

































































 medicine
medicineSimilar presentations:










Медицинская генетика понятие о наследственных болезнях. Определение и развитие пола
1.
ФГБОУ ВО СЗГМУ ИМ. И.И. МЕЧНИКОВА МИНЗДРАВА РОССИИкафедра медицинской биологии
2.
Раздел антропогенетики,разрабатывающий методы
диагностики, лечения
и профилактики
наследственной патологии
человека
3.
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ(известно более 2 тыс.)
Структурные или функциональные
нарушения в организме, вызванные
качественным или количественным
изменением наследственного
материала
! Не все наследственные болезни наследуются
Часть появляется вновь в результате мутаций
4.
По уровню нарушения наследственногоматериала:
o Молекулярные (генные)
o Хромосомные
По способу наследования признака:
o Моногенные (генные)
o Полигенные (мультифакториальные)
5.
ОбобщённаяКлассификация НБ
Генные (молекулярный
уровень - структура ДНК)
Хромосомные (изменение
структуры хромосом)
Геномные (изменение
количества хромосом)
Генные
(молекулярные)
Хромосомные
Мультифакториальные
(с наследственным
предрасположением)
6.
МЕХАНИЗМ РАЗВИТИЯ ГЕННЫХ(МОЛЕКУЛЯРНЫХ) БОЛЕЗНЕЙ
Изменение ДНК (мутация гена)
Изменение структуры РНК
Изменение структуры Белка
Появление патологического признака
7.
Характеристика Генных болезнейНаследуются моногенно
Характер расщепления в потомстве
определяется по законам Менделя
Между здоровыми и больными имеются
четкие фенотипические различия
Болезни являются
истинно
наследственными (1-2 % от всех болезней)
8.
КЛАССИФИКАЦИЯ ГЕННЫХ БОЛЕЗНЕЙДЕФЕКТЫ
СТРУКТУРНЫХ БЕЛКОВ
Тип наследования А-Д
(сцепленно с полом - Д)
генотип АА, Аа
Результат: нарушение
структуры белка коллагена или
гемоглобина. Интеллект - норма
Классификация:
- Коллагенозы
- Гемоглобинопатии
Диагностика - по
фенотипическим
особенностям, микроскопия
ФЕРМЕНТОПАТИИ
Тип наследования А-Р
(сцепленно с полом - Р)
генотип аа
Результат: нарушение реакций
метаболизма.
Нарушение интеллекта!
Классификация:
по типу нарушенного
обмена веществ
Диагностика -
биохимический,
микробиологический методы
9.
Генные болезниПо доминантному типу наследуются болезни,
связанные с нарушением
o Структурных белков
o Транспортных белков
o Белков - рецепторов
Это болезни костной системы и соединительной ткани
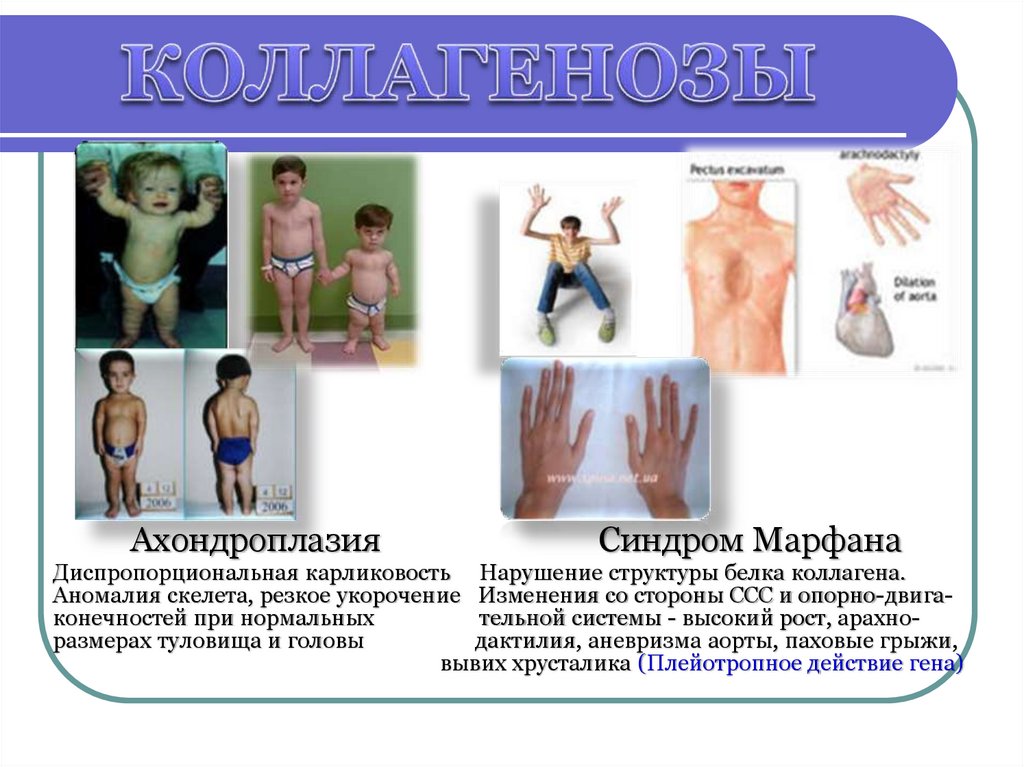
Примеры: Ахондроплазия, синдром Марфана,
нейрофиброматоз
Диагностика: данные электронной микроскопии изменение мышечных волокон, клеток эпидермиса,
хондроцитов.
10.
АхондроплазияСиндром Марфана
Диспропорциональная карликовость Нарушение структуры белка коллагена.
Аномалия скелета, резкое укорочение Изменения со стороны ССС и опорно-двигаконечностей при нормальных
тельной системы - высокий рост, арахноразмерах туловища и головы
дактилия, аневризма аорты, паховые грыжи,
вывих хрусталика (Плейотропное действие гена)
11.
12.
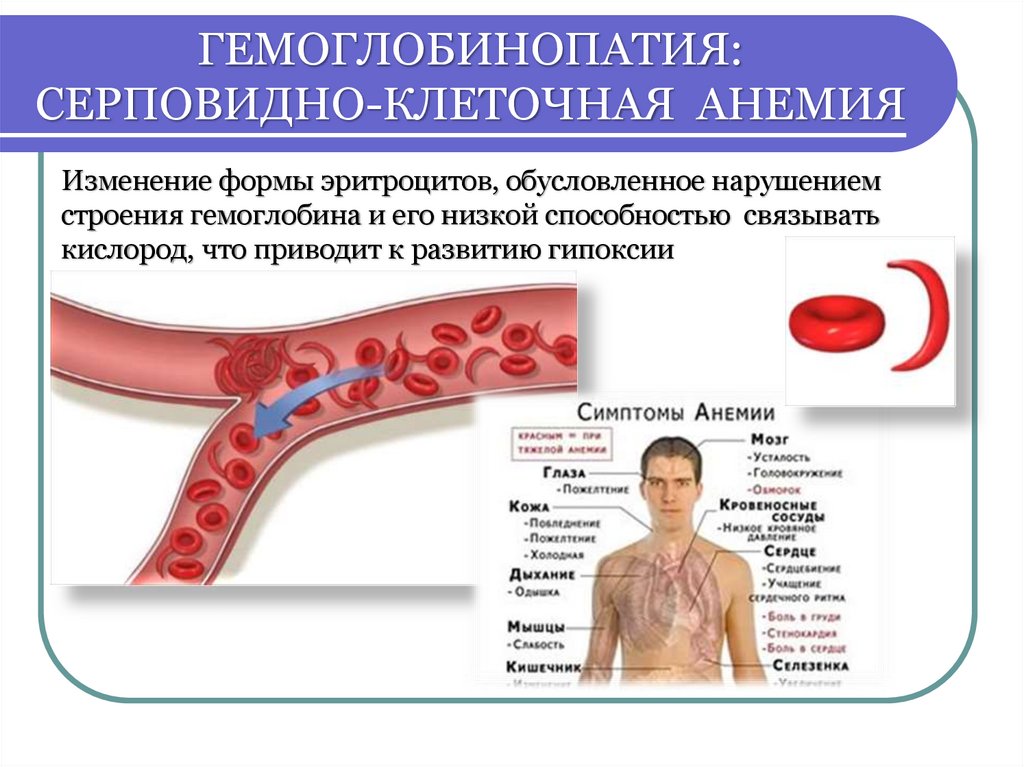
ГЕМОГЛОБИНОПАТИЯ:СЕРПОВИДНО-КЛЕТОЧНАЯ АНЕМИЯ
Изменение формы эритроцитов, обусловленное нарушением
строения гемоглобина и его низкой способностью связывать
кислород, что приводит к развитию гипоксии
13.
СЕРПОВИДНОКЛЕТОЧНАЯ АНЕМИЯИзвестен механизм возникновения на уровне гена, белка, фенотипа
Мутация структурного гена (замена Т на А ) приводит к изменению
β - цепи гемоглобина, замене глутаминовой кислоты на валин
В условиях гипоксии полипептидные цепи глобина
полимеризуются, образуя длинные тяжи, и эритроциты
принимают форму серпа
Изменение их формы
приводит к
изменению функции -
способность связывать
кислород
14.
СЕРПОВИДНОКЛЕТОЧНАЯ АНЕМИЯУ гомозигот (аа) с раннего возраста
развивается характерная картина
хронической гипоксии и анемии
Гетерозиготные носители (Аа) в обычных
условиях клинически здоровы
Гетерозиготы невосприимчивы к малярии
15.
ФЕРМЕНТОПАТИИПо рецессивному типу (аутосомному или
сцепленному с Х-хромосомой) наследуются многие
болезни обмена веществ - энзимопатии или
ферментопатии - вызванные нарушением
активности ферментов
Они возникают в результате мутаций генов,
ответственных за синтез ферментов
У человека большинство ферментопатий вызвано
мутациями структурных генов. Это приводит к
качественному, а не количественному изменению
ферментов
У гетерозигот активность фермента – 50%
(биохимический метод). Фенотип – норма
16.
ХАРАКТЕРИСТИКАФЕРМЕНТОПАТИЙ
Часто – задержка физического и
умственного развития
Непереносимость отдельных продуктов
питания
Некоторые поддаются коррекции с
помощью диет
Могут возникать при нарушении всех видов
обмена веществ
Классификация - по типу обмена веществ
17.
