






")








")

")






")
")


 в прион РгРSc (б)")









 medicine
medicineSimilar presentations:










Дифференциальная диагностика прионных болезней
1. Дифференциальная диагностика прионных болезней
Выполнила студентка 7 курса лечебного факультета МГМСУим.А.И. Евдокимова Калинина Ольга Ивановна
2. Типы прионных болезней с учётом причины их возникновения
ТипыКлинический синдром
Причина заболевания
Наследственные
(семейные)
Наследственная БКЯ
СГШШ
ФСИ
Различные атипичные
деменции
Мутации PRNP
Приобретенные
(инфекционные)
Куру
Ятрогенная БКЯ
Новый вариант БКЯ
Каннибализм
Инокуляция
Алиментарный путь
Спорадические
(случайные)
БКЯ
Атипичные варианты БКЯ
Спородическая фатальная
инсомния
Возможно, соматическая
мутация PRNP или
спонтанная конверсия
PrPc в PrPsc
3. ВОЗ была предложена классификация, включающая 3 основных типа развития БКЯ:
I типНарушения зрения, прогрессирующая
деменция, афазия, миоклонические судороги,
приступами подавленности. Заболевание имеет
наименьшую длительность, равную 1 или
нескольким месяцам.
II тип Ригидность мышц, прогрессирующая
деменция, тремор, развивающаяся атаксия.
Заболевание продолжительнее (до 9 месяцев)
III тип Признаки Паркинсонизма и (или)
амиотрофического бокового склероза (БАС) +
признаки I и II типа. Заболевание затяжное
(до 1-2 лет)
4. Эпидемиологические особенности
Убиквитарный (повсеместно) характер распространениязаболеваний (некоторые европейские страны, Австралия, страны
Северной и Южной Америки)
Примерно равномерная пораженность лиц обоего пола, хотя
мужчины чаще, чем женщины болеют (1,5 : 1,0)
Преимущественная заболеваемость взрослых, главным образом лиц
пожилого возраста
(30-70 лет – 90%)
Покровский Валентин Иванович
академик РАМН,
Президент Российской академии медицинских наук,
директор ЦНИИ эпидемиологии МЗ РФ
5.
В эпидемиологическом отношении БКЯможет проявляться в спорадической,
семейной (наследственной) и ятрогенной
формах
Ганс Герхард
Крейтцфельдт
(1885—1964)
6. Куру является эндемической медленной инфекцией, встречающейся в восточной части о. Новая Гвинея и выявлена у племен народности
7. 1. Неврологические осложнения при системных васкулитах
Васкулит – это аутоиммунное воспаление стенки сосуда (артерии,вены, артериолы, венулы, капилляры, аорта). Проявляется в виде
кровотечений на коже, затем в суставы, мышцы и нервные
окончания)
Ступенеобразное нарастание клинических
симптомов
Изменение цереброспинальной жидкости
Ишемические очаги, обнаруженные
при КТ- или МРТ-исследовании
Выраженная головная боль
Нередко генерализованные
судорожные припадки
Резкое наличие миоклоний
Присоединение системных проявлений
8. 2.Антифосфолипидный синдром (АФС)
это аутоиммунное заболевание, преимущественноженщин молодого возраста, характеризующееся
клинико-лабораторным симптомокомплексом,
включающим рецидивирующие тромбозы, различные
формы акушерской патологии (в первую очередь
привычное невынашивание беременности) и
тромбоцитопению при наличии в крови
антифосфолипидных антител
волчаночный антикоагулянт (ВАК)
антитела к кардиолипину (АКЛ)
бета 2-гликопротеин-1 (АБ2ГП)
9.
Антифосфолипидный синдром (АФС) –аутоиммунная, невоспалительная тромботическая
васкулопатия.
Впервые был описан в 1986 г. английским
ревматологом G. Hughes при СКВ.
