



















































 medicine
medicineSimilar presentations:










Podstawowe wiadomości z farmakodynamiki i farmakokinetyki leków
1.
Podstawowe wiadomościz farmakodynamiki i farmakokinetyki
leków
Dr Marcin Delijewski
Katedra i Zakład Farmakologii Wydziału Nauk Medycznych w Zabrzu
2.
FARMAKOLOGIAFarmakologia – nauka interdyscyplinarna – łączy wiedzę z chemii, biochemii,
fizjologii, mikrobiologii
Główne obszary:
Projektowanie leków (drug design) – badania in silico (chemoinformatics)
Badania in vitro oraz in vivo
Repozycjonowanie leków (repurposing, repositioning)
Badania kliniczne (fazy I-IV)
Bezpieczeństwo farmakoterapii (pharmacovigilance)
3.
LEKLek – to substancja lub jej metabolit, wywołująca
w organizmie określony efekt terapeutyczny, profilaktyczny lub diagnostyczny.
leczenie przyczynowe - usuwanie przyczyn choroby
np. antybiotyki w infekcjach bakteryjnych, witaminy
w awitaminozie
leczenie objawowe – usuwanie objawów choroby lub nie dopuszczają do ich
powstania
np. leki przeciwgorączkowe
4.
Leczenie objawowe:Leczenie homeopatyczne – podawanie małych dawek takiej substancji, która w
większych dawkach wywołuje objawy podobne do objawów danej choroby
„podobne leczymy podobnym” (similia similibus curantur)
Leczenie allopatyczne – stosowanie leków, które wykazują działanie przeciwne
do objawów danej choroby
„przeciwne leczymy przeciwnym” (contraria contraribus curantur)
5.
Postacie lekówPostacie płynne:
- roztwory
- zawiesiny
- emulsje
Postacie półstałe:
- maści
- kremy
- pasty
Postacie stałe:
- stosowane doustnie: tabletki, tabletki powlekane, kapsułki, proszki, granulaty
- stosowane doodbytniczo lub dopochwowo: czopki, globulki
Systemy terapeutyczne:
- systemy transdermalne, implantacyjne, do oczu
6.
Wprowadzenie leku do ustrojuDroga doustna: tabletki, kapsułki, zawiesiny, syropy, płyny
Iniekcje: jałowe roztwory, zawiesiny, implanty
Transdermalnie: plastry, maści, kremy, żele
Przez błony śluzowe: aerozole, gazy, czopki, tabletki podjęzykowe
7.
Przestrzeń poszukiwania lekówLista leków zatwierdzonych przez FDA (Food and Drug
Administration) = ok 1700 cząsteczek
Lista związków badanych na różnych fazach (potencjalne leki) =
6798 cząsteczek
Przestrzeń „NPs” Natural Products = ok 400 000 cząsteczek
Przestrzeń chemiczna = 111 M cząsteczek
8.
Kandydaci na leki („druggability”)Lipinski’s Rule of Five (RO5)
Masa molowa < 500 Da
logP < 5
Liczba donorów wiązania wodorowego < 5
Liczba akceptorów wiązania wodorowego < 10
9.
Kandydaci na leki („druggability”)Reguły Vebera
Liczba wiązań rotowalnych < 10
Pole powierzchni polarnej < 140
10.
Dawkato ilość podanego leku
określona: wagowo, objętościowo, procentowo lub w jednostkach
aktywności biologicznej (enzymy, hormony, witaminy, niektóre
antybiotyki)
najczęściej w μg i mg lub w przeliczeniu na 1 kg masy ciała (np. mg/kg
m.c.)
dobór dawki zależy od: wieku, płci, stanu fizjologicznego, stanów
patologicznych, zmian genetycznych
11.
Dawkawspółczynnik terapeutyczny - stosunek największej dawki tolerowanej
przez organizm do dawki terapeutycznej (im większa wartość
współczynnika, tym lek jest bezpieczniejszy)
wskaźnik bezpieczeństwa leku określa się, badając stosunek LD50 (dawki
śmiertelnej dla połowy badanej
populacji zwierząt) do DE50
(dawki wywołującej efekt
terapeutyczny w połowie
badanej populacji zwierząt).
12.
Rodzaje dawekDawka superminimalna (dosis subminimalis) nie wywiera
obserwowalnego działania
Dawka graniczna (dosis minimalis) to najmniejsza ilość leku, która
wywołuje zauważalne reakcje
Dawka terapeutyczna (dosis therapeutica) to ilość leku, która
wywiera działanie lecznicze
Dawka maksymalna (dosis maximalis) to największa dawka, która
nie wywołuje jeszcze zatrucia
13.