Классификация ФЕРМЕНТОПАТИЙНарушение аминокислотного обмена - фенилкетонурия,
алкаптонурия, тирозиноз
Углеводного - галактоземия, фруктозурия, муковисцедзы,
полисахаридоз
Липидного - б-нь Тея-Сакса, гиперхолестеринемия
Пуринового и пиримидинового - с-м Леш-Нихана
Нуклеинового - прогерия
Минерального - нарушение обмена Сu - б-нь ВильсонаКоновалова (гепато-церебральная дегенерация)
- гипофосфатемия - витамин-D-резистентный
рахит
Описаны нарушения обмена гормонов, витаминов, дефекты
ферментов эритроцитов и т.д.
18.
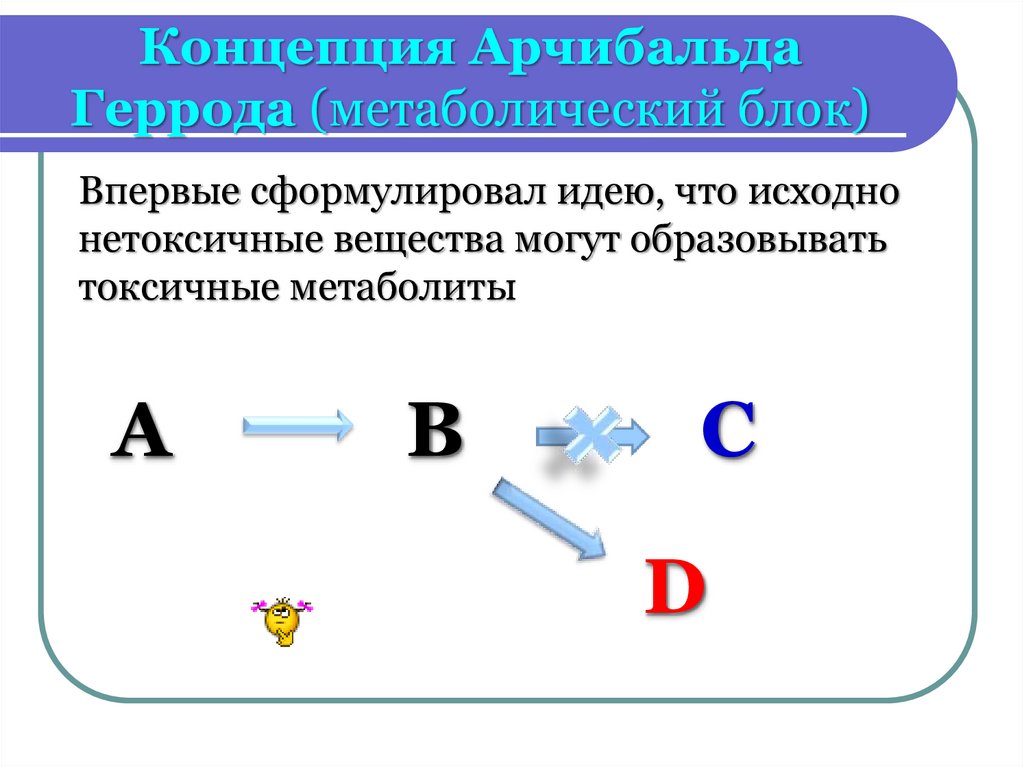
Концепция АрчибальдаГеррода (метаболический блок)
Впервые сформулировал идею, что исходно
нетоксичные вещества могут образовывать
токсичные метаболиты
А
В
С
D
19.
ФЕНИЛКЕТОНУРИЯМоделью для изучения молекулярных болезней может служить
фенилкетонурия:
o
o
o
o
o
известна локализация гена в 12 хромосоме (определяющего
синтез фермента фенилаланингидроксилазы)
тип наследования (аутосомно-рецессивный)
частота распространения мутантного гена в популяциях
метаболизм фенилаланина у здоровых (превращение
фенилаланина в тирозин под влиянием фермента
фенилаланингидроксилазы)
метаболизм фенилаланина у больных людей (некоторая часть
фенилаланина из-за изменения фермента превращается в
токсическую фенилпировиноградную кислоту - ФПК)
20.
ФЕНИЛКЕТОНУРИЯразработана диагностика (выявление
фенилаланина в крови и фенилпировиноградной
кислоты в моче)
o лечение (специальная диета и использование
гидролизата белка без фенилаланина)
o профилактика (предупреждение браков между
гетерозиготами)
o описаны четкие фенотипические проявления
мутантного гена у гомозигот (выраженная
умственная отсталость, изменение пигментации
волос и кожи - голубоглазые блондины со светлой
кожей)
o
21.
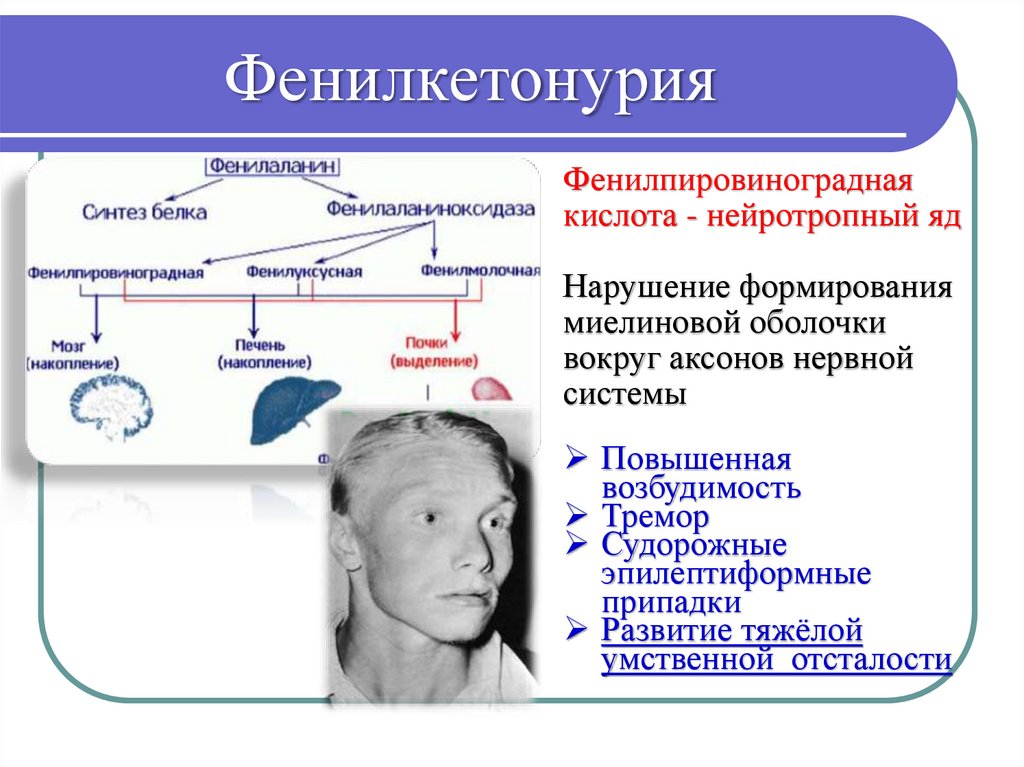
ФенилкетонурияФенилпировиноградная
кислота - нейротропный яд
Нарушение формирования
миелиновой оболочки
вокруг аксонов нервной
системы
Повышенная
возбудимость
Тремор
Судорожные
эпилептиформные
припадки
Развитие тяжёлой
умственной отсталости
22.
Преждевременное старение, связанное с нарушениемпроцессов репликации и репарации ДНК
23.
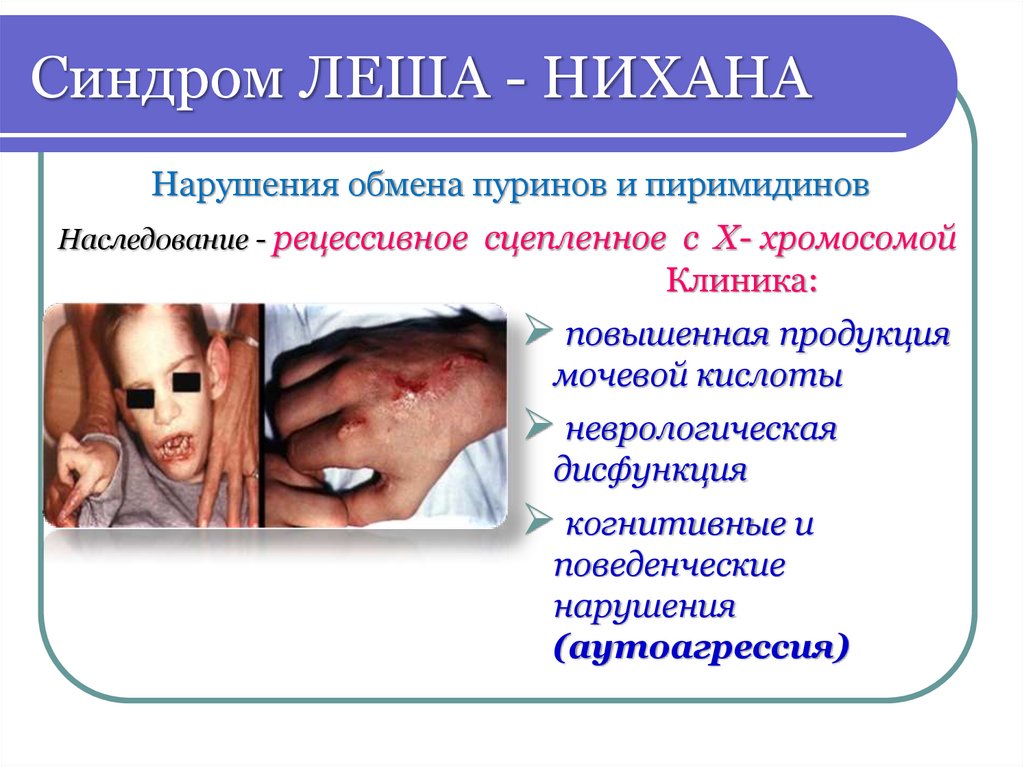
Синдром ЛЕША - НИХАНАНарушения обмена пуринов и пиримидинов
Наследование - рецессивное сцепленное с X- хромосомой
Клиника:
повышенная продукция
мочевой кислоты
неврологическая
дисфункция
когнитивные и
поведенческие
нарушения
(аутоагрессия)
24.