В 1994 г. на международном
симпозиуме по АФС было
предложено также использовать
термин «синдром Хьюза»
Graha Hughes
10. Кожные проявления 40%
Поражения глазНеврологические
проявления 66%
Акушерская
патология 80%
Поражение легких 20%
Периферический
тромбоз 64%
11. 3.Нейросифилис Проникновение бледных трепонем в клетки головного мозга с последующим их разрушением. Усиливается разрушение
12.
4.Криптококковая инфекцияЗаболевание начинается, если организм ослаб
(снижение иммунитета, глубокий
иммунодефицит, когда число CD меньше 50100/мкл):
Пациенты со СПИДом
Онкологические больные
Пациенты после пересадки органов
Пациенты длительное время получающие
глюкокортикоидные гормоны
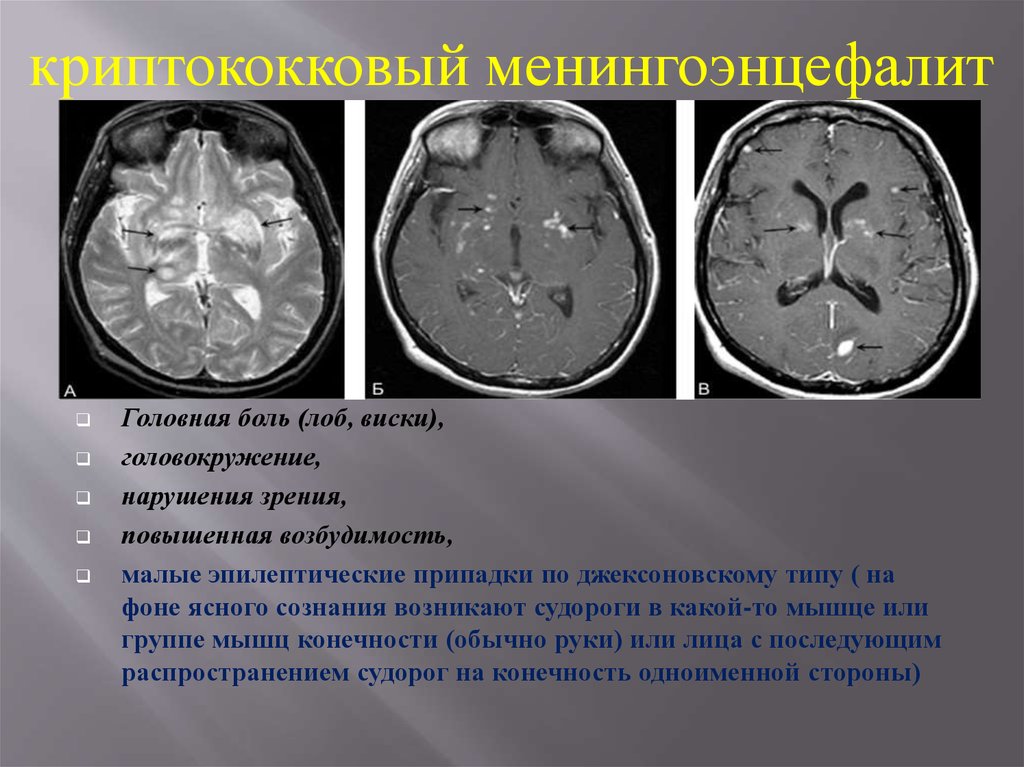
13.
криптококковый менингоэнцефалитГоловная боль (лоб, виски),
головокружение,
нарушения зрения,
повышенная возбудимость,
малые эпилептические припадки по джексоновскому типу ( на
фоне ясного сознания возникают судороги в какой-то мышце или
группе мышц конечности (обычно руки) или лица с последующим
распространением судорог на конечность одноименной стороны)
14. Нейросифилис и криптококковый менингоэнцефалит
Могут проявляться деменцией и миоклониями,нарастающими довольно быстро.
Решающими в данном случае являются данные
исследования цереброспинальной жидкости
15. 3.1. 5.Группа болезней, проявляющихся миоклонус-эпилепсией, мнестико-интеллектуальными нарушениями и атаксией может напоминать
16. 5.1.Митохондриальная энцефаломиопатия с синдромом «рваных» красных волокон
(синдром MERRF)Наследуется по материнской линии
Клиника:
Низкорослость, снижение толерантности к
физическим нагрузкам, снижение памяти,
миопатия, деменция, периферическая
нейропатия
Диагностика:
Биопсия мышц по типу Гомори
17. 5.2. Болезнь Лафора
Наследственная миоклоническая эпилепсия, прикоторой наблюдается отложения полисахаридных
веществ в различных тканях (церебральных
структур)
Миоклонические пароксизмы
Генерализованные эпиприступы
Деменция
Психические нарушения и расстройства зрения
Атаксия
Тельца Лафоры в протоках потовых желез (биопсия
кожи, мышц, печени)
18. 5.3. Болезнь Унферрихта- Лундборга (Семейная миоклония, миоклонус-эпилепсия, прогрессирующий миоклонус)
Хроническое прогрессирующее наследственноезаболевание ЦНС с аутосомно-рецессивным
механизмом наследования.
Миоклонические гиперкинезы
Эпилептиформные припадки
Возможно атакия ходьбы
Дизартрия
Тремор
Деменция
19.
ДиагностикаКлиника в виде тяжёлого стимул-сенситивного
миоклонуса
Возраст
ЭЭГ (фотосенситивность, замедление фоновой
ритмики, генерализованные высокоамплитудные
полиспайки, спайк- или полиспайк-волна (35ГЦ)
20. 5.4. Нейрональный цероидный липофусциноз (восковидные липофусцинозы нейронов)
Нейродегенеративные наследственныезаболевания, относящиеся к лизосомным
болезням накопления (накопление
пигмента липофусцина в лизосомах
нервных клетках и других тканях (печень,
селезенка, миокард, почки)
21.
Клиника:Атрофия головного мозга
Судороги (миоклонические)
Диагностика:
Электронная микроскопия (включения пигмента
липофусцина в лизосомах тканях (печень,
мышцы, лейкоциты)
22. 6. СПИД-деменция
Начальные проявления СПИД-деменция такжемогут напоминать БКЯ по началу заболевания,
клиническим признакам, отсутствию
отклонений при рутинном исследовании
цереброспинальной жидкости и по КТ- и МРТкартине.
10% больных со СПИДом не имеют других
системных проявлений этого заболевания на его
ранних этапах.
23. Дифференциальных диагноз СГШШ проводится с:
Оливопонтоцеребеллярной атаксиейГепатоцеребральной дегенерацией
Рассеянным склерозом
Семейной формой болезни
Альцгеймера
Метахроматической лейкодистрофией
Болезнью Рефсума
24. 1. Оливопонтоцеребеллярная атаксия
Наследственное заболевание, связанное сдефектом фермента дегидрогеназы
глутамата в фибробластах и лейкоцитах.
Глутаминовая
Кислота
глутамат
глутамат дегидрогеназа
25. В норме глутамат – это нейромедиатор, который передает нервный импульс от оливы, ядер моста, спинного мозга и вестибулярных
26.
ДеменцияНарушение психики и поведения
Атаксия мозжечка
Двигательные нарушения (экстрапирамидные
расстройства, поражение черной субстанции и
подкорковых ганглий)
Мышечная атония и атрофия (поражение двигательных
нейронов периферической нервной системы –
недержание мочи, кала, газов, нарушение глотания)
Поражение глазодвигательных мышц
КТ (уменьшение толщины мозжечка (средней его
ножки), увеличение подпаутинного пространства и
желудочков головного мозга
МРТ (атрофия церебрального моста, продолговатого
мозга)
27. 2. Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация или болезнь Вильсона-Коновалова)
Наследственный дефект метаболизма меди (20 хр), связанный снехваткой фермента церулоплазмина (белок, связывающий
медь в сыворотке крови)
Прогрессирующая деменция
Тремор
мышечная ригидность
Диагностика
Кровь - снижение количества церулоплазмина
Моча - увеличение количества меди в моче
Кольца Кайзера – Флейшера
МРТ: «глаз тигра»
28. 3. Болезнь Рефсума (полиневритоподобная гемератюпическая гередоатаксия)
Редкое наследственное заболевание с накоплением фитановых кислотв центральной и периферической нервных системах и внутренних
органах.
Мозжечковая атаксия
Полинейропатия с амиотрофиями и парезами
Глухота
Аносмия
Снижение остроты зрения
Ночная слепота
Снижение полей зрения
Пигментный ритинит
Определение уровня фитановых кислот в моче и крови
29. Поскольку было показано, что инфекционный прионный белок отличается от обычного третичной структурой, т.е. конформационно,
“Конформационные белки – белки, у которых в результате измененийтретичной и даже четвертичной структуры меняются некоторые
свойства “
(Prusiner S.,1996)
30.
31. Предполагаемое изменение характера укладки полипептидной цепи при превращении белка РгРC (а) в прион РгРSc (б)
32. Микрофотография конечной фазы губкообразных изменений в коре большого мозга:
33. Конформационные болезни человека
Белки, с которыми связано развитиеконформационных болезней
Конформационные болезни
Прионные инфекции – прионы, PrPc
БКЯ, нвБКЯ, ГШШ, ССБ, куру
β- амилоид, APP, PS-1, PS-2, убиквитин БА, синдром Дауна, семейная БА
α- Синуклеин
Болезнь Паркинсона
Амилин
Диабет II типа
Супероксиддисмутаза
Боковой амиотрофический склероз
PrP
Наследственные формы БКЯ, ГШШ,
ССБ
Глутаминовые повторы
Врождённые нейродегенеративные
заболевания. Болезнь Хантингтона.
Спиноцеребральная атаксия. Атрофия
субстанции Рубро. Атрофия МачадоДжозефа
34.
Белковая природаамилоида была
установлена М. М.
Рудневым вместе с
Кюне в 1865г.
В 1854г. Р. Вирхов показал, что изменения
эти связаны с появлением в органах особого
вещества, которое под действием йода и
серной кислоты окрашивается в синий цвет.
Поэтому он назвал его амилоидом, а
«сальную болезнь» - амилоидозом.
35.
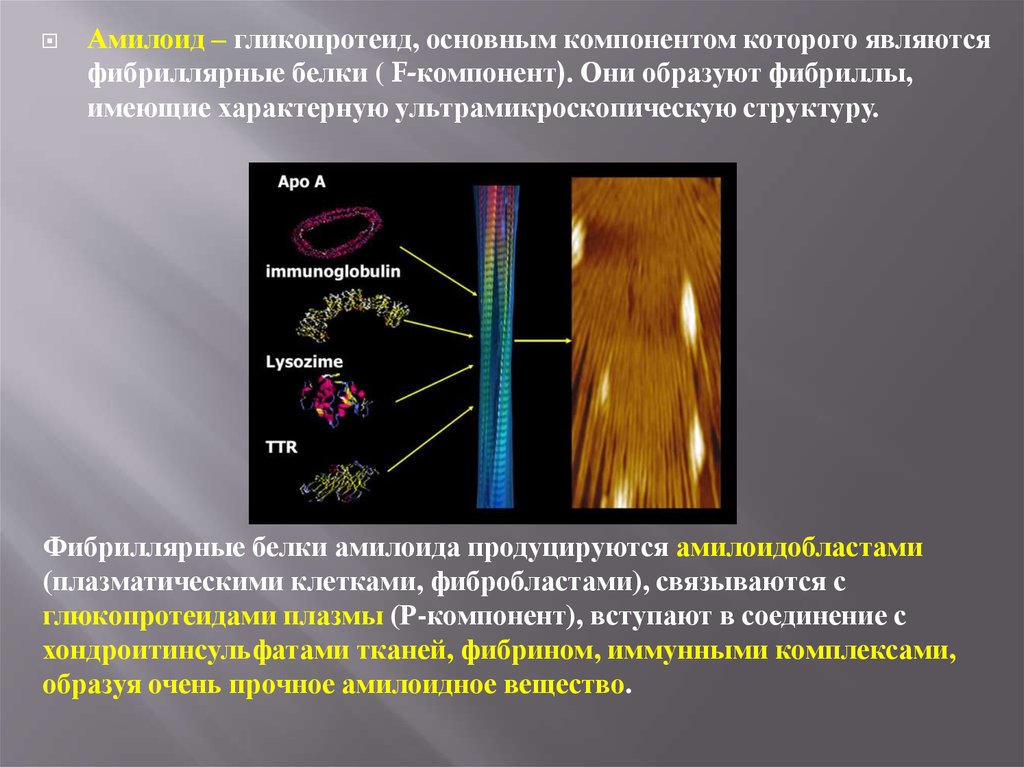
Амилоид – гликопротеид, основным компонентом которого являютсяфибриллярные белки ( F-компонент). Они образуют фибриллы,
имеющие характерную ультрамикроскопическую структуру.
Фибриллярные белки амилоида продуцируются амилоидобластами
(плазматическими клетками, фибробластами), связываются с
глюкопротеидами плазмы (Р-компонент), вступают в соединение с
хондроитинсульфатами тканей, фибрином, иммунными комплексами,
образуя очень прочное амилоидное вещество.
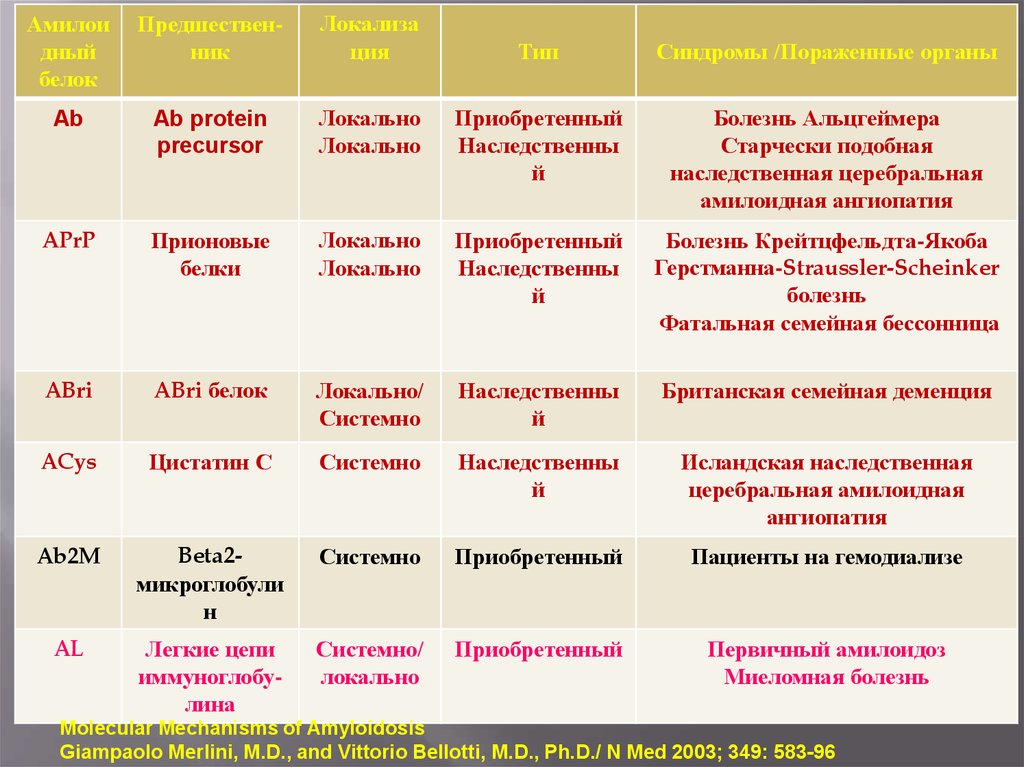
36.
Амилоидный
белок
Предшественник
Локализа
ция
Ab
Ab protein
precursor
APrP
Тип
Синдромы /Пораженные органы
Локально
Локально
Приобретенный
Наследственны
й
Болезнь Альцгеймера
Старчески подобная
наследственная церебