




")















")











 medicine
medicine biology
biologySimilar presentations:





")




Проблемы медицинской генетики
1. Проблемы медицинской генетики
2.
• Примерно 5-6 детей из 100рождаются с какими-нибудь
генетически обусловленными
заболеваниями. В
большинстве — это заболевания
с генетическими
предрасположенностями. Это
могут быть пороки развития,
нарушения в интеллектуальном
развитии ребенка. В эти 5-6
процентов входят
наследственные заболевания,
возникшие впервые или
унаследованные от одного из
родителей. Наследственные
болезни многочисленны
(известно свыше 6000) и
разнообразны по проявлениям.
3.
• Насле́дственные заболева́ния — заболевания,возникновение и развитие которых связано с дефектами
в наследственном аппарате клеток, передаваемыми по
наследству через гаметы. Термин употребляется в
отношении полиэтиологических заболеваний, в отличие от
более узкой группы — генные болезни. Наследственные
заболевания обусловлены нарушениями в процессах
хранения, передачи и реализации генетической
информации.
4.
Классификация наследственных заболеваний• В основе наследственных
заболеваний лежат
нарушения (мутации)
наследственной
информации —
хромосомные, генные и
митохондриальные.
Отсюда — классификация
наследственных
заболеваний.
5. Болезни, обусловленные дефектами ядерной ДНК
Генные болезни,связанные с точечными
мутациями ядерной
ДНК:
– Моногенные
заболевания, связанные с
мутациями ядерной ДНК;
Причиной моногенных наследственных заболеваний является
мутация в каком-нибудь одном единственном гене. Те или иные
гены ответственны за продукцию определенного белка.
Следовательно, если есть нарушение в каком-нибудь гене, значит
нарушается структура необходимого белка, либо он не продуцируется
вовсе. Из-за нехватки ключевого белка в организме человека
начинают страдать те физиологические процессы, в которых белок
должен быть вовлечен. Так развивается болезнь. К моногенным
болезням относят серповидноклеточную анемию, а также ряд
ферментопатий (энзимопаталогий) – заболеваний обмена веществ,
возникающих вследствие отсутствия фермента, либо ограничения его
функций.
6.
• Моногенныеболезни наследуются в
соответствии с законами
классической
генетики Менделя.
Соответственно этому, для
них генеалогическое
исследование позволяет
выявить один из трёх
типов наследования:
• аутосомнодоминантный,
• аутосомнорецессивный и
• сцепленное с полом
наследование.
7.
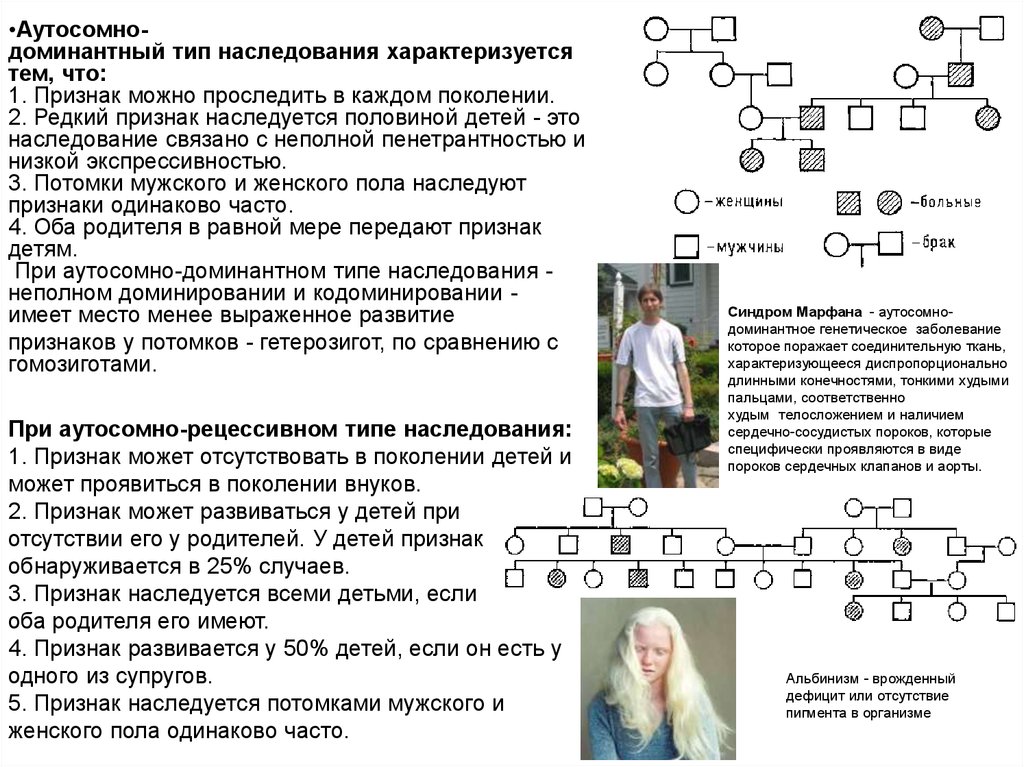
•Аутосомнодоминантный тип наследования характеризуетсятем, что:
1. Признак можно проследить в каждом поколении.
2. Редкий признак наследуется половиной детей - это
наследование связано с неполной пенетрантностью и
низкой экспрессивностью.
3. Потомки мужского и женского пола наследуют
признаки одинаково часто.
4. Оба родителя в равной мере передают признак
детям.
При аутосомно-доминантном типе наследования неполном доминировании и кодоминировании имеет место менее выраженное развитие
признаков у потомков - гетерозигот, по сравнению с
гомозиготами.
При аутосомно-рецессивном типе наследования:
1. Признак может отсутствовать в поколении детей и
может проявиться в поколении внуков.
2. Признак может развиваться у детей при
отсутствии его у родителей. У детей признак
обнаруживается в 25% случаев.
3. Признак наследуется всеми детьми, если
оба родителя его имеют.
4. Признак развивается у 50% детей, если он есть у
одного из супругов.
5. Признак наследуется потомками мужского и
женского пола одинаково часто.
Синдром Марфана - аутосомнодоминантное генетическое заболевание
которое поражает соединительную ткань,
характеризующееся диспропорционально
длинными конечностями, тонкими худыми
пальцами, соответственно
худым телосложением и наличием
сердечно-сосудистых пороков, которые
специфически проявляются в виде
пороков сердечных клапанов и аорты.
Альбинизм - врожденный
дефицит или отсутствие
пигмента в организме
8.
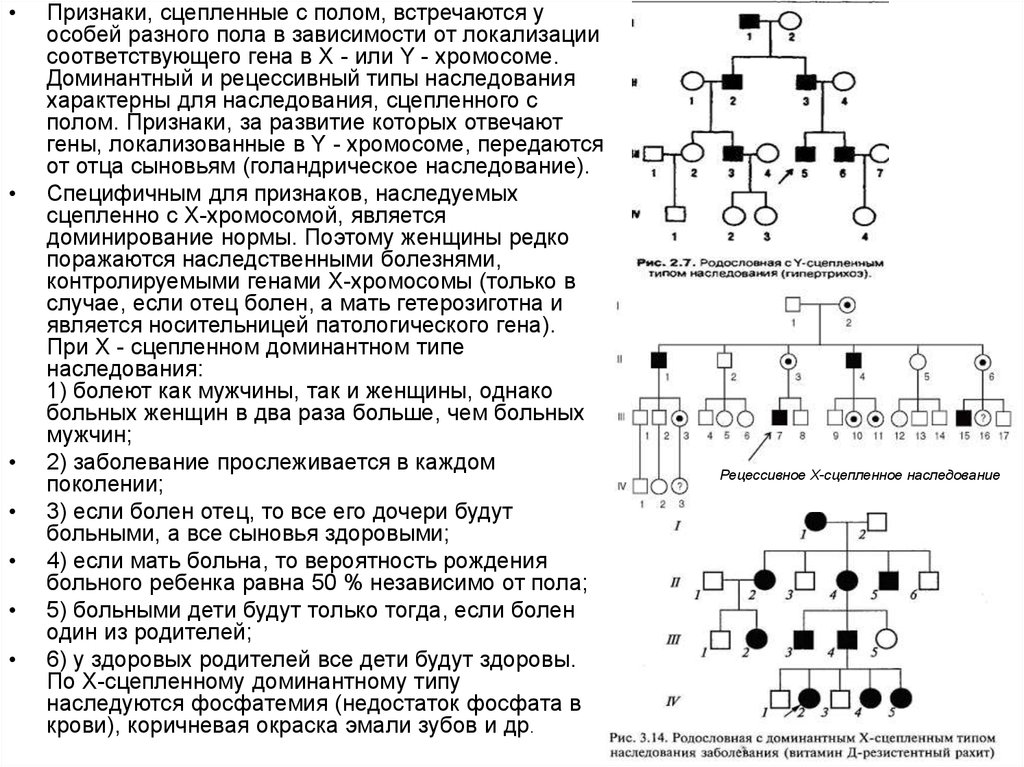
Признаки, сцепленные с полом, встречаются у
особей разного пола в зависимости от локализации
соответствующего гена в X - или Y - хромосоме.
Доминантный и рецессивный типы наследования
характерны для наследования, сцепленного с
полом. Признаки, за развитие которых отвечают
гены, локализованные в Y - хромосоме, передаются
от отца сыновьям (голандрическое наследование).
Специфичным для признаков, наследуемых
сцепленно с X-хромосомой, является
доминирование нормы. Поэтому женщины редко
поражаются наследственными болезнями,
контролируемыми генами X-хромосомы (только в
случае, если отец болен, а мать гетерозиготна и
является носительницей патологического гена).
При Х - сцепленном доминантном типе
наследования:
1) болеют как мужчины, так и женщины, однако
больных женщин в два раза больше, чем больных
мужчин;
2) заболевание прослеживается в каждом
поколении;
3) если болен отец, то все его дочери будут
больными, а все сыновья здоровыми;
4) если мать больна, то вероятность рождения
больного ребенка равна 50 % независимо от пола;
5) больными дети будут только тогда, если болен
один из родителей;
6) у здоровых родителей все дети будут здоровы.
По Х-сцепленному доминантному типу
наследуются фосфатемия (недостаток фосфата в
крови), коричневая окраска эмали зубов и др.
Рецессивное Х-сцепленное наследование
9.
• - Полигенные болезни, связанные с мутациями ядерной ДНК.Эта группа наследственных
заболеваний, которые обусловлены
воздействием многих генов. Часто
полигенные заболевания реализуются
при определенных условиях внешней
среды. К полигенным наследственным
заболеваниям относятся цирроз печени,
ревматоидный артрит, псориаз,
шизофрения, гипертоническая болезнь,
ишемическая болезнь сердца и другие.
Стоит отметить, что полигенные
наследственные заболевания
составляют более 90% всех
наследственных заболеваний.
Полигенные болезни наследуются сложно. Для них вопрос о наследовании не может быть решён на
основании законов Менделя. Ранее такие наследственные заболевания характеризовались как болезни с
наследственной предрасположенностью. Однако сейчас о них идёт речь как о мультифакториальных
заболеваниях с аддитивно-полигенным наследованием с пороговым эффектом.
10. Хромосомные болезни
• Обусловлены грубыми нарушениями наследственного аппарата —изменением числа или структуры хромосом.
К хромосомным заболеваниям
относятся синдром Дауна,
синдромы Клайнфельтера,
Эдвардса, Шерешевского-Тернера и
другие патологии.
11. Triple X syndrome (also known as triplo-X, trisomy X, XXX syndrome, 47,XXX aneuploidy)
Triple X syndrome (also known as triplo-X, trisomy X, XXXsyndrome, 47,XXX aneuploidy)
XXX синдром из-за проблем в М
II стадии овогенеза.
Синие круги мужские клетки.
Синие полоски Y-хромосомы
Розовые круги женские клетки.
Розовые полоски X-хромосомы.
МI соответствует первом этапе
в мейоза
MII соответствует второй стадии
мейоза
Проблема связана с
неправильным расхождением
хромосом в стадии II М, в связи
с чем образуется яйцеклетка с 2
Х-хромосом. Когда эта гамета
сливается с мужской гаметой с
Х-хромосомой, потомство
будет иметь 3 X-хромосомы
(розовая клетка в центре).
12. Болезни, обусловленные дефектами митохондриальной ДНК
Болезни, обусловленныедефектами митохондриальной ДНК
Генные болезни, связанные
с точечными мутациями мтДНК
– это группа моногенных заболеваний,
которая связана с нарушением работы
митохондрий – главных энергетических
«станций» клеток. При мутациях в
некоторых генах нарушается
функционирование митохондрий, что
влечет за собой развитие целого ряда
патологий, в частности нарушений
тканевого дыхания. Примером
митохондриальных заболеваний может
быть митохондриальный сахарный
диабет и рассеянный склероз.
• Болезни, обусловленные грубыми
структурными нарушениями мтДНК
К числу болезней, связанных с грубыми
перестройками мтДНК, относятся:
синдром Кернса—Сэйра, обусловленный
делециями больших участков мтДНК;
синдром Пирсона
13. Наследование митохондриальных болезней
Синдром Вольфа-Паркинсона-УайтаНаиболее частый синдром
преждевременного возбуждения
желудочков (его наблюдают у 0,1 — 0,3 %
населения в общей популяции),
возникающий при наличии
дополнительного пучка Кента.
Большинство людей при этом не имеют
признаков заболевания сердца. У части
больных может не выявляться
клинических проявлений. Основное
проявление синдрома ВольфаПаркинсона-Уайта — аритмии. Более чем
в 50 % случаев возникают
пароксизмальные тахиаритмии:
наджелудочковые реципрокные,
фибрилляция предсердий, трепетание
предсердий. Довольно часто синдром
возникает при заболеваниях сердца —
аномалии Эбштайна, гипертрофической
кардиомиопатии, пролапсе митрального
клапана.
Митохондрии наследуются иначе, чем ядерные гены.
Ядерные гены в каждой соматической клетке обычно
представлены двумя аллелями (за исключением
большинства сцепленных с полом генов у гетерогаметного
пола ). Один аллель унаследован от отца, другой от матери.
Однако митохондрии содержат собственную ДНК, причем в
каждой митохондрии человека обычно содержится от 5 до 10
копий кольцевой молекулы ДНК, и все они наследуются от
матери. Когда митохондрия делится, копии ДНК случайным
образом распределяются между ее потомками, а затем
происходит удвоение ДНК. Если только одна из исходных
молекул ДНК содержит мутацию, в результате случайного
распределения такие мутантные молекулы могут накопиться
в некоторых митохондриях. Митохондриальная болезнь
начинает проявляться в тот момент, когда заметное число
митохондрий во многих клетках данной ткани приобретают
мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным
причинам, намного чаще, чем в ядерной. Это означает, что
митохондриальные болезни достаточно часто проявляются
из-за спонтанных вновь возникающих мутаций. Иногда темп
мутирования увеличивается из-за мутаций в ядерных генах,
кодирующих ферменты, которые контролируют репликацию
ДНК митохондрий. Изначально мутации мтДНК считались
крайне редкими, однако исследование 3000 здоровых
новорожденных на 10 наиболее известных патогенных
мутаций, проведённое в 2008 году, выявило таковые у одного
человека из 200. «Горячей точкой» в мтДНК оказалась
позиция 3243, здесь часто происходит замена A-G,
изменяющая функционирование гена MT-TL1.
14. Методы диагностики наследственных болезней
• ЦитогенетическиеЦитогенетический метод используют для
изучения нормального кариотипа человека, а
также при диагностике
наследственных заболеваний, связанных с
геномными и хромосомными мутациями. Кроме
того, этот метод применяют при исследовании
мутагенного действия различных химических
веществ, пестицидов, инсектицидов,
лекарственных препаратов и др.
В период деления клеток на стадии метафазы
хромосомы имеют более четкую структуру и
доступны для изучения. Диплоидный набор
человека состоит из 46 хромосом: 22 пар
аутосом и одной пары половых хромосом (XX —
у женщин, XY — у мужчин). Обычно исследуют
лейкоциты периферической крови человека,
которые помещают в специальную
питательную среду, где они делятся. Затем
готовят препараты и анализируют число и
строение хромосом. Разработка специальных
методов окраски значительно упростила
распознавание всех хромосом человека, а в
совокупности с генеалогическим методом и
методами клеточной и генной инженерии дала
возможность соотносить гены с конкретными
участками хромосом. Комплексное
применение этих методов лежит в основе
составления карт хромосом человека.
15.
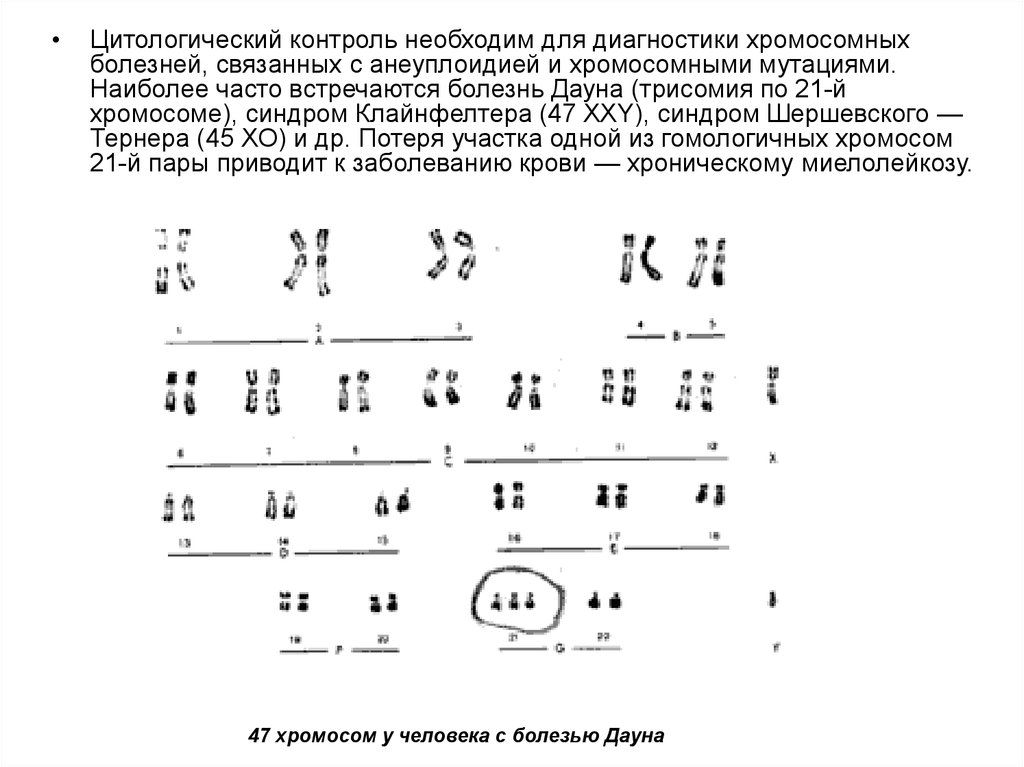
Цитологический контроль необходим для диагностики хромосомных
болезней, связанных с анеуплоидией и хромосомными мутациями.
Наиболее часто встречаются болезнь Дауна (трисомия по 21-й
хромосоме), синдром Клайнфелтера (47 XXY), синдром Шершевского —
Тернера (45 ХО) и др. Потеря участка одной из гомологичных хромосом
21-й пары приводит к заболеванию крови — хроническому миелолейкозу.
47 хромосом у человека с болезью Дауна
16. Возможные наборы половых хромосом при нормальном и аномальном течении мейотического деления гаметогенеза
ОтецМать
У
X
ХУ норма XX норма ХХУ*
Х0**
XX
ХХУ*
XX норма?
У0
леталь
X
XXX
ХУ
ХХХУ*
ХУ
ХО леталь леталь
00 леталь
* — синдром Клайнфельтера
** — синдром Шерешевского—Тернера
•При цитологических исследованиях
интерфазных ядер соматических клеток можно
обнаружить так
называемое тельце Барри, или половой
хроматин. Оказалось, что половой хроматин
в норме есть у женщин и отсутствует
у мужчин. Он представляет собой результат
гетерохроматизации одной из двух Ххромосом у женщин. Зная эту особенность,
можно идентифицировать половую
принадлежность и выявлять аномальное
количество Х-хромосом.
