любом из этапов товародвижения ЛС
нарушениями условий реализации ЛС.
15.
2. Нормативное правовое регулирование вопросов обеспечениякачества ЛС
• Законодательные акты
• Нормативные правовые акты федеральных органов
исполнительной власти
• - Постановления Правительства РФ
• - Приказы Минздрава России
• - Приказы Росзравнадзора
• Нормативные акты органов исполнительной власти
субъектов РФ
• Нормативные акты (приказы) на уровне
организации
16.
Законодательные акты1. Конституция Российской Федерации
2. «О защите прав потребителей» (от 07.02.1992 № 2300-1);
«О защите прав юридических лиц и индивидуальных предпринимателей
при проведении государственного контроля» (от 08.08.2001 № 134-ФЗ, ст.7 12);
«О лицензировании отдельных видов деятельности» (от 04.05.2011 № 99);
«О техническом регулировании» (от 27.12.2002 № 184-ФЗ, ст.32 -40);
«О наркотических средствах и психотропных веществах»
(от 08.01.1998 № 3-ФЗ)
«О рекламе» от 13.03.2006 № 38-ФЗ;
3.«Основы законодательства РФ об охране здоровья граждан» (закон от
21.11.2011 № 323);
«Об обращении лекарственных средств» (от 12.04.2010 № 61);
17.
Законодательные акты в рамках ЕАЭС1. "Соглашение о единых принципах и правилах обращения
лекарственных средств в рамках Евразийского экономического союза"
(Заключено в г. Москве 23.12.2014)
2. Решение Коллегии ЕАЭК от 11.08.2020 N 100 (ред. от 25.10.2022) "О
Фармакопее Евразийского экономического союза»;
3. Решение Совета ЕАЭК от 03.11.2016 N 78 (ред. от 29.11.2024) "О
Правилах регистрации и экспертизы лекарственных средств для
медицинского применения"
4. Решение Коллегии ЕАЭК от 22.12.2015 N 172 (ред. от 21.11.2023) "Об
утверждении Номенклатуры лекарственных форм"
5. Проект Постановления Правительства РФ "О порядке ввода в
гражданский оборот лекарственных средств для медицинского
применения« (по состоянию на 14.01.2025)
18.
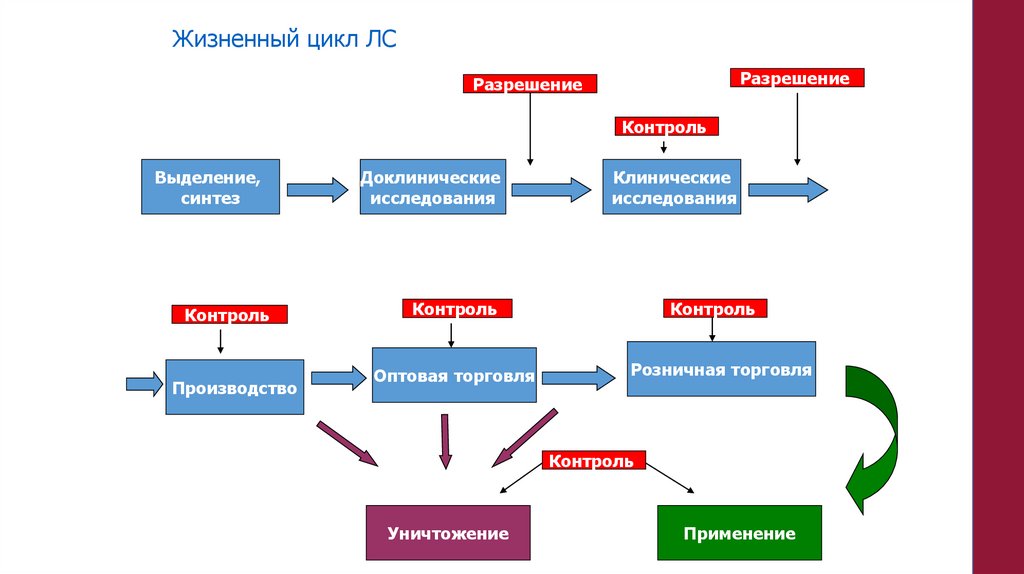
Жизненный цикл ЛСРазрешение
Разрешение
Контроль
Выделение,
синтез
Доклинические
исследования
Контроль
Контроль
Контроль
Оптовая торговля
Розничная торговля
Производство
Клинические
исследования
Контроль
Уничтожение
Применение
19.
Система качества, эффективности ибезопасности ЛС
функционирует на трех уровнях:
федеральном,
территориальном,
производственном.
20.
Федеральный уровень• МЗ РФ - Департамент государственного регулирования
обращения ЛС
• МЗ РФ - Департамент лекарственного обеспечения и
регулирования обращения МИ
• Научный центр экспертизы средств медицинского
применения
• Федеральная служба по надзору в сфере здравоохранения
(Росздравнадзор)
21.
Задачи федерального уровня• государственная регистрация и регистрация ЛП
• организация экспертизы ЛС
• организация этической экспертизы возможности
проведения клинического исследования ЛП
• государственный контроль
• мониторинг безопасности ЛП (фармаконадзор)
22.
Регистрация ЛСРегистрация ЛП - процесс получения разрешения для
медицинского применения ЛП на территориях одного или
нескольких государств-членов, осуществляемый в соответствии с
настоящими Правилами;
В Российской Федерации допускаются производство, хранение,
перевозка, ввоз в Российскую Федерацию, вывоз из Российской
Федерации, реклама, отпуск, реализация, передача, применение
лекарственных препаратов, если они зарегистрированы
уполномоченным федеральным органом исполнительной власти в
соответствии с настоящим Федеральным законом либо в
соответствии с актами, составляющими право Союза.
23.
Регистрация ЛСРегистрации в соответствии с настоящими
Правилами подлежат лекарственные препараты,
предназначенные для обращения на общем рынке
лекарственных средств в рамках Союза или на
территории одного из государств-членов.
24.
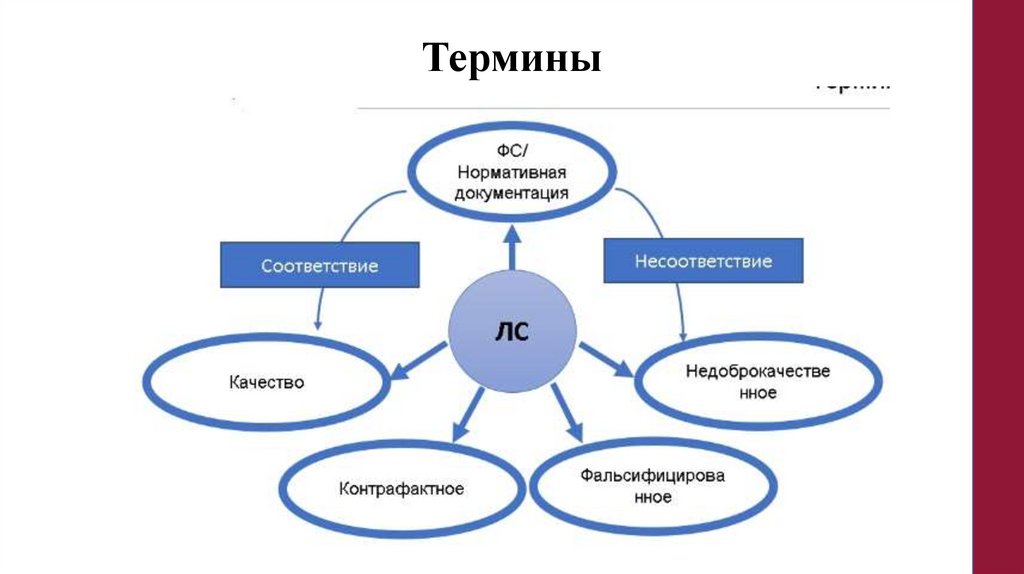
Основные понятия, используемые в рамках системыкачества, эффективности и безопасности ЛС
Безопасность ЛП - оценка положительных терапевтических
эффектов ЛП по отношению к рискам, связанным с его
применением (понятие риска включает любой риск, связанный
с качеством, безопасностью или эффективностью ЛП по
отношению к здоровью пациента или населения)
Качество ЛС - совокупность свойств и характеристик
фармацевтической субстанции и ЛП, обеспечивающая их
соответствие целевому назначению согласно требованиям актов
органов Союза;
Эффективность
ЛП
совокупность
характеристик,
обеспечивающих
достижение
профилактического,
диагностического или лечебного эффекта либо восстановление,
коррекцию или модификацию физиологической функции.
25.
Оригинальный ЛПРешение 78, 85
ЛП с новым действующим веществом, который
был первым зарегистрирован и размещен на
мировом фармацевтическом рынке на основании
досье,
содержащего
результаты
полных
доклинических (неклинических) и клинических
исследований, подтверждающих его качество,
безопасность и эффективность;
26.
Воспроизведенный ЛП (дженерик)Решение 78
ЛП, который имеет такой же количественный и качественный состав
действующих веществ и ту же ЛФ, что и оригинальный препарат, и
биоэквивалентность которого оригинальному ЛП подтверждается
соответствующими исследованиями биодоступности. Различные
соли, эфиры, изомеры, смеси изомеров, комплексы или производные
действующего вещества признаются одним и тем же действующим
веществом, если их безопасность и эффективность существенно не
отличаются. Различные ЛФ для приема внутрь с немедленным
высвобождением признаются в рамках исследований биодоступности
одной и той же ЛФ
27.
