













































































































 medicine
medicine biology
biologySimilar presentations:


")







Медицинская цитогенетика
1.
Медицинская цитогенетикаСамойлова Л.Р., врач-лабораторный
генетик,
преподаватель кафедры НППиМГ
ИФМиБ
Казань, 2023
2.
3.
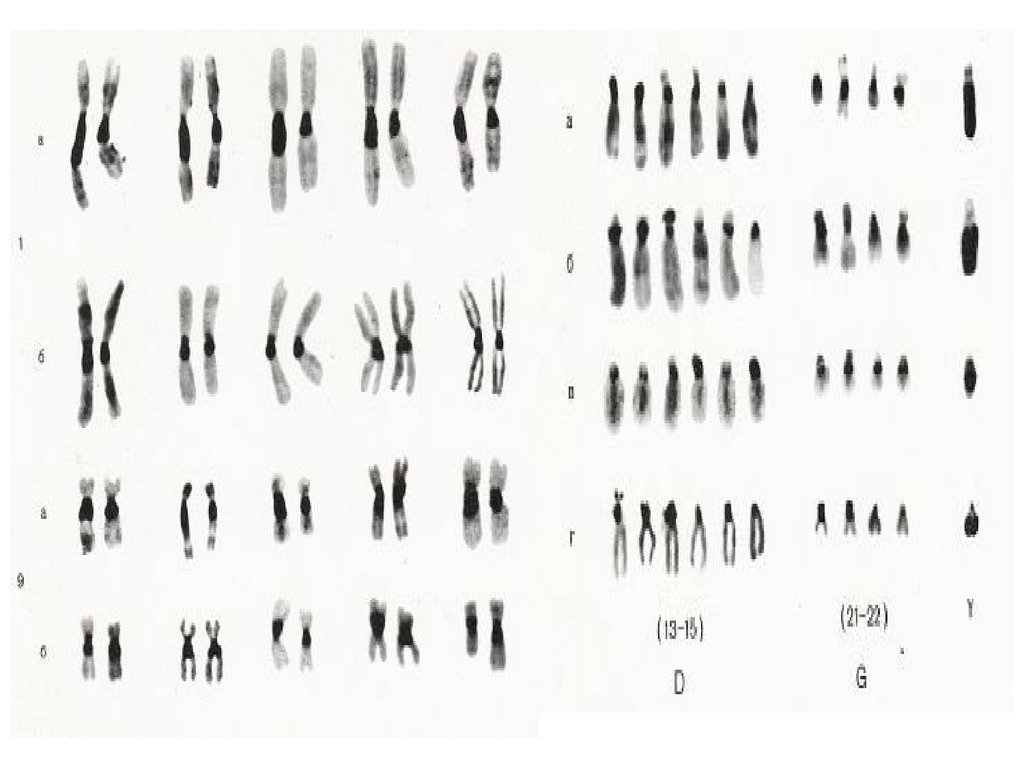
1956г.-46 хромосом1959г.-47 хромосом
1959 г. – синдром Клайнфельтера,
синдром Шерешевского-Тернера
1960 г. – синдром Патау
синдром Эдвардса
4.
Кариотип – систематизированныйнабор хромосом ядра клетки с его
количественными и качественными
характеристиками
Хромосома –
дезоксирибонуклеопротеидный
комплекс, образованный молекулой
ДНК и белками (гистонами и
негистонами)
5.
Расположение хромосом в интерфазном ядре6.
Показания к кариотипированию лимфоцитовпериферической крови
•рождение/наличие ребенка с МВПР,
задержкой психомоторного развития,
установленным хромосомным синдромом
•бесплодие женское и мужское
•аменорея
•мертворождения, внутриутробная гибель
плода
•привычное невынашивание беременности
•неудачные протоколы ЭКО
7.
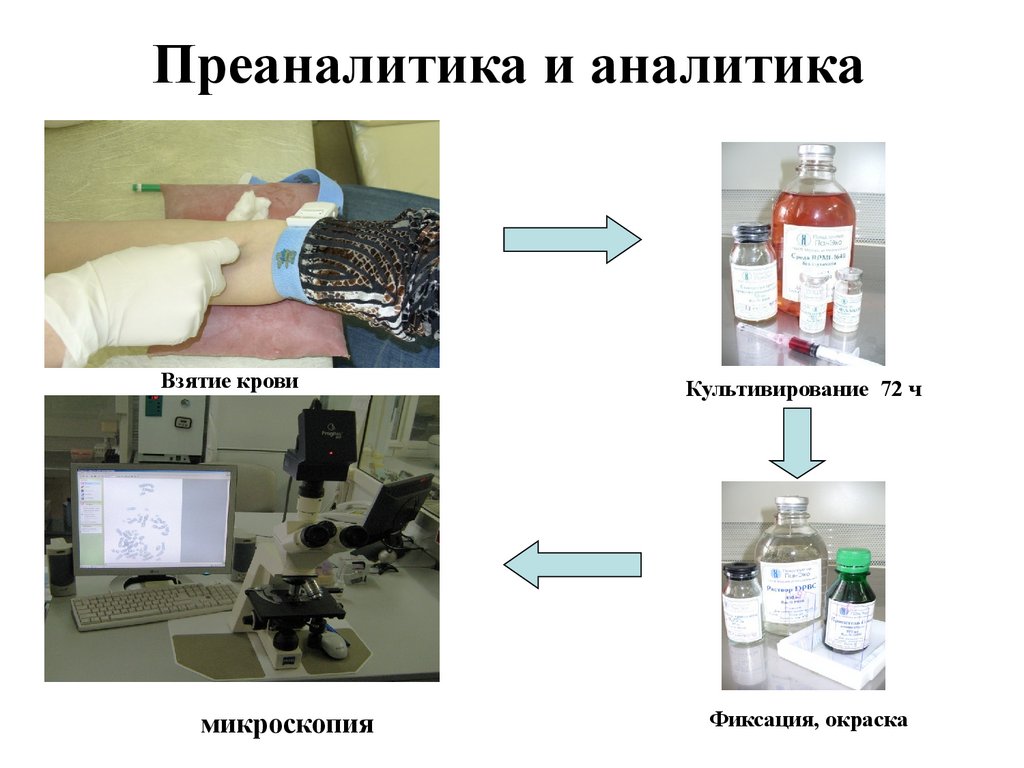
Преаналитика и аналитикаВзятие крови
микроскопия
Культивирование 72 ч
Фиксация, окраска
8.
Как рассмотреть хромосомы в клетке?Окуляр 10х
Объектив 90х-100х
Бинокулярная насадка 1.5х
9.
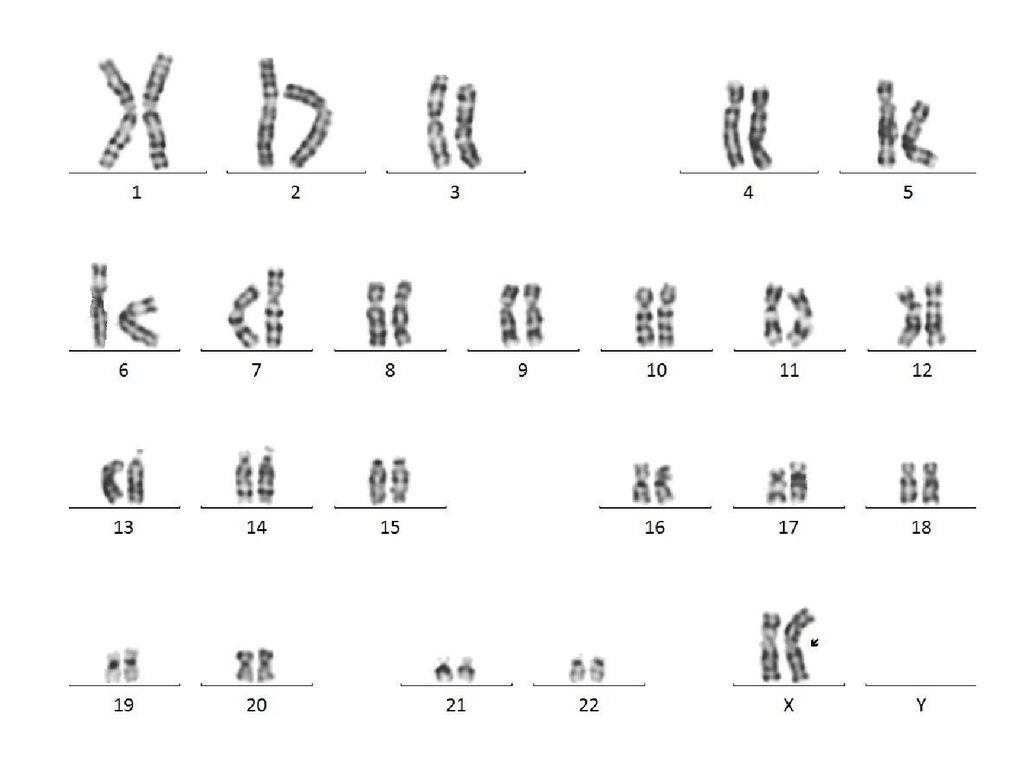
Анализ метафазной пластинкиКариотип 46,XY
10.
RV1960г. – Дэнвер
1963г. – Лондон
1966г. - Чикаго
11.
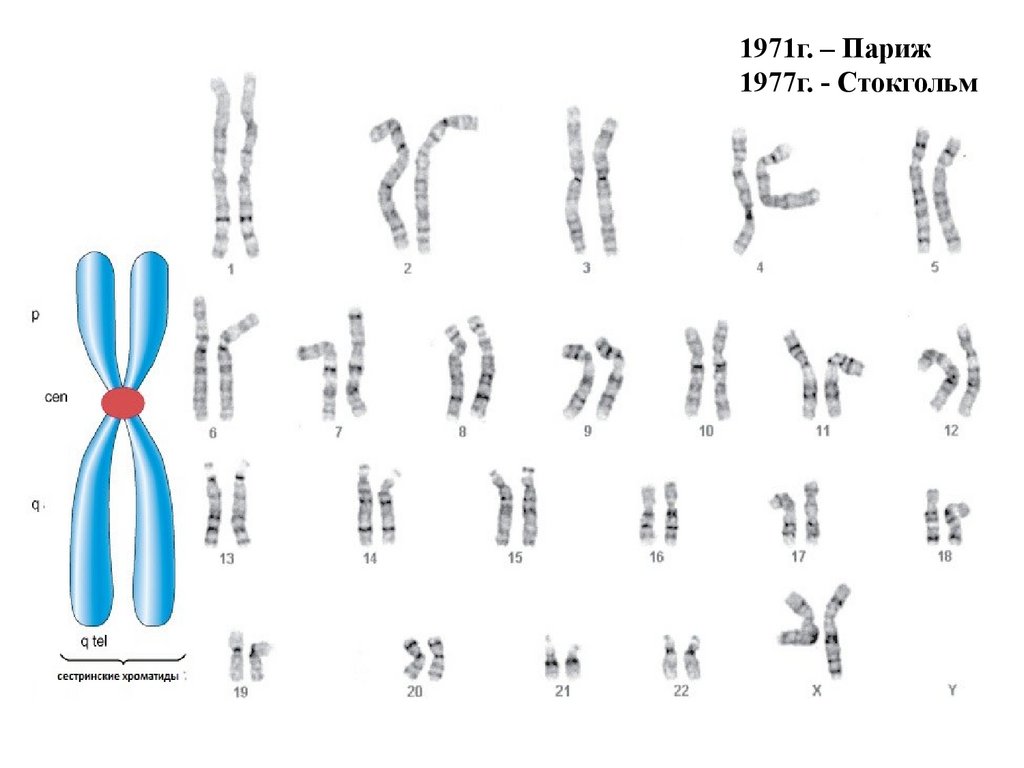
1971г. – Париж1977г. - Стокгольм
12.
короткоеплечо
центромера
длинное плечо
теломера
12
15
13.
• Центромера (первичная перетяжка)-местоскрепления хроматид
• Необходима для правильной сегрегации
хромосом
• Состоит из сателлитной ДНК (классическая и
альфоидная), не транскрибируется
• Альфоидная ДНК-роль в функционировании
центромеры
• Простые (классические) сателлиты
(тандемные повторы АТССС) формируют
гетерохроматиновые участки 1,9,16 (вторичные
перетяжки), короткие плечи акроцентриков,
длинное плечо Y
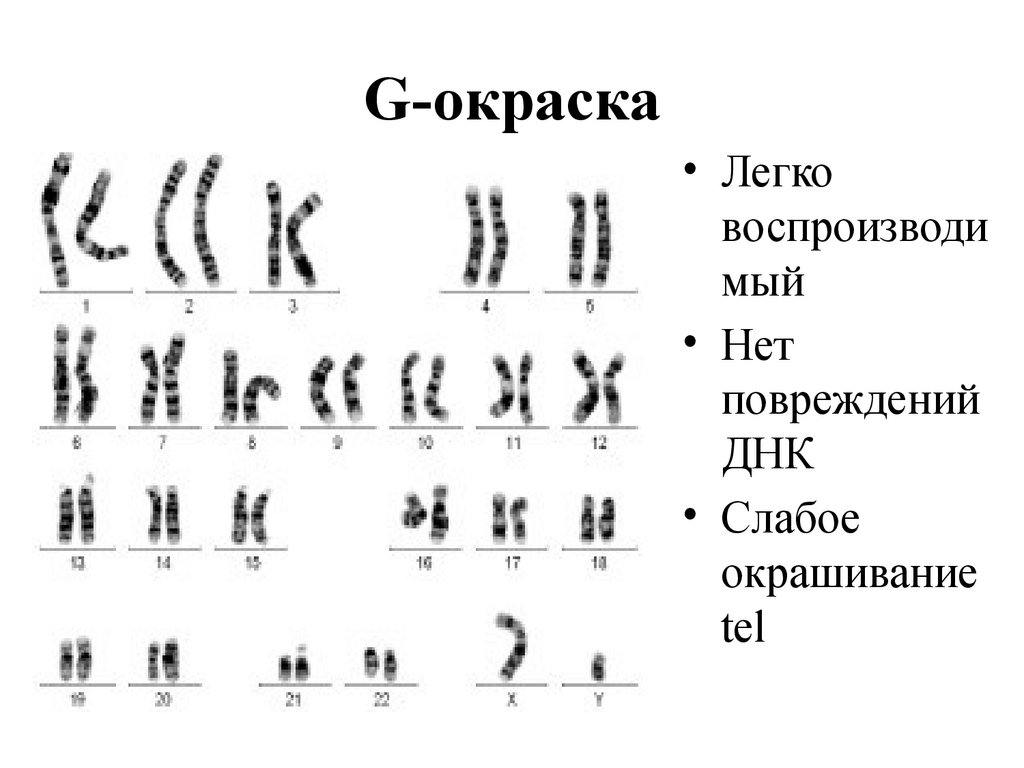
14.
G-окраска• Легко
воспроизводи
мый
• Нет
повреждений
ДНК
• Cлабое
окрашивание
tel
15.
Идиограммы хромосомчеловека
1
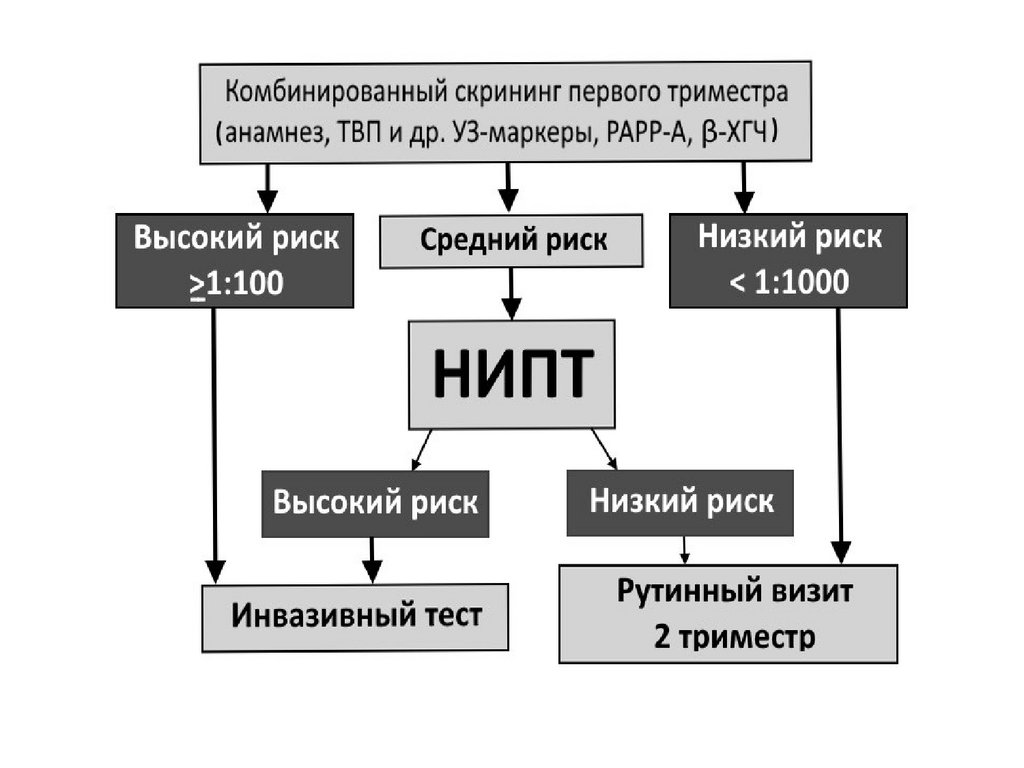
2
3
4
5
6
7
8
9
10
11
12
13
14
15 16
17 18 19
20
21
22
X
Y
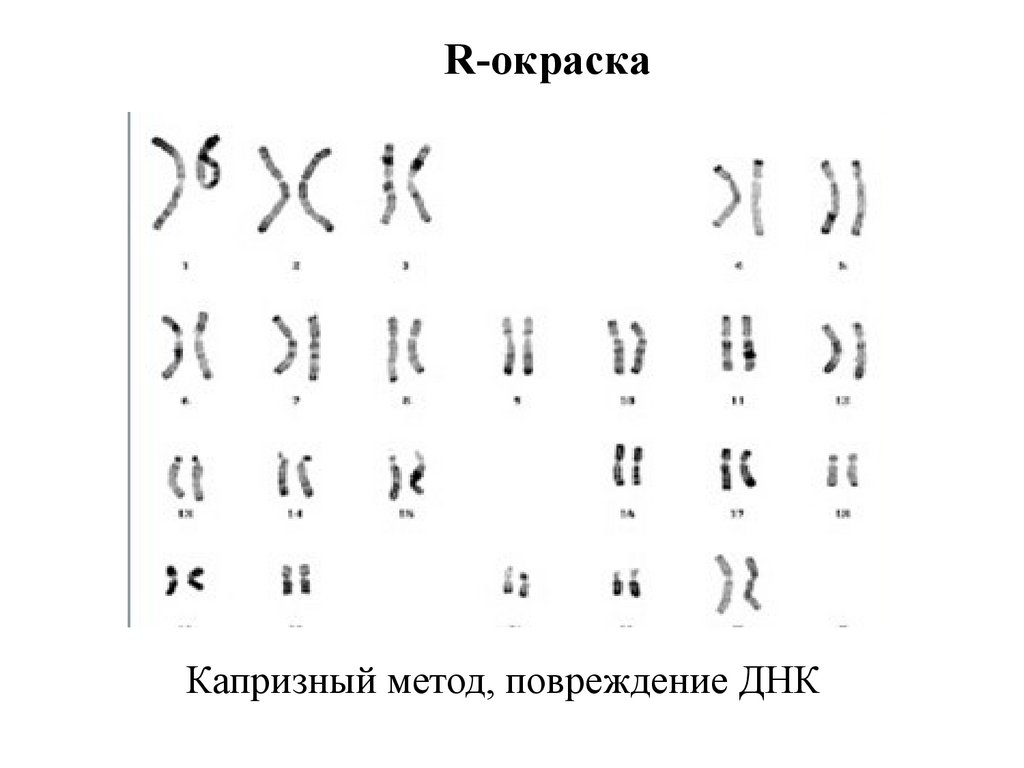
16.
R-окраскаКапризный метод, повреждение ДНК
17.
18.
Q - окраска19.
Изменения кариотипа• Числовые – геномные мутации,
количество хромосом отличается от
2n=46
• Структурные – внутри- и
межхромосомные перестройки,
количество хромосом 2n=46, изменены
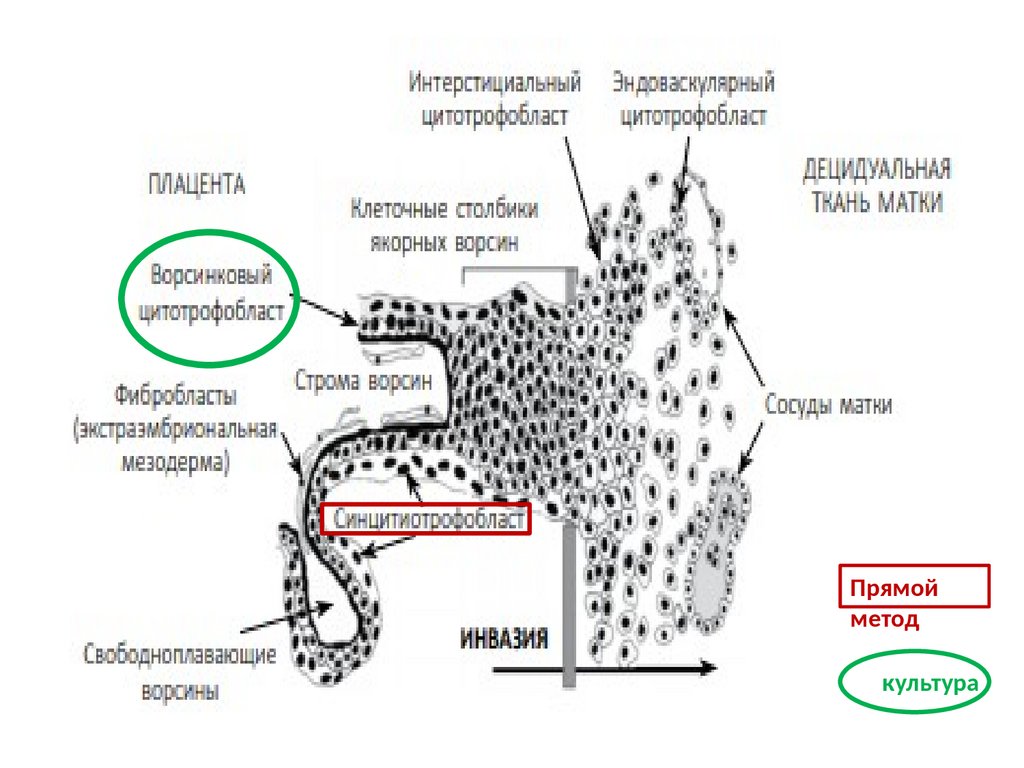
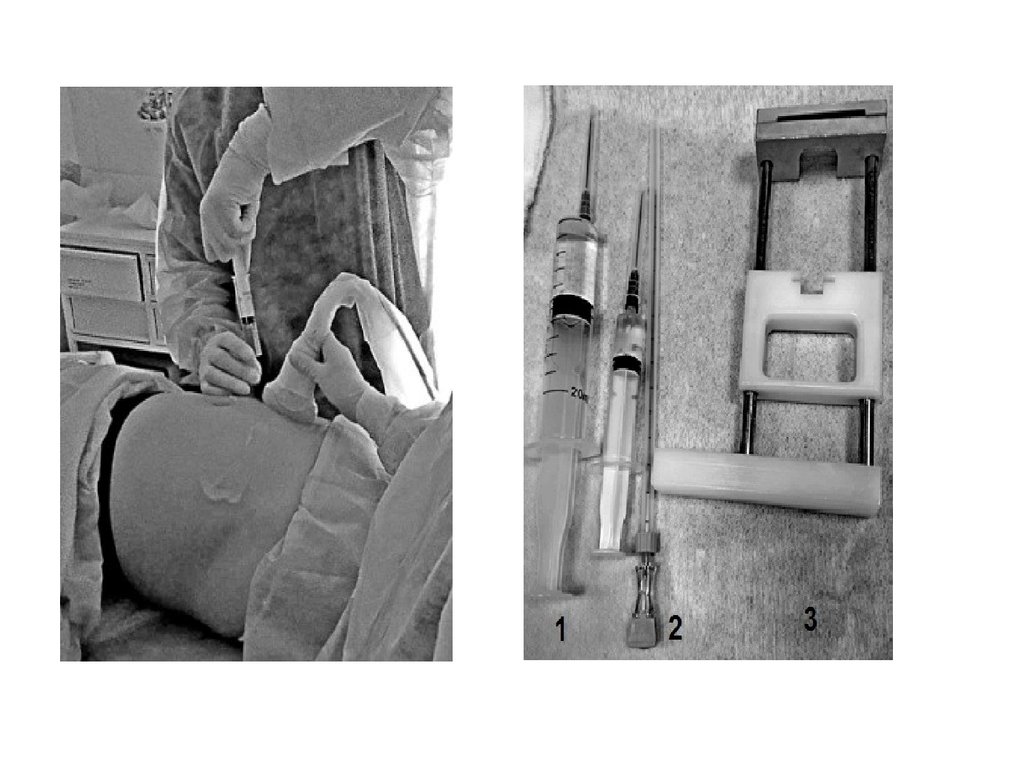
или морфология хромосом, и/или
порядок генетического материала
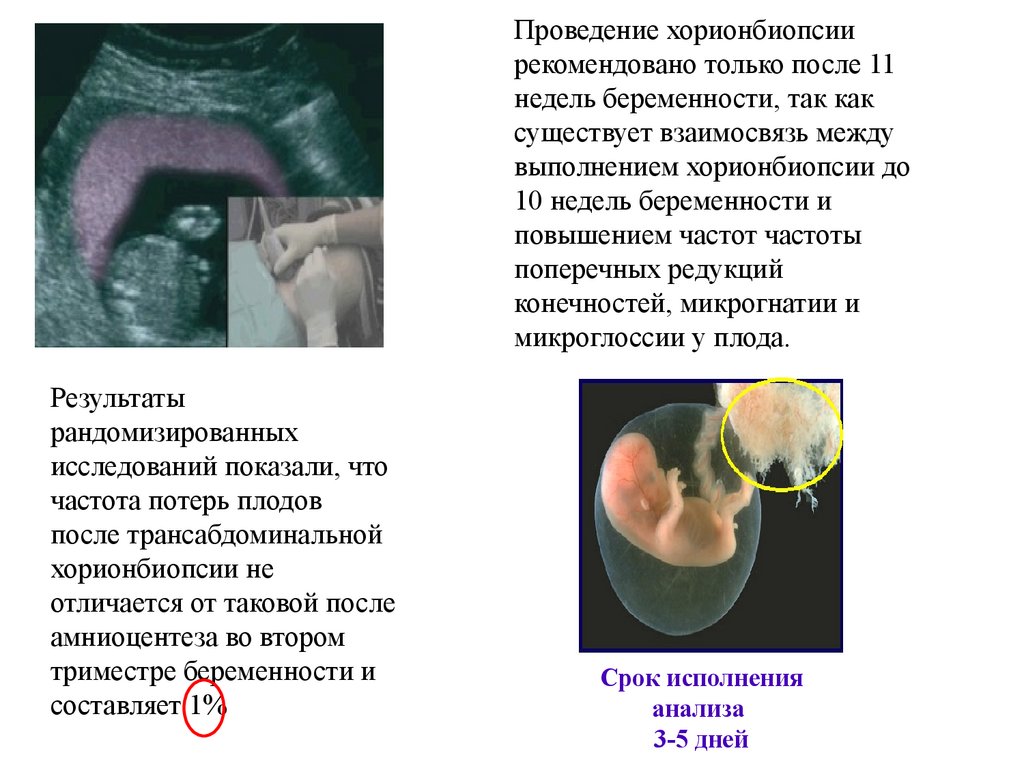
20.
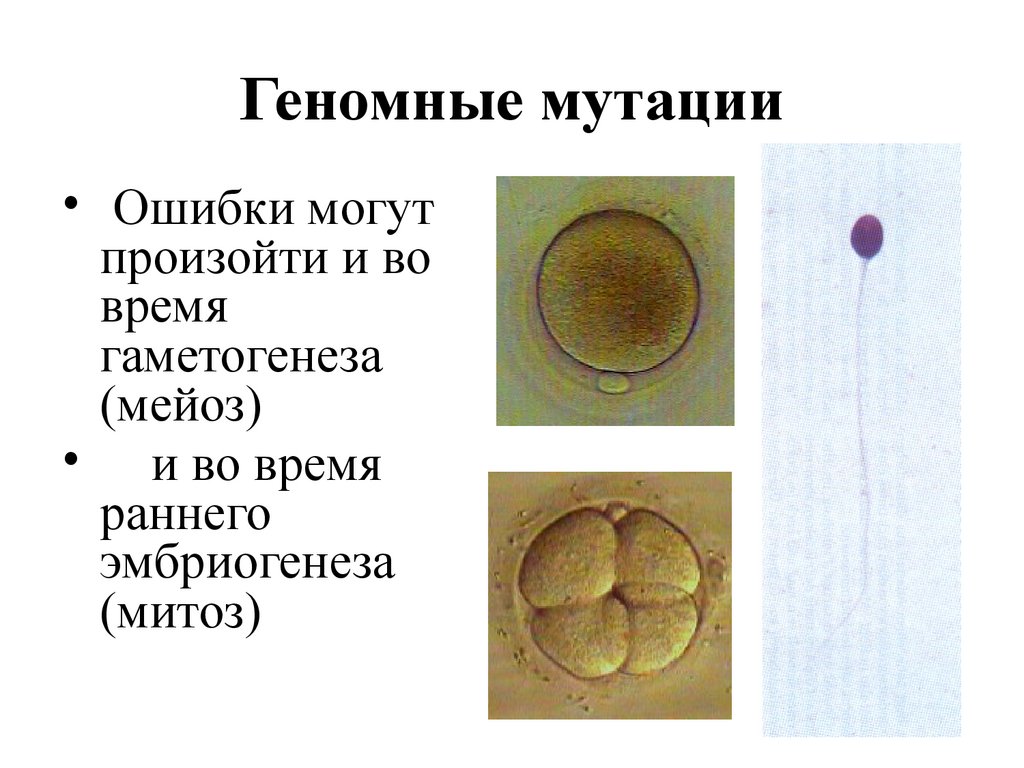
Геномные мутации• Ошибки могут
произойти и во
время
гаметогенеза
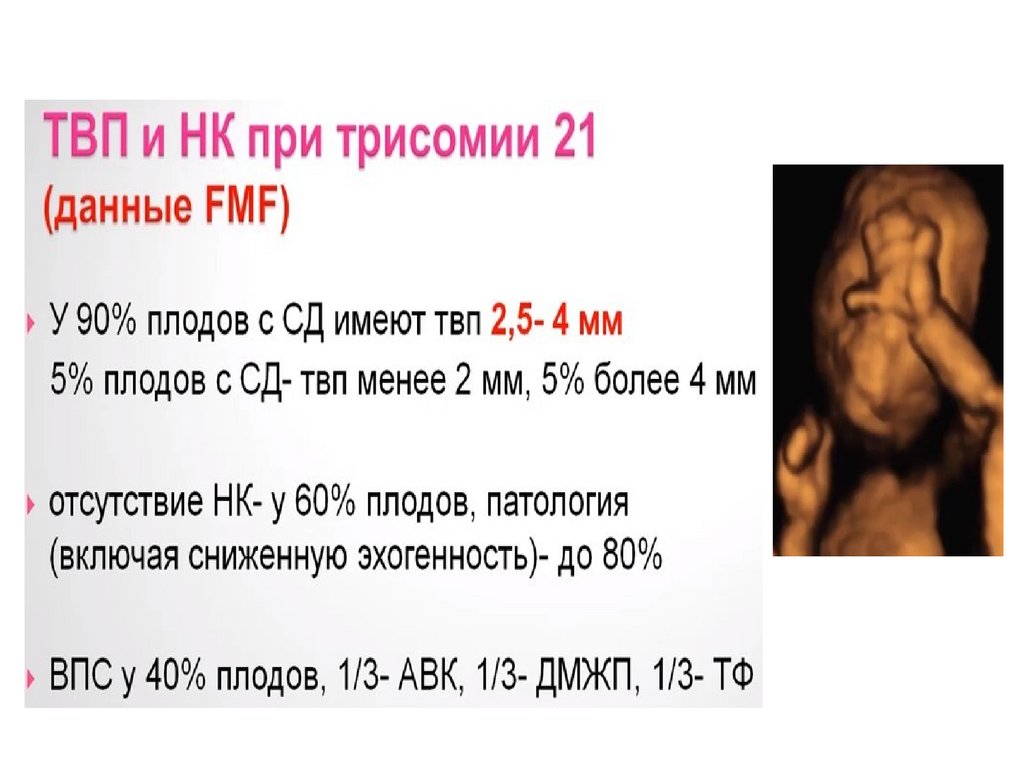
(мейоз)
• и во время
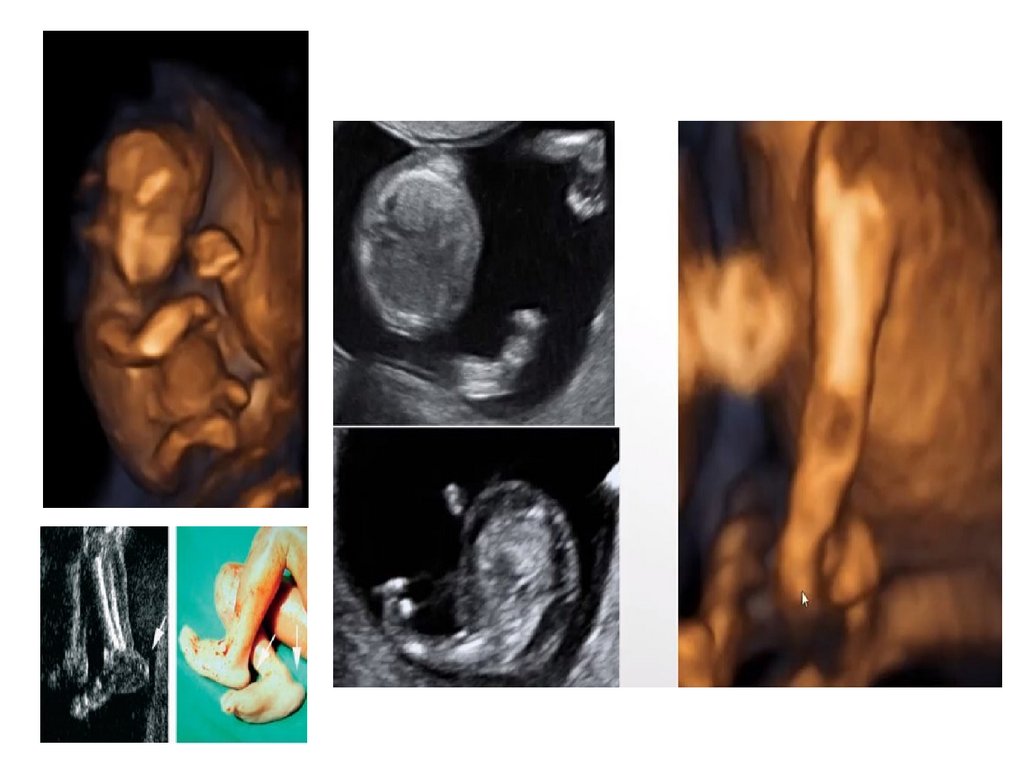
раннего
эмбриогенеза
(митоз)
21.
Примеры патологии22.
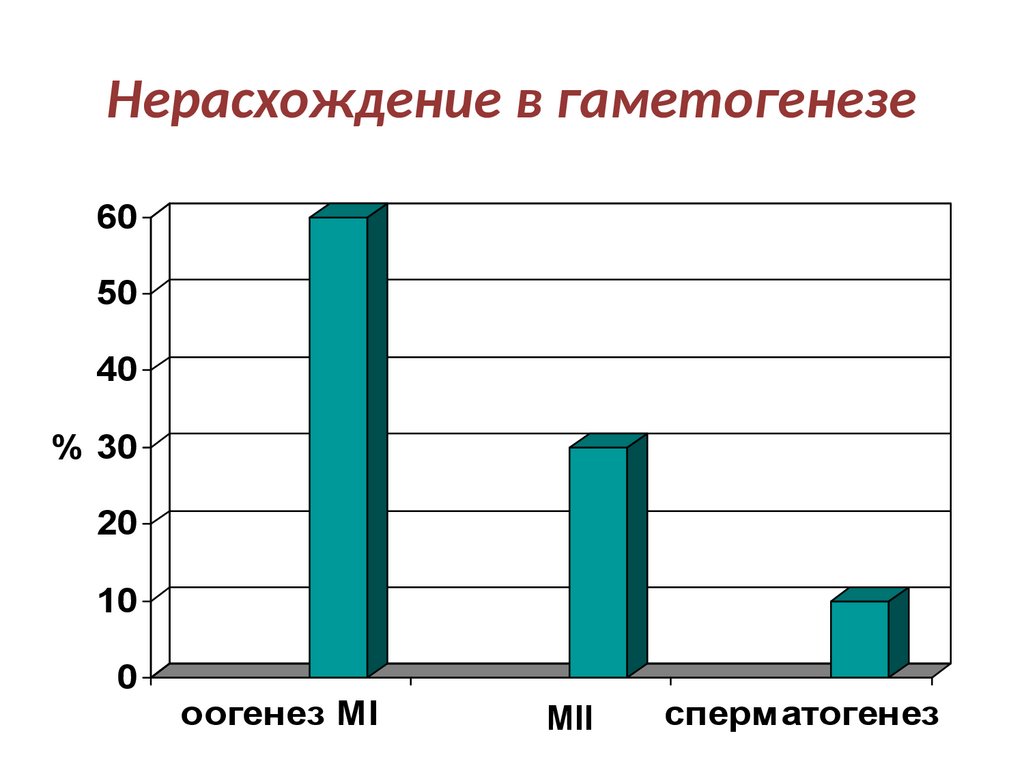
Нерасхождение в гаметогенезе60
50
40
% 30
20
10
0
оогенез MI
MII
сперматогенез
23.
24.
Ооциты в 1 делении мейозаБлок мейоза в лептотене
отставание\задержка темпов внутриутробного
развития при отсутствии внешних пороков
Ускорение темпов прохождения начальных
стадий профазы
формирование МВПР,
особенно пороки нервной и сердечнососудистой систем
25.
Ооцит во 2 делении мейоза• 4-58% хромосомных нарушений
обнаруживается в ооцитах MII
• 30% анеуплоидии обусловлено
нерасхождением во 2 делении созревания
ооцитов
• в 30% причина анеуплоидии –
преждевременное разделение центромер
(у женщин младше 35 лет)
26.
Хромосомы в сперматогенезе• Частота структурных перестроек
в сперматогенезе здорового
мужчины – около 6-7%,
анеуплоидий – 1-4%
• Наиболее подвержены
нерасхождению 21, X, Y , 1,9,16
хромосомы
27.
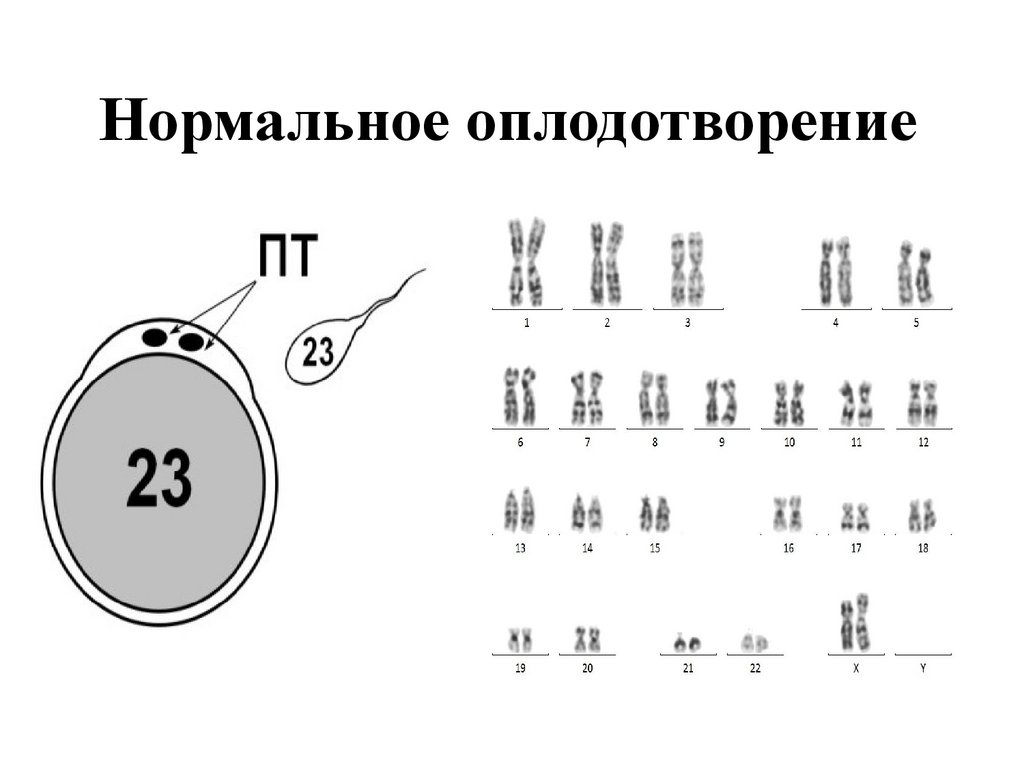
Нормальное оплодотворение28.
Нормальное оплодотворение29.
Нерасхождение в MI30.
Генетическими мозаиками называютособей — продуктов одной зиготы, в организме
которых сосуществуют две или более популяции
клеток с различным генотипом.
Цитогенетический мозаицизм - сочетание
в тканях индивидуума клеточных линий с
различным хромосомным набором. При этом
смесь клеток с нормальным и аномальным
кариотипами может быть представлена во всех
тканях организма (так называемый истинный,
или генерализованный, мозаицизм), или
ограничена клетками какой-либо одной ткани
(ограниченный мозаицизм).
31.
Мозаичные формы32.
33.
34.
В 1993 году была создана Европейскаяассоциация по
исследованию ограниченного плацентой
хромосомного
мозаицизма (EUCROMIC), основная цель
которой создание коллекции мозаичных
случаев, установление причин их
возникновения,
а также исследование влияния хромосомного
мозаицизма на развитие плода .
35.
Синдром Дауна (+21)36.
Описан в 1866 г.
Частота 1:600-700
Критический сегмент 21q22
М1:Ж1
УО, мышечная гипотония, плоское лицо, монголоидный
разрез,плоская переносица, пятна Брушфилда,
диспластичные уши, аркообразное небо, зубные
аномалии, короткая шея, кожная складка на шее,
короткие конечности, клинодактилия V пальца, ВПС,
четырехпальцевая складка
Предрасположенность к лейкозам
94% регулярная трисомия
2% мозаичная форма
4% транслокационная (центрические слияния)
37.
38.
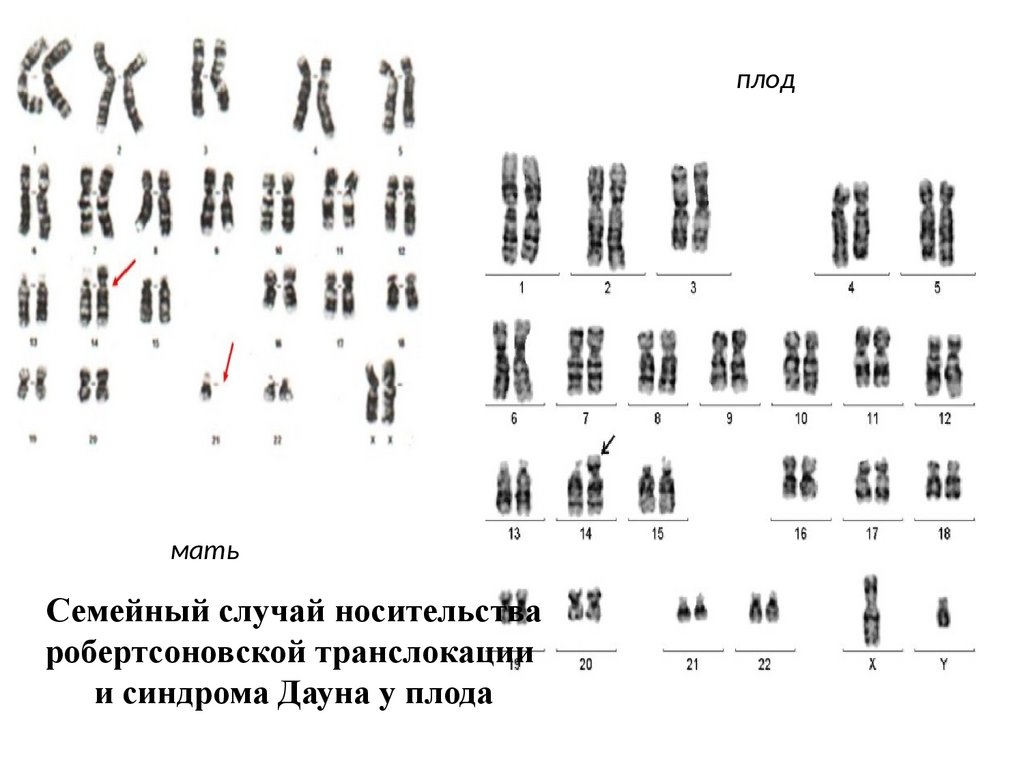
плодмать
Семейный случай носительства
робертсоновской транслокации
и синдрома Дауна у плода
39.
Синдром Патау(+13)40.
41.
• Описан в 1960 г.• Частота 1:8000, с учетом мертворождений 1:6500
• Критический сегмент 13q21-13q32
• М1:Ж1
• Гипотрофия плода, дискрания черепа (брахицефалия с
выраженными лобными буграми), дефекты скальпа
(алопеция участков без костной ткани), гипотелоризм,
неполная голопрозэнцефалия, анофтальмия, страбизм,
нистагм, катаракта, колобома, полидактилия, пороки почек,
аномалии мозга, ДМЖП и ДМПП, патологии гениталий.
• 94% регулярная трисомия
• 2% мозаичная форма
• 4% транслокационная (центрические слияния) Пол
носителя Ж2:М1
42.
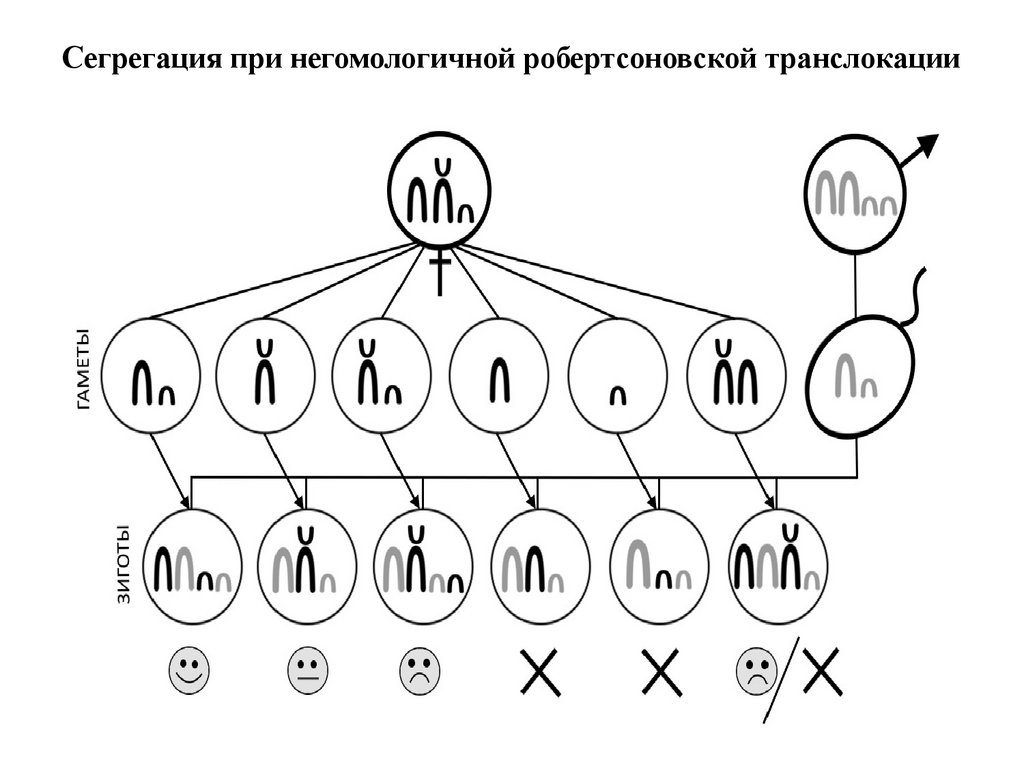
Сегрегация при негомологичной робертсоновской транслокации43.
Сегрегация при гомологичной робертсоновской транслокации44.
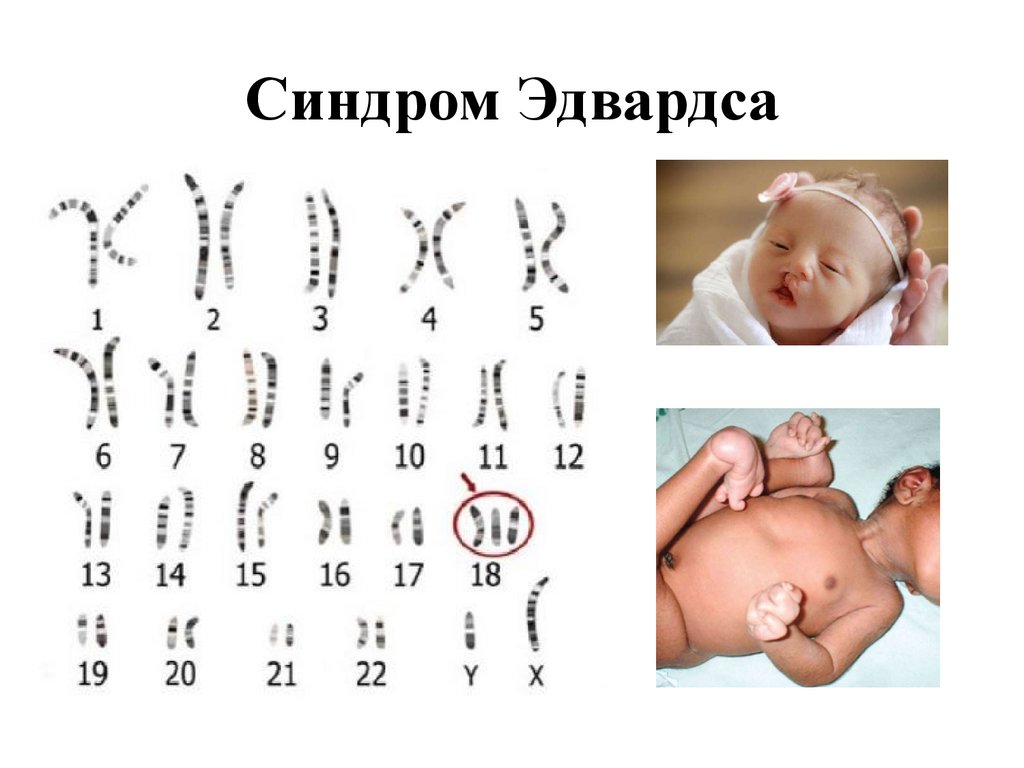
Синдром Эдвардса45.
Описан в 1960 г.
Частота 1:5000-7000
Критический сегмент 18q11
М1:Ж3
Нарушение сроков беременности, многоводие, слабая
активность плода, маленькая плацента,омфалоцеле,
единственная артерия пуповины, УО, гипоплазия
скелетной мускулатуры и подкожной жировой клетчатки
ДМЖП, низкопосаженные уши, высокое небо,
микрогнатия, микростомия. Короткие глазные щели,
сжатые пальцы рук с перекрыванием IVго пальца
Vм ,стопа-качалка, паховая или пупочная грыжа, пороки
почек, ЖКТ, аномалии наружных гениталий
• 96% регулярная трисомия, 4 % мозаичная форма
46.
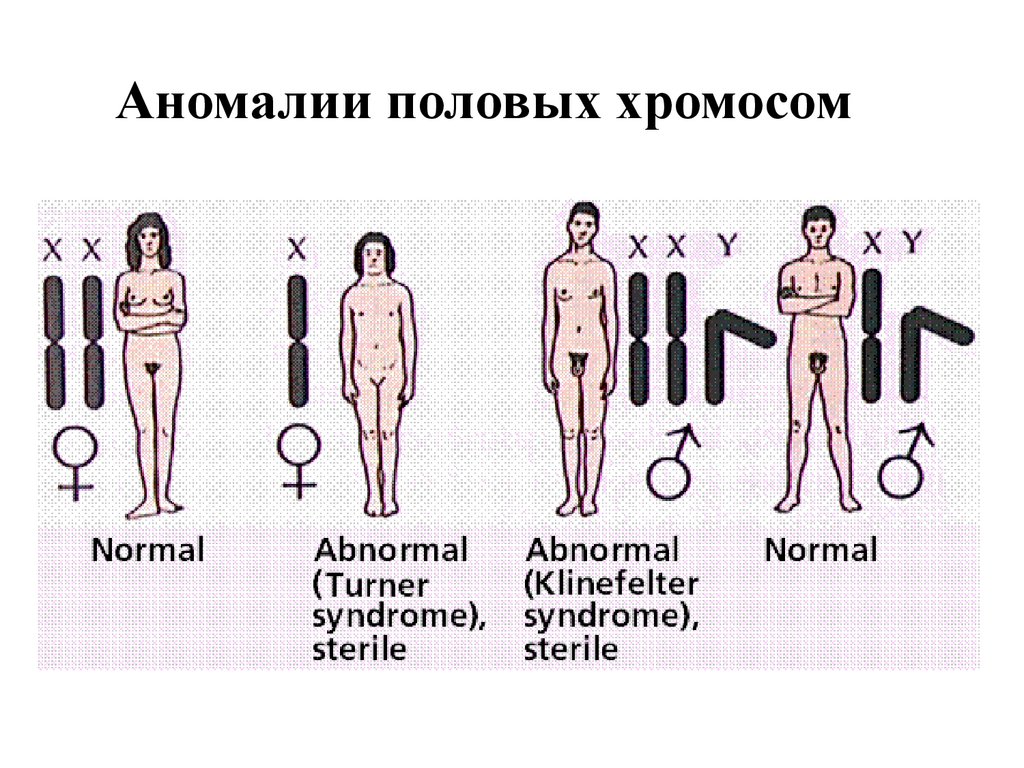
Аномалии половых хромосом47.
синдром Клайнфельтера48.
• Описан в 1942 г.• Частота 1:500
• Гипогенитализм, гипогонадизм,
оволосение по женскому типу
• Высокого роста с непропорционально
длинными конечностями, у взрослых
развивается ожирение, лабильная
психика
• 90% «чистых» форм
• 10% мозаичных
49.
Синдром Шерешевского-Тернера• Описан в 1925 г. Шерешевским, в
1938 г – Тернером
• Отек кистей и стоп, кожные складки
на шее (крыловидные складки),
низкий рост, широкая грудная
клетка, первичная аменорея
! Самый разнообразный
по цитогенетическим
проявлениям
синдром !
50.
51.
52.
53.
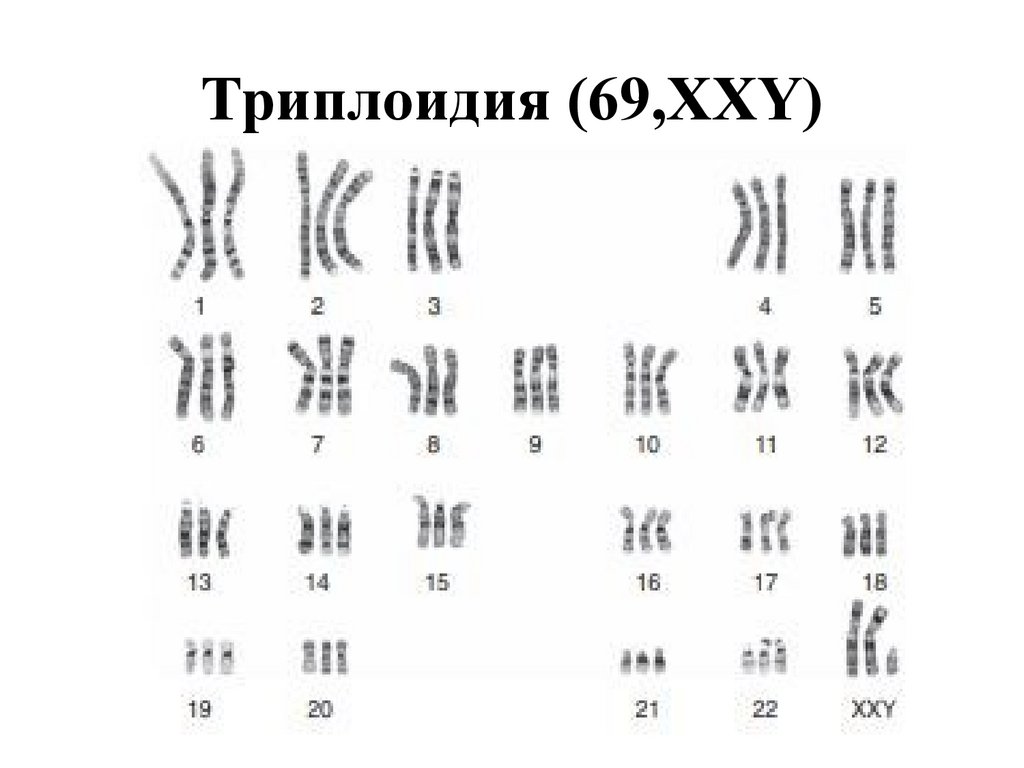
Триплоидия (69,XXY)54.