ДИАГНОСТИКА ФЕРМЕНТОПАТИЙВАЖНО!!!
РАННЯЯ ДИАГНОСТИКА
У индивидуумов – биохимический и
микробиологический методы
амниоцентез (пренатальная диагностика)
На уровне популяций изучение
характера наследования –
генеалогический метод
25.
ПРОФИЛАТИКА ФЕРМЕНТОПАТИЙo Медико-генетическое консультирование
o Возможное исключение браков между
гетерозиготами
o Выявление гетерозигот (биохимический метод)
В настоящее время проводится СКРИНИНГ
всех новорождённых на:
ФЕНИЛКЕТОНУРИЮ
ГИПОТИРЕОЗ
МУКОВИСЦЕДОЗ
26.
Хромосомные болезниПричины
Хромосомные болезни возникают при
нарушениях кариотипа (нормальный
кариотип человека 46,ХХ, 46,ХУ),
которые могут быть вызваны изменением:
o числа (геномные мутации) или
o структуры (хромосомные мутации)
аутосом и половых хромосом
27.
Хромосомные болезниПри хромосомных болезнях имеется определенный
комплекс стабильных аномальных признаков симптомов, который входит в понятие синдром.
Синдромы характеризуются:
определенной частотой проявления,
продолжительностью жизни детей
средним весом при рождении
внешними морфологическими признаками,
пороками развития внутренних органов,
функциональными симптомами,
дерматоглификой
определенным кариотипом
28.
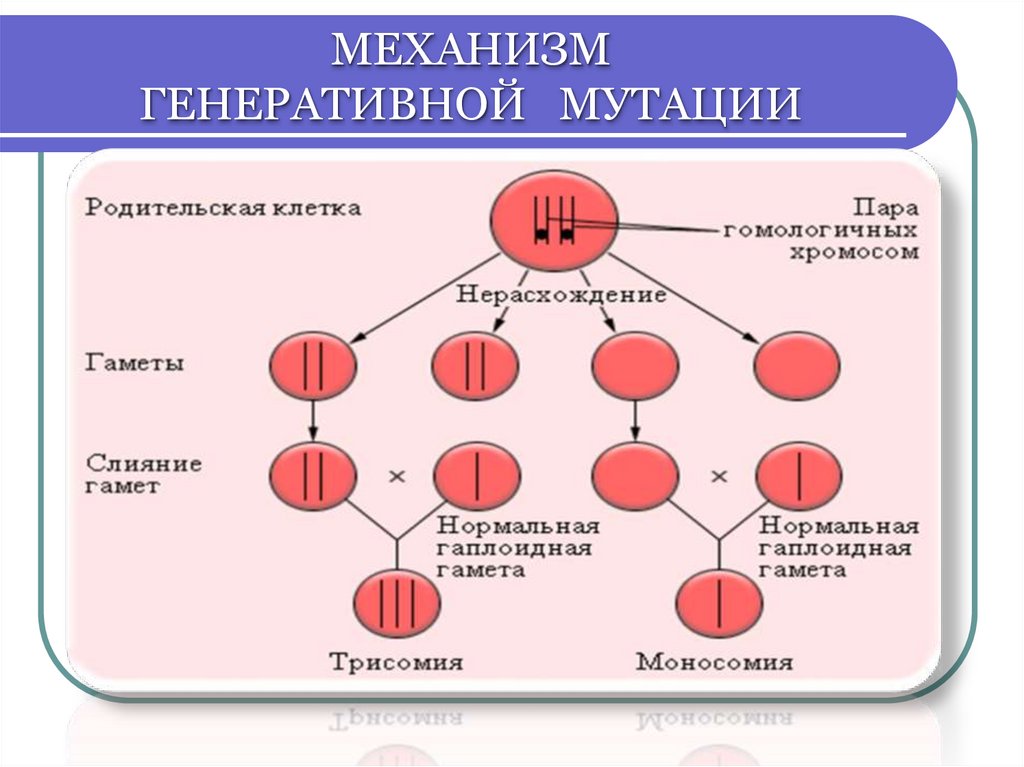
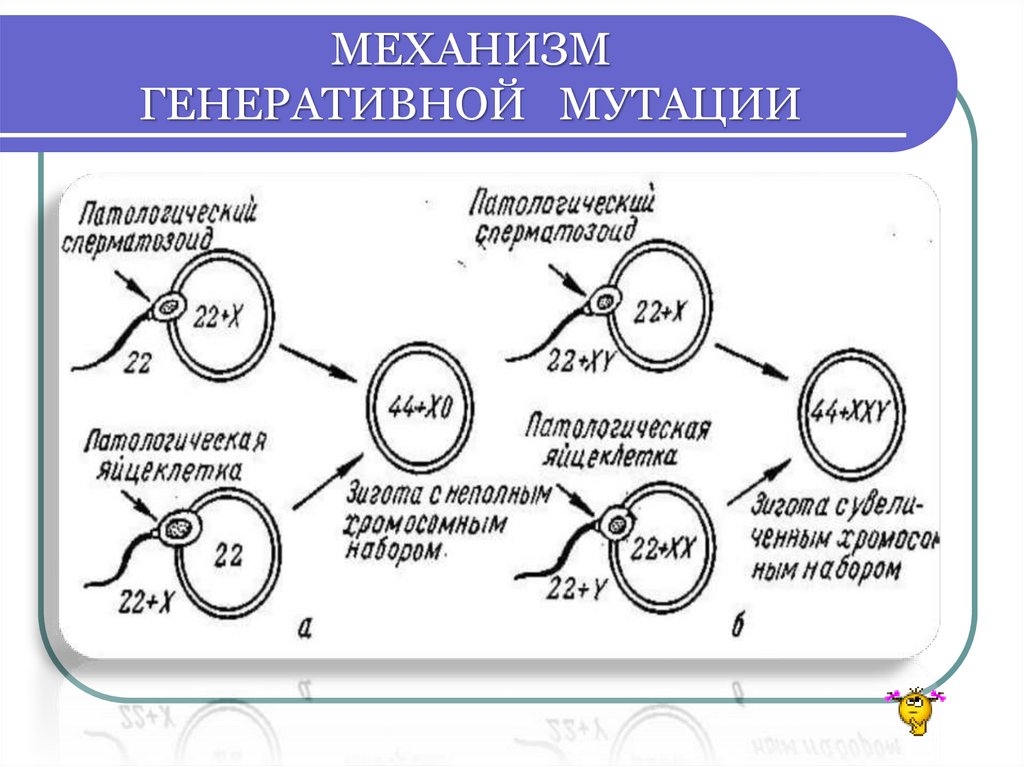
ХРОМОСОМНЫЕ БОЛЕЗНИБольшинство ХБ - не наследуются!
У больных нарушена репродуктивная функция
(стабилизирующий естественный отбор)
Это результат вновь возникших
генеративных мутаций
у здоровых людей
Поэтому ХБ появляются в каждом поколении с
определённой частотой
29.
МЕХАНИЗМГЕНЕРАТИВНОЙ МУТАЦИИ
30.
МЕХАНИЗМГЕНЕРАТИВНОЙ МУТАЦИИ
31.
ХАРАКТЕРИСТИКА ХБМасса и длина тела при рождении – ниже
средних значений
Дизморфические (диспластические)
признаки развития
Пороки развития внутренних органов
(при аномалиях аутосом)
Отставание в физическом и умственном
развитии (при аномалиях аутосом)
Задержка и аномалии полового развития
(при аномалиях половых хромосом)
32.
Геномные мутацииПолиплоидия (3n, 4n)–
зиготы нежизнеспособны
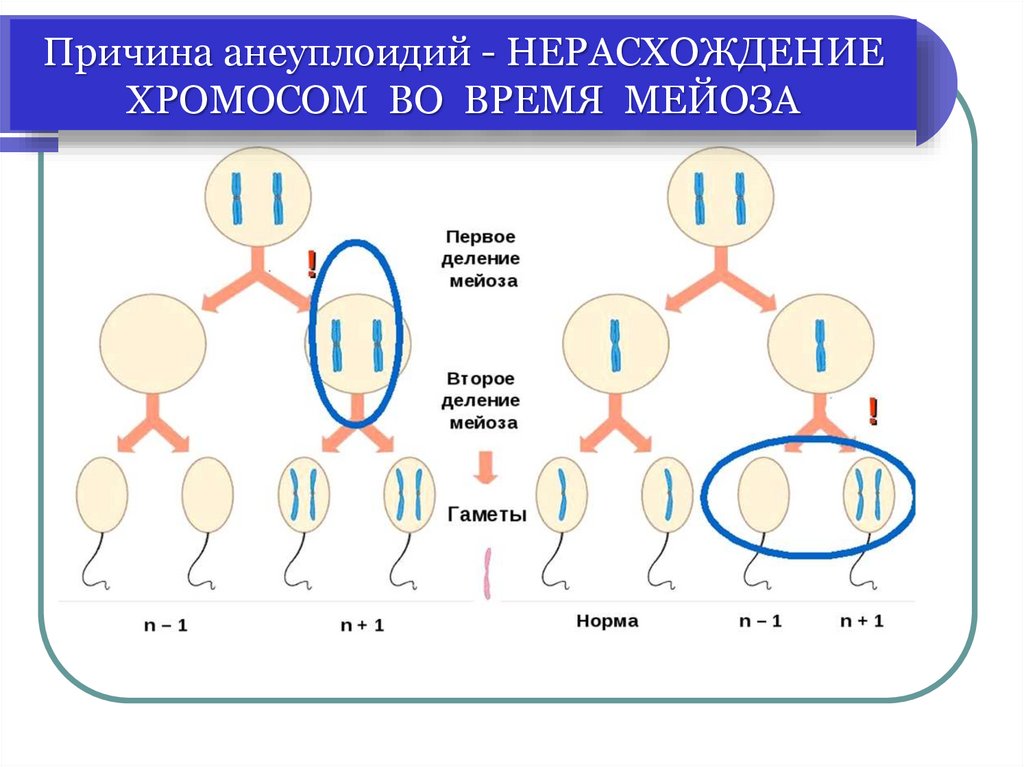
Анеуплоидия
трисомии (наиболее часто)
моносомии (только по Х-хромосоме)
полисомии (только половых хромосом)
33.
Причина анеуплоидий - НЕРАСХОЖДЕНИЕХРОМОСОМ ВО ВРЕМЯ МЕЙОЗА
34.