2728.
Воспроизведенный лекарственныйпрепарат (дженерик)
Решение 85
ЛП, имеющий такой же качественный и
количественный состав действующих веществ
(активных фармацевтических субстанций) и ту же
ЛФ, что и референтный ЛП, и биоэквивалентность
которого
референтному ЛП подтверждается
соответствующими исследованиями биодоступности
29.
Гибридный лекарственный препаратРешение 78
ЛП, не подпадающий под определение воспроизведенного
ЛП
при
невозможности
подтверждения
его
биоэквивалентности
с
помощью
исследований
биодоступности, а также в случае, если в данном
препарате произошли изменения действующего вещества
(веществ), показаний к применению, дозировки, ЛФ или
пути введения по сравнению с оригинальным препаратом.
30.
Гибридный лекарственный препаратРешение 85
ЛП, не подпадающий под определение воспроизведенного ЛП,
приведенного в настоящих Правилах, или, при невозможности
подтверждения его биоэквивалентности с помощью исследований
биодоступности, а также если действующее вещество
(действующие вещества), показания к применению, дозировка, ЛФ
или путь введения такого ЛП отличаются от таковых референтного
ЛП, что требует представления результатов доклинических и (или)
клинических исследований
31.
Референтный лекарственный препаратРешение 78, 85
ЛП, который используется в качестве препарата
сравнения и является эталоном, по которому
определяются (нормируются) свойства ЛП
32.
Биоаналогичный (биоподобный) ЛП(биоаналог)
биологический
лекарственный
препарат,
который
содержит
версию
действующего
вещества
зарегистрированного
биологического
оригинального
(референтного)
препарата
и
для
которого
продемонстрировано сходство (подобие) на основе
сравнительных исследований с референтным препаратом
по показателям качества, биологической активности,
эффективности и безопасности
33.
Орфанный (редкий) лекарственныйпрепарат
лекарственный препарат, предназначенный для
диагностики,
этиопатогенетического
или
патогенетического лечения (лечения, направленного
на механизм развития заболевания) редких
(орфанных) заболеваний, частота которых не
превышает официально определенного уровня в
государстве-члене
34.
Взаимозаменяемый ЛП (ФЗ №61)ЛП
с
доказанной
терапевтической
эквивалентностью или биоэквивалентностью в
отношении референтного
ЛП, имеющий
эквивалентные ему качественный состав и
количественный состав действующих веществ,
состав вспомогательных веществ, лекарственную
форму и способ введения.
35.
Единый реестр зарегистрированных ЛССоюза
общий
информационный
ресурс,
формируемый в рамках интегрированной
системы и содержащий сведения о ЛС,
зарегистрированных или прошедших иные
процедуры, связанные с регистрацией, в
соответствии с настоящими Правилами;
36.
ЕАЭС: процедуры регистрации препаратовОсновная процедура первичной регистрации (выбор – за заявителем)
Локальная
РФ – Национальная (160 рабочих дней или 80 рабочих дней (ускоренная) + 90
рабочих дней – ответ на каждый запрос)
ЕАЭС
ЕАЭС –децентрализованная (210 календарных дней + 180 суммарно
календарных дней – ответы на запросы)
ЕАЭС – взаимного признания (210 календарных дней +100 календарных дней в
каждой стране)
до 31.12.2020
с 2016 г
с 2016 г
Временная процедура
Переходный
Приведение досье в соответствие для ранее зарегистрированных по
период:
от локальной
национальной процедуре (100 календарных дней)
регистрации к ЕАЭС
с 2016 по 31.12.2025
Подтверждение регистрации
ЕАЭС
Для лекарственных препаратов, зарегистрированных по процедурам ЕАЭС (120
календарных дней)
с 2016 г
37.
ЕАЭС: процедуры регистрации (с 2021 г )Децентрализованная
Этапы
«Одновременная» подача в РГ и ГП
Документация
в составе досье
В Референтное государство
Заявление
Оплаченная пошлина
Досье М1, 2, 3, 4, 5 (на электронном носителе)
Образцы ЛП* (*если дорогостоящие, относятся
к наркотическим, психотропным – организуется
выездная ФЭ)
В Государство признания
(в течение 14 дней)
Заявление
Оплаченная пошлина
Досье М1 (на бумажном и/или электронном
носителе)
Взаимное признание
Последовательная подача досье: в референтное
государство, и после получения РУ – в государства
признания.
Возможно получение РУ только в референтной
стране, но в соответствии с Правилами ЕАЭС
В Референтное государство
Заявление
Оплата пошлины
Досье М1, 2, 3, 4, 5 (на электронном носителе)
Модуль М1 на бумажном носителе
Образцы ЛП*(*если дорогостоящие, относятся к
наркотическим, психотропным – организуется выездная ФЭ)
В Государство признания
Заявление Оплаченная пошлина
Досье М1 (на электронном носителе) +
ОХЛП, ЛВ, макеты – на государственном языке страны
признания
38.
ЕАЭС: виды экспертиз и инспекцийРеферентное государство
Государство признания
Оценка полноты и комплектности
(валидация досье)
Рассмотрение документов досье
Экспертная
организация
Оценка документов (экспертиза)
безопасности, эффективности, качества
Рассмотрение документов досье
Лаборатория
экспертной
организации
Фармацевтическая Экспертиза
-
Инспекторат
Инспектирование производства на
соответствие GMP (в течение 30-90 дней
после принятия решения уполномоченным
органов об инспектировании производства)
-
Регуляторный орган
Регуляторный орган Составление Экспертного Отчета
Рассмотрение Экспертного отчета
Референтного Государства
39.
Требования к представлениюдокументов регламентируют:
Решение Евразийской экономической комиссии №78
от 03.11.2016 г. «О правилах регистрации и
экспертизы лекарственных средств для медицинского
применения»
II. Специальные требования к модулям
регистрационного досье лекарственного препарата
6. Требования к документам регистрационного досье
воспроизведенных лекарственных препаратов
40.
Требования к представлению документоврегламентируют Приложения 1-5:
• Требования к документам регистрационного досье (в
формате ОТД) – Приложение №1 к Правилам
• Требования к формату и расположению документов
регистрационного досье лекарственного препарата в
формате ОТД - Приложение №4 к Правилам
• Структура общего технического документа для
регистрации лекарственных препаратов медицинского
назначения - Приложение №5 к Правилам
41.
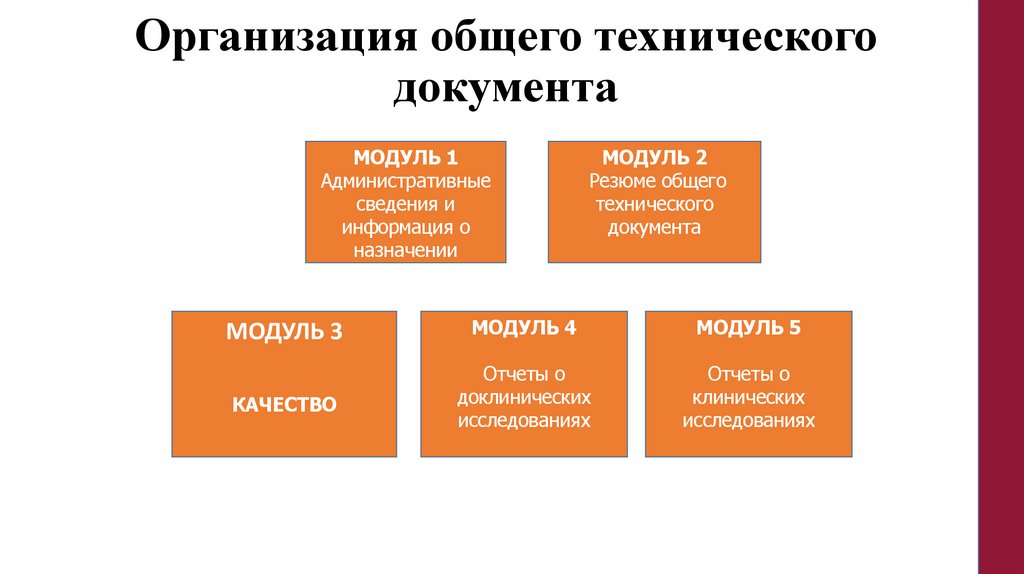
Организация общего техническогодокумента
МОДУЛЬ 1
Административные
сведения и
информация о
назначении
МОДУЛЬ 2
Резюме общего
технического
документа
МОДУЛЬ 3
МОДУЛЬ 4
МОДУЛЬ 5
КАЧЕСТВО
Отчеты о
доклинических
исследованиях
Отчеты о
клинических
исследованиях
42.
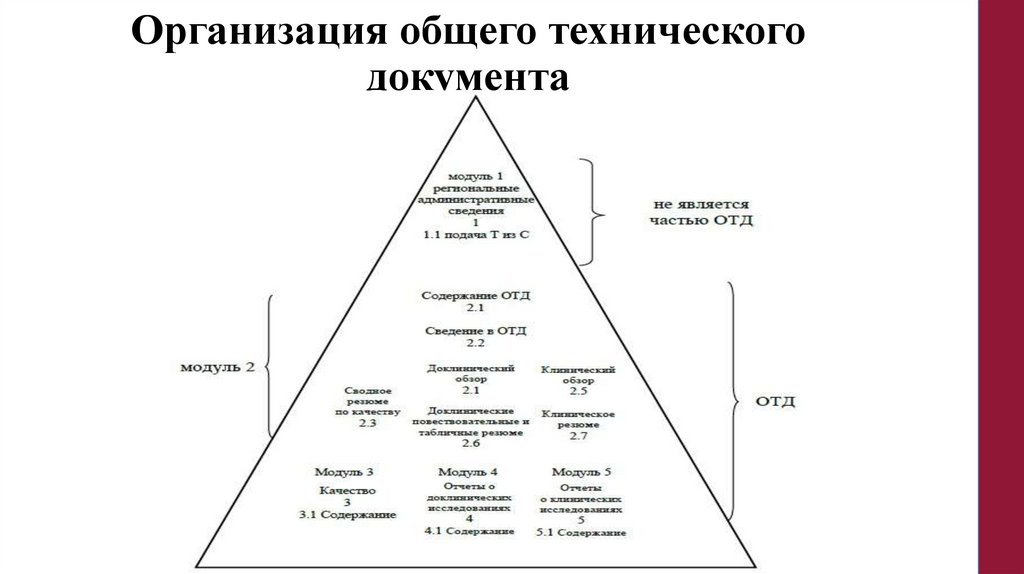
Организация общего техническогодокумента
43.
МОДУЛЬ 1Административные сведения и информация о назначении
1.0. Сопроводительное письмо
1.1. Содержание досье, необходимо представить полное содержание модулей 1 – 5
регистрационного досье, включая модуль 1
1.2. Общая документация (заявление на регистрацию, документы об оплате, информация о
назначении препарата, регистрационное удостоверение, заключение о научном
консультировании, копия ЭО страны-производителя)
1.3. Общая характеристика лекарственного препарата (ОХЛП), инструкция по медицинскому
применению (ИМП), результаты пользовательского тестирования текста ИМП, маркировка.
1.4. Информация по регуляторному статусу лекарственного препарата в других странах (при
наличии)
1.5. Документы по качеству
1.6. Документы по производству
1.7. Информация о специалистах
1.8. Специфические требования для различных типов заявлений
1.9. Документы заявителя об оценке потенциальной опасности для окружающей среды
1.10. Информация относительно фармаконадзора заявителя в государстве-члене
1.11 Копии документов, подтверждающих регистрацию товарного знака (при наличии).
44.
Общая характеристика лекарственного препарата,инструкция по медицинскому применению, маркировка
• Проекты ОХЛП, ИМП (на русском языке)
• Макеты (полноцветные копии плоского оригинала-макета, обеспечивающие
воспроизведение как вторичной (потребительской), так и первичной (внутренней)
упаковки и маркировки лекарственного препарата в двухмерном исполнении,
называемые «бумажной копией» или «компьютерной версией») вторичной
(потребительской), первичной (внутренней) и промежуточной упаковок. Макеты
промежуточной упаковки, этикеток, стикеров представляются при наличии.
• Результаты пользовательского тестирования текста ИМП (при наличии)
• Копии ОХЛП и ИМП, одобренных уполномоченным органом страны-производителя
и (или) страны – держателя регистрационного удостоверения лекарственного
препарата с датой последнего пересмотра, заверенные уполномоченным лицом
держателя регистрационного удостоверения лекарственного препарата (при наличии).
45.
Пользовательское тестированиеПри представлении результатов пользовательского тестирования
необходимо кратко обобщить, как было проведено тестирование и каким
образом в окончательную редакцию ИМП внесены все необходимые
изменения.
Резюме необходимо представить в данном разделе модуля, по следующей
форме:
• краткое описание лекарственного препарата;
• краткое описание проведенного тестирования или изучения отдельных
элементов ИМП (использованная методика, пояснения по критериям
выбора участников для тестирования, язык тестирования);
• использованные анкеты (опросные листы, в том числе инструкции по их
заполнению и формы наблюдения);
• исходная и пересмотренная редакция ИМП;
• краткое описание и обсуждение результатов тестирования (ответы
субъектов, выявленные проблемы и изменения, внесенные в
соответствующие разделы ИМП);
• заключение
46.
1.3 ОХЛП и ИМПОбщая характеристика лекарственного препарата и
инструкция по медицинскому применению
воспроизведенного лекарственного препарата должны
соответствовать общей характеристике
лекарственного препарата и инструкции по
медицинскому применению оригинального
лекарственного препарата
47.
В случае отличия показаний к применению в сторону расширения илирежима дозирования либо пути введения в инструкции по
медицинскому применению воспроизведенного лекарственного
препарата от оригинального лекарственного препарата следует
представить результаты соответствующих клинических исследований.
48.
1.4.Информация по регуляторному статусулекарственного препарата в других странах (при
наличии)
Перечень стран, в которых лекарственный препарат:
• подан на регистрацию,
• зарегистрирован,
• получил отказ в регистрации или
• его обращение на рынке было приостановлено;
с указанием:
• наименования лекарственного препарата,
• номера и даты регистрационного удостоверения,
• срока его действия или даты принятия решений об отказе в
регистрации,
• приостановлении действия регистрационного удостоверения.
49.
1.7. Информация о специалистах• Информация о специалисте, подготовившем резюме по качеству.
• Информация о специалисте, подготовившем резюме по доклиническим
данным.
• Информация о специалисте, подготовившем резюме по клиническим данным.
*Информация о специалистах по качеству, доклиническим и клиническим
данным включает сведения об их образовании, специализации и
профессиональном опыте, должна быть подписана специалистами, составившими
резюме и обзор по качеству, доклиническим, клиническим данным.
Данные специалисты должны иметь соответствующую квалификацию.
Следует указать наличие профессиональных отношений между специалистом,
составившим резюме, и заявителем.
50.
1.8 Специфические требования для различных типовзаявлений
1.8.1. Письмо держателя регистрационного удостоверения о
дополнительном торговом наименовании лекарственного
препарата представляется, если заявитель планирует
регистрировать лекарственный препарат под разными
торговыми наименованиями в стране-производителе, в
референтом государстве и государстве признания (если
применимо).
В письме должны быть указаны гарантии того, что в этих
целях используется одно регистрационное досье.
Письмо должно быть подписано держателем
регистрационного удостоверения и датировано.
51.
1.8.2. Документы по клиническимисследованиям (если применимо):
• Разрешение уполномоченного органа на проведение клинического исследования, в том
числе на внесенные поправки.
• Перечень проведенных инспекций на соответствие надлежащей клинической практике
(GCP).
• Копии отчетов о проведении GCP-инспекций (при наличии).
• Копии договоров между спонсором клинического исследования и исследовательским
центром.
• Таблица с перечнем клинических исследований (если применимо).
• Письмо держателя регистрационного удостоверения о соответствии клинических
исследований заявленного на регистрацию лекарственного препарата требованиям Правил
надлежащей клинической практики Евразийского экономического союза, утверждаемых
Комиссией
52.
1.8 Специфические требования для различных типовзаявлений/воспроизведенные
1.8.2 Документы по клиническим исследованиям (если применимо)
Резюме (до 5 страниц) обоснований и фактов, показывающих, что
лекарственный препарат является воспроизведенным лекарственным
препаратом соответствующего оригинального лекарственного препарата:
• качественном, количественном составе содержании в нем активного вещества
• лекарственной форме
• профиле безопасности и (или) эффективности его активного вещества по
сравнению с активным веществом оригинального препарата
• при необходимости сведения о биологической доступности и
биоэквивалентности данного препарата
• в частных случаях может потребоваться план управления рисками.
При отсутствии в резюме некоторых из перечисленных в настоящем пункте
элементов следует представить обоснование их отсутствия в соответствующем
разделе регистрационного досье лекарственного препарата.
53.
1.8 Специфические требования для различных типов заявлений/гибридные1.8.2 Документы по клиническим исследованиям
Резюме (до 5 страниц) обоснований и фактов, показывающих, что лекарственный препарат
является гибридным лекарственным препаратом по отношению к соответствующему
оригинальному лекарственному препарату:
• препарате
• активной фармацевтической субстанции
• лекарственной форме
• дозировках
• показаниях к применению
• способе применения по сравнению с оригинальным препаратом
• при необходимости сведения о биоэквивалентности и биодоступности данного препарата
• в частных случаях может потребоваться план управления рисками.
При отсутствии в резюме некоторых из перечисленных в настоящем пункте элементов
следует представить обоснование их отсутствия в соответствующем разделе
регистрационного досье лекарственного препарата.
54.
Информация относительно фармаконадзоразаявителя в государстве-члене
• Мастер-файл системы фармаконадзора держателя РУ представляется в случае,
когда держатель РУ впервые подает заявку на регистрацию ЛП на рынок Союза.
• При последующих заявках на регистрацию ЛП от имени данного держателя РУ
представляется краткая характеристика системы фармаконадзора держателя РУ
• Письменное подтверждение держателем РУ факта наличия уполномоченного
лица, ответственного за фармаконадзор на территории государства-члена.