ПРИЧИНЫ АНОМАЛИЙ кариотипа: триплоидия (материнскийтип)
•тонкий «лысый» хорион
•ассиметрия плода
•вынужденное положение
лицевого угла
плода,
изменение
55.
ПРИЧИНЫ АНОМАЛИЙ кариотипа: триплоидия (отцовскийтип)
•пузырный занос
•изобилие хориональной ткани
•отставание в размерах плода
56.
Структурные перестройки• Возникают часто de novo
• Внутрихромосомные (делеции,
дупликации, инверсии)
• Межхромосомные (транслокации)
• Носители имеют высокий риск рождения
ребенка с патологией
• Высока вероятность возникновения при
сперматогенезе
57.
делеции58.
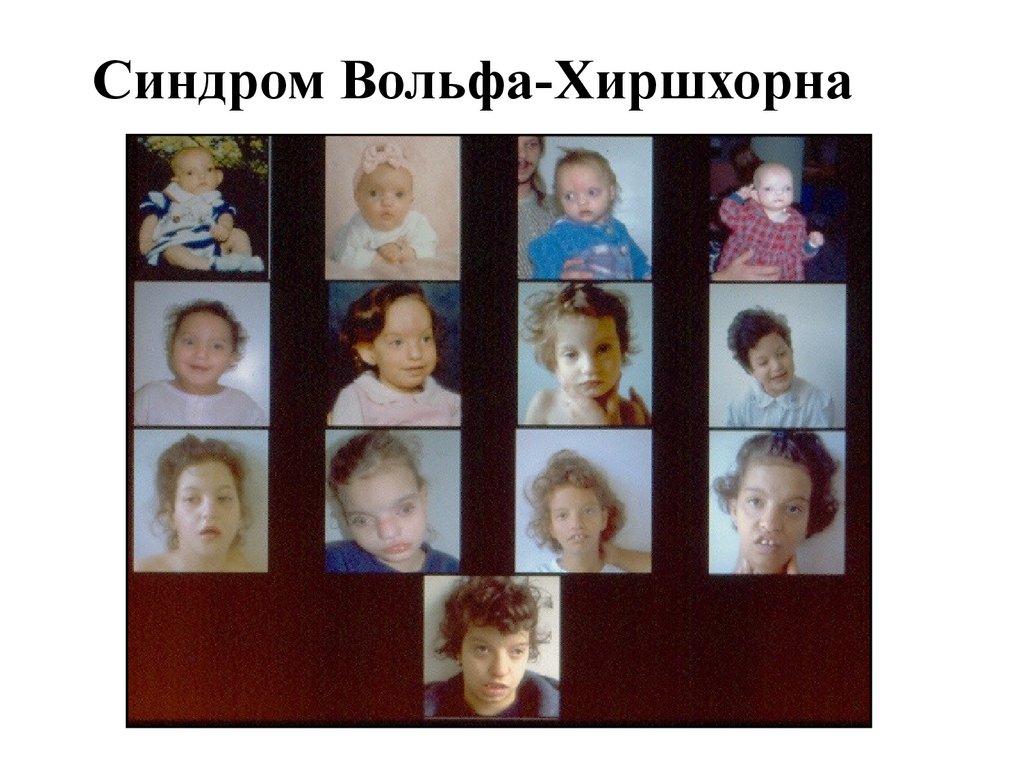
Синдром Вольфа-Хиршхорна59.
60.
• Описан в 1965 г.• Критический сегмент 4р16
• Частота 1:100 000
• М1:Ж1
• Гипертелоризм, широкий или клювовидный нос,
микроцефалия, ассимметрия черепа,
низкорасположенные диспластичные уши, задержка
психомоторного развития, гипотония мышц, часто расщелины губы и неба, экзофтальм, воронкообразная
грудь, косолапость, патологии почек и половых
органов
• Дерматоглифика – четырехпальцевая складка,
увеличение количества дуг
61.
кольцевая хромосома62.
дупликацияУдвоение одного
фрагмента
внутри
хромосомы
63.
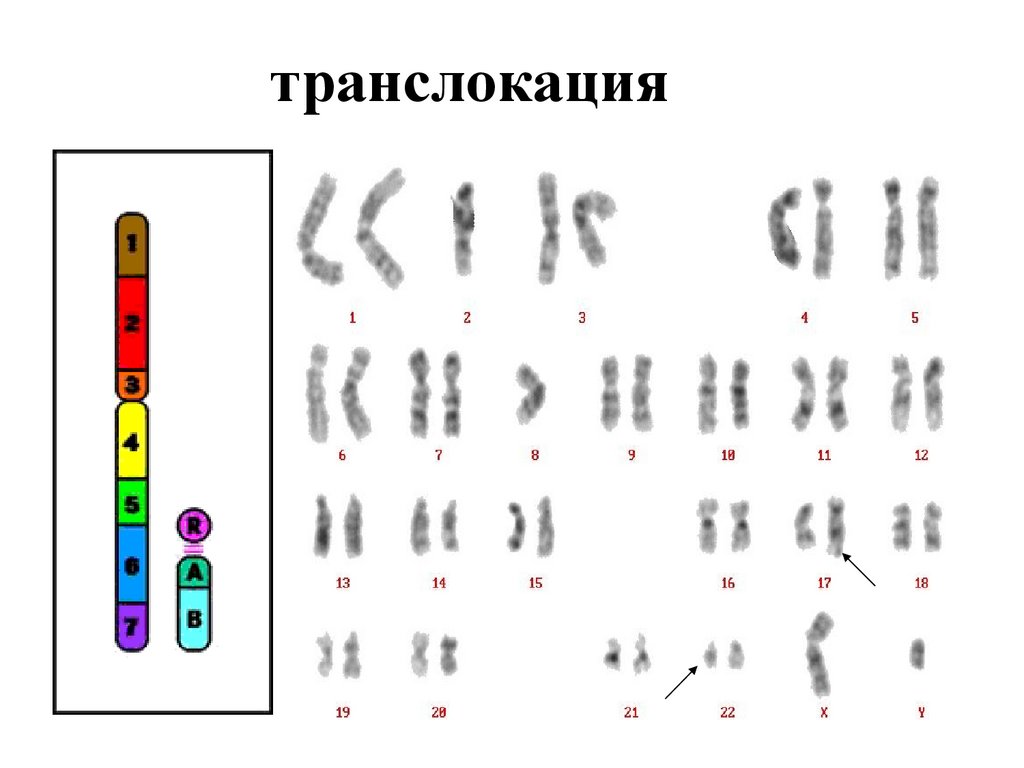
транслокация64.
65.
робертсоновское(центрическое) слияние
66.
Сегрегация при сбалансированной реципрокнойтранслокации
67.
Синдром Прадера-Вилли68.
отсутствие отцовской копии участка хромосомы 15q11-13(или однородительская дисомия (upd15 mat), или del(15)(q11-13))
Частота встречаемости — 1 : 12 000-15 000
Дисплазия тазобедренных суставов; ожирение; склонность к
перееданию (чаще проявляется к 2-м годам); пониженный
мышечный тонус (гипотонус); пониженная координация
движений; маленькие кисти и стопы, низкий рост; повышенная
сонливость; страбизм (косоглазие); сколиоз (искривление
позвоночника); пониженная плотность костей; густая слюна;
плохие зубы; сниженная функция половых желёз
(гипогонадизм); в результате, как правило, бесплодие; речевая
задержка, задержка психического развития; отставание в
освоении навыков общей и мелкой моторики. Более позднее
половое созревание
69.
Синдром Ангельмана70.
Отсутствие материнской копии участка хромосомы 15q11-13(или однородительская дисомия (upd15 рat), или del(15)(q11-13))
Частота встречаемости 1:10000-20000
В 75 % случаев — проблемы с питанием, особенно с грудным
вскармливанием, такие младенцы плохо набирают вес; задержка в
развитии навыков общей моторики (умение сидеть, ходить);
задержка речевого развития, неразвитая речь (у всех детей); дети
больше понимают, чем могут сказать или выразить; сложности с
обучением; эпилепсия (в 80 % случаев), нарушения выявляются
также при ЭЭГ; необычные движения (мелкий тремор, хаотические
движения конечностей); частый смех без повода; ходьба на
негнущихся ногах — из-за этой особенности детей с данным
синдромом иногда сравнивали с марионетками; размер головы
меньше среднего, нередко с уплощением затылка; иногда
характерные черты лица — широкий рот, редко расположенные
зубы, выдающийся вперед подбородок, высунутый язык; нарушения
сна; страбизм в 40 % случаев; сколиоз в 10 % случаев; повышенная
чувствительность к высокой температуре
71.
Флуоресцентная гибридизацияin situ (FISH)
• В 1970г. – радиоактивные метки
• В 1985г. - флуорохромы
72.
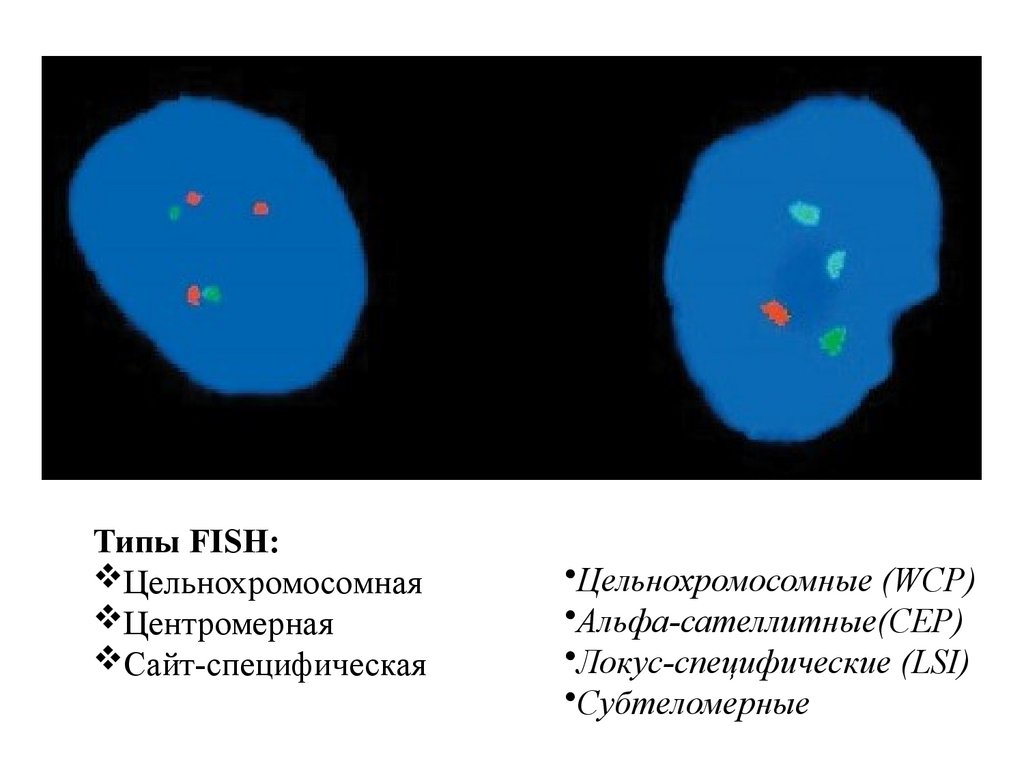
Типы FISH:Цельнохромосомная
Центромерная
Сайт-специфическая
•Цельнохромосомные (WCP)
•Альфа-сателлитные(CEP)
•Локус-специфические (LSI)
•Субтеломерные
73.