Геномные мутации АУТОСОМЭффект различен:
Моносомии аутосом - не совместимы с жизнью
Возможны только трисомии
Трисомии 1-12 пары– летальные мутации
Трисомии 13 – 18 пары – полулетальные
мутации, их проявления:
o Спонтанные аборты, множественные уродства,
незначительная продолжительность жизни от нескольких недель до нескольких лет
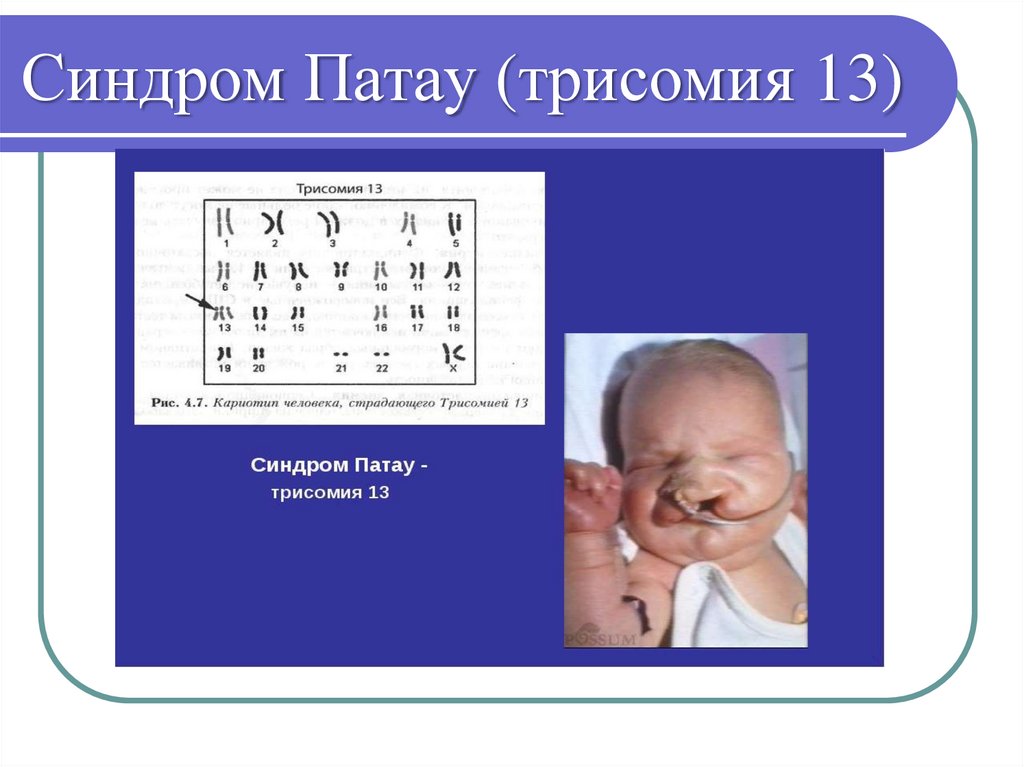
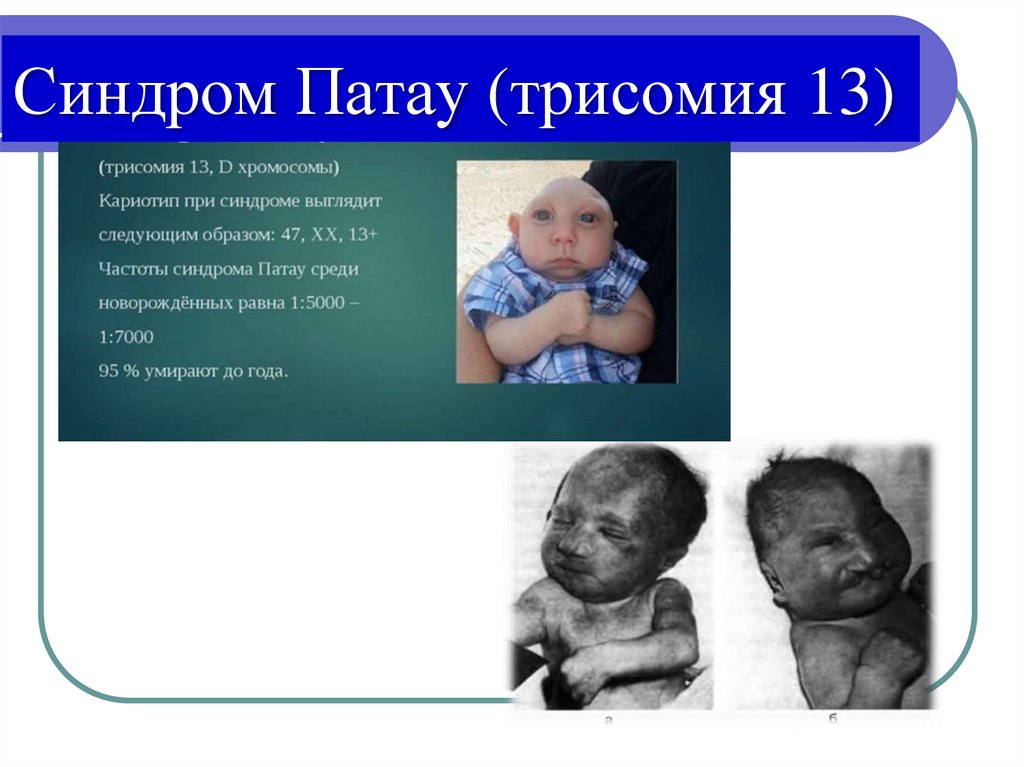
o Синдром Патау - 47, XX/XY +13
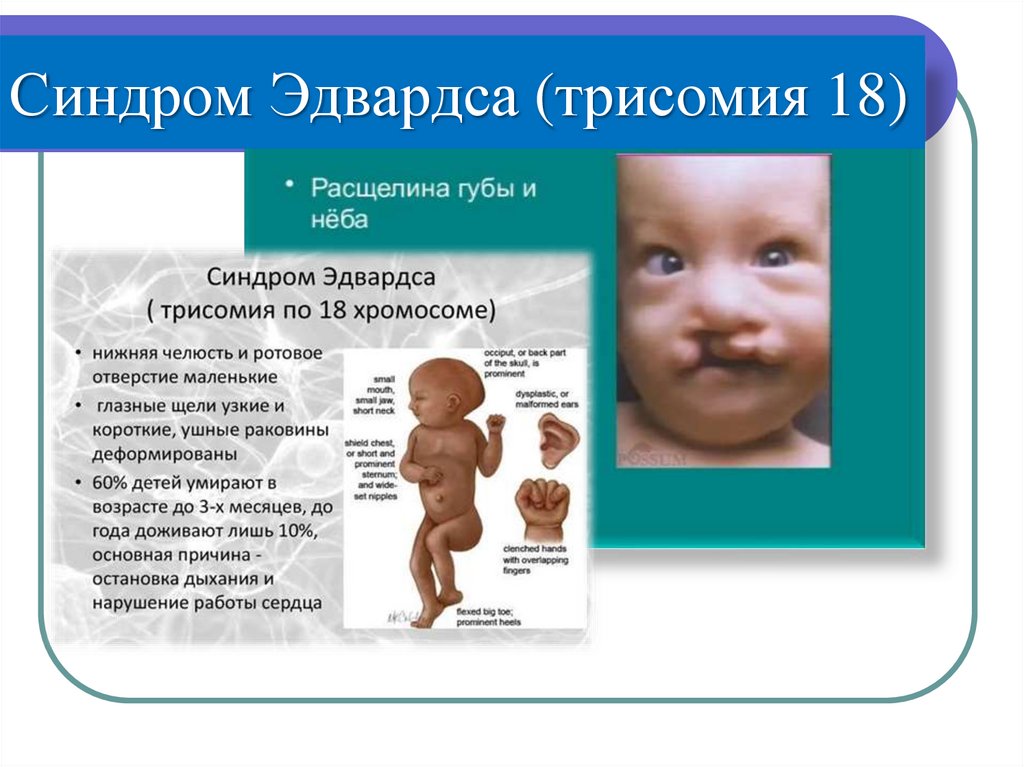
o Синдром Эдвардса - 47, XX/XY +18
35.
Синдром Патау (трисомия 13)36.
Синдром Патау (трисомия 13)37.
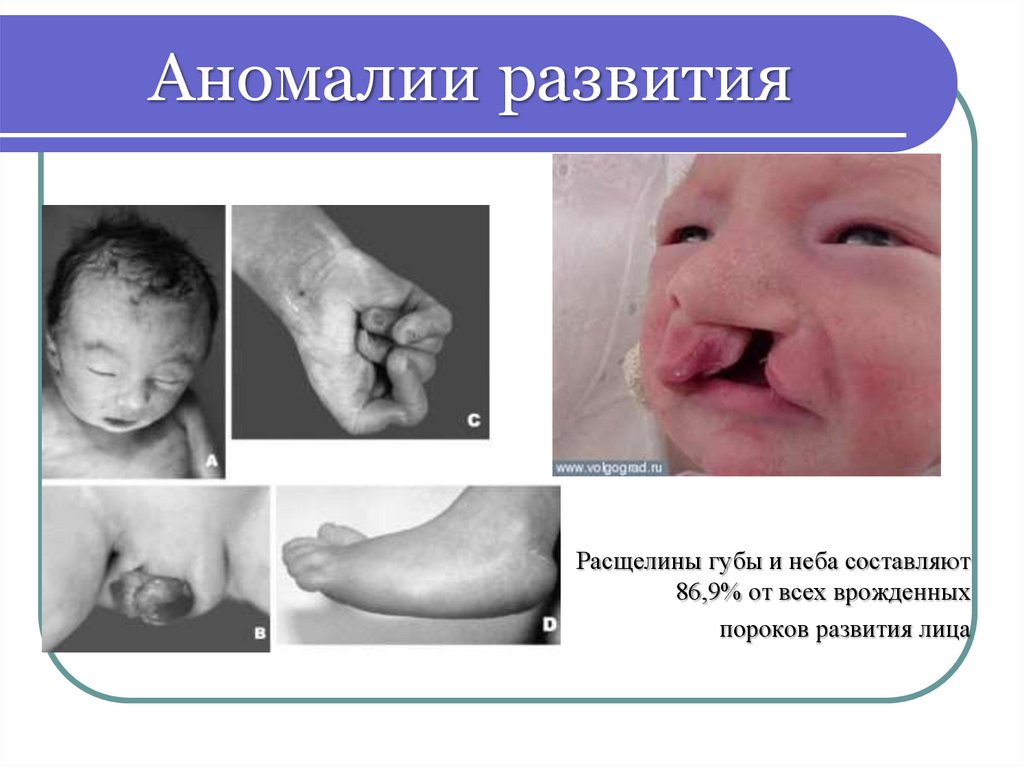
Аномалии развитияРасщелины губы и неба составляют
86,9% от всех врожденных
пороков развития лица
38.