• План управления рисками на ЛП, заявляемый на регистрацию
• Документы, заверенные надлежащим образом, подтверждающие наличие
взаимодействия, обеспечивающего надлежащее выполнение несколькими
юридическими лицами всех обязанностей держателя РУ, в случае если
держателями РУ лекарственного препарата, выданных референтным
государством и государствами признания, являются разные юридические лица
(если применимо).
55.
МОДУЛЬ 2Резюме общего технического документа
Резюме химической и биологической документации,
доклинических и клинических данных, представленных в
модулях 3 – 5 регистрационного досье лекарственного
препарата и заключениях специалистов, подготовивших
резюме по качеству, доклиническим и клиническим данным.
Представляются обобщенные фактические данные, включая
материалы в виде таблиц.
56.
МОДУЛЬ 2Резюме общего технического документа
2.1 Сводное содержание ОТД (модули 2 – 5)
2.2 Введение (информация о фармакологической группе, механизме
действия и клиническом применение лекарственного препарата)
2.3 Сводное резюме по качеству (обзор информации, связанной с
химическими, фармацевтическими и биологическими данными)
2.4 Доклинический обзор
2.5 Клинический обзор
2.6 Резюме по доклиническим исследованиям
2.7 Резюме по клиническим данным
57.
2.4. Доклинический обзор2.5. Клинический обзор
- резюме профиля примесей активного вещества (и в соответствующих случаях –
возможные продукты разложения, образующиеся при хранении лекарственного
препарата) в сериях лекарственного препарата, который подлежит реализации на
фармацевтическом рынке;
- оценка исследований биоэквивалентности или объяснение причины, по которой
исследования биоэквивалентности не проводились;
- обновление литературных публикаций об активном веществе данного
лекарственного препарата (данное требование может выполняться посредством
указания ссылок на публикации в рецензируемых журналах);
58.
2.4. Доклинический обзор2.5. Клинический обзор
- ранее неизвестные или следующие из характеристик препарата и (или)
его терапевтической группы пункты в общей характеристике ЛП,
которые следует проанализировать в доклинических и клинических
обзорах (резюме) и подкрепить доказательствами из научной литературы
и (или) доказательствами, полученными в результате проведения
дополнительных исследований;
- дополнительная информация, доказывающая, что профили
безопасности и (или) эффективности заявленного препарата не
отличаются от таковых у референтного препарата в случае различия
химических форм активного вещества (солей, эфиров, изомеров, смеси
изомеров, комплексов или производных от активного вещества
референтного препарата).
59.
2.6. Резюме по доклиническимисследованиям
Резюме доклинических данных нужно представлять на
основе фактических результатов фармакологических,
фармакокинетических и токсикологических
исследований, проведенных на животных in vitro, в
текстовом формате и в виде таблиц в представленной
ниже последовательности, с вводной частью.
60.
2.7. Резюме клинических данныхНеобходимо представить подробное с приведением фактических
данных резюме клинической информации по изучению
лекарственного препарата, включенного в модуль 5. Резюме
должно включать результаты всех биофармацевтических
исследований, исследований по клинической фармакологии, а
также исследований по клинической эффективности и
безопасности. Необходимо представить краткий обзор
индивидуальных исследований. Клиническая информация в
виде резюме должна представляться в определенной
последовательности частей (с перечнем использованных
научных источников).
61.
МОДУЛЬ 3. Качество.(представляется полностью)
3.1. Содержание
3.2. Основные сведения
3.2.S. Активная фармацевтическая субстанция.
3.2.S.1. Общая информация об исходных материалах и сырье
3.2.S.2. Процесс производства активной фармацевтической
субстанции
3.2.S.3. Описание характеристик активной фармацевтической
субстанции
3.2.S.4. Контроль качества активной фармацевтической субстанции
3.2.S.5. Стандартные образцы или материалы
3.2.S.6. Система упаковки (укупорки)
3.2.S.7. Стабильность
62.
3.2.P. Лекарственный препарат3.2.P.1.Описание и состав лекарственного препарата
3.2.P.2. Фармацевтическая разработка
3.2.P.3. Процесс производства лекарственного препарата
3.2.P.4. Контроль качества вспомогательных веществ
3.2.P.5. Контроль качества лекарственного препарата
3.2.P.6. Стандартные образцы и материалы
3.2.P.7. Система упаковки (укупорки)
3.2.P.8. Стабильность лекарственного препарата
63.
3.2.А. Дополнения.3.2.А.1. Производственные помещения и оборудование.
3.2.А.2. Оценка безопасности лекарственных препаратов относительно
наличия посторонних агентов.
3.2.А.3. Новые вспомогательные вещества.
3.2.R.1. Досье производственного участка.
3.2.R.2. Валидационный мастер-план.
3.2.R.3. Последний обзор по качеству лекарственного препарата.
3.2.R.4. Руководство по качеству или лабораторное руководство
лаборатории контроля качества производителя.
3.2.R.5. Список аналитических методик, которые выполняет лаборатория
контроля качества производителя.
64.
МОДУЛЬ 4Отчеты о доклинических (неклинических)
исследованиях
4.1. Содержание модуля 4
4.2. Отчеты об исследованиях
4.2.1. Фармакология
4.2.2.Фармакокинетика
4.2.3.Токсикология
4.3. Ссылки на литературу
65.
Доклинические исследованияДоклинические исследования безопасности лекарственных
средств, проведенные в государствах, не являющихся
членами Союза, рассматриваются в процессе экспертизы
лекарственных препаратов при условии, что они
спланированы, проведены и описаны в отчете о
доклиническом исследовании в соответствии с
требованиями надлежащей лабораторной практики,
эквивалентными требованиям Союза (или не ниже).
66.
4.2. Отчеты о доклинических(неклинических) исследованиях
В отдельных случаях в соответствии с требованиями по
исследованию отдельных групп препаратов части II
настоящих Требований и Правил регистрации и экспертизы
лекарственных средств для медицинского применения,
утверждаемых Комиссией в данном разделе может быть
приведен обзор данных научной литературы вместо
результатов собственных проведенных доклинических
исследований
67.
МОДУЛЬ 5Отчеты о клинических испытаниях
5.1 Содержание модуля 5
5.2 Табличный перечень всех клинических
исследований
5.3 Отчеты о клинических исследованиях
5.4 Ссылки на литературу
68.
5.3 Отчеты о клинических исследованиях5.3.1. Отчеты о биофармацевтических исследованиях – должны
быть
включены
результаты
исследований
биоэквивалентности (проведенных в случаях, когда это
необходимо), отчет о валидации биоаналитического метода,
данные о концентрациях, фармакокинетике, статистическому
анализу
5.3.1.2. результаты доказательства эквивалентности по процедуре
биовейвер.
В случае применения процедуры биовейвер в
регистрационном
досье
лекарственного
препарата
необходимо представить отчет о проведении исследований in
vitro
69.
Отчеты о проведении исследованийбиоэквивалентности
- сведения об исследователе (с указанием его рабочего места),
- организации, в которой проводились исследования,
- сроке проведения исследований
- сертификаты аудитов
- подтверждение выбора референтного препарата (или в отдельном
официальном письме)
- рекомендация Экспертного комитета по ЛС по выбору референтного
препарата (при наличии)
- сертификаты анализа серии референтного и тестируемого препарата
- официальное письмо, подтверждающее соответствие количественного
состава и производства исследуемого препарата регистрируемому
70.
Сведения о референтном препаратеторговое наименование;
дозировка;
лекарственная форма;
держатель регистрационного удостоверения;
дата регистрации;
номер регистрационного удостоверения;
государство-член, на территории которого зарегистрирован
референтный препарат;
номер серии;
наименование производителя;
срок годности;
страна приобретения
71.
Сведения о тестируемом препарате• наименование
• состав
• размер серии
• дату производства
• дату окончания срока годности (по возможности)
72.
К отчету по проведенномуисследованию необходимо приложить
• данные лабораторных и инструментальных методов
исследования,
• Данные статистической обработки результатов
клинических исследований
• Отчет о валидации
• Аналитический отчет
• Отчет о фармакокинетике (данные о концентрации)
73.
Следует представить дополнительную информацию профили безопасности и(или) эффективности заявленного лекарственного препарата не отличаются от
таковых у референтного препарата в случае различия химических форм
активного вещества (солей, эфиров, изомеров, смеси изомеров, комплексов или
производных активного вещества референтного препарата).
В случае если активное вещество воспроизведенного лекарственного препарата
представлено другой солью, эфиром или производным активного вещества
зарегистрированного препарата, представляются дополнительная информация
(библиографические обзоры) или отчеты соответствующих доклинических и
(или) клинических исследований (исследований сравнительной биодоступности),
доказывающие отсутствие изменений в фармакокинетике, фармакодинамике
и (или) токсичности воспроизведенного лекарственного препарата. При
непредставлении таких доказательств данное вещество рассматривается в
качестве нового активного вещества.
74.
Проведение исследованийбиоэквивалентности не требуется
• Лекарственные препараты для приема внутрь (I, III класс по БКС)
• Растворы для приема внутрь (вспомогательные вещества!)