Rodzaje dawekDawka toksyczna (dosis toxica) to dawka wywołująca objawy zatrucia
Dawka śmiertelna (dosis lethalis) to najmniejsza ilość leku powodująca
śmierć
14.
Definicje:Działanie uboczne: to przewidywalny lecz niepożądany efekt
terapeutyczny występujący w zakresie dawek leczniczych
Przeciwwskazanie: okoliczność stanowiąca przeszkodę w leczeniu
chorego danym lekiem, gdy lek ten zaostrza stany patologiczne
współistniejące u pacjenta
15.
Definicje:Działanie embriotoksyczne: prowadzi do obumarcia zarodka w
pierwszych 2 tygodniach ciąży
Działanie teratogenne: prowadzi do zaburzeń w 1-4 miesiącu ciąży, do
uszkodzenia płodu lub jego narządów
Działanie karcynogenne: rakotwórcze
16.
Definicje:Tolerancja: to zmniejszenie efektu terapeutycznego kolejnych dawek leku
na skutek zmniejszenia liczby receptorów lub zwiększenia aktywności
enzymów mikrosomalnych wątroby
Tachyfilaksja: zmniejszenie reakcji na kolejne dawki leków o pośrednim
mechanizmie działania (np. na skutek wyczerpania się zapasów
neuroprzekaźnika)
17.
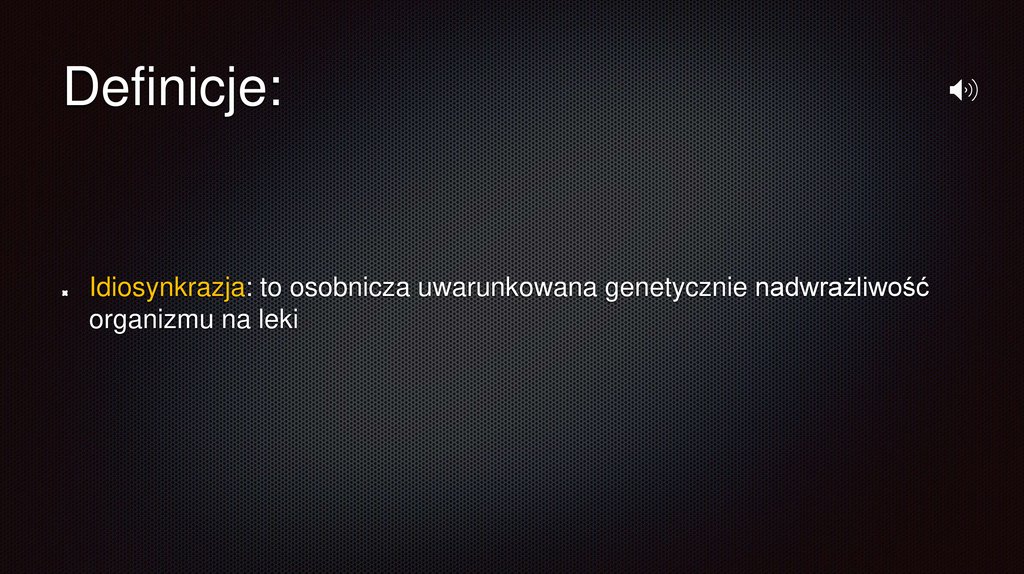
Definicje:Idiosynkrazja: to osobnicza uwarunkowana genetycznie nadwrażliwość
organizmu na leki
18.
FarmakodynamikaOpisuje mechanizmy działania leków
i ich wpływ na procesy fizjologiczne
w ustroju:
- działanie ogólne i/lub miejscowe
- działanie ośrodkowe i/lub obwodowe
- działanie przyczynowe
(etioterapeutyczne) lub objawowe
- działanie substytucyjne (np. insulina)
19.
Mechanizmy działania lekówpoprzez wpływ na receptory
bezpośrednie działanie na enzymy (zwiększenie lub zmniejszenie
aktywności enzymów)
działanie pośrednie, gdy lek jest prekursorem związku o aktywności
biologicznej
poprzez mechanizmy fizykochemiczne (np. zmiana pH środowiska)
20.
ReceptoryLigand – to substancja wiążąca się
z receptorem, która wywołuje efekt pobudzenia lub hamowania jego
czynności
Pobudzenie receptorów związanych
z błoną komórkową wpływa na procesy metaboliczne w cytoplazmie, a
także może wpływać na transkrypcję
i translację
21.
Wpływ na receptoryLek może pobudzać receptor (agonizm) lub hamować jego aktywność
(antagonizm)
Agonizm – ligand posiada powinowactwo do receptora i aktywność
wewnętrzną
Antagonizm – ligand posiada powinowactwo do receptora ale nie posiada
aktywności wewnętrznej
22.
Wpływ na receptoryAntagonizm kompetycyjny: gdy dwa ligandy konkurują o to samo
miejsce wiązania na receptorze – z receptorem zwiąże się ligand,
który posiada większe powinowactwo, a jeśli mają jednakowe
powinowactwo, to ten, którego stężenie jest większe
Antagonizm niekompetycyjny: połączenie liganda z receptorem
zmienia konformację miejsca wiązania na receptorze
23.
Rodzaje receptorówTyp I: jonotropowe – przewodzenie sygnałów odbywa się przy pomocy
kanałów jonowych
Typ II: metabotropowe – przewodzenie sygnałów odbywa się przy udziale
białka G
Typ III: przewodzenie sygnałów odbywa się przy pomocy kinaz
tyrozynowej
Typ IV: jądrowe (steroidowe) – wpływają na transkrypcję genów
24.
LADME – droga lekuw organizmie
L (liberation) – uwolnienie
A (absorption) – absorpcja
D (distribution) – dystrybucja
M (metabolism) – metabolizm
E (elimination) – wydalanie
25.
Absorpcja (wchłanianie)Przemieszczanie z miejsca podania do krążenia ogólnego
Jest poprzedzona uwolnieniem substancji czynnej z postaci leku i jej
przejściem do roztworu
26.
DystrybucjaProces przemieszczania się leku z krwi do tkanek, dotarcie leku do
miejsca działania
Zależy od wiązania z białkami osocza
Dystrybucja zachodzi dopiero po przekroczeniu stanu nasycenia (to
graniczna wartość stężenia leku, przy której wszystkie miejsca
wiązania
w białkach osocza są wysycone)
Tylko wolna frakcja leku ulega dyfuzji do tkanek
27.
DystrybucjaPenetracja do tkanek może być ograniczona np. przez barierę krew-mózg
Dystrybucja do OUN zależy od lipofilności, określonej przez współczynnik
podziału woda- tłuszcz
Redystrybucja – to uwalnianie leku
z tkanek po zmniejszeniu jego stężenia we krwi (np. z tkanki tłuszczowej,
z połączeń z melaniną)
28.
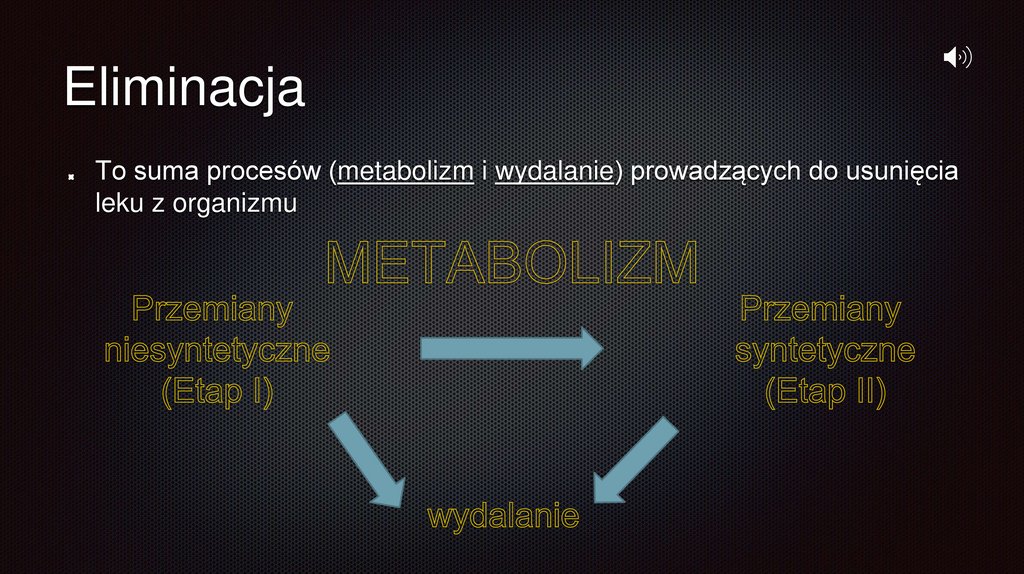
EliminacjaTo suma procesów (metabolizm i wydalanie) prowadzących do usunięcia
leku z organizmu
29.
Metabolizm (biotransformacja)Zmiany chemiczne i strukturalne
w cząsteczce leku
2/3 dawki ulega przekształceniom
w wątrobie
Przekształcenie niepolarnej i lipofilowej formy leku w postać polarną
i hydrofilową
30.
Przemiany niesyntetyczne (Etap I)Enzymy frakcji mikrosomalnej wątroby (cytochrom P-450, reduktazy)
Utlenianie: hydroksylacja pierścieni aromatycznych, dealkilacja,
deaminacja
Redukcja (grupy nitrowe, azotowe)
Hydroliza (estry)
31.
Przemiany syntetyczne (Etap II)Sprzęganie z substancjami endogennymi:
- glukoronidacja
- łączenie z aminokwasami
- acetylacja
- alkilacja
32.
WydalanieMoże nastąpić także z pominięciem biotransformacji
Z moczem (przez nerki)
Z żółcią przez wątrobę
Z potem (przez skórę)
Z mlekiem, ze śliną
Przez płuca
33.
WydalanieStopień dysocjacji związku zależy od pH moczu (4,5 – 8,0)
Zakwaszenie moczu
wydalanie słabych zasad i
zwrotne słabych kwasów
Alkalizacja moczu
słabych zasad
wydalanie słabych kwasów i
wchłanianie
wchłanianie zwrotne
34.
WydalanieWydalanie leku z żółcią zachodzi tylko
w przypadku przekroczenia wydolności krążenia wątrobowo-jelitowego
Transport do dróg żółciowych –
w sposób aktywny
Wydalanie leku z żółcią
dla związków o dużej masie cząsteczkowej
Wydalanie leku z żółcią
glukuronowym
dla leków sprzężonych z kwasem
35.
36.
Interakcje farmaceutyczne(chemiczne)
występowanie reakcji między substancjami leczniczymi
obejmują interakcje recepturowe
mogą prowadzić do powstania toksycznych produktów
mogą prowadzić do dezaktywacji substancji czynnych
37.
Interakcje farmakokinetycznedotyczą one każdego etapu metabolizmu leków w organizmie człowieka,
czyli absorpcji, dystrybucji, biotransformacji (obu faz) oraz eliminacji leku
z ustroju
wypieranie leków z połączeń z białkami
indukcja lub inhibicja enzymów mikrosomalnych wątroby (CYP-450)
wpływ na eliminację leków poprzez zmianę ich stopnia dysocjacji
38.
Interakcje farmakodynamiczneSynergizm addycyjny: efekt wspólnego działania leków jest sumą efektów
działania każdego osobno.
Synergizm hiperaddycyjny: efekt łącznego podania dwóch leków jest
większy niż suma efektów po podaniu każdego z nich osobno =
potencjalizacja działania
Antagonizm – gdy lek znosi działanie innego równocześnie podanego
leku
39.
Interakcje farmakodynamiczneAntagonizm funkcjonalny: jednoczesne oddziaływanie na odrębne
receptory regulujące czynności tego samego narządu.
40.
Dostępność biologicznaTo szybkość z jaką określona liczba aktywnych cząsteczek leku przechodzi do
krążenia ogólnego, uzyskując dostęp do miejsca działania. Określają ją:
Równoważność farmaceutyczna (chemiczna) – wyznacza tożsamość i ilość
substancji czynnej, dopuszcza różnice w występowaniu biologicznie
nieczynnych dodatków
Równoważność biologiczna – ocenia działanie chemicznych równoważników
leku w różnych stężeniach we krwi tej samej osoby
Równoważność terapeutyczna – określa efekt leczniczy po podaniu
jednakowych dawek różnych leków u tej samej osoby
41.
Farmakokinetykabada przebieg zmian stężenia leku
i jego metabolitów w czasie i ocenia te zmiany matematycznie
bada związek pomiędzy podaniem leku, czasem trwania jego dystrybucji
i eliminacji, a stężeniem osiągniętym
w różnych częściach ustroju.
42.