Синдром Эдвардса (трисомия 18)39.
Синдром Эдвардса (трисомия 18)40.
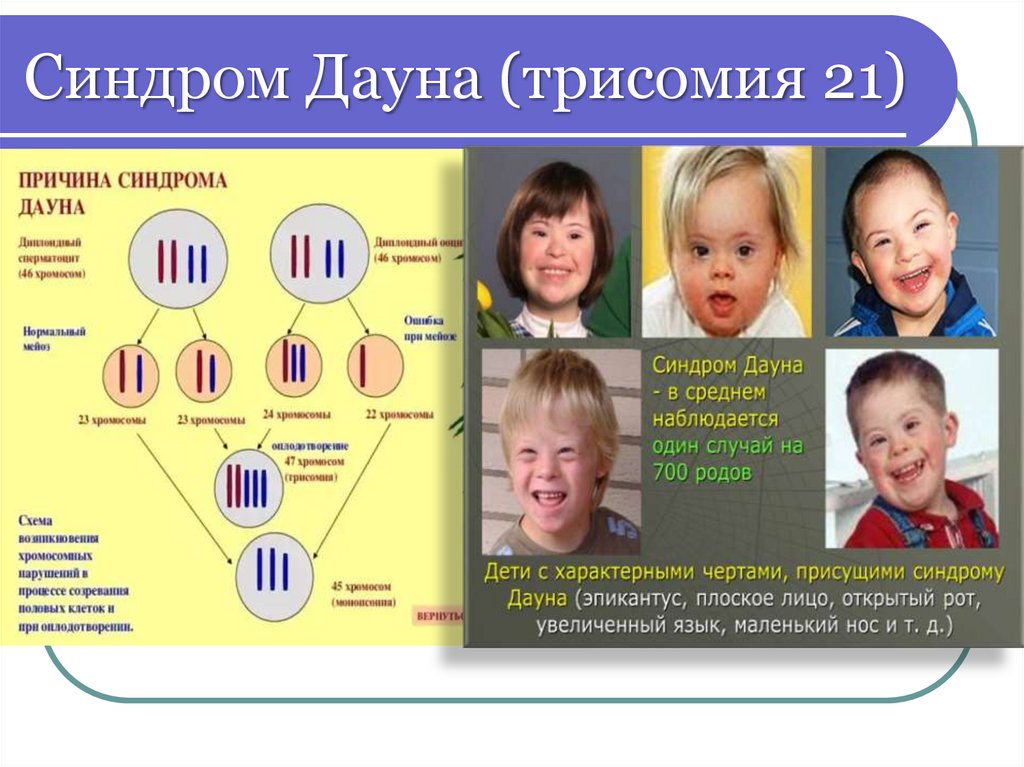
Геномные мутации АУТОСОМТрисомия по 21 хромосоме
синдром Дауна – 47, XX/XY +21
единственная совместимая с жизнью
трисомия аутосом
Описан Джоном Дауном в 1866 году
41.
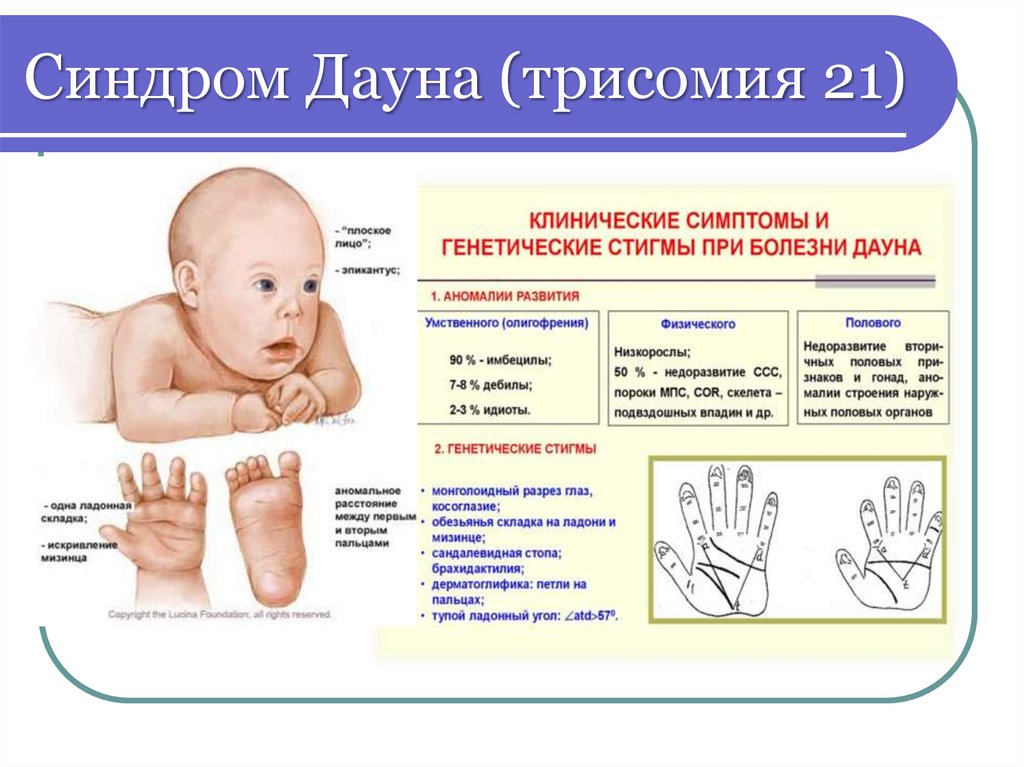
Синдром Дауна (трисомия 21)42.
Синдром Дауна (трисомия 21)43.
Аномалии половых хромосомПри аномалиях по половым
хромосомам часто сохраняется
нормальный интеллект или
отмечается его снижение
Но в большей степени нарушается
развитие половых органов и
гормонозависимый рост
(выше или ниже средней нормы)
44.
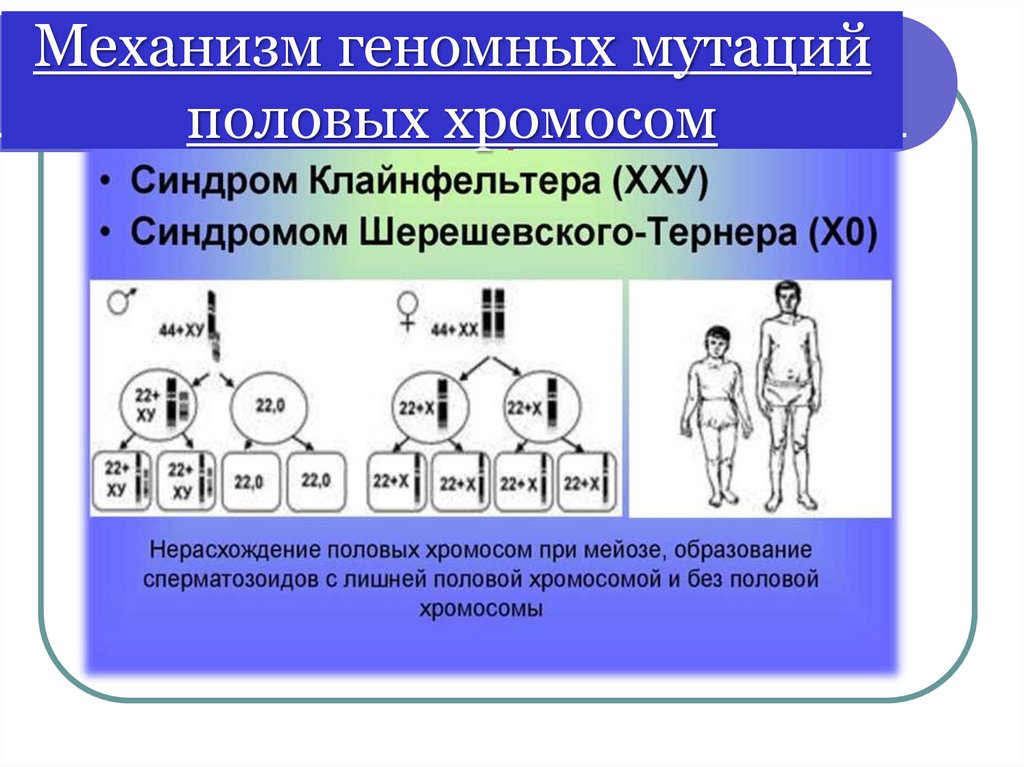
Геномные мутации половых хромосомМоносомии Х
(с-м Шерешевского-Тернера)
Полисомии (трисомии):
- 47, ХХХ – трисомия по Х хромосоме
- 47, ХYY – с-м Вай-Вай
- 47, ХХY – с-м Клайнфельтера
- 48, ХХYY - полисомия по Х- и Y-хромосомам
Моносомия Х встречается реже (1:2500),
чем полисомии ХХY (1:700) и ХХХ (1:1000)
45.
Механизм геномных мутацийполовых хромосом
46.