• Лекарственный препарат является водным раствором для внутривенного
введения
• Парентеральные растворы (например, для внутримышечного и
подкожного введения), имеющие одинаковые типы растворителя,
действующее вещество в той же концентрации и те же вспомогательные
вещества в схожих количествах
• Лекарственный препарат представляет собой раствор для наружного и
местного применения (например, капли глазные, спрей назальный или
раствор для наружного применения) с той же концентрацией
действующего вещества
• Лекарственный препарат является газом
• Эмульсии (не предназначены для контролируемого высвобождения,
способ и скорость введения совпадают)
75.
Дополнительные данные длягибридных ЛП
Характеристика лекарственных
препаратов или заявлений на
регистрацию
Требуемые дополнительные данные
Различные соли, сложные эфиры,
комплексы, их производные (с одной и
той же активной частью молекулы)
доказательства того, что нет никаких
изменений в фармакокинетике
активной части молекулы,
фармакодинамике и (или) токсичности,
которые могут существенно повлиять
на профиль безопасности и (или)
эффективности (иначе активное
вещество следует рассматривать в
качестве нового активного вещества)
76.
Дополнительные данные длягибридных ЛП
Другой способ применения или другая
лекарственная форма: новый путь введения (для
парентерального введения, необходимо
проводить различия между
внутриартериальным, внутривенным,
внутримышечным, подкожным и другими
методами введения) иная лекарственная форма
(при том же способе введения)
клинические данные (безопасность и
эффективность), фармакокинетика, а также
соответствующие доклинические данные
(например, местная переносимость) (при
наличии)
Другая дозировка при тех же пути введения
(лекарственной форме) и показаниям к
применению
данные сравнительной биодоступности в
соответствии с правилами проведения
исследований биоэквивалентности
лекарственных препаратов в рамках
Евразийского экономического союза,
утверждаемыми Комиссией
77.
Дополнительные данные длягибридных ЛП
Сверхбиодоступные препараты
при сохранении интервала
дозирования, но со снижением
дозы, предназначенные для
достижения сходной
концентрации в плазме (крови)
в отдельных случаях достаточно
исследований сравнительной
биодоступности в соответствии с
правилами проведения
исследований
биоэквивалентности
лекарственных препаратов в
рамках Евразийского
экономического союза,
утверждаемыми Комиссией
78.
Экспертиза• Оценка полноты, комплектности и правильности
оформления документов;
• Оценка представленных данных по аспектам качества,
безопасности и эффективности;
• Проведение лабораторных испытаний на соответствие
требованиям НД по качеству и воспроизводимости методик
контроля качества;
• Инициирование при необходимости внеплановой или
плановой фармацевтической инспекции;
• Составление экспертного отчета
79.
• По результатам регистрации лекарственного препаратауполномоченный орган каждого государства-члена,
зарегистрировавшего лекарственный препарат, выдает
регистрационное удостоверение лекарственного препарата,
подтверждающее факт его регистрации.
• Регистрационное удостоверение лекарственного препарата
выдается по единой форме и в соответствии с правилами
заполнения регистрационного удостоверения
лекарственного препарата для медицинского применения
согласно приложению № 17 к настоящим Правилам
уполномоченным органом, зарегистрировавшим
лекарственный препарат.
80.
ПРИЛОЖЕНИЕ № 17к Правилам регистрации и экспертизы
лекарственных средств для
медицинского применения
ФОРМА
регистрационного удостоверения лекарственного
препарата для медицинского применения
(форма)
РЕГИСТРАЦИОННОЕ УДОСТОВЕРЕНИЕ
лекарственного препарата для медицинского применения
ЛП-№ (XXXXXX)-(YY-ZZ)
___________________________________________________
____
(номер регистрационного удостоверения)
В соответствии с Правилами регистрации и экспертизы
лекарственных средств для медицинского применения,
настоящее регистрационное удостоверение выдано:
81.
Регистрация и экспертиза лекарственногопрепарата в референтном государстве
• заявление на бумажном носителе и (или) в виде электронного
документа по установленной форме в соответствии с
приложением № 2 к настоящим Правилам;
• документы, подтверждающие оплату сбора (пошлины);
• регистрационное досье в соответствии с приложениями № 1 – 5
к настоящим Правилам на электронном носителе
(дополнительно модуль 1 регистрационного досье
представляется на бумажном носителе (за исключением плана
управления рисками, основного досье (мастер-файла)
производственной площадки (производственных площадок) и
мастер-файла по фармаконадзору));
• образцы лекарственных препаратов.
82.
Экспертиза лекарственного препарата вреферентном государстве
а) оценку полноты, комплектности и правильности оформления
документов, представленных в регистрационном досье;
б) оценку документов и сведений, представленных заявителем в
регистрационном досье лекарственного препарата, на предмет
безопасности, эффективности и качества;
в) проведение лабораторных испытаний на соответствие требованиям
нормативного документа по качеству и воспроизводимости заявленных
методик контроля качества, осуществляемых в аккредитованных
испытательных лабораториях;
г) инициирование при необходимости внеплановой или плановой
фармацевтической инспекции в случаях, установленных настоящими
Правилами;
д) составление референтным государством экспертного отчета по
оценке лекарственного препарата.
83.
Положительное решение• регистрационное удостоверение лекарственного препарата по
форме согласно приложению № 17
• инструкцию по медицинскому применению
• нормативный документ по качеству, макеты упаковок,
экспертный отчет по оценке
• согласованный план управления рисками при применении
лекарственного препарата
• размещает сведения о лекарственном препарате и входящих в
его состав активных фармацевтических субстанциях в едином
реестре (с приложением соответствующих материалов после
изъятия конфиденциальных данных и данных об экспертах, )
84.
По результатам экспертизы• заключительный экспертный
отчет по форме согласно
приложению № 16 к
настоящим Правилам
• Экспертные отчеты по
аспектам качества,
доклиническим,
клиническим аспектам,
заключительный экспертный
отчет по оценке
составляются в соответствии
с приложениями № 13 – 15 и
23 к настоящим Правилам
• При непредставлении
заявителем в установленный
срок запрошенных
документов и сведений
экспертиза и регистрация
лекарственного препарата
прекращаются
• О принятом решении
уполномоченный орган
(экспертная организация)
извещает заявителя в
письменном и (или)
электронном виде в течение
14 рабочих дней со дня
принятия этого решения
85.
Отказа) соотношение ожидаемой пользы к возможным рискам, связанным
с применением лекарственного препарата, не является
благоприятным;
б) эффективность лекарственного препарата не подтверждена
представленными заявителем сведениями;
в) качество лекарственного препарата не подтверждено;
г) предложенные методы и методики контроля качества не
воспроизводимы;
д) заявителем представлены недостоверные сведения;
е) по результатам назначенной инспекции в период регистрации
лекарственного препарата не подтверждено соответствие
надлежащим фармацевтическим практикам Союза.
86.
Регистрационное удостоверение ЛПСрок действия регистрационного удостоверения на впервые
регистрируемый ЛП в референтном государстве составляет 5 лет. По
истечении указанного срока выдается бессрочное регистрационное
удостоверение лекарственного препарата при условии подтверждения
его регистрации (перерегистрации).
В случаях, связанных с вопросами фармаконадзора, уполномоченный
орган может повторно выдать регистрационное удостоверение со
сроком действия 5 лет по итогам подтверждения регистрации
(перерегистрации).
87.
Условная регистрация– неудовлетворенные медицинские потребности для ЛП
Регистрация, осуществленная в соответствии с настоящим разделом,
действительна в течение 1 года и требует подтверждения регистрации
(перерегистрации) с переоценкой соотношения "польза - риск" на
ежегодной основе. Если особые условия, указанные в пункте 120.5
настоящих Правил, выполнены, уполномоченный орган государствачлена вправе по заявлению держателя регистрационного удостоверения
и после получения положительного заключения экспертной организации
выдать регистрационное удостоверение, действительное в течение 5 лет
и подлежащее перерегистрации в соответствии с разделом VIII
настоящих Правил.
88.
Требования Правил регистрации и экспертизы ЛСдля медицинского применения не применяются в
отношении:
а) лекарственных препаратов, которые предназначены для применения в
условиях военных действий, чрезвычайных ситуаций, для профилактики и
лечения заболеваний и поражений, полученных в результате воздействия
химических, биологических, радиационных факторов, разработаны по
заданию уполномоченных в области безопасности и обороны органов
государственной власти государств-членов и обращение которых регулируется
законодательством государств-членов;
б) ветеринарных лекарственных препаратов, обращение которых регулируется
иными актами, входящими право Союза.
89.
Регистрации в рамках Союза не подлежат:а) лекарственные препараты, изготовленные в аптеках;
б) фармацевтические субстанции;
в) лекарственные препараты, предназначенные для проведения доклинических и
клинических исследований;
г) лекарственные средства, ввезенные физическими лицами для личного
применения;
д)
радиофармацевтические
лекарственные
препараты,
изготовленные
непосредственно в медицинских организациях в порядке, установленном
уполномоченными органами государств-членов;
е) лекарственные препараты, не предназначенные для реализации на таможенной
территории Союза;
ж) образцы лекарственных препаратов, предназначенные для регистрации, и
стандартные образцы;
з) лекарственные препараты, предназначенные для использования в качестве
выставочных образцов.