Рис. Х.10. Кариотип больного с синдромом Клайнфельтера (47, XXY)
17.
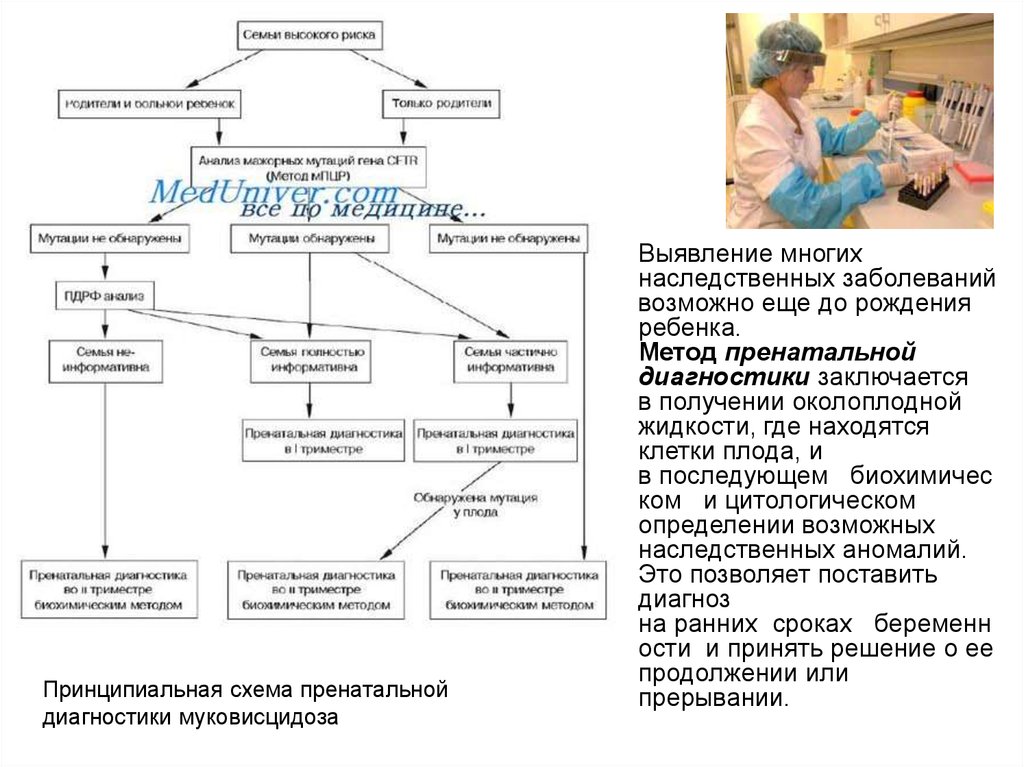
Принципиальная схема пренатальной
диагностики муковисцидоза
Выявление многих
наследственных заболеваний
возможно еще до рождения
ребенка.
Метод пренатальной
диагностики заключается
в получении околоплодной
жидкости, где находятся
клетки плода, и
в последующем биохимичес
ком и цитологическом
определении возможных
наследственных аномалий.
Это позволяет поставить
диагноз
на ранних сроках беременн
ости и принять решение о ее
продолжении или
прерывании.
18. Биохимические
БиохимическиеНекоторые из наследственных болезней
характеризуются выраженными биохимическими
изменениями, которые связаны с нарушениями
определенного метаболического пути. Эта группа
заболеваний объединена в особый класс,
называемый наследственные нарушения обмена
веществ (НБО).
» Энзимопатия (enzymopathia;
энзим + греч. pathos страдание,
болезнь; син. ферментопатия) общее название болезней или
патологических состояний,
развивающихся вследствие
отсутствия или нарушения
активности каких-либо
ферментов.
Диагностика НБО включает качественный и
количественный анализ различных метаболитов в
образцах биологических жидкостей, определение
активности ферментов в культуре клеток или
лейкоцитах периферической крови. Многие из этих
исследований довольно сложные и проводятся с
помощью таких высокотехнологичных методов как
высокоэффективная жидкостная хроматография,
хроматомасс-спектрометрия, тандемная массспектрометрия и т.д.
19.
Биохимические методы в
пренатальной диагностике
Биохимические и некоторые другие методы
исследования в отдельных случаях
оказываются полезными в системе ПД, хотя
чаще всего играют вспомогательную роль.
Нельзя, однако, исключить, что по мере все
более глубокого познания функции генов, в
том числе идентифицированных в процессе
выполнения программы «Геном человека»,
роль биохимических методов в ПД будет
возрастать. Наиболее часто материалом для
биохимических исследований в ПД является
амниотическая жидкость (АЖ), получаемая
путем амниоцентеза. АЖ образуется за
счет секреторной активности клеток амниона
на ранних эмбриональных стадиях развития
и за счет первичной мочи плода — в более
поздние сроки. Количественный и
качественный состав околоплодных вод
регулируется компонентами системы амнион
- мать - плод; нарушение любого из них
приводит к избытку или недостатку вод,
влияет на биохимический и клеточный состав
АЖ.
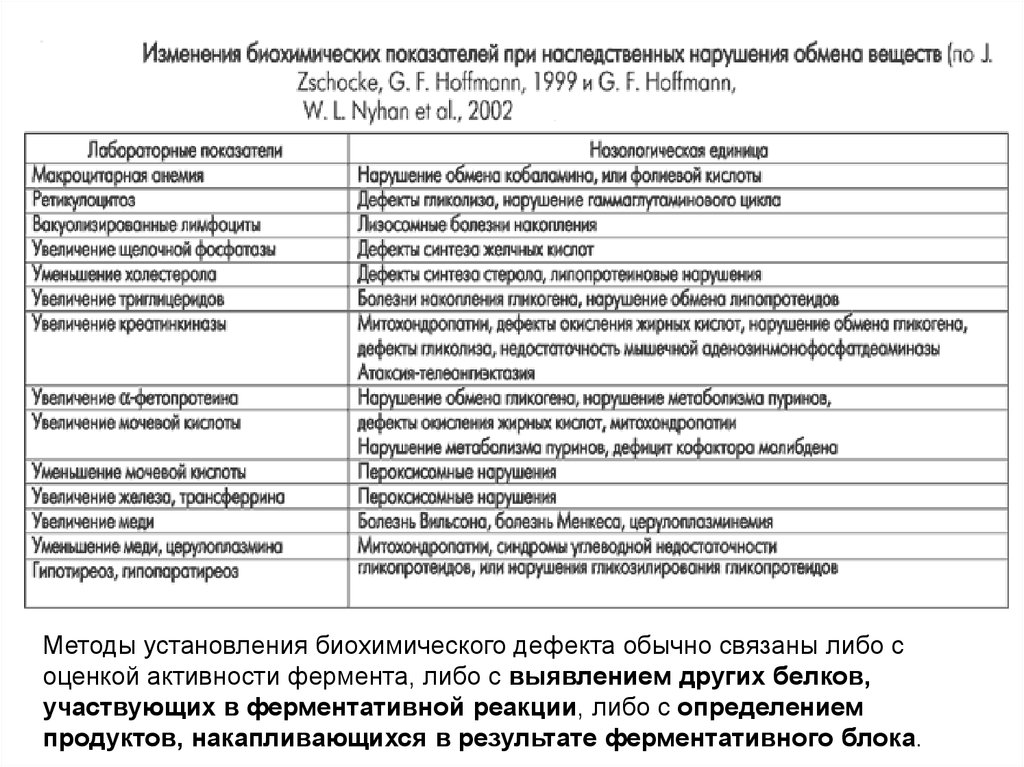
Методы установления биохимического
дефекта обычно связаны либо с оценкой
активности фермента, либо с выявлением
других белков, участвующих в
ферментативной реакции, либо с
определением продуктов, накапливающихся
в результате ферментативного блока.
20. Энзимопатологии.
Синдромологические методыБольшинство наследственных заболеваний характеризуется поражением многих
органов и систем и разнообразной клинической симптоматикой. Кроме того, некоторые
признаки у больных с наследственными заболеваниями не являются собственно
патологическими и, обычно врачи не обращают на них внимание.
Основания для подозрения на наличие энзимопатологии:
наличие кровного родства;
наличие сибсов с необъяснимыми болезнями (энцефалопатия, сепсис, синдром внезапной
смерти);
наследственные заболевания (прогрессирующие неврологические заболевания, фенилкетонурия у
матери, невынашивание и т.д.);
появление симптомов заболевания в связи с изменениями в диете;
остановка или задержка физического развития;
сильное пристрастие или отвращение к некоторым продуктам;
задержка умственного развития;
судороги;
неустановленная причина смерти;
необычный запах тела, выдыхаемого воздуха;
необычный запах и цвет мочи;
микро- или макроцефалия;
нарушения мышечного тонуса;
органомегалия;
грубые черты лица, толстая кожа, ограничения движений в суставах, гирсутизм;
икота и рвота;
опрелости.
21. Классификация энзимопатий по принципу ведущих нарушений обмена веществ
I. ферментопатии обмена аминокислот (алкаптонурия, альбинизм, гипервалинемия,
гистидинемия, гомоцистинурия, гиперлизинемия, лейциноз, тирозиноз, фенилкетонурия,
цистатионинурия, цистиноз);
II. обмена углеводов (галактоземия, гликогенозы, лактат-ацидоз, непереносимость
фруктозы);
III. обмена липидов (липидозы) — плазматические (наследственная гиперлипидемия,
гиперхолестеринемия, недостаточность лецитин-холестеринацилтрансферазы) и
клеточные (ганглиозидозы, муколипидозы, сфингомиелинозы, цереброзидозы);
IV. обмена пуринов и пиримидинов (подагра, синдром Леша—Найхана, оротовая
ацидурия);
V. биосинтеза кортикостероидов (адреногенитальный синдром, гипоальдостеронизм);
VI. порфиринового (порфирии) и билирубинового) обмена (гепатозы);
VII. соединительной ткани (Марфана синдром, Элерса—Данлоса синдром)',
VIII. обмена металлов — гепатоцеребральная дистрофия и болезнь Менкеса (обмен
меди), гемохроматоз (обмен железа), семейный периодический паралич (обмен калия);
IX. ферментопатии эритрона — гемолитические анемии, недостаточность глюкозо-6фосфатдегидрогеназы и глютатионредуктазы в эритроцитах, анемия Фанкони
(недостаточность супероксиддисмутазы);
X. ферментопатии лимфоцитов и лейкоцитов— иммунодефицитные состояния при
недостаточности аденозин-деаминазы, пурин-нуклеотид-фосфорилазы, септический
гранулематоз;
XI. ферментопатии транспортных систем почек (тубулопатии)—почечный канальцевый
ацидоз, болезнь де Тони—Дебре—Фанкони, фосфат-диабет (рахитоподобные болезни),
XII. ферментопатии желудочно-кишечного тракта— мальабсорбции синдром при
недостаточности дисахаридаз, патология кишечного транспорта глюкозы и галактозы,
врожденная хлоридная диарея.