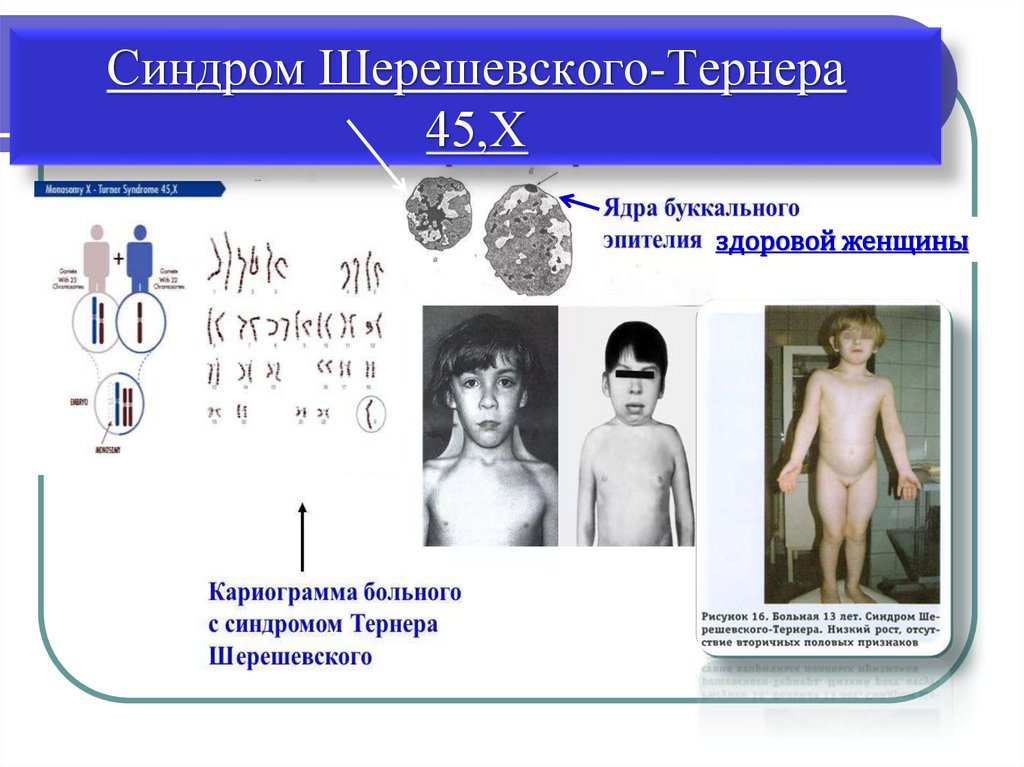
Синдром Шерешевского-Тернера45,Х
здоровой женщины
47.
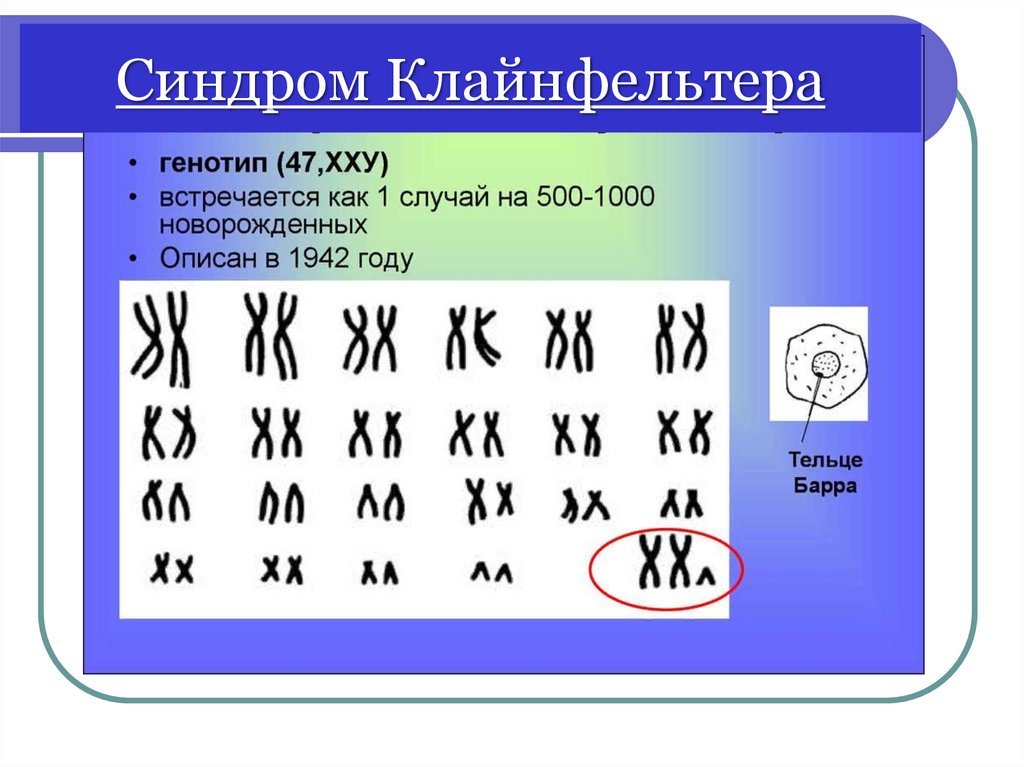
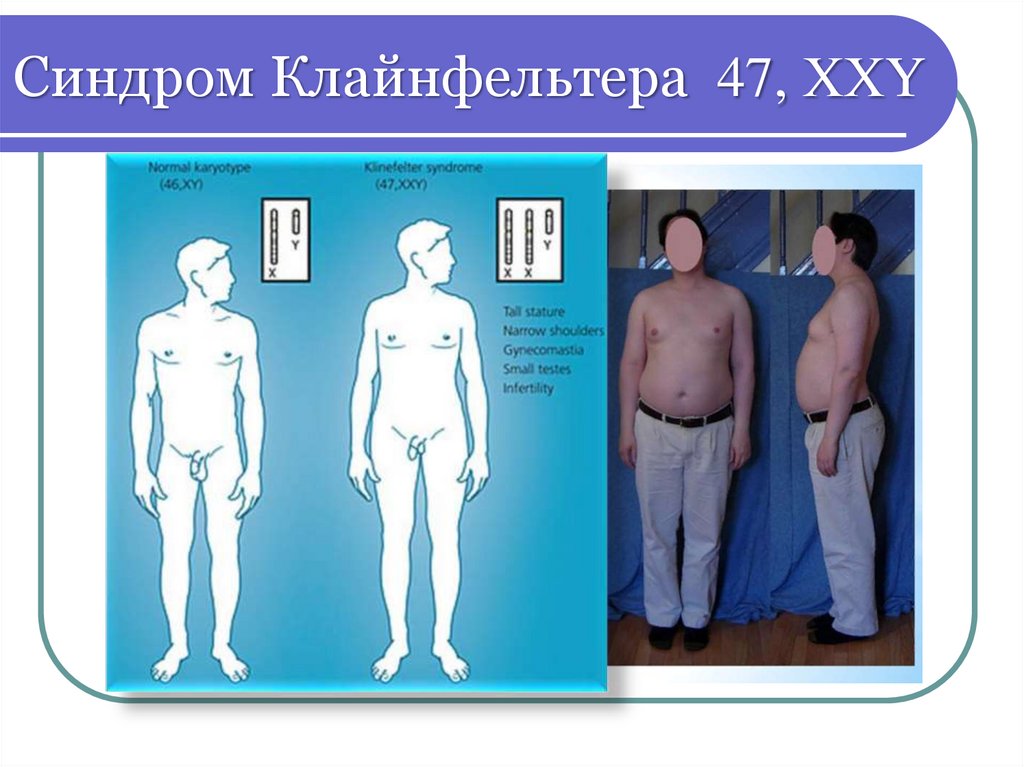
Синдром Клайнфельтера48.
Синдром Клайнфельтера 47, ХХYДобавочная Х- хромосома в 60% случаев
наследуется от матери
В период полового созревания проявляется
фенотипические особенности: высокий рост
(пик прибавки - 5-8 лет), нарушений пропорций
тела - длинные ноги, высокая талия, узкая
грудная клетка, оволосение по женскому типу,
гинекомастия, ожирение
Клиника проявляется после полового
созревания: недоразвитие первичных и
вторичных половых признаков, бесплодие
Умственная отсталость
49.
Синдром Клайнфельтера 47, ХХY50.
Синдром Вай-Вай 47, XYYПолисомия по У-хромосоме - частота 1:1000
Впервые аномалия была описана Хаушком в 1962 г.
В 1965 г. были опубликованы данные обследования
мужчин со сниженным интеллектом, находящихся в
специальном учреждении, вследствие их склонности
к преступлениям.
Высокий рост. Иногда - увеличение нижней челюсти,
кистей, стоп, грубые черты лица, выступающие
надбровные дуги
У 30-40% наблюдается легкая умственная отсталость,
снижение критики, агрессивность, взрывчатость.
51.
Синдром Вай-Вай 47, XYYРепродуктивная функция не нарушена. На фото - 2 брата
Слева - здоров, справа - с-м Вай-Вай
52.
Трисомия по Х-хромосоме 47, ХХХВстречается с частотой 1:1000
Редко диагностируется в раннем детстве
Взрослые имеют нормальный женский фенотип
Фенотипических изменений может не быть,
т.к. лишние Х-хромосомы спирализованы
(Х-половой хроматин - гетерохроматин)
Могут отмечаться: высокий рост, незначительное
снижение интеллекта
Репродуктивная функция чаще не нарушена
Риск рождения ребёнка с трисомией
по Х-хромосоме повышен у пожилых матерей
53.
Хромосомные аберрацииДелеции - по 5, 13, 18, 21, 22 хромосмам
(с-м «Кошачьего крика», с-м Орбели)
Транслокации – 15/21 – с-м Дауна
9/22 – хронический миелолейкоз
Инверсии
Дубликации
Кольцевые хромосомы
54.
Синдром “Кошачьего крика”Делеция короткого плеча 5-й хромосомы - 46,ХХ,del (5p-) или 46,ХY,del (5p-).
Описал Дж. Лежен в 1963 году . Частота 1:45 000.
У детей отмечается необычный плач, напоминающий требовательное кошачье
мяуканье или крик. Причина сходства плача ребёнка с мяуканьем кошки -
нарушение ЦНС, а не аномалия голосового аппарата.
Низкая масса тела при рождении, мышечная гипотония.
Лунообразное лицо с широко расставленными глазами.
Задержка умственного и физического развития.
55.
Диагностика хромосомных болезнейЦитогенетические методы
- кариотипирование
- определение полового хроматина
Методы пренатальной
диагностики
- амниоцентез
Дерматоглифика
56.
МУЛЬТИФАКТОРИАЛЬНЫЕБОЛЕЗНИ
Болезни с наследственным предрасположением.
Зависят как от генотипа, так и от внешней среды
Наследуются не сами болезни,
а предрасположенность к ним
57.
МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ– это заболевания, возникающие при неблагоприятном
сочетании ряда факторов: генетических особенностей
(генетической предрасположенности) и влияния
«внешней среды» - вредных привычек, образа жизни,
профессиональной деятельности и других.
Для реализации наследственной
предрасположенности необходимы
определённые условия
Характерно изменение нормы реакции на действие факторов
внешней среды.
Пример: при предрасположенности к диабету изменяется норма
реакции на крахмал и сахар - уровня глюкозы в крови
58.