90.
Допускается предоставление пациентам иприменение в отношении их
незарегистрированных ЛП
• ЛП, ввозимые в государство-член для оказания медицинской
помощи по жизненным показаниям конкретного пациента либо
для оказания медицинской помощи ограниченной группе
пациентов с редкой и (или) особо тяжелой патологией, на
основании заключения (разрешительного документа), выданного
уполномоченным органом государства-члена;
• высокотехнологичные ЛП, изготавливаемые на
нестандартизированной (нерутинной) основе и применяемые на
территории того же государства-члена в стационаре в целях
исполнения индивидуального медицинского назначения ЛП,
специально произведенного для отдельного пациента.
91.
Регистрация ЛСЗапрещается регистрация под одним
торговым наименованием лекарственных
препаратов, имеющих различный
качественный состав действующих
веществ.
92.
Регистрация одного ЛП с различными торговыминаименованиям в разных государствах-членах:
а) использование предложенного торгового наименования может
противоречить нормам права и морали или иным образом не учитывает
национальные культурные и (или) языковые особенности;
б) интеллектуальные права на торговое наименование в виде товарного знака
принадлежат лицу, отличающемуся от лица, подавшего заявление на
регистрацию лекарственного препарата (далее - заявитель), или держателя
регистрационного
удостоверения,
и
заявитель
или
держатель
регистрационного удостоверения не могут предоставить соответствующего
лицензионного договора о предоставлении права использования товарного
знака;
в) лекарственный препарат был зарегистрирован под разными торговыми
наименованиями в соответствии с законодательством государств-членов до
31 декабря 2020 г.
93.
Ускоренная экспертиза ЛП• орфанных лекарственных препаратов;
• ЛП,
предназначенных
исключительно
для
применения
несовершеннолетними;
• ЛП, представляющих особую значимость для здоровья населения,
в частности, при отсутствии эффективных методов оказания
медицинской помощи в государствах-членах, определяемых
Экспертным комитетом по лекарственным средствам на основании
обращения уполномоченного органа государства-члена, в котором
подано обращение заявителя об особой значимости лекарственного
препарата до подачи заявления на регистрацию.
Срок проведения регистрации и экспертизы ЛП в референтном
государстве не должен превышать 100 рабочих дней с даты подачи
заявления на регистрацию лекарственного препарата до дня выдачи
регистрационного удостоверения.
94.
ИЗМЕНЕНИЯ, КОТОРЫЕ ТРЕБУЮТ НОВОЙРЕГИСТРАЦИИ
Изменения активных фармацевтических субстанций, которые не расцениваются как новая АФС:
• замена химической АФС другой солью (эфиром, комплексом, производным) с той же самой активной
функциональной частью молекулы действующего вещества, отвечающей за терапевтический эффект, при
отсутствии значимых различий в эффективности/безопасности;
• замена другим изомером, иной смесью изомеров, смесью отдельных изомеров (например, рацемата на
единственный энантиомер) при отсутствии значимых различий в эффективности/безопасности;
• замена биологической АФС на другую с несколько измененной молекулярной структурой при
отсутствии существенных различий по эффективности и (или) безопасности, за исключением изменений
АФС сезонной, препандемической или пандемической вакцины для профилактики гриппа человека;
• модификации вектора, используемого для получения антигена или исходного материала, включая новый
главный банк клеток из другого источника при отсутствии значимых различий в
эффективности/безопасности;
• новый лиганд или связывающий механизм радиофармацевтического препарата при отсутствии значимых
различий в эффективности/безопасности;
• изменение экстрагента (растворителя) или соотношения лекарственного растительного сырья и
фармацевтической субстанции растительного происхождения при отсутствии значимых различий в
эффективности/безопасности;
95.
ИЗМЕНЕНИЯ, КОТОРЫЕ ТРЕБУЮТНОВОЙ РЕГИСТРАЦИИ
Изменения дозировки, лекарственной формы и способа
применения:
• изменение биодоступности;
• изменение фармакокинетики;
• изменение или добавление новой дозировки/активности;
• изменение или добавление новой лекарственной формы;
• изменение или добавление нового пути введения
96.
Фармаконадзорвид деятельности по мониторингу эффективности
и безопасности лекарственных препаратов,
направленный на выявление, оценку и
предотвращение нежелательных последствий
применения лекарственных препаратов
97.
ФармаконадзорФЗ №61 от 12.04.2010 «Об обращении ЛС» ст. 64
Решение
Совета
Евразийской
экономической
комиссии
от
03.11.2016
N
87
"Об утверждении Правил надлежащей практики
фармаконадзора
Евразийского
экономического
союза"
Приказ
Росздравнадзора от 17.06.2024 N 3518
"Об
утверждении
Порядка
фармаконадзора
лекарственных
препаратов
для
медицинского
применения"
98.
Фармаконадзор (ст. 64 ФЗ «Об обращении ЛС»ЛП, находящиеся в обращении в РФ, подлежат
мониторингу эффективности и безопасности в
целях
выявления
возможных
негативных
последствий их применения, индивидуальной
непереносимости, предупреждения медицинских
работников,
ветеринарных
специалистов,
пациентов или владельцев животных и их защиты
от применения таких лекарственных препаратов.
99.
Фармаконадзоросуществляется путем анализа предоставляемой
субъектами обращения ЛС информации о побочных
действиях, нежелательных реакциях, серьезных
нежелательных реакциях, непредвиденных нежелательных
реакциях при применении ЛП, об индивидуальной
непереносимости, отсутствии эффективности ЛП, а также
об иных фактах и обстоятельствах, представляющих угрозу
жизни или здоровью человека либо животного при
применении ЛП и выявленных на всех этапах обращения
ЛП в РФ и других государствах.
100.
При выявлении информации о серьезных нежелательныхреакциях и непредвиденных нежелательных реакциях при
применении ЛП, об особенностях их взаимодействия с
другими ЛП, индивидуальной непереносимости, и т.д.,
держатели или владельцы регистрационных удостоверений
ЛП, обязаны принять меры, направленные на устранение
негативных последствий применения таких
ЛП,
предупреждение причинения вреда жизни или здоровью
человека , защиту их от применения таких
ЛП,
на
дополнительный сбор данных об эффективности и
безопасности таких ЛП.
101.
Территориальный уровень (уровеньсубъектов РФ)
территориальные управления Росздравнадзора
центры контроля качества ЛС, осуществляющие
экспертизу качества ЛС на основании договоров с
субъектами обращения лекарственных средств
федеральные лабораторные комплексы, на базе
которых проводится контроль качества ЛС, находящихся
в обращении, в рамках мероприятий по
государственному контролю качества.
102.
Задача территориального уровня - этогосударственный контроль обращения лекарственных
средств:
организация мероприятий по выявлению и
предотвращению попадания в обращение
субстандартных ЛС
организация проведения выборочного контроля качества
ЛС
инспектирование организаций здравоохранения,
аптечных учреждений по вопросам изготовления ЛС
организация проведения экспертизы качества ЛС.
103.
Производственный уровеньпредставлен
системами обеспечения качества
предприятий – производителей ЛС,
дистрибьюторов ЛС и аптечных
организаций.
104.
Задачапроизводственного уровня
обеспечение возможности приобретения
потребителем качественных ЛС, что включает
гарантирование сохранности качества ЛС,
поступающих в аптечную организацию, и качества
услуг по реализации ЛС.
105.
Система обеспечения качестваэто совокупность организационных мер, принятых в
целях
гарантии
качества
ЛП
и
качества
фармацевтической деятельности.
В аптечных организациях она включает:
помещение
и оборудование для обеспечения условий хранения и
изготовления,
документацию
для идентификации происхождения, количества и
качества поступающих ЛП,
входной контроль поступающего товара,
персонал,
систему непрерывного обучения персонала,
должностные инструкции,
фонд нормативных и справочных материалов.
Положение о системе обеспечения качества утверждается приказом по
аптечной организации.
106.
Государственный информационный стандарт ЛС(ГИСЛС)(пр. МЗ РФ № 88 от 26. 03. 2001г.)
требования и структура официальной информации о ЛС
ГИСЛС состоит из 4 элементов:
1. фармакопейная статья ЛС
2. формулярная статья ЛС
3. клинико-фармакологическая статья ЛП
• типовая клинико-фармакологическая статья ЛС (ТКФС)
• клинико-фармакологическая статья ЛП ( КФС)
4. паспорт ЛП.
107.
1.2.
3.
4.
Формулярная статья - это нормативный документ, содержащий
стандартизированные по форме и содержанию сведения о применении
ЛС при определенном заболевании.
ТКФС - это официальный документ, содержащий сведения об основных
свойствах ЛС под международным непатентованным названием (МНН),
разрабатывается экспертным органом и утверждается МЗ РФ.
КФС - это официальный документ, который отражает совокупность
клинико-фармакологических данных конкретного ЛП под определенным
торговым наименованием и разрабатывается на основе
соответствующей типовой клинико-фармакологической статьи ЛС
производителем, проходит экспертизу при регистрации и утверждается
МЗ РФ.
Паспорт лекарственного препарата - это официальный документ,
содержащий обобщенную информацию о ЛП, имеющую юридическое
значение в сфере обращения ЛС, в том числе - идентифицирующую
отличительные свойства упаковки.