22.
Методы установления биохимического дефекта обычно связаны либо соценкой активности фермента, либо с выявлением других белков,
участвующих в ферментативной реакции, либо с определением
продуктов, накапливающихся в результате ферментативного блока.
23. Некоторые энзимопатии аминокислотного обмена
ЗаболеваниеФермент, активность
которого снижена или
отсутствует
Накапливающийся
метаболит
Фенилкетонурия
Фенилаланин-4гидроксилаза
Фенилаланин
Алкаптонурия
Оксидаза гомогентизиновой Гомогентизиновая
кислоты
кислота
Гистидинемия
Гистидин аммиак лиаза
Гистидин
Гомоцистинурия
Цистатионинсинтетазы
Метионин, гомоцистин
Гипераммониемия I типа
Карбамоилфосфатсинтетаза
Аммиак, глутамин,
оротовая кислота
Гипераммониемия II типа
Орнитинтранскарбамоилаза
Аммиак, глутамин,
оротовая кислота
Методы установления биохимического дефекта обычно связаны либо с
оценкой активности фермента, либо с выявлением других белков,
участвующих в ферментативной реакции, либо с определением
продуктов, накапливающихся в результате ферментативного блока.
24. Нарушения в фенилаланиновом метаболическом пути
БелокФерменты
Фенилпируват
Фенилаланин
Фенлиаланингидроксилаза
Фенилкетонурия
Меланин
Тирозин
Альбинизм
Трансаминаза
Кретинизм
Гидроксифенилпируват
Гидроксифенилпируватоксидаза
Тирозиноз
Тироксин
Гомогентизиновая
кислота
Гомогентизиноксидаза
Алкаптонурия
Малеилацетоуксусная
кислота
СО2 + Н2О
25. Реакция превращения фенилаланина в тирозин
26. Некоторые энзимопатии углеводного обмена
ЗаболеваниеФермент, активность
которого снижена или
отсутствует
Накапливающийся
метаболит
Алактазия, гиполактазия
Лактаза
Лактоза
Галактоземия
Галактозофосфатуридилтрансфераза
Галактоза, галактозо-1фосфат
Непереносимость
фруктозы
Альдолаза,
фруктозобисфосфатаза
Фруктозофосфаты
Непереносимость
сахарозы
Сахараза (инвертаза)
Сахароза
Гликогеноз I типа
(болезнь Гирке)
Глюкозо-6-фосфатаза
Гликоген
Гликогеноз II типа
(болезнь Помпе)
Кислая альфа-1,4глюкозидаза
Гликоген
Гаргоилизм (синдром
Херлера)
Альфа-L-идуронидаза
Мукополисахариды
27. Некоторые энзимопатии липидного обмена
ЗаболеваниеФермент, активность
которого снижена или
отсутствует
Накапливающийся
метаболит
Болезнь Тея-Сакса
N-ацетилгексозаминидаза
Ганглиозид Gm2
Болезнь Нимана-Пика
Сфигномиелиназа
Сфингомиелины
Болезнь Гоше
Бета-глюкоцереброзидаза
Глюкоцереброзиды
Болезнь Тея-Сакса
Основные симптомы болезни: постепенно нарастающее снижение интеллекта до степени
идиотии и падение зрения до полной слепоты (амавроз). Наряду с этим развиваются параличи
конечностей вначале с гипо-, а затем с гипертонией мышц. Вызывается мутацией гена HEXA,
ответственного за синтез фермента гексозоаминидазы A — фермента, принимающего участие в
метаболизме ганглиозидов. В результате, ганглиозиды накапливаются в нервных клетках,
нарушая их работу. Наследуется по аутосомно-рецессивному типу наследования.
это наследственное заболевание, вызванное нарушением липидного метаболизма и
Болезнь Нимана-Пика накоплением липидов в первую очередь в печени, селезёнке, лёгких, костном мозге и головном
мозге. Заболевание относится к лизосомным болезням накопления и характеризуется
аутосомально-рецессивным наследованием. Различают три типа заболевания: типы A, B и C.
Тип А — самый тяжёлый тип, который начинается у грудных детей и характеризуется
увеличением печени и селезёнки (гепатоспленомегалия) и прогрессивным поражением нервной
системы. При этом дети не переживают раннего детского периода.
является самой распространённой из лизосомных болезней накопления. Развивается в результате
Болезнь Гоше недостаточности фермента глюкоцереброзидазы, которая приводит к накоплению глюкоцереброзида во
многих тканях, включая селезёнку, печень, почки, лёгкие, мозг и костный мозг. Болезнь Гоше
подразделяется на три основных типа. Одна из главных причин инвалидизации при 1 и 3 типе болезни
Гоше —- поражение костной ткани. Нарушение нормальных физиологических процессов происходит из-за
накопления липидов в остеокластах и замещении инфильтратами клеток Гоше нормальных элементов
костного мозга. Несмотря на увеличение печени и её дисфункцию, случаи тяжелой печеночной
недостаточности встречаются редко.
28. Классификация лизосомных болезней
• мукополисахаридозы (синдром Гурлера тип I H, синдром Шейе тип I S, синдром Хантера - тип II, синдром Сан-Филиппо - тип IIIA, III B , III C, III D, синдром Моркио - тип IY A, IY B, синдром
Морото -_Лами - тип YI, синдром Слая - тип YII);
• сфинголипидозы (ганглиозидоз GM1, болезнь Гоше,
ганглиозидоз GM2 (Тея-Сакса), метахроматическая
лейкодистрофия, болезнь Крабе, болезнь Фарбера, болезнь
Шиндлера, болезнь Фабри, Болезнь Нимана-Пика А,В, болезнь
Нимана-Пика С);
• муколипидозы, гликопротеинозы и другие ЛБ (болезнь
Вольмана, цероидный липофусциноз, муколипидоз І типа
(сиалидоз), муколипидоз ІІ типа, муколипидоз ІІІ типа (псевдо
Гурлер), маннозидоз).
29. Молекулярно-генетические методы
Молекулярно-генетические методыМолекулярно-генетические методы это самые современные методы
исследования генетического материала клеток человека, который
представлен дезоксирибунуклеиновой (ДНК) и рибунонуклеиновой (РНК)
кислотами.
30. ДНК-фингерпринтинг
ДНК-фингерпринтирование(DNA fingerprinting, DNA fingerprint
technique) [англ. finger — палец и print —
печать, оттиск, отпечаток] — метод создания
генетических «отпечатков пальцев» (см. ДНКфингерпринт), основанный на анализе
полиморфизма ДНК (см. ДНК полиморфизм).
Первоначально геномная ДНК рестрицируется
эндонуклеазами, затем образующиеся
фрагменты разделяются при помощи
электрофореза в геле и переносятся на
фильтры (напр., нитроцеллюлозные фильтры).
После этого фильтры гибридизуют
(см. Саузерн-блоттин, Саузерн-блотгибридизаци, Саузерн-блот-анализ) со
специфическими мечеными зондами.
Фрагменты ДНК, гомологичные зондам,
образуют полиморфные полосы гибридизации,
отдельные из которых специфичны для
каждого индивидуума (см. ДНК-фингерпринт),
поэтому метод может быть использован для
генетической идентификации разных
индивидуумов одного вида. ДНК-ф.
применяется при картировании генов,
определении отцовства и материнства, в
криминалистике. Метод ДНК-ф. предложен А.
Рестриктазные полосы - паттерны
Джеффрисом в 1985 г.
ДНК-фингерпринт (DNA fingerprint) — генетический «отпечаток пальцев», представляющий собой
высокоспецифичные гибридизационные полосы на электрофореграммах, полученный с помощью ДНКфингерпринтирования; отражает индивидуальный полиморфизм длин рестрикционных фрагментов геномной ДНК,
причиной которого могут быть мутации в пределах сайта рестрикции, изменение числа повторов в ДНК и др.
31. Метод "разветвленной" ДНК (bDNA)
Метод "разветвленной" ДНК (bDNA)•Метод разветвленной ДНК-гибридизации (bDNA assay — branched DNA assay), в
отличие от других методов, основан не на амплификации ДНК, а на амплификации
сигнала гибридизации исследуемой нуклеиновой кислоты с синтетической
разветвленной детектирующей молекулой ДНК.
•амплификация (англ. amplification) — (от лат. amplificatio – усиление,
увеличение) — в молекулярной биологии — увеличение числа копий ДНК в
результате ее репликации.
1.
2.
3.
4.
5.
разрушение организма и денатурация нуклеиновой кислоты
гибридизация зонда с мишенью
гибридизация bДНК (амплификационный мультимер)
гибридизация меченных ферментом зондов
детекция: добавление субстрата и измерение хемилюминесценции
32. Использование принципа иммунологического захвата гибрида ДНК/РНК с последующей иммунохимической детекцией
В нанобиотехнологии и генной инженерии амплификацияшироко используется для получения больших количеств
специфических последовательностей ДНК для различного
применения (создание зондов, векторов, чипов, молекул
ДНК заданной геометрической формы и др.).
33.
Определение нуклеотидной последовательности(секвенирование) ДНК
Секвенирование позволяет довольно быстро определить полную нуклеотидную
последовательность сегмента длиной 100 - 500 нуклеотидных пар, образующегося при
расщеплении ДНК рестрикционными эндонуклеазами.
Полимера́зная цепна́я реа́кция (ПЦР) —
экспериментальный метод молекулярной
биологии,
позволяющий
добиться
значительного
увеличения
малых
концентраций определённых фрагментов
ДНК в биологическом материале (пробе).
В основе метода ПЦР лежит многократное
удвоение определенного участка ДНК амплификация.
В
результате
нарабатываются
количества
ДНК,
достаточные для визуальной детекции.