Болезни с наследственной предрасположенностьюБолезни с наследственной
предрасположенностью
Частота(%)
Ишемическая болезнь сердца
5-10
Гипертоническая болезнь
Язвенная болезнь желудка и
двенадцатиперстной кишки
Диабет
Бронхиальная астма
Шизофрения
Эпилепсия
10-20
2-5
1-2
0,2-0,5
1-2
1
Маниакально-депрессивный психоз
0,5
Алкоголизм
1,4-10
Олигофрения (недифференцированная)
0,5-0,05
Расщелина губы и нёба
Вывих бедра
Косолапость
Анэнцефалия и черепномозговая грыжа
0,1-0,2
0,5
0,5
0,1
59.
Причины мультифакториальныхболезней
ГЕНЕТИЧЕСКИЕ – «главный ген» или
полигенный комплекс
НЕГЕНЕТИЧЕСКИЕ:
- средовые – факторы среды, аллергены
- поведенческие – пристрастие к определенной пище,
вредные привычки
- социальные – влияние окружения, родителей,
школы
Мультифакториальные болезни являются ответом
организма с определенной генетической
конституцией на воздействие средовых факторов
60.
МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИГенетическая компонента может быть
обусловлена:
суммарным (аддитивным) действием
специфических комбинаций аллелей
нескольких генов с незначительным
влиянием каждого, либо
один ген является главным, а остальные
будут иметь модифицирующее влияние
61.
Мультифакториальные болезниВысокая распространенность в популяциях (90%)
Могут возникать на разных этапах онтогенеза -
отсроченная наследственная патология
Отсутствие типичного расщепления, генетический
риск определяется по специальным эмпирическим
таблицам с учётом семейного анамнеза
Различие в частоте заболевания у родственников 1-й
и 2-й степени родства. Если болезнь у родственников
1 степени, то вероятность составляет 10% или 40%
Зависимость от возраста, пола, питания и т.д.
Нет четких различий между больными и здоровыми
62.
Мультифакториальные болезниНовые возможности в изучении генетики мультифакториальных
заболеваний появились в результате успешной реализации
программы «Геном человека».
Возможности новых технологий позволяют определять
предрасположенность к заболеваниям на молекулярногенетическом уровне. Это даёт возможность раннего и
рационального лечения заболевания и его профилактики.
Болезни мультифакториальной природы можно рассматривать
как экогенетические болезни, так как для их развития
необходимо взаимодействие генов предрасположенности и
средовых факторов риска.
Экогенетика является научной основой для обеспечения
адаптивной среды для каждого человека: подбор
индивидуального рациона и климата, профессиональный отбор.
63.
Профилактика НБПричиной возникновения наследственной патологии являются
мутации (вновь возникающие или унаследованные от
предыдущих поколений), поэтому одной из важнейших задач
является уменьшение числа мутаций в популяциях
Профилактика может быть:
Первичной - предупреждение зачатия или
рождения больного ребенка
Вторичной - разнообразные способы коррекции
проявления патологического генотипа
Может проводиться на разных этапах онтогенеза
человека: преэмбриональном, эмбриональном,
постэмбриональном
64.
Преэмбриональный период(гаметогенез)
Мероприятия
по улучшению среды обитания
и уменьшению контакта с мутагенами
Применение
антимутагенов
(витамины,
аминокислоты, радиопротекторы и др.)
Химический скрининг с использованием
специальных тест-систем (контроль за содержанием
мутагенов в продуктах питания, лекарствах и др.)
Планирование семьи
Медико-генетическое консультирование
Пропаганда
здорового
образа
жизни
(вред алкоголя, никотина, наркотиков)
65.
Эмбриональный периодПри подозрении на наследственные болезни
и с целью их ранней диагностики:
Определение генотипа и кариотипа плода
на
ранних
сроках
беременности
с
использованием
инвазивных
и
неинвазивных методов
Обследование беременной женщины
(цитогенетический анализ, определения
альфа-фетопротеина
и
хорионического
гонадотропина, как маркеров некоторых
наследственных заболеваний)
66.
Постэмбриональный периодМассовое скринирование новорожденных на
наследственные болезни с использованием скринингтестов.Скрининг-диагностика – бесплатное тотальное
обследование всех новорожденных на фенилкетонурию,
врожденный гипотиреоз, муковисцидоз.
Применяется для диагностики заболеваний, которые
имеют высокую распространенность в популяциях и
приводят к ранней инвалидизации
При раннем выявлении и лечении симптомы заболеваний
не проявляются
67.
Профилактика НБo На уровне популяций:
Уменьшение действия мутагенных факторов, применение
антимутагенов, разработка и использование специальных
просеивающей (скрининг) программ проверки факторов
и веществ на мутагенность.
Диагностика и профилактика болезней обмена,
хромосомных болезней.
o На уровне семей:
Медико – генетическое консультирование
(проспективное и ретроспективное)
68.
МЕДИКО – ГЕНЕТИЧЕСКОЕКОНСУЛЬТИРОВАНИЕ (МГК)
Цель: предупреждение рождения ребенка с тяжелыми
наследственными заболеваниями
МГК может проводиться на разных этапах онтогенеза: плод,
ребенок, взрослый.
Обследование вступающих в брак (проспективное)
Обследование во время беременности - пренатальная
диагностика (проспективное)
Обследование ребенка после рождения (ретроспективное)
МГК состоит из нескольких этапов и имеет определенную
специфику для каждого типа наследственных заболеваний:
моногенных, хромосомных, мультифакториальных
69.
МЕДИКО – ГЕНЕТИЧЕСКОЕКОНСУЛЬТИРОВАНИЕ
Наиболее эффективным является проспективное
консультирование – риск рождения больного
ребенка определяется до наступления беременности
или в ранние ее сроки (показания - кровное родство
родителей,
отягощенная
наследственность,
воздействие вредных факторов на родителей до
наступления беременности)
Ретроспективное консультирование проводится
после рождения больного ребенка относительно
здоровья будущих детей
70.
ПЛАНИРОВАНИЕ СЕМЬИОдним из направлений в работе МГК является
планирование семьи.
Термин «планирование семьи» по определению
обозначает виды деятельности, целью которых является:
ВОЗ
Предотвращение нежелательной беременности
Появление на свет желанных детей
Достаточные интервалы между беременностями
Выбор времени деторождения в зависимости от
возраста родителей и количества детей в семье
71.
ПЛАНИРОВАНИЕ СЕМЬИСледует придерживаться определенных правил:
Оптимальный репродуктивный возраст женщины (до 35 лет) с
целью предупреждения рождения ребенка с хромосомной или
врожденной патологией.
Решение вопроса об искусственном прерывании беременности
при высоком риске рождения ребенка с наследственной
патологией (больше 20%)
Отказ от браков с близкими родственниками (1, 2 степень
родства) и гетерозиготными носителями патологического гена
Соблюдение
здорового образа жизни (отказ будущих
родителей от употребления алкоголя, никотина, наркотиков)
Правильное пользование средствами контрацепции
72.
РАЗВИТИЕ ПОЛАПОЛ – совокупность свойств и признаков
организма, обеспечивающих воспроизведение
потомства и передачу наследственной
информации
ПОЛОВЫЕ ПРИЗНАКИ:
Первичные – признаки организма, которые
обеспечивают образование гамет и их соединение
(половая система)
Вторичные – фенотипические различия между
полами (особенности скелета, мускулатуры,
подкожного жира, развития психики)
73.
Дифференцировка пола в онтогенезеГенетический (хромосомный)
Гонадный
Гормональный
Фенотипический (акушерский)
Гражданский
74.
ХРОМОСОМНЫЙ ПОЛ1 этап
Определение пола происходит в момент
оплодотворения и зависит от сочетания
половых хромосом в зиготе (46,ХХ/46,ХУ)
Он формирует будущую генетическую программу
организма, в частности, дифференцировку половых
желёз - гонадный пол.
75.
Дифференцировка мужского полаДля понимания особенностей развития по мужскому типу важен
“принцип Адама” или дополнительной маскулинной дифференцировки.
Природа в первую очередь заботится о создании самки.
На всех критических стадиях развития, если организм не получает
каких-то дополнительных команд, половая дифференцировка
автоматически идёт по женскому типу.
Чтобы получить самца, нужно обязательно “прибавить”
нечто, способное подавить исходное фемининное начало:
Сначала это H-Y антиген, затем фетальный андроген.
При отсутствии андрогенов в соответствующей стадии развития, у
плода независимо от его генетического пола будут формироваться
женские гениталии, а при частичном недостатке андрогенов мужские
гениталии будут недоразвиты.
76.
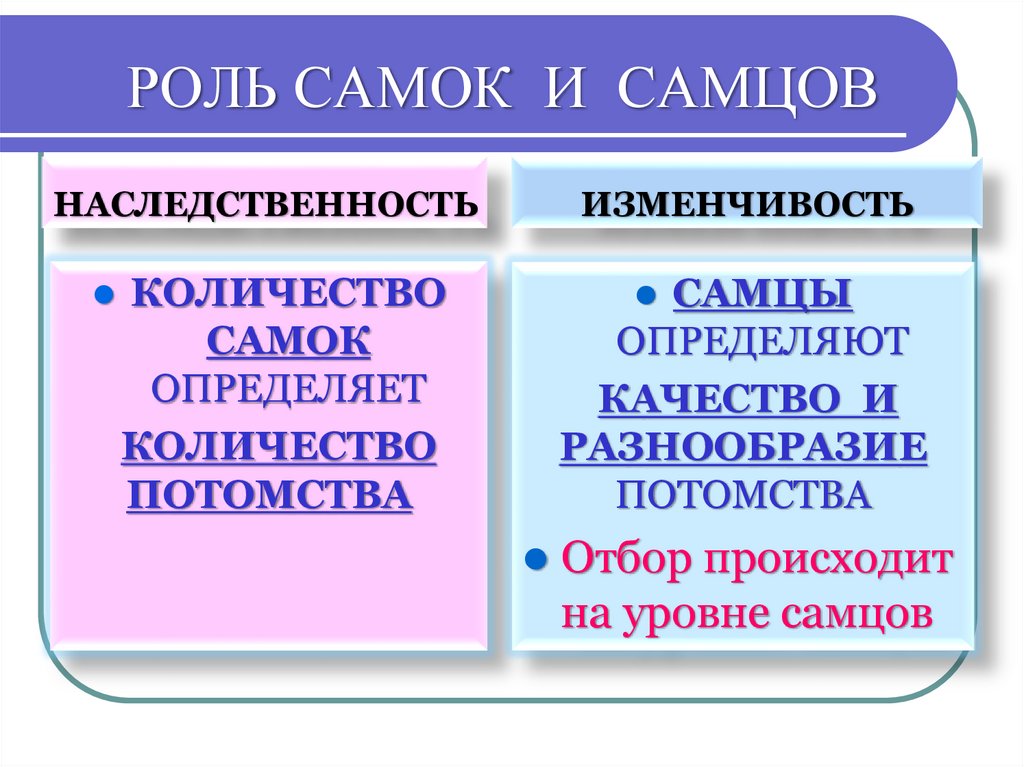
РОЛЬ САМОК И САМЦОВНАСЛЕДСТВЕННОСТЬ
ИЗМЕНЧИВОСТЬ
КОЛИЧЕСТВО
САМЦЫ
САМОК
ОПРЕДЕЛЯЕТ
КОЛИЧЕСТВО
ПОТОМСТВА
ОПРЕДЕЛЯЮТ
КАЧЕСТВО И
РАЗНООБРАЗИЕ
ПОТОМСТВА
Отбор происходит
на уровне самцов
77.