Показания для FISH• Выявление анеуплоидии в интерфазных
ядрах некультивированных клеток
• Выявление анеуплоидии в интерфазных
ядрах при низкой/отсутствующей
митотической активности
• Установление уровня мозаицизма
• Установление природы маркерной
хромосомы
74.
Области применения FISHПреимплантационная генетическая диагностика
(определение анеуплоидий, диагностика дериватных
хромосом у носителей перестроек)
Пренатальная генетическая диагностика (скрининг
анеуплоидий на некультивированных амниоцитах,
идентификация маркерных хромосом, выявление
мозаицизма, верификация результатов цитогенетического
исследования)
Постнатальная генетическая диагностика
(определение степени мозаицизма, верификация
результатов кариотипирования, установление природы
маркерных хромосом,диагностика солидых опухолей,
выявление криптических транслокаций при лейкозах,
скрининг анеуплоидий в сперматозоидах)
75.
Кошачьего крикаДи Джорджи
Каллмана
Смита-Магениса
Прадера-Вилли
Вильямса
Вольфа-Хиршхорна и др.
76.
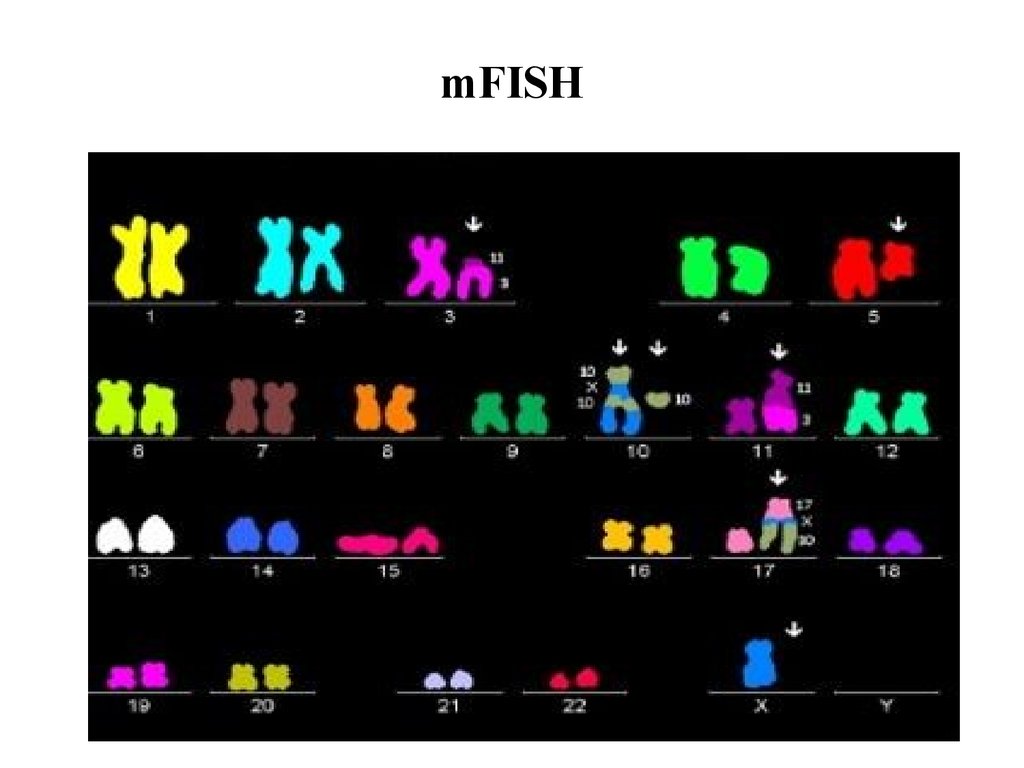
mFISH77.
78.
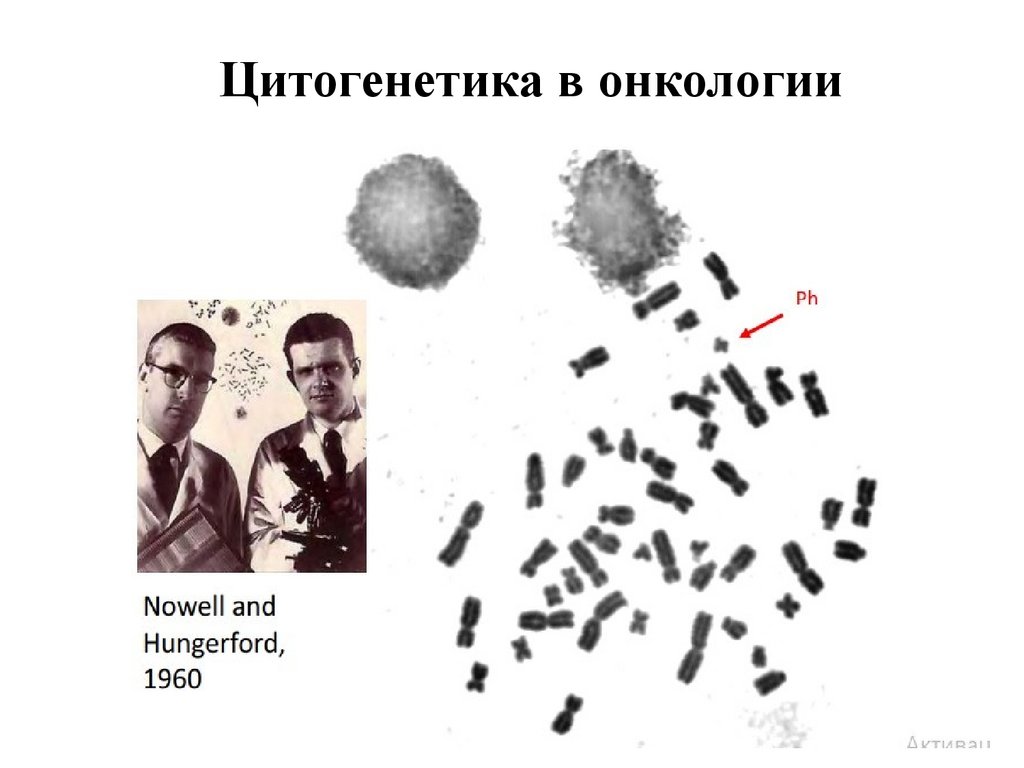
Цитогенетика в онкологии79.
80.
Исследование мутагенезаРазрыв со смещением
Дицентрическая хромосома
81.
Кольцевая хромосомаУтрата части хроматиды
82.
ПРЕНАТАЛЬНАЯДИАГНОСТИКА
Самойлова Л.Р., врач-лабораторный
генетик,
преподаватель кафедры НППиМГ
ИФМиБ
Казань, 2023
83.
частота врожденной и наследственнойпатологии составляет 5%
от общего числа новорожденных
Хромосомные болезни встречаются у 4-7 на 1000
новорожденных
84.
Пренатальная диагностика (ПД) –комплекс мер по выявлению
патологических состояний у плода.
Конечная цель ПД – это раннее
выявление патологии у плода и
предупреждение рождения
ребенка с тяжелыми
некорригируемыми пороками.
85.
Задачи ПД:•Предоставление семье в доступном для
понимания виде объективной информации о
степени риска рождения ребенка с патологией
•Разработка стратегии и тактики ведения
беременности и родов при выявлении патологии
•Прогноз для будущих беременностей
86.
87.
88.
Современная концепция ПДдо 2010 г
после 2010 г.
89.
Ультразвуковая диагностика 1 триместр• КТР-копчико-теменной размер. (в 11 недель 45-
84мм)
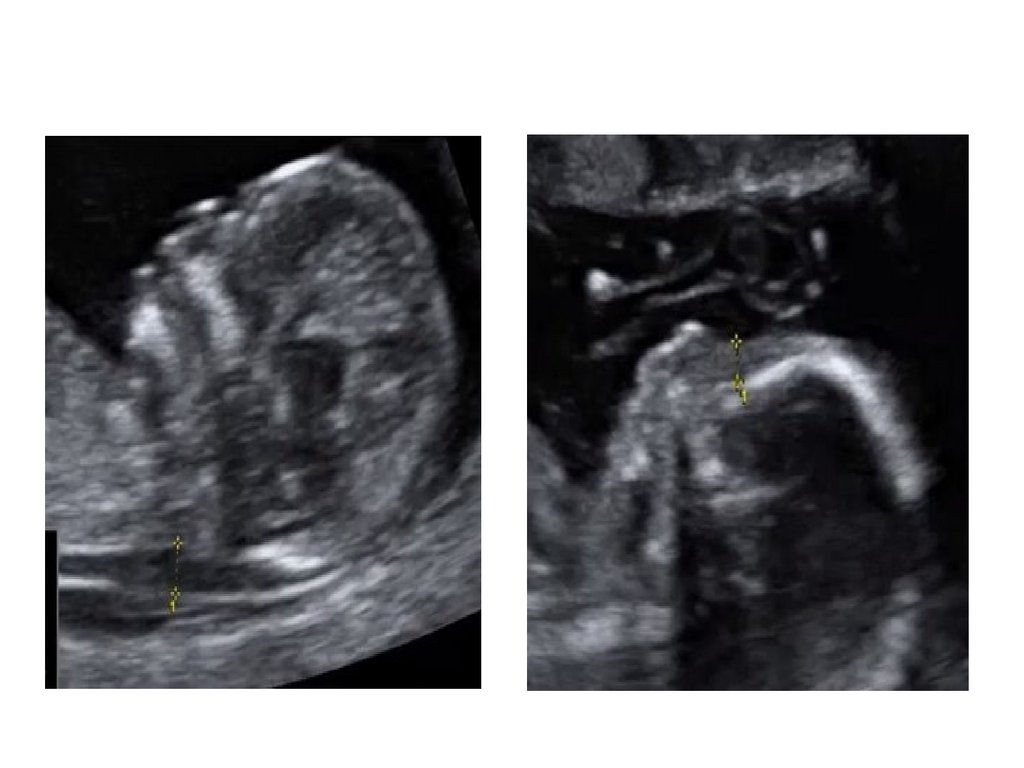
• ТВП-толщина воротникового пространства
• НК-носовая кость
• ЧСС – частота сердечных сокращений. В норме с
ростом плода чсс увеличивается со 110
уд/мин (5 н.б.) до 170 уд/мин (10 н.б.),
затем снижается до 150 уд/мин (14 н.б.)
90.
91.
92.
93.
94.
95.
Отсутствие носовой кости96.
Аплазия НКN
97.
98.
99.
100.
101.
102.
Увеличение ЧСС –маркер трисомии 1
103.
104.
Реверсная а-волна105.
Биохимический скрининг 1 триместра• РАРР-А – белок, ассоциированный с беременностью;
протеиназа, расщепляющая инсулиноподобный
фактор роста 1. При различных патологических состояниях
(неразвивающаяся беременность, угроза прерывания,
хромосомное заболевание у плода) уровень РАРР-А снижается
• ХГЧ-гормон с лютеинезирующей гонадотропной
активностью. При тр21 у плода уровень β-хгч повышен. Так
же повышается он при многоплодной беременности,
преэклампсии. А при тр13 и тр18 уровень наоборот, снижен.