34. Полимеразная цепная реакция
35.
Основные термины и понятия:1. Праймер
2. Отжиг праймера
3. Термостабильная
ДНК-полимераза
4. Амплификация ДНК
5. Ампликон
36.
Критерии подбора праймеров для ПЦР1. Длина праймера должна быть 15 - 30 нуклеотидов. Уменьшение длины
может привести к снижению специфичности связывания праймера и матрицы. Длинные
праймеры увеличивают специфичность системы, но в них сильно возрастает
вероятность образования вторичных структур.
2. Праймеры должны быть специфичны. Особое внимание уделяют 3'-концам
праймеров, т.к. именно с них Taq-полимераза начинает достраивать комплементарную
цепь ДНК. Если их специфичность недостаточна, то, вероятно, что в пробирке с
реакционной смесью будут происходить синтез неспецифической ДНК (коротких или
длинных фрагментов). Это значительно усложняет оценку результатов реакции, т.к.
легко перепутать специфический продукт амплификации с синтезированной
посторонней ДНК. Часть праймеров и дНТФ расходуется на синтез неспецифической
ДНК, что приводит к значительной потере чувствительности;
3. Праймеры не должны образовывать димеры и петли, т.е. не должно
образовываться устойчивых двойных цепей в результате отжига праймеров самих на
себя или друг с другом;
4. Область отжига праймеров должна находиться вне зон мутаций, делеций
или инсерций в пределах видовой или иной, взятой в качестве критерия при выборе
праймеров, специфичности. При попадании на такую зону, отжига праймеров
происходить не будет, и как следствие - ложноотрицательный результат.
5. Содержании ГЦ-пар в праймере должно быть около 50 %. Чем ниже
содержание ГЦ-пар в праймере, тем длиннее он должен быть.
37.
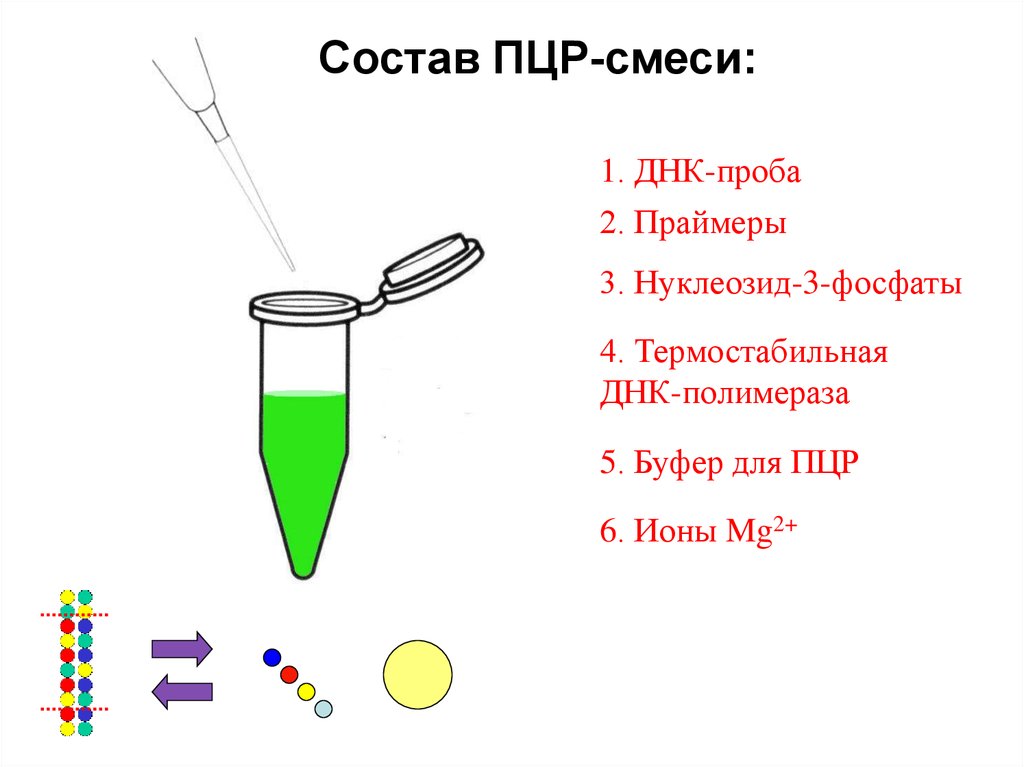
Основные компоненты, влияющие на ПЦР1. Концентрация Магния (для работы Taq ДНК-полимераза оптимальной
является концентрация 1 - 3 мМ).
2. Концентрация фермента (Рекомендуется использование 1 - 1.25 единицы Taq
ДНК-полимеразы в 50 мл реакционной смеси).
3. Качество и количество матрицы (Зависит от качества состава матрицы).
4. Усиливающие агенты ПЦР (бетаина, диметилсульфоксида и формамида,
сывороточный альбумин, желатин и неионогенные детергенты).
5. Концентрация праймеров (избыток праймеров может приводить к появлению
неспецифических продуктов реакции, оптимальным является концентрация 10
пМ/мкл)
38.
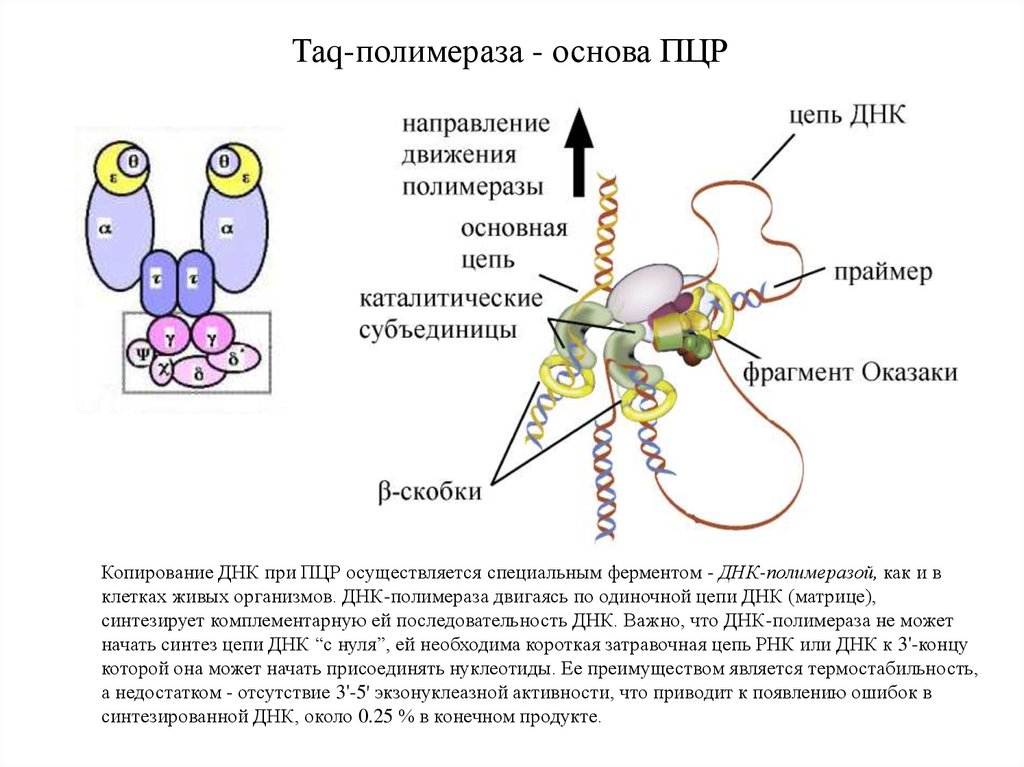
Таq-полимераза - основа ПЦРКопирование ДНК при ПЦР осуществляется специальным ферментом - ДНК-полимеразой, как и в
клетках живых организмов. ДНК-полимераза двигаясь по одиночной цепи ДНК (матрице),
синтезирует комплементарную ей последовательность ДНК. Важно, что ДНК-полимераза не может
начать синтез цепи ДНК “с нуля”, ей необходима короткая затравочная цепь РНК или ДНК к 3'-концу
которой она может начать присоединять нуклеотиды. Ее преимуществом является термостабильность,
а недостатком - отсутствие 3'-5' экзонуклеазной активности, что приводит к появлению ошибок в
синтезированной ДНК, около 0.25 % в конечном продукте.
39. Типы Taq-полимераз
С 3‘-5‘- эндонуклеазной активностьюБез этой функции
Pfu-полимераза
Tht-полимераз
Taq-полимераза
Tfl-полимераза
Tli-полимераз
Klenov fragment
40.
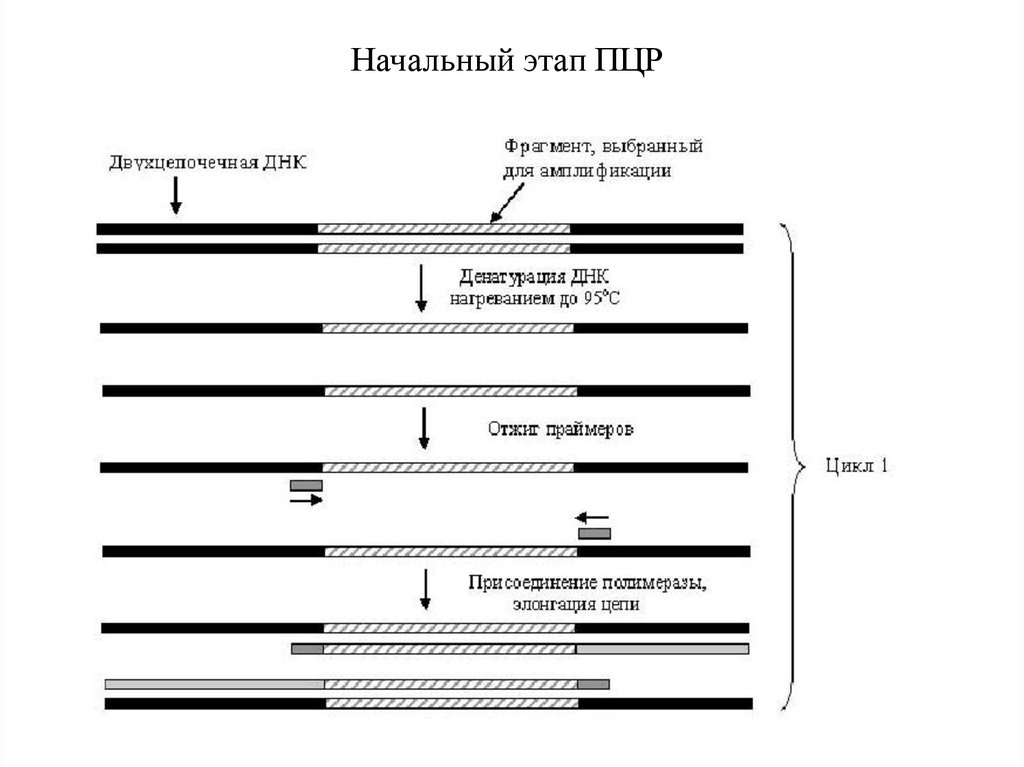
Стадии полимеразной цепной реакции1) денатурации, или "плавления" ДНК, когда двух цеп
очечная ДНК под действием высокой температуры переходит в
одноцепочечное состояние;
2) связывания (отжига) праймеров с матричной ДНК;
3) элонгации, или удлинения цепи.