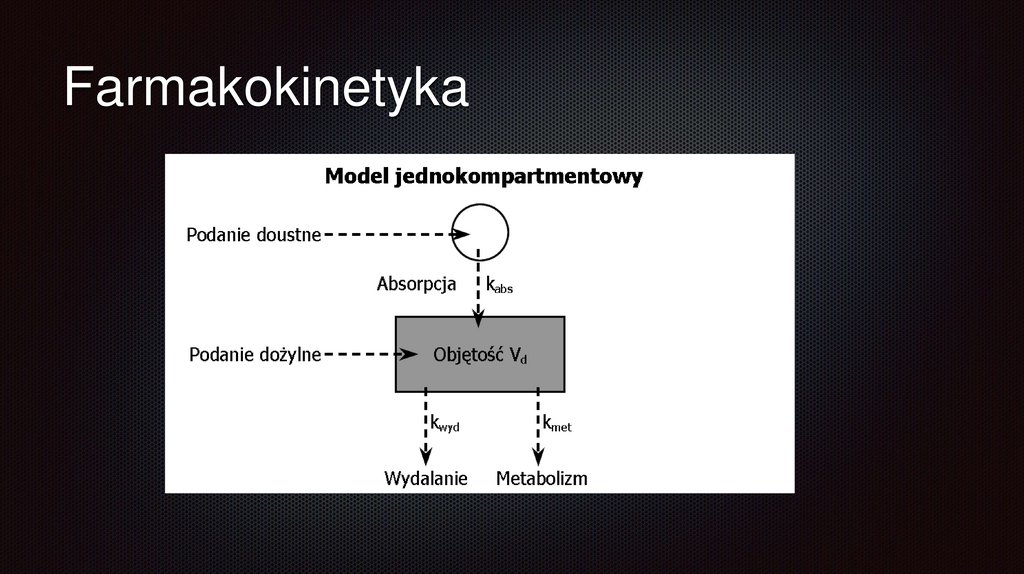
Farmakokinetyka43.
FarmakokinetykaUproszczony model jednokompartmentowy zakłada istnienie w
organizmie jednego kompartmentu o objętości Vd.
Jeśli podamy do niego, za pomocą szybkiej iniekcji dożylnej, dawkę
leku Q, to ulegnie ona z czasem procesom metabolizmu i
wydalania.
Większość leków wykazuje farmakokinetykę pierwszorzędową
(czyli szybkość eliminacji leku jest wprost proporcjonalna do
stężenia).
44.
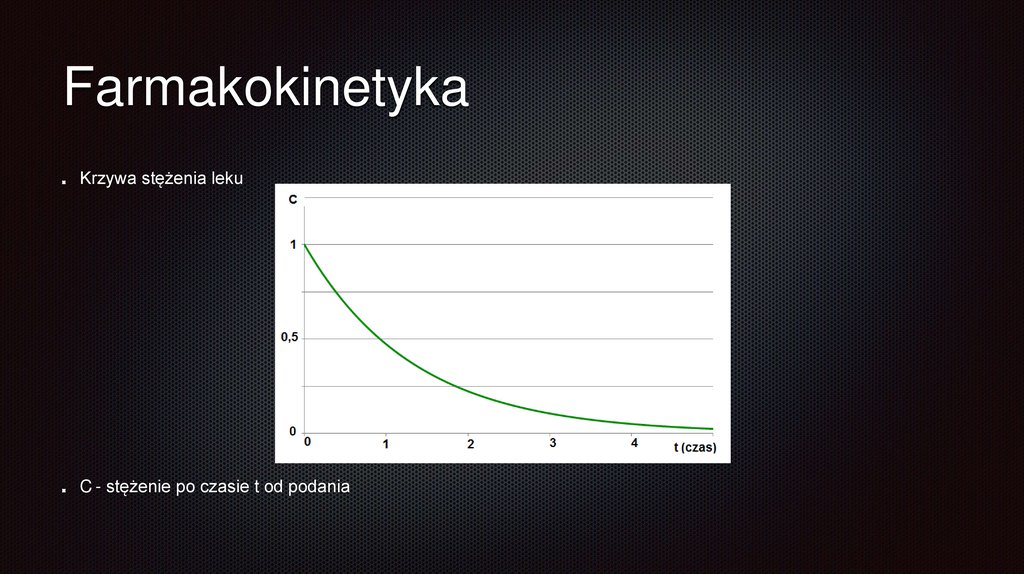
FarmakokinetykaStężenie leku obniża się wykładniczo, zgodnie ze wzorem:
C(t) - stężenie po czasie t od podania
C(0) - stężenie początkowe
e - stała logarytmów naturalnych
K - stała eliminacji (wydalanie + metabolizm)
45.
FarmakokinetykaKrzywa stężenia leku
C - stężenie po czasie t od podania
46.
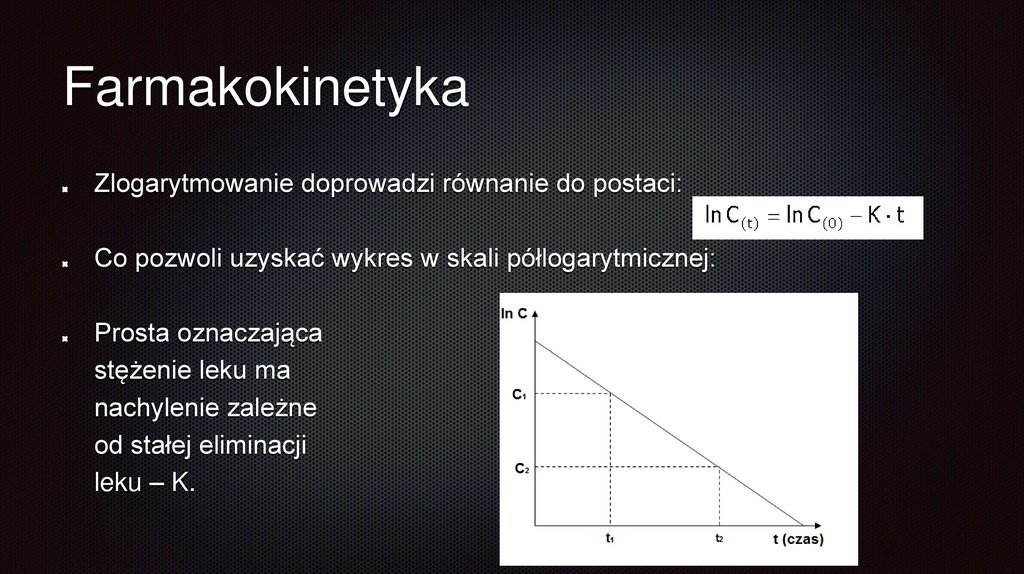
FarmakokinetykaZlogarytmowanie doprowadzi równanie do postaci:
Co pozwoli uzyskać wykres w skali półlogarytmicznej:
Prosta oznaczająca
stężenie leku ma
nachylenie zależne
od stałej eliminacji
leku – K.
47.
Okres półtrwania lekuBiologiczny okres półtrwania (t0,5) jest to czas wymagany do eliminacji
połowy wprowadzonej dawki leku.
Wzór na t0,5 wyprowadzamy z równania:
więc:
48.
Okres półtrwania lekuAby otrzymać równanie na t0,5,
w miejsce C(t), wstawiamy połowę stężenia początkowego, czyli C(0)/2:
ln 2 = 0,693, więc
49.
Okres półtrwania lekuokres półtrwania leku można obliczyć również znając jego klirens i objętość
dystrybucji:
Stałą eliminacji K oblicza się ze wzoru:
CL – klirens leku
Vd – objętość dystrybucji
50.
Objętość dystrybucjiTo teoretyczna objętość potrzebna do rozmieszczenia się leku w
organizmie,
w której stężenie leku jest takie samo jak oznaczone w surowicy
Vd < 7 l: lek ulega dystrybucji jedynie
w łożysku naczyniowym
7 < Vd < 40 l: lek ulega rozmieszczeniu poza łożyskiem naczyniowym
Vd > 40 l (powyżej 100% masy ciała): lek kumuluje się w organizmie
51.
Objętość dystrybucjiOkreśla rozmieszczenie leku między krwią a tkankami:
D – dawka leku podanego dożylnie
C0 – stężenie leku we krwi bezpośrednio po podaniu
52.
KlirensTo objętość osocza oczyszczonego
z substancji leczniczej w jednostce czasu.
K – stała eliminacji
Vd – objętość dystrybucji
klirens podawany jest zwykle w ml/min
D – dawka
AUC – pole powierzchni pod krzywą zależności stężenia leku od
czasu
53.
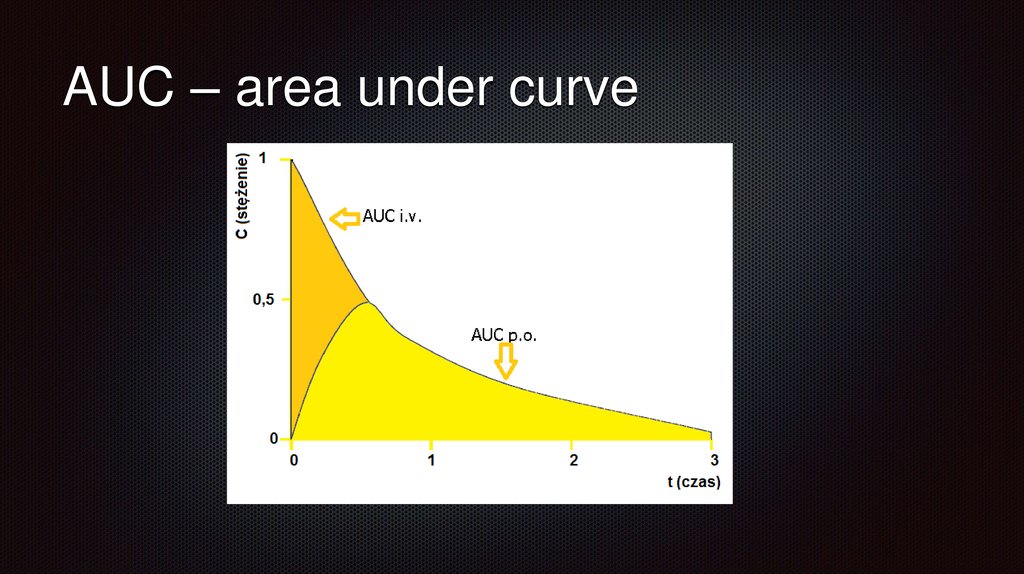
AUC – area under curveTo pole pod krzywą zależności stężenia leku w osoczu do czasu.
więc
K – stała eliminacji
Vd – objętość dystrybucji
D – dawka
Cl - klirens
54.
AUC – area under curve55.
AUC – area under curvejest wprost proporcjonalne do całkowitej dawki leku wprowadzonej do
kompartmentu centralnego
Jest odwrotnie proporcjonalne do klirensu i stałej eliminacji
56.
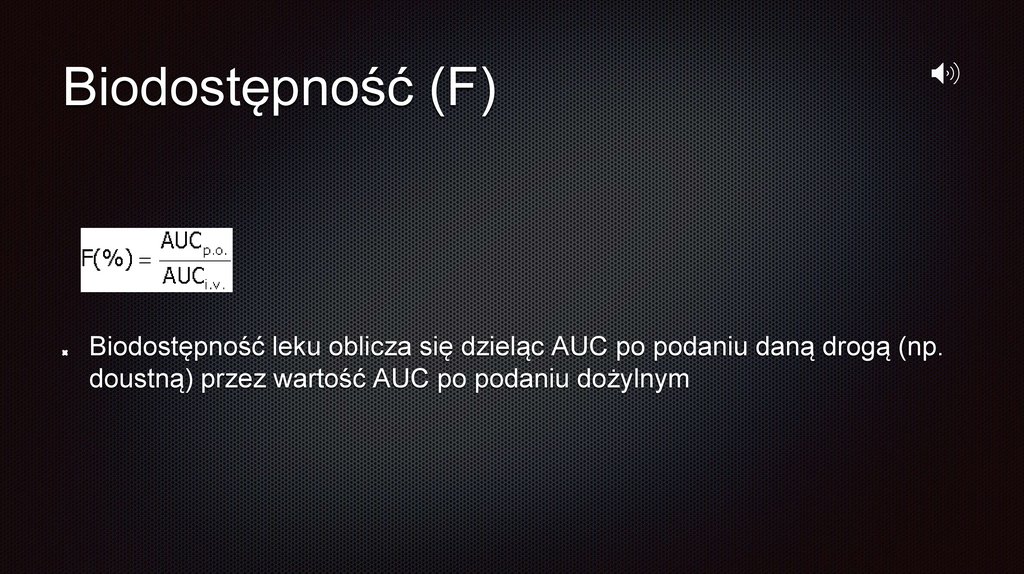
Biodostępność (F)Biodostępność (F) jest to odsetek podanej dawki leku, który osiąga
krążenie systemowe
Przy podaniu dożylnym biodostępność wynosi 100%
Przy podaniu innymi niż dożylne biodostępność zależy od absorpcji oraz
efektu pierwszego przejścia
57.
Biodostępność (F)Biodostępność leku oblicza się dzieląc AUC po podaniu daną drogą (np.
doustną) przez wartość AUC po podaniu dożylnym
58.
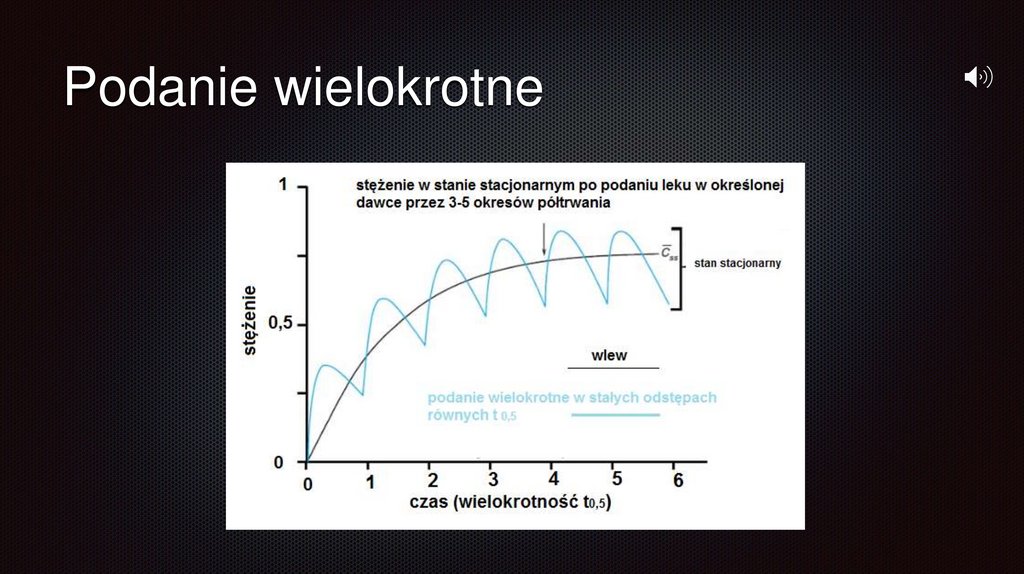
Podanie wielokrotnew przypadku podawania leku w infuzji, stężenie leku w osoczu rośnie do
chwili uzyskania stanu nasycenia, gdy szybkość infuzji jest równa
szybkości eliminacji
w przypadku podania wielokrotnego stężenie rośnie skokowo do
momentu uzyskania stanu stacjonarnego (wartość średnią stanowi
krzywa stężenia leku podawanego w infuzji)
stan stacjonarny dla danego leku osiąga się po podawaniu go w
określonej dawce przez trzy do pięciu okresów półtrwania
59.
Podanie wielokrotne60.
Dawka podtrzymującapo osiągnięciu stanu stacjonarnego szybkość podawania leku jest
równa szybkości jego eliminacji
Dawka podtrzymująca zależy od klirensu leku:
Cp – pożądane stężenie w osoczu
CL – klirens leku
F – biodostępność leku
61.
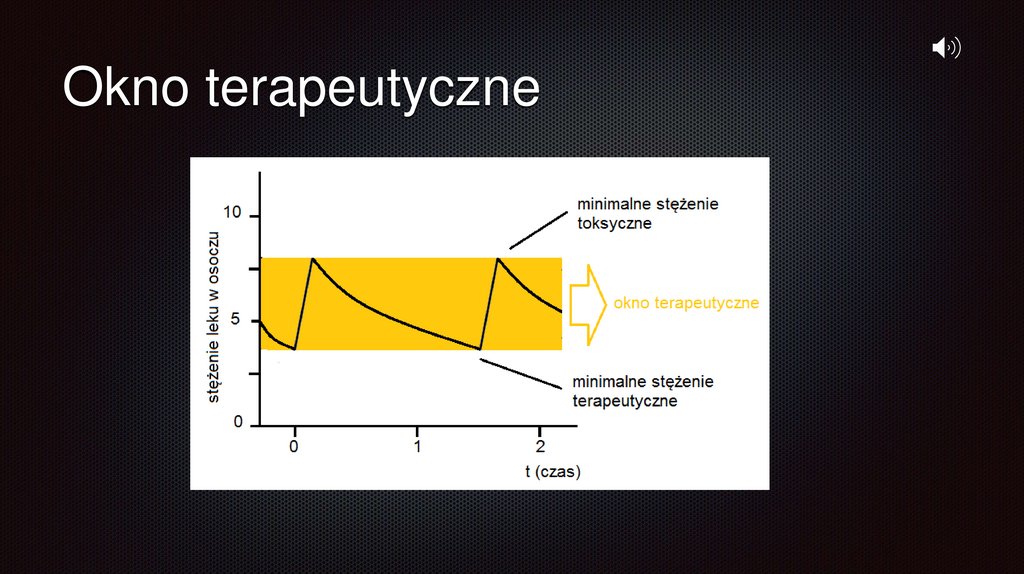
Okno terapeutyczneto bezpieczna „przestrzeń” pomiędzy minimalnym stężeniem
terapeutycznym leku
w osoczu a minimalnym stężeniem toksycznym
minimalne stężenie terapeutyczne to najniższe stężenie leku we krwi
wywołujące efekt leczniczy
minimalne stężenie toksyczne to najwyższe dopuszczalne stężenie leku
we krwi
62.
Okno terapeutyczne63.
w przypadku pytań zapraszam dokontaktu pod adresem:
mdelijewski@sum.edu.pl
Dziękuję za uwagę