• Сроки проведения : 10нед 6 дней - 12 недель
• Данные переводятся в медиану от множеств (МоМ).
Нормальный показатель – 1МоМ.
106.
повышение РАРР-Амногоплодная
синдромы Дауна,
беременность
низкое
Эдвардса, Патау, триплоидия
расположение
плаценты
плаценты
фетоплацентарная
недостаточность
крупный плод
увеличенная
понижение РАРР-А
гипотрофия плода
масса
угроза
беременности
прерывания
107.
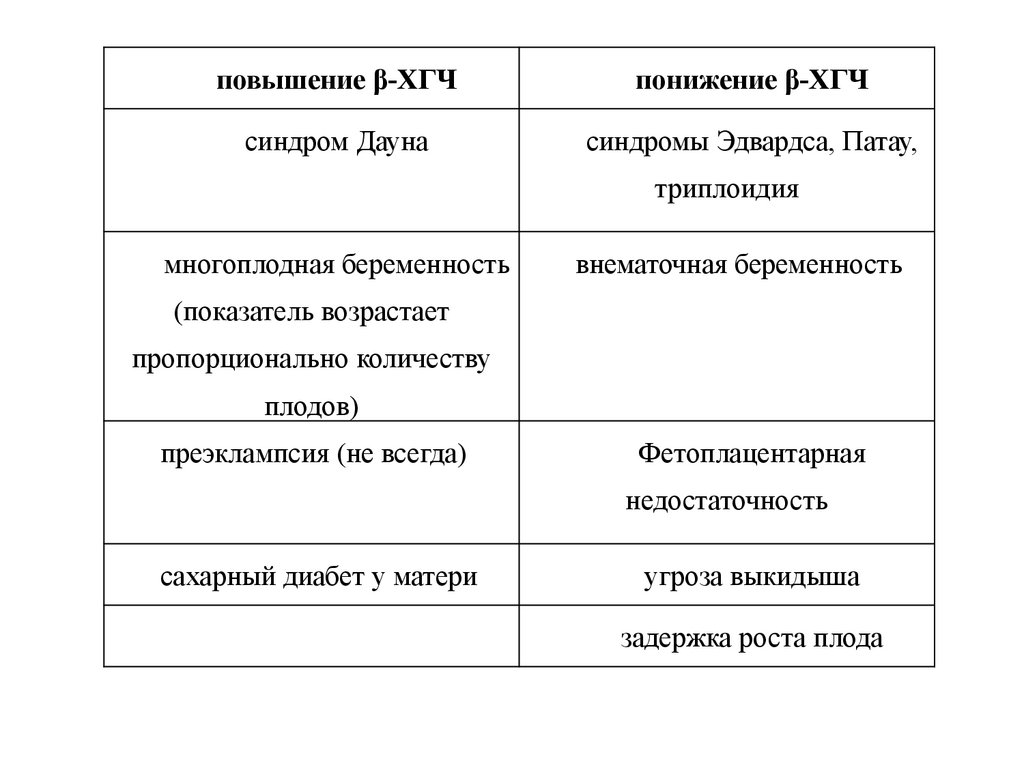
повышение β-ХГЧпонижение β-ХГЧ
синдром Дауна
синдромы Эдвардса, Патау,
триплоидия
многоплодная беременность
внематочная беременность
(показатель возрастает
пропорционально количеству
плодов)
преэклампсия (не всегда)
Фетоплацентарная
недостаточность
сахарный диабет у матери
угроза выкидыша
задержка роста плода
108.
109.
110.
111.
Этапы расчета риска112.
113.
114.
ПРИЧИНЫЛОЖНОПОЛОЖИТ.
РЕЗУЛЬТАТОВ
НИПТ
• мозаицизм,
ограниченный
плацентой
• тканевой мозаицизм
матери
• «исчезающий» близнец
• онкологическое
заболевание у матери
ПРИЧИНЫ
ЛОЖНООТРИЦАТ.
РЕЗУЛЬТАТОВ
НИПТ
• истинный плодный
мозаицизм
• множественные
анеуплоидии
115.
Ограничения и противопоказания для нипт• злокачественные новообразования у матери,
• трансплантация органов и тканей у матери
• радио- или иммунотерапия у матери (в том числе лечение
гемопоэтическими стволовыми клетками)
• замершая беременность в течение 3 мес. перед прохождением
исследования
• синдром «исчезнувшего близнеца» (беременность двойней с
потерей одного из плодов)
• относительные противопоказания — лечение
низкомолекулярными гепаринами (необходима отмена препаратов
гепаринов на 3–4 дня или взятие крови для НИПТ перед
следующим введением гепарина), лечение препаратами
антиретровирусной терапии (необходимо взятие крови перед
следующим приемом антиретровирусного препарата),
переливание крови в течение последних 6 месяцев, хирургические
вмешательства и травмы менее 3 месяцев назад.
116.
Биохимический скрининг 2 триместра117.
УЗ-маркеры во 2 триместре• аномальная форма головки плода
• дефекты лица и шеи
• вентрикуломегалия
• кисты сосудистых сплетений
• гиперэхогенные включения в желудочках сердца
• гиперэхогенный кишечник
• пиелоэктазия
• укорочение трубчатых костей
• задержка развития плода
• генерализованный отек
• единственная артерия пуповины
118.
Показания кинвазивной ПД
1. возраст беременной
старше 35 лет
2. наличие УЗ и/или
биохимических
маркеров
3.установленное
носительство
хромосомной
перестройки
4. рождение ребенка с
МВПР, хромосомным
синдромом
мертворождение в
анамнезе
119.
120.
Биопсия ворсин хориона121.
Прямойметод
культура
122.
123.
Проведение хорионбиопсиирекомендовано только после 11
недель беременности, так как
существует взаимосвязь между
выполнением хорионбиопсии до
10 недель беременности и
повышением частот частоты
поперечных редукций
конечностей, микрогнатии и
микроглоссии у плода.
Результаты
рандомизированных
исследований показали, что
частота потерь плодов
после трансабдоминальной
хорионбиопсии не
отличается от таковой после
амниоцентеза во втором
триместре беременности и
составляет 1%
Срок исполнения
анализа
3-5 дней
124.
Амниоцентез10-21 день
7-8
день
3
день
в среднем, от момента
взятия материала до
получения результата
проходит 12-21 дней
125.
Амниоцентез раньше 16недели
чреват повышением
риска выкидыша
(до 2%) или деформацией
стоп плода
Nicolaides,20
10
(до 1,5%)
Срок исполнения
анализа
10-21 дней
Harper ,2007
126.
ВидСрок
исследован проведени
Оптимальн
ый срок
ия
я
проведения
хорионбиоп
11-14
11
Срок
получения
результата
Специфические
осложнения
(дни)
3-5
Кровотечение 10%
Ретрохориальная
сия
гематома
Подтекание вод 0,5%
Редукция конечностей на
раннем сроке (<11)
амниоценте
17-20
18
10-21
Хориоамнионит 0,1%
з
кордоцентез
Подтекание вод 1-2%
>18-20
18
7-10
Кровотечение из места
пункции
Гематома пуповины
Брадикардия у плода
127.
у 90% ТВП=2,6-4мму
60%
аплазия
НК,
нарушение эхогенностиНК-у
80%
у 40% врожденный порок
сердца
гипоплазия верхней челюсти
преназальный отек
брахицефалия
вентрикуломегалия
клинодактилия мизинцев
128.
плодмать
Семейный случай носительства
робертсоновской транслокации
и синдрома Дауна у плода
129.
особенностистроения
лица
(расщелина, гипоплазия нижней
челюсти)
флексорное
положение
кисти
и/или приведенный первый палец,
лучевая косорукость
пороки
развития центральной
нервной системы (spina bifida,
вентрикуломегалия)
пороки сердца
омфалоцеле
мегацистис
130.
голопрозэнцефалиясрединная расщелина лица
пороки сердца
омфалоцеле
тахикардия
гиперэхогенные почки
«рука акушера»
полидактилия кистей и/или
стоп
мегацистис
131.
Кистознаягигрома, отек
водянка
дефект
межжелудочковой
перегородки
132.
Хромосомный микроматричный анализ(ХМА)
• в основе лежит ПЦР с последующей гибридизацией фрагментов
ДНК с SNP-олигонуклеотидными микроматрицами высокой
плотности
• решается проблема не только высокоточного полногеномного
анализа вариаций числа копий (CNVS), но и выявления участков
отсутствия гетерозиготности, однородительских дисомий, а
также определения происхождения хромосомного дисбаланса
• выявление субмикроскопических хромосомных перестроек
позволяет уточнить этиологию каждого конкретного случая и,
благодаря возможности точного установления границ дисбаланса,
решить вопросы о причинах клинико-генетической
гетерогенности ряда наследственных болезней
133.
• Таргетный• Стандартный
• Расширенный
134.
• основное отличие - разрешающая способность, котораярегулируется количеством используемых ДНКмаркеров (плотность матрицы)
• в таргетном-350 тысяч, это самая низкая плотность,
выявляются самые распространенные микроделеции и
микродупликации
• в стандартном используются 750 тысяч маркеров.
позволяет выявлять не только самые распространенные
хромосомные нарушения, но и неспецифические
аномалии
• самое глубокое исследование генома – при помощи
матрицы в 2,67 миллионов ДНК-маркеров, это
расширенный тип ХМА
135.
136.
Ограничения метода (невыявляемые случаи):•Реципрокные транслокации
•Роберсоновские транслокации
•Инверсии
•Инсерции
•Триплоидия
•Несбалансированные перестройки
меньше 5 млн.н.п.
137.
ПРЕНАТАЛЬНЫЙ КОНСИЛИУМ (функции)• проведение экспертизы случаев выявленной патологии плода с вынесением
рекомендаций о сохранении или прерывании беременности при выявлении у плода
врожденных пороков развития или наследственных заболеваний.
• определение целесообразности применения методов внутриутробной терапии и
хирургии при выявлении патологии плода.
• определение учреждения здравоохранения для родоразрешения беременной женщины
с выявленной пренатальной патологией плода.
• информирование беременной и членов ее семьи о характере и тяжести поражения
плода, возможных исходах беременности, прогнозе для жизни и здоровья ребенка,
возможных методах лечения, о возможной степени утраты физических и психических
параметров здоровья (инвалидизации)
.
• направление беременных женщин с выявленной пренатальной патологией плода в
специализированные учреждения здравоохранения для родоразрешения или лечения.
• рассмотрение жалоб и иных вопросов, связанных с проведением пренатальной
диагностики.
138.
ПРЕНАТАЛЬНЫЙ КОНСИЛИУМ (показания для направления)• пренатально выявленные врожденные пороки
развития у плода.
• пренатально выявленные хромосомные
заболевания, подтвержденные
кариотипированием.
• высокий риск рождения ребенка с
наследственным заболеванием, приводящим к
инвалидности и невозможности его
пренатальной диагностики.
• тяжелые фетопатии (водянка, задержка
развития плода).
139.
Критерии пренатального прерывания беременности• болезнь должна быть достаточно тяжелой, чтобы было
оправдано прерывание беременности;
• лечение болезни плода невозможно и
неудовлетворительно;
• семья, которая консультируется, должна быть согласна
на прерывание беременности;
• существует точный тест для постановки
пренатального диагноза;
• достаточно высокий генетический риск
неблагоприятного исхода беременности;
• послетестовое консультирование должно включать
описание всего диапазона тяжести расстройства, от
наименее до наиболее выраженного