41.
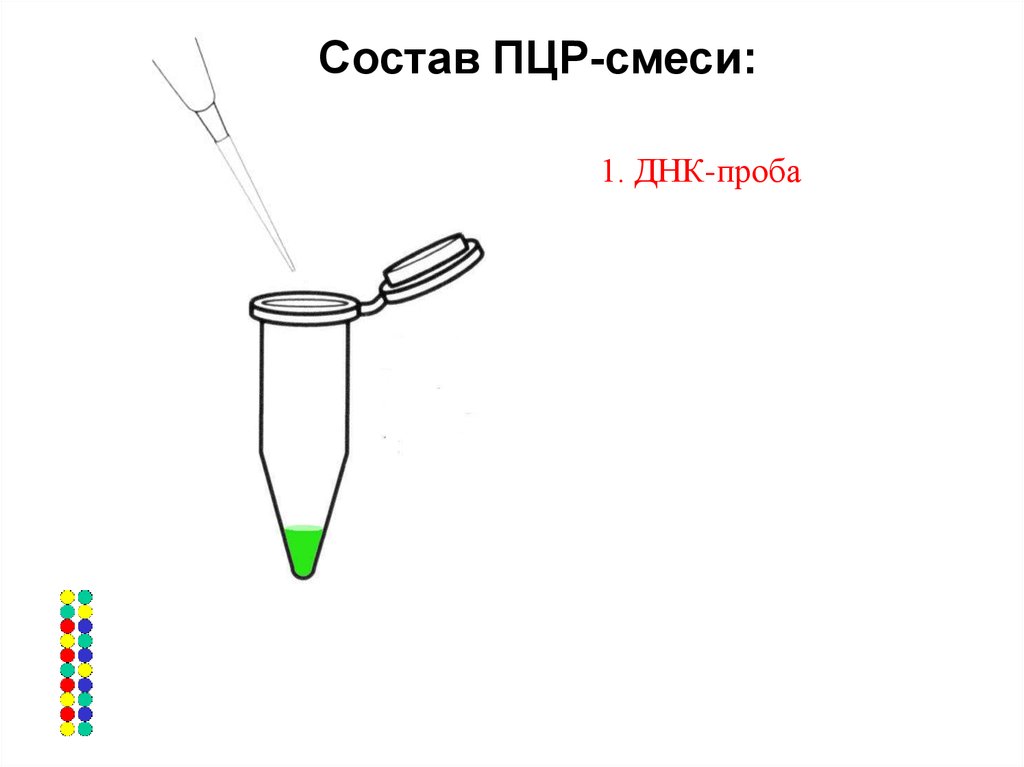
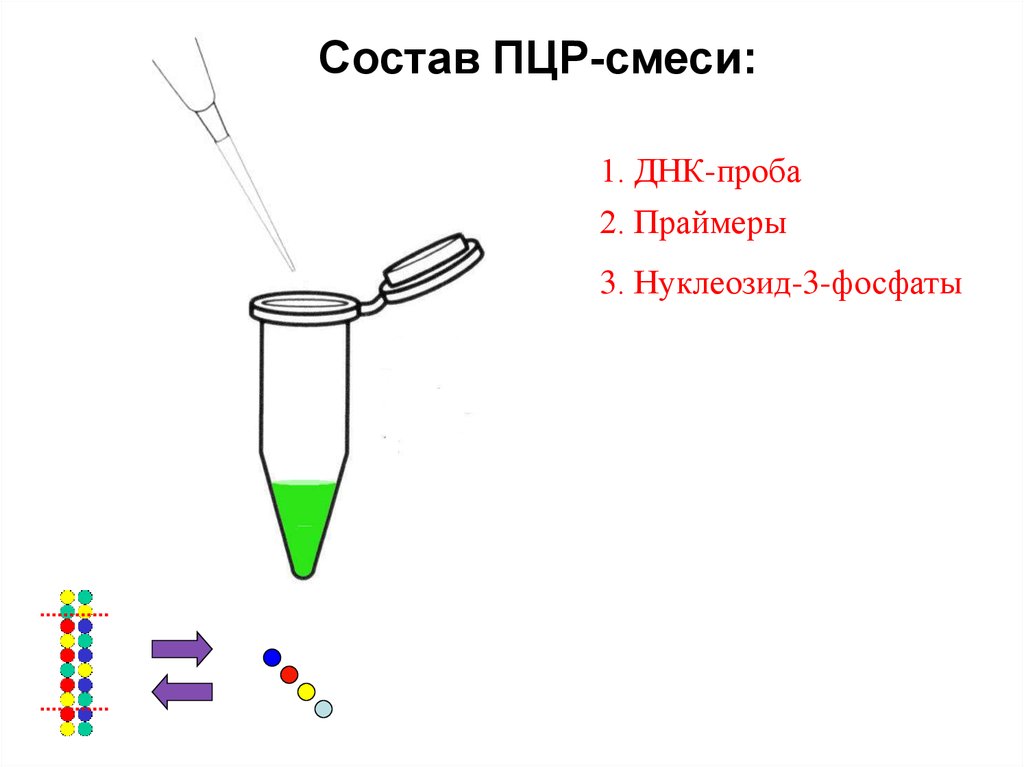
Состав ПЦР-смеси:1. ДНК-проба
42.
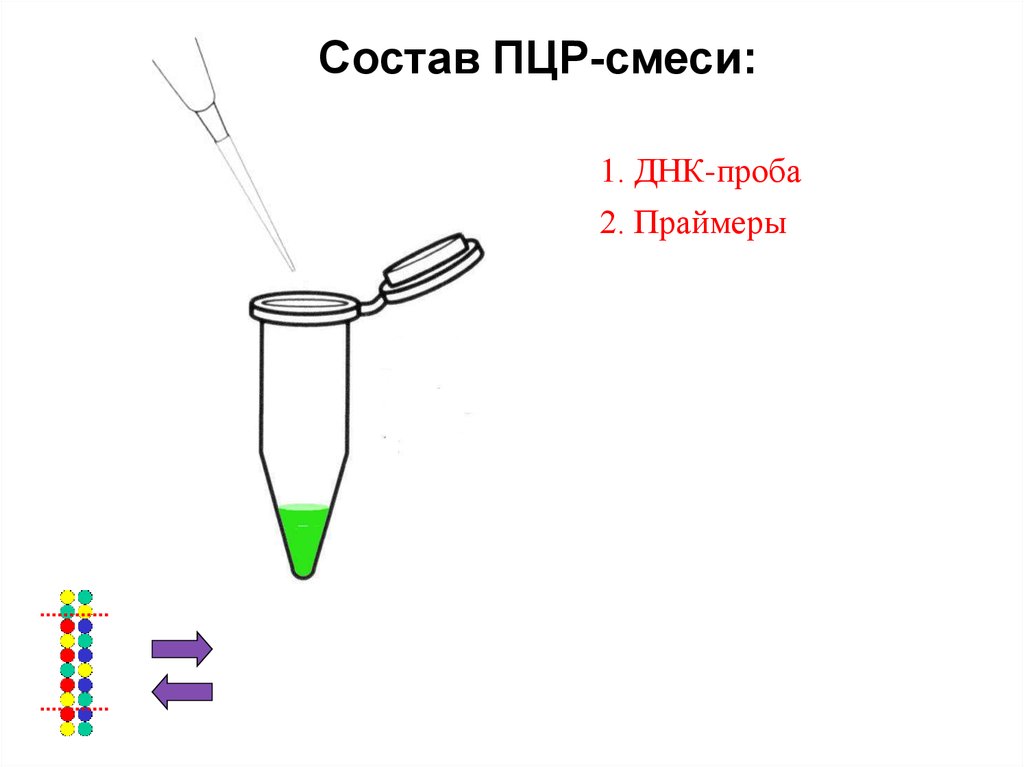
Состав ПЦР-смеси:1. ДНК-проба
2. Праймеры
43.
Состав ПЦР-смеси:1. ДНК-проба
2. Праймеры
3. Нуклеозид-3-фосфаты
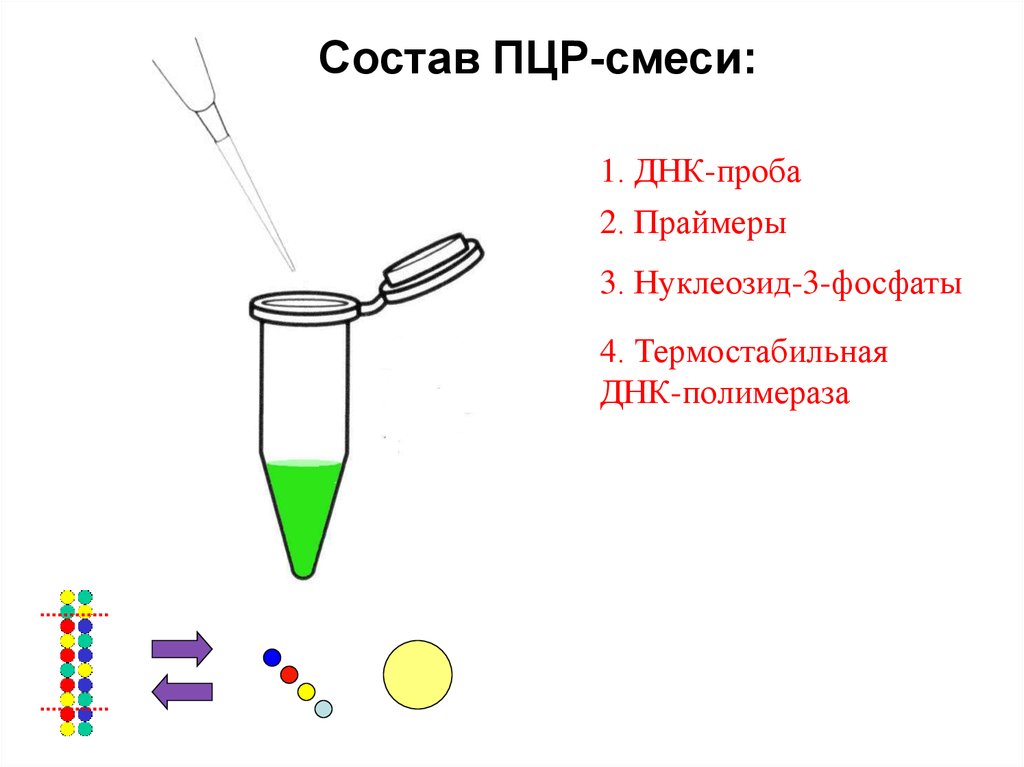
44.