108.
На основе ГИСЛС разрабатываетсяГосударственный реестр ЛС
Инструкции по применению ЛП
Перечень жизненно необходимых и важнейших ЛП
Список льготного отпуска ЛС
Список ЛС, отпускаемых без рецепта врача
Обязательный ассортимент ЛС для аптечных
организаций, обслуживающих амбулаторных больных
Федеральное руководство для врачей по использованию
ЛС.
109.
Лекция 5-7. Система качества,эффективности и безопасности
лекарственных средств
(продолжение)
110.
Термины111.
Система ввода лекарственных препаратов вгражданский оборот
Сертификация и декларирование соответствия более
не применяются - данные процедуры
заменены другими, а именно:
• процедурой предоставления определенных
документов и сведений в Росздравнадзор - в
отношении обычных (т.е. не иммунобиологических) ЛП
(далее – обыкновенные ЛП);
• процедурой получения разрешения Росздравнадзора в отношении иммунобиологических ЛП.
112.
Иммунобиологические лекарственныепрепараты
• Под иммунобиологическими ЛП понимаются ЛП,
предназначенные для формирования активного или пассивного
иммунитета либо диагностики наличия иммунитета или
диагностики специфического приобретенного изменения
иммунологического ответа на аллергизирующие вещества. К
иммунобиологическим ЛП относятся вакцины, анатоксины,
токсины, сыворотки, иммуноглобулины и аллергены (п. 7 ст. 4
Федерального закона от 12.04.2010 № 61-ФЗ «Об обращении
лекарственных средств» (далее – ФЗ № 61-ФЗ)).
113.
Не нужно вводить в гражданскийоборот
• ЛП, предназначенные для проведения клинических
исследований ЛП;
• ЛП, предназначенные для проведения экспертизы
лекарственных средств для осуществления
государственной регистрации ЛП;
• незарегистрированные ЛП, предназначенные для
оказания медицинской помощи по жизненным
показаниям конкретного пациента, ввозимых в РФ в
соответствии с ч. 3 ст. 47 ФЗ № 61 (ч. 9 ст. 52.1 ФЗ № 61ФЗ).
114.
Регламентации новых правил• ФЗ № 61-ФЗ;
• ФЗ № 184-ФЗ;
• Федеральный закон от 17.09.1998 № 157-ФЗ «Об
иммунопрофилактике инфекционных болезней»;
• Постановлением Правительства РФ «О порядке
ввода в гражданский оборот лекарственных
препаратов для медицинского применения»
115.
ПП РФ «О порядке ввода в гражданский оборотлекарственных препаратов для медицинского
применения» утверждены:
• Правила представления документов и сведений о ЛП для медицинского
применения, вводимых в гражданский оборот;
• Правила выдачи протокола испытаний о соответствии первых трех
серий или партий ЛП для медицинского применения (за исключением
иммунобиологического ЛП), впервые произведенного в РФ или
впервые ввозимого в РФ, показателям качества, предусмотренным
нормативной документацией;
• Правила выдачи разрешения на ввод в гражданский оборот серии или
партии иммунобиологического ЛП, выдачи заключения о соответствии
серии или партии иммунобиологического ЛП требованиям,
установленным при его государственной регистрации;
• Правила принятия решения о прекращении гражданского оборота
серии или партии ЛП для медицинского применения.
116.
Правила ввода в гражданский оборот обыкновенных ЛППроизводители перед вводом в гражданский оборот серии или партии ЛП представляют в АИС
через личный кабинет:
• для каждой серии или каждой партии ЛП:
документ производителя, подтверждающий качество ЛП*;
подтверждение уполномоченного лица производителя соответствия ЛП требованиям,
установленным при его государственной регистрации;
• для первых трех серий или партий ЛП, впервые произведенного в РФ, - протокол испытаний о
соответствии серии или партии ЛП показателям качества, предусмотренным нормативной
документацией (далее - протокол испытаний), проводимых аккредитованными федеральными
государственными бюджетными учреждениями, подведомственными Минздраву РФ или
Росздравнадзору**.
• представление протокола испытаний допускается только в отношении первой серии или первой
партии ЛП, впервые произведенного в РФ в случае предоставления сведений о дате выдачи и
регистрационном номере заключения соответствия производства ЛС требованиям правил
надлежащей производственной практики или сертификата соответствия производства ЛС
требованиям правил надлежащей производственной практики ЕЭС, выданного
уполномоченным органом государства - члена ЕЭС и отсутствия в 3 лет, предшествующих дате
выдачи протокола испытаний, случаев выявления несоответствия установленным требованиям
качества ЛС
Росздравнадзор в течение 3 рабочих дней со дня поступления в АИС необходимых документов и
сведений размещает на своем официальном сайте в сети «Интернет» сведения о сериях или
партиях ЛП, вводимых в гражданский оборот (с соблюдением требований законодательства РФ о
коммерческой и иной охраняемой законом тайне).
117.
Правила ввода в гражданский оборот обыкновенных ЛПИмпортеры перед вводом в гражданский оборот ЛП представляют в АИС через личный
кабинет:
• для каждой серии или каждой партии ЛП:
документ производителя, подтверждающий качество ЛП;
подтверждение представителя импортера, уполномоченного иностранным производителем
лекарственных средств, соответствия ввозимого ЛП, установленным при его государственной
регистрации;
• для первых трех серий или партий ЛП, впервые ввозимого в РФ, - протокол испытаний,
проводимых аккредитованными федеральными государственными бюджетными учреждениями,
подведомственными Минздраву РФ или Росздравнадзору**.
• представление протокола испытаний допускается только в отношении первой серии или первой
партии ЛП для впервые ввозимого в РФ в случае предоставления сведений о дате выдачи и
регистрационном номере заключения соответствия производства ЛС требованиям правил
надлежащей производственной практики или сертификата соответствия производства ЛС
требованиям правил надлежащей производственной практики ЕЭС, выданного
уполномоченным органом государства - члена ЕЭС и отсутствия в 3 лет, предшествующих дате
выдачи протокола испытаний, случаев выявления несоответствия установленным требованиям
качества ЛС
Росздравнадзор в течение 3 рабочих дней со дня поступления в АИС необходимых документов и
сведений размещает на своем официальном сайте в сети «Интернет» сведения о сериях или
партиях ЛП, вводимых в гражданский оборот (с соблюдением требований законодательства РФ о
коммерческой и иной охраняемой законом тайне).
118.
Правила ввода в гражданский оборот обыкновенных ЛПНе требуется представление протокола испытаний в отношении орфанных ЛП,
высокотехнологичных ЛП, полученных из биологического материала определенного
человека и предназначенных для применения этому же человеку.
В отношении одной серии или одной партии высокотехнологичного, впервые
произведенного в РФ или впервые ввозимого в РФ, представляется протокол
испытаний, проведенных с использованием средств дистанционного
взаимодействия в течение первых трех лет после ввода такого препарата в
гражданский оборот.
Ежегодно не позднее 1 апреля производители ЛС или организации,
осуществляющие ввоз ЛП в РФ, представляют в федеральный орган
исполнительной власти, осуществляющий функции по контролю и надзору в сфере
здравоохранения, протокол испытаний поступившего в течение предшествующего
календарного года в гражданский оборот ЛП согласно регистрационному
удостоверению (на одну серию торгового наименования с учетом ЛФ одной
дозировки), проводимых аккредитованными испытательными лабораториями
(центрами).
119.
Правила ввода в гражданский оборот иммунобиологических ЛП• Так, в соответствии с указанными правилами ввод иммунобиологических
ЛП в гражданский оборот осуществляется на основании выданного
Росздравнадзором разрешения на ввод в гражданский оборот в РФ серии
или партии произведенного в РФ или ввозимого в РФ иммунобиологического
ЛП (далее - разрешение).
• Разрешение же выдается на основании заключения о соответствии серии
или партии иммунобиологического ЛП требованиям, установленным при его
государственной регистрации, которое, в свою очередь, выдается теми
же ФГБУ, которые выдают протокол испытаний для первых трех серий или
партий обыкновенных ЛП, впервые произведённых или впервые ввозимых в
РФ (далее – заключение).
• Заключение выдается по результатам проведенных ФГБУ испытаний
качества иммунобиологического ЛП.
• При этом, в отличие от процедуры испытаний обыкновенных ЛП,
установлено, что объем проводимых испытаний качества
иммунобиологического ЛП конкретного наименования и производителя (с
учетом лекарственной формы и дозировки) должен определяться
специально созданной для этого Росздравнадзором Комиссией по качеству
иммунобиологических ЛП (далее - Комиссия)
120.
ПП РФ• целью установления новой системы ввода ЛП в гражданский оборот
является создание эффективного механизма «выпускного» контроля ЛП,
поступающих в гражданский оборот.
• Эффективность такой системы выражается в том, что теперь вместо
обязательного подтверждения ЛП посредством процедур декларирования
или сертификации, проводимых с участием третьей стороны –
аккредитованных испытательных лабораторий, достаточно будет лишь
предоставления производителями или импортерами ЛП определенных
сведений о ЛП в Росздравнадзор.
• фактически «выпускной» контроль будет действовать лишь в отношении
обыкновенных ЛП, которые ранее уже были введены в оборот.
• В отношении же обыкновенных ЛП, которые вводятся в гражданский оборот
впервые, а также в отношении всех иммунобиологических ЛП, как и ранее
потребуется предварительно получать разрешительные документы, только
теперь это будут не сертификат соответствия или декларация соответствия, а
протокол испытаний (для обыкновенных ЛП) или разрешение, выдаваемое
Росздравнадзором (для иммунобиологических ЛП).
121.
Система мониторинга движениялекарственных препаратов
Требования касаются всех без исключения субъектов обращения
лекарственных препаратов – юридических лиц и ИП, которые
осуществляют с лекарствами какие-либо из следующих действий:
• производство;
• хранение;
• ввоз в Россию;
• отпуск;
• реализацию;
• передачу;
• применение;
• уничтожение.
122.
«Автоматизированная система мониторингадвижения лекарственных препаратов…»
123.
Новые термины• ИС «Маркировка» - государственная информационная
система, создаваемая в целях информационного
обеспечения маркировки товаров КИЗ.
• Мониторинг движения ЛП - проведение анализа
движения ЛП в Компоненте МДЛП на основании
сведений, зарегистрированных субъектами обращения
ЛП в соответствии с настоящими Методическими
рекомендациями.
• Компонент МДЛП - функциональная подсистема ИС
«Маркировка», создаваемая в целях информационного
обеспечения маркировки ЛПКИЗ.
124.
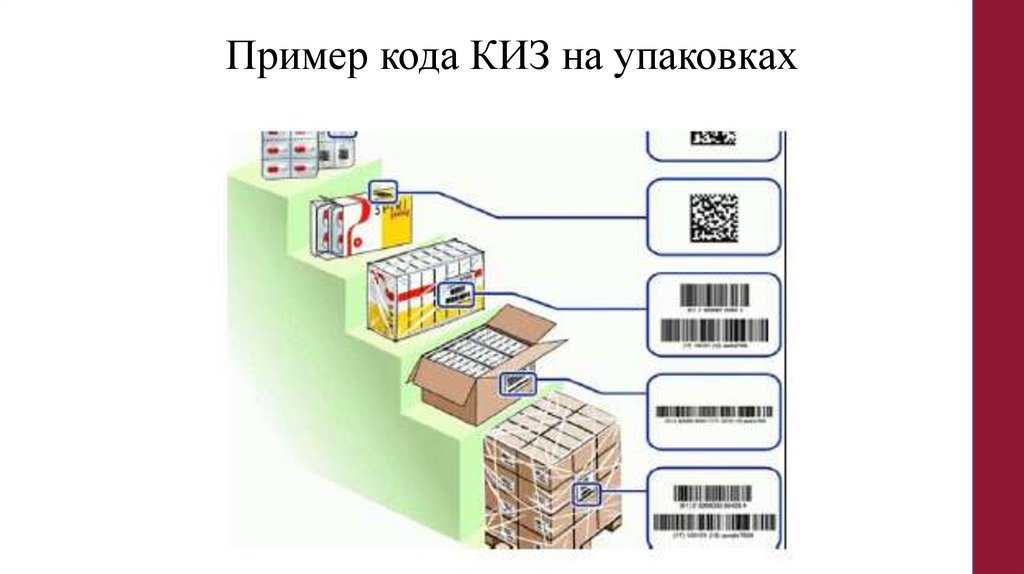
Новые термины• Контрольный (идентификационный) знак (КИЗ) - информационный
носитель, формируемый в рамках эксперимента для нанесения на
вторичную (потребительскую) упаковку ЛП для нанесения на
третичную (заводскую, транспортную) упаковку ЛП.
• Идентификационный номер ЛП - уникальный код, позволяющий
идентифицировать как минимум производителя, торговое наименование
ЛП, лекарственную форму, дозировку лекарственного средства и
комплектность упаковки ЛП. В рамках Эксперимента в качестве
идентификационного номера ЛП используется глобальный номер
предмета торговли (GTIN, Global Trade Item Number).
• Индивидуальный серийный номер - цифровая или буквенно
цифровая последовательность, которая для целей идентификации
вторичных (потребительских) упаковок ЛП составляется в соответствии
с пунктом 9 настоящих методических рекомендаций, а для целей
идентификации третичных (заводских, транспортных) упаковок.
125.
Информация в КИЗ• На вторичную (потребительскую) упаковку наносится КИЗ в виде
двумерного штрихового кода, пригодный для машинного
считывания, а его функция распознавания и коррекции ошибок
должна быть эквивалентна или выше, чем у Data Matrix ЕСС200
(далее - DataMatrix). Соответствует ГОСТ Р ИСО/МЭК 16022-2008
126.
Пример кода КИЗ на упаковках127.
Требования к информационной системе в аптеке• идентификация ЛП в ИС «Маркировка» производится на
основании уникальных идентификаторов вторичной
(потребительской) упаковки;
• •полная прослеживаемость движения серии ЛП от производителя
до конечного потребителя обеспечивается за счет внесения в ИС
«Маркировка» соответствующей информации в форме
электронных документов, подписанных усиленной
квалифицированной электронной подписью, передаваемых
участниками информационного взаимодействия в связи с
изменениями состояния и/или местоположения ЛП на
протяжении всего жизненного цикла ЛП;
• •информация о каждом перемещении ЛП от одного субъекта
обращения к другому должна быть акцептована каждым
участником взаимодействия.
128.
Требования к обмену информацией с ИС «Маркировка»• Разрабатываемые информационные электронные сервисы,
осуществляют автоматизированный обмен должны
обеспечивать выполнение следующих функций:
• формирование, подписание электронной подписью документов
в систему;
• получение ответа на направленный ранее документ;
• сохранение содержимого направляемых документов и
получаемых ответов на них, а также информации о фактах
направления документов (номер документа; дата и время
отправки; информация об уполномоченном лице, подписавшем
и направившем документ; дата и время получения ответа).
129.
Варианты информационного взаимодействия сИС «Маркировка»
• Ручная работа в личном кабинете ИС
«Маркировка»
• Полуавтоматическая работа с использованием
универсальной системы обмена (УСО).
Интеграция через обмен файлами.
• Автоматическая работа с использованием
протокола обмена интерфейсного уровня. Полная
интеграция.
130.
131131.
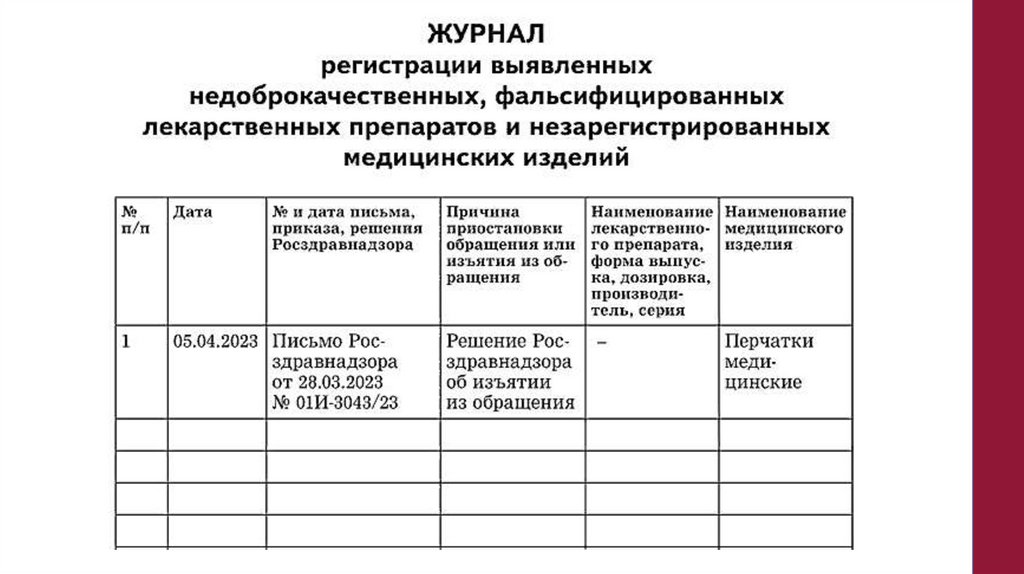
Работа с письмами о выявлении ЛП,имеющих признаки фальсификации
• При выявлении серии ЛП, указанной в письме Росздравнадзора:
1. Переместить всю серию выявленного ЛП в карантинную зону
2. Создать комиссию минимум из трех человек для проведения сравнительного
анализа отличительных признаков фальсификации (если это возможно при
визуальном осмотре)
3. Провести визуальный осмотр для выявления признаков подделки, сравнить
полученные данные с приложением к письму
4. Зарегистрируйте
ЛП
в
Журнале
регистрации
выявленных
недоброкачественных, фальсифицированных лекарственных препаратов и
незарегистрированных медицинских изделий
5. Составить акт – указать состав комиссии, наименование ЛП, дозировку,
фасовку, форму выпуска, производителя, серию, количество упаковок в
наличии, параметры сравнительного анализа, количество проверенных
упаковок, выявлены ли признаки фальсификации.
132.
133133.
134134.
135135.
136136.
Передача информации в Территориальный орган137
137.
138138.
Архивация информационных писем – разрабатывается иутверждается организацией самостоятельно
139