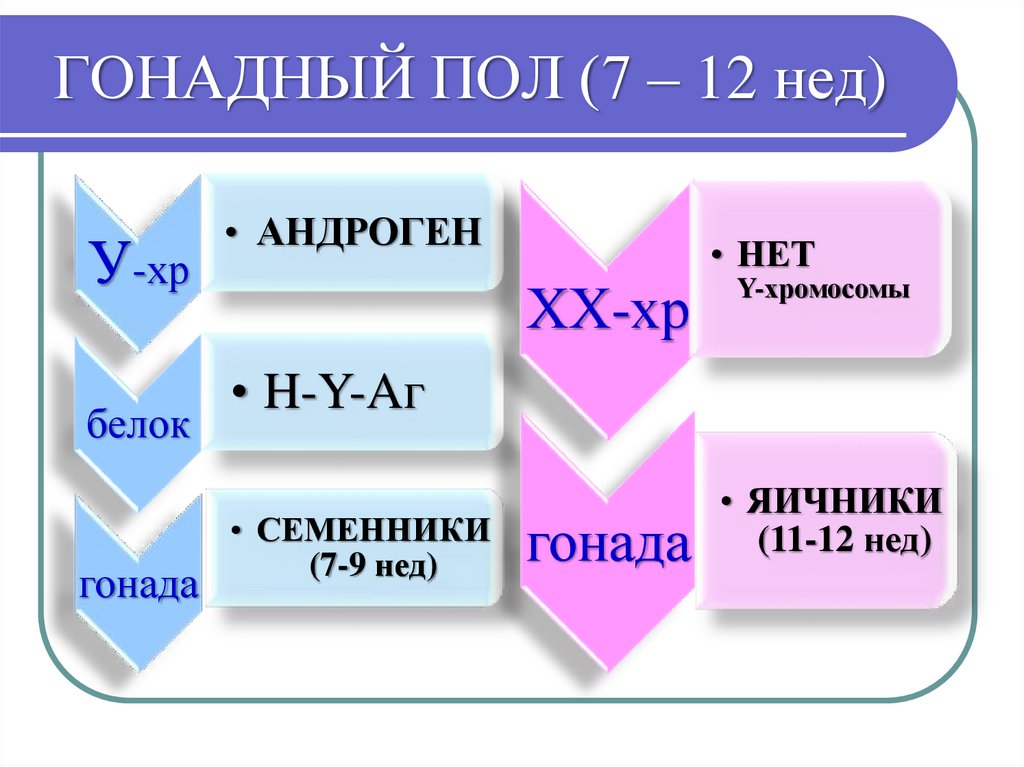
ГОНАДНЫЙ ПОЛПервоначальные зародышевые гонады не
дифференцированы по полу.
В дифференцировке мужского пола участвует андроген
(находится в Y-хромосоме), который детерминирует
образование специфического белка Н-Y антигена , а он,
в свою очередь, определяет превращение зачаточных
гонад в семенники - на 7-й нед эмбриогенеза.
Наряду с андрогеном, в этом участвует ген TFM
(локализован в Х-хромосоме), который определяет
образование в цитоплазме клеток-мишеней белковрецепторов, взаимодействующих с тестостероном.
78.
ГОНАДНЫЙ ПОЛ (7 – 12 нед)У-хр
белок
гонада
• АНДРОГЕН
• НЕТ
ХХ-хр
Y-хромосомы
• H-Y-Aг
• СЕМЕННИКИ
(7-9 нед)
гонада
• ЯИЧНИКИ
(11-12 нед)
79.
ГОРМОНАЛЬНЫЙ ПОЛ (15 нед)Гонады плода вырабатывают гормоны, в клетках-
мишенях происходит взаимодействие молекул гормона с
белками рецепторами - формируется гормональный пол
СЕМЕННИКИ
ЯИЧНИКИ
ТЕСТОСТЕРОН
В Х-хромосоме ген-трансформер (TFM):
формирование рецепторов
к тестостерону
ЭСТРОГЕНЫ
При мутации гена TFM наблюдается синдром тестикулярной
феминизации (синдром Морриса) - женский фенотип при
мужском кариотипе
80.
ГОРМОНАЛЬНЫЙ ПОЛ (15 нед)Половые гормоны определяют:
формирование полового диморфизма:
Особенности половой системы
Дифференцировка нервных путей, определённых
отделов головного мозга, регулирующих половые
различия в поведении
Формируется триада:
ГОРМОНЫ
МОЗГ
ПОВЕДЕНИЕ
Половая дифференцировка мозга связана не только с
гормональными, но и с информационными процессами,
связывающими организм со средой (стресс беременной
женщины), а также с обменов веществ в организме
матери и плода
81.
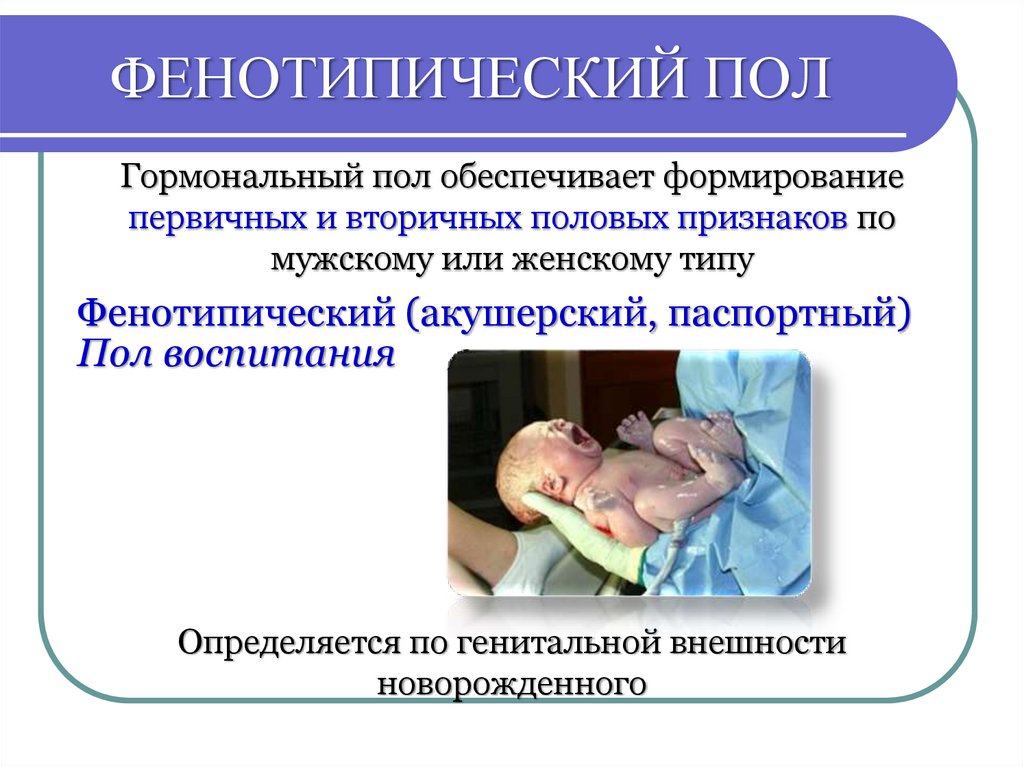
ФЕНОТИПИЧЕСКИЙ ПОЛГормональный пол обеспечивает формирование
первичных и вторичных половых признаков по
мужскому или женскому типу
Фенотипический (акушерский, паспортный)
Пол воспитания
Определяется по генитальной внешности
новорожденного
82.
ГРАЖДАНСКИЙ ПОЛВ постнатальном онтогенезе биологические
факторы половой дифференцировки
дополняются социальными
Большую роль играют условия
индивидуального развития
Формируется в период полового созревания
Определяет поведение человека в обществе
83.
Дифференцировка пола в онтогенезеРазвитие пола - многоступенчатый процесс
Нарушение в любом его звене может иметь
серьёзные, часто необратимы, последствия
При столкновении мужского и женского начал в
дифференцировке пола обычно побеждает первое
Поскольку для создания самца природе требуются
дополнительные усилия, она чаще при этом делает
ошибки:
o повышенная смертность мужчин
o восприимчивость мужчин к ряду болезней
o нарушение половой дифференцировки
84.
ПЕРЕОПРЕДЕЛЕНИЕ ПОЛАГенотип мужской – 46,XY
фенотип женский
1. Мутация андрогена в Y-хромосоме –
не оформляются семенники,
формируются женские гениталии
(но они недоразвиты – т.к. нет второй
Х-хромосомы)
85.
ПЕРЕОПРЕДЕЛЕНИЕ ПОЛАГенотип мужской – 46,XY
фенотип женский
2. Мутация гена TFM в Х-хромосоме,
детерминирующего формирование
рецепторов к тестостерону -
синдром Морриса
(с-м тестикулярной феминизации)
генотип ХУ, семенники, тестостерон
86.
Дифференцировка пола в онтогенезеДля нормального развития пола необходимо
наличие двух половых хромосом (ХХ или ХУ).
При геномных мутациях (хромосомные
болезни) наблюдается их увеличение или
уменьшение, что приводит к недоразвитию
первичных и вторичных половых признаков.
87.
Благодарюза внимание!
Вернитесь в moodle и выполните
«Тест по лекционному
материалу»