Состав ПЦР-смеси:1. ДНК-проба
2. Праймеры
3. Нуклеозид-3-фосфаты
4. Термостабильная
ДНК-полимераза
45.
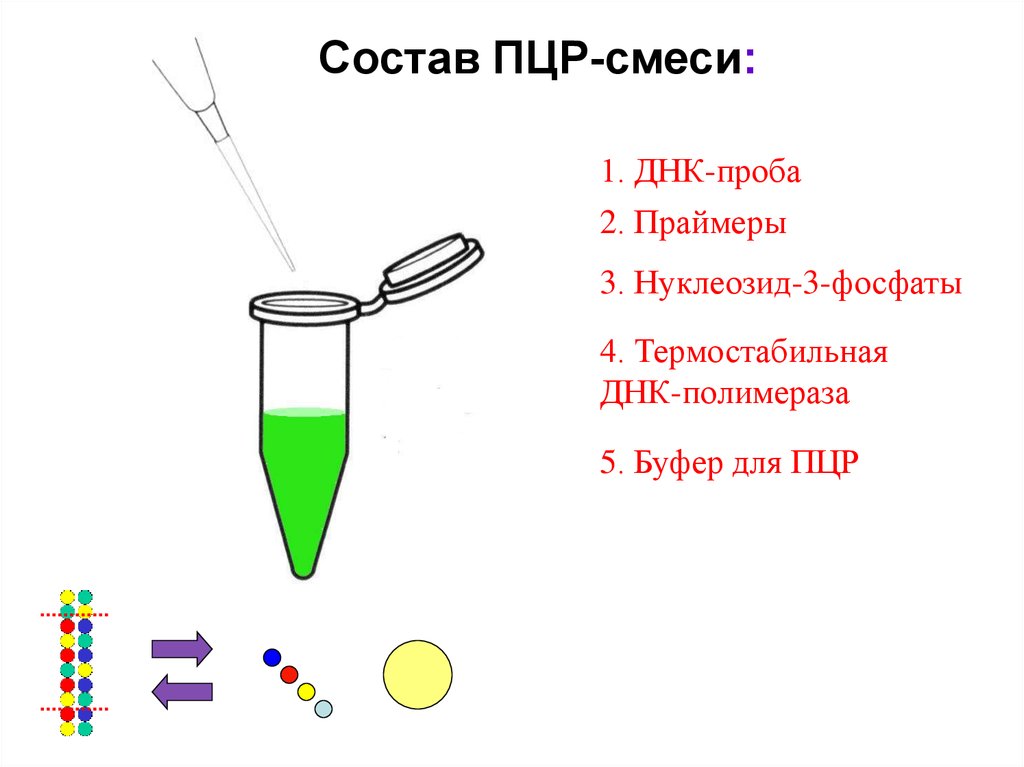
Состав ПЦР-смеси:1. ДНК-проба
2. Праймеры
3. Нуклеозид-3-фосфаты
4. Термостабильная
ДНК-полимераза
5. Буфер для ПЦР
46.
Состав ПЦР-смеси:1. ДНК-проба
2. Праймеры
3. Нуклеозид-3-фосфаты
4. Термостабильная
ДНК-полимераза
5. Буфер для ПЦР
6. Ионы Mg2+
47.
Начальный этап ПЦР48.
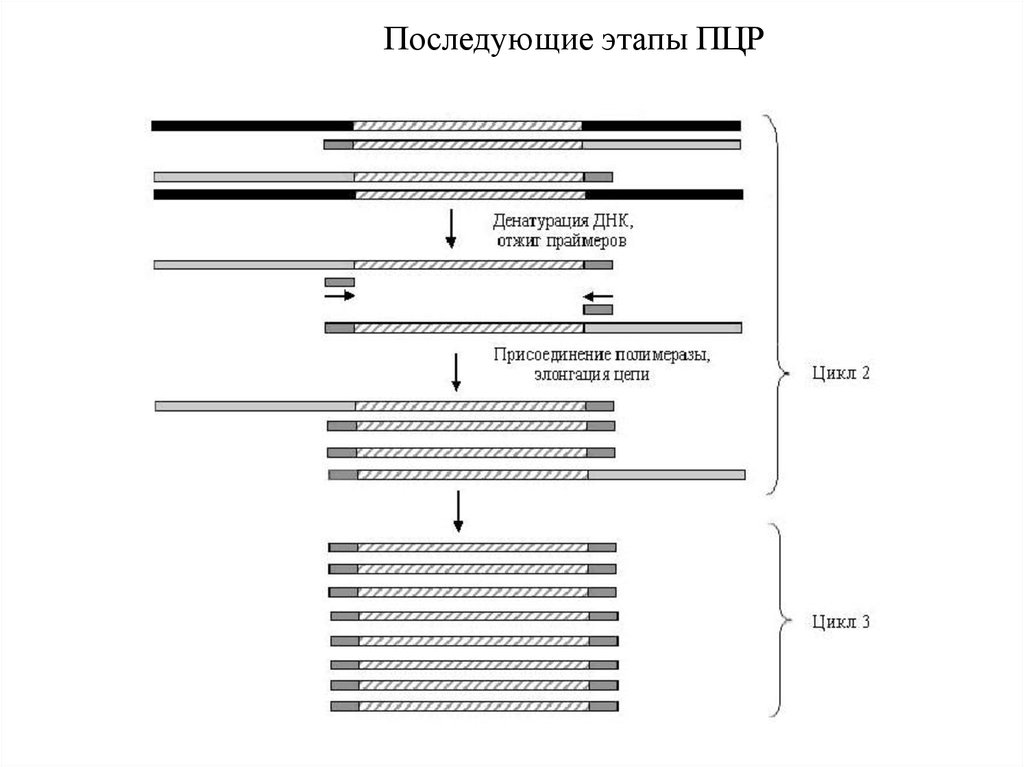
Последующие этапы ПЦР49.
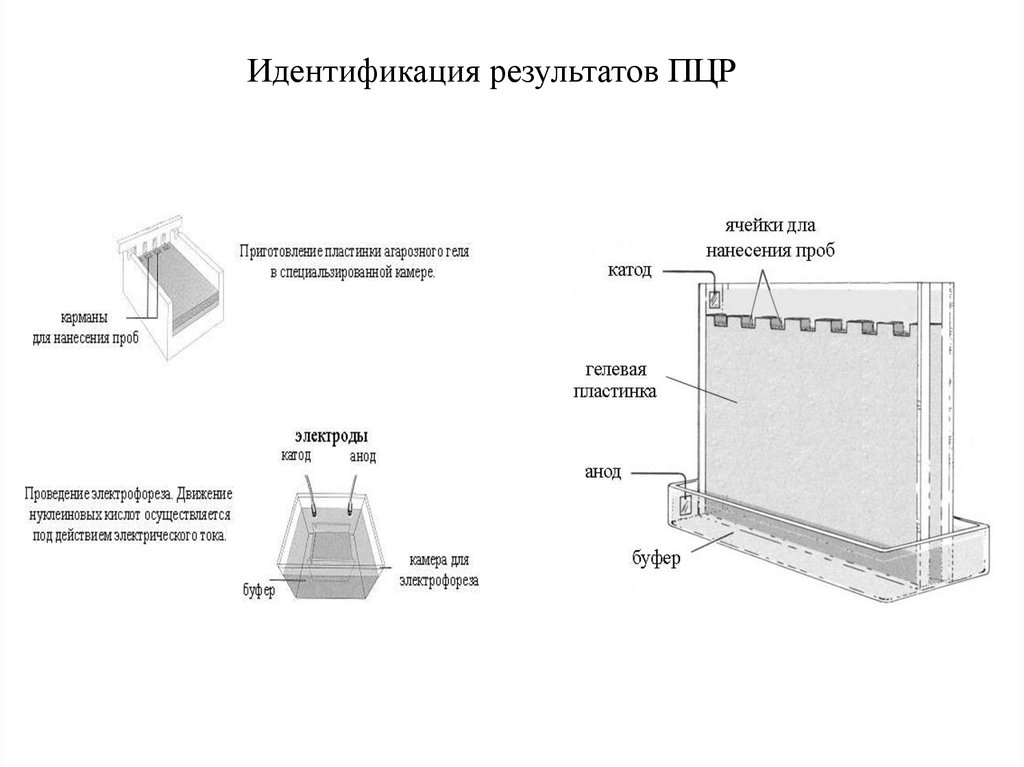
Идентификация результатов ПЦР50.
—Пластинку геля помещают в
буфер и подают напряжение
от 60 до 130 В.
+
51.
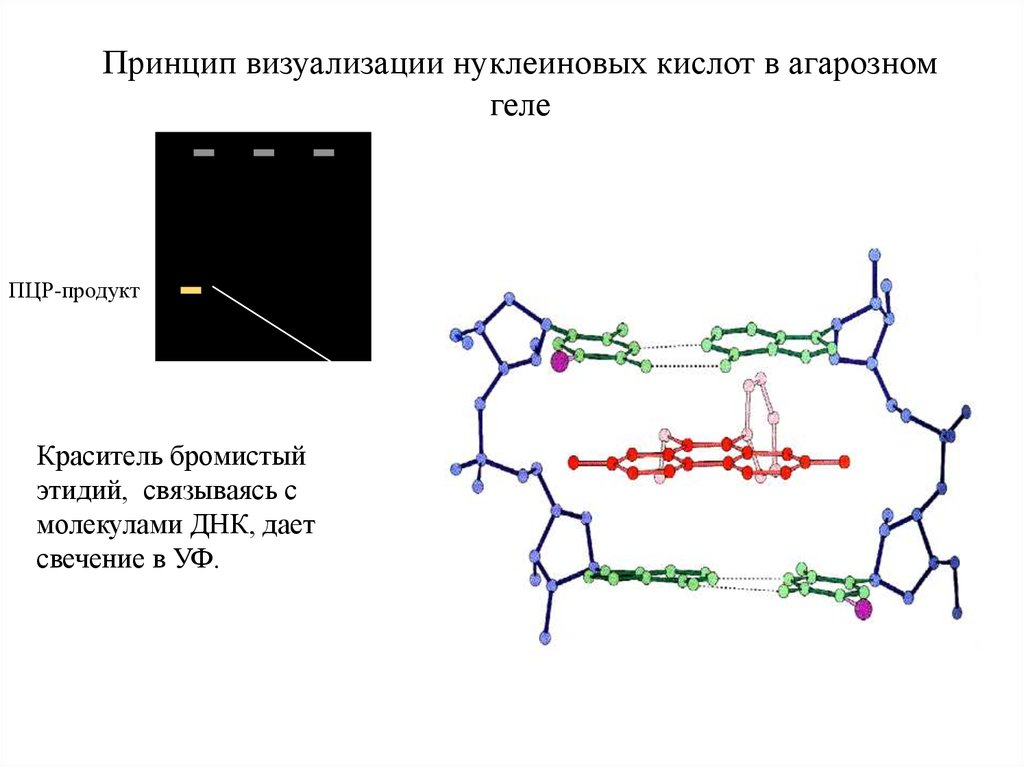
Принцип визуализации нуклеиновых кислот в агарозномгеле
ПЦР-продукт
Краситель бромистый
этидий, связываясь с
молекулами ДНК, дает
свечение в УФ.
52.
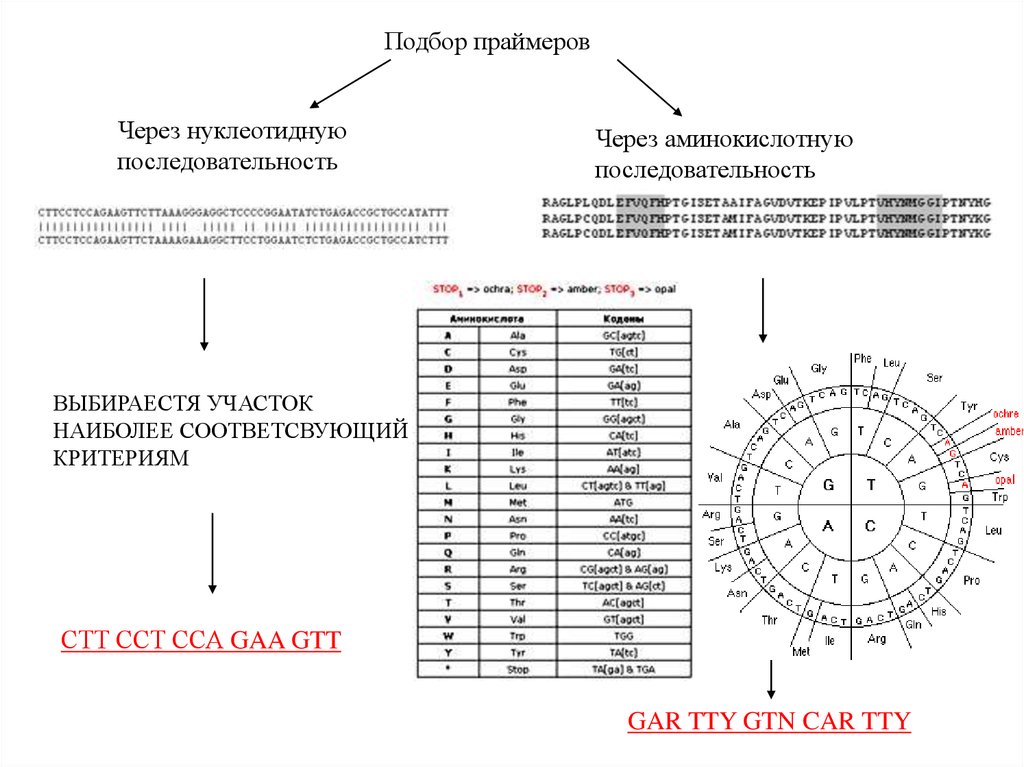
Подбор праймеровЧерез нуклеотидную
последовательность
Через аминокислотную
последовательность
ВЫБИРАЕСТЯ УЧАСТОК
НАИБОЛЕЕ СООТВЕТСВУЮЩИЙ
КРИТЕРИЯМ
СТТ ССТ ССА GAA GTT
GAR TTY GTN CAR TTY
53.
Применение ПЦРДиагностический метод молекулярной биологии,
молекулярной генетики (поиск и идентификация генов и
нуклеотидных
последовательностей);
клинической
лабораторной диагностики, позволяющим выявлять в тканях и
биологических жидкостях организма единичные клетки
возбудителей многих инфекционных заболеваний (тестсистемы позволяют за короткий промежуток времени
произвести обнаружение и идентификацию возбудителей
заболеваний);
выявлять
и
идентифицировать
даже
однонуклеотидные изменения в структуре генов (анализ и
детекция различных мутационных изменений в структуре
генома); клинической диагностике и судебно-медицинской
практике (идентификация личности человека по останкам,
образцам ткани и др., установление отцовства).
54.
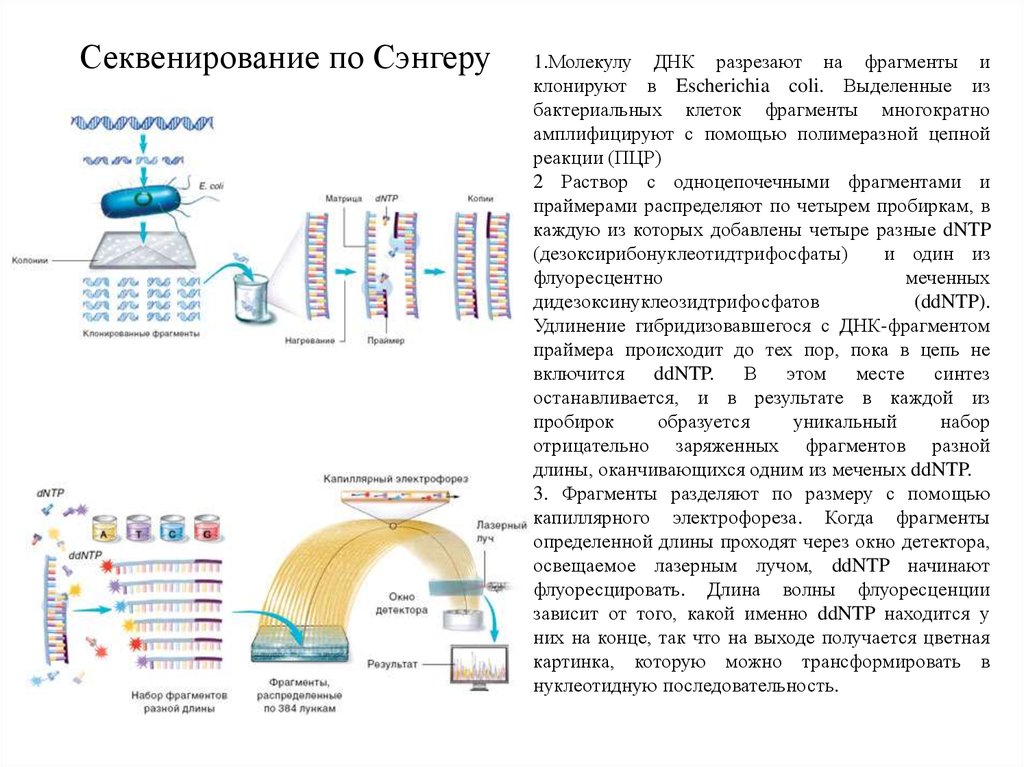
Секвенирование по Сэнгеру1.Молекулу ДНК разрезают на фрагменты и
клонируют в Escherichia coli. Выделенные из
бактериальных клеток фрагменты многократно
амплифицируют с помощью полимеразной цепной
реакции (ПЦР)
2 Раствор с одноцепочечными фрагментами и
праймерами распределяют по четырем пробиркам, в
каждую из которых добавлены четыре разные dNTP
(дезоксирибонуклеотидтрифосфаты)
и один из
флуоресцентно
меченных
дидезоксинуклеозидтрифосфатов
(ddNTP).
Удлинение гибридизовавшегося с ДНК-фрагментом
праймера происходит до тех пор, пока в цепь не
включится ddNTP. В этом месте синтез
останавливается, и в результате в каждой из
пробирок
образуется
уникальный
набор
отрицательно заряженных фрагментов разной
длины, оканчивающихся одним из меченых ddNTP.
3. Фрагменты разделяют по размеру с помощью
капиллярного электрофореза. Когда фрагменты
определенной длины проходят через окно детектора,
освещаемое лазерным лучом, ddNTP начинают
флуоресцировать. Длина волны флуоресценции
зависит от того, какой именно ddNTP находится у
них на конце, так что на выходе получается цветная
картинка, которую можно трансформировать в
нуклеотидную последовательность.
55. Медико-генетическое консультирование — вид медицинской помощи, направленной на профилактику наследственных болезней
• Основные задачи:• определение прогноза в отношении будущего потомства в
семьях, где имеется больной с наследственной патологией или
предполагается рождение ребенка с такой патологией;
• уточнение диагноза наследственного заболевания с помощью
специальных методов исследования;
• объяснение обратившимся за консультацией в доступной
форме смысла медико-генетического заключения и помощь в
принятии правильного решения относительно дальнейшего
планирования семьи;
• пропаганда медико-генетических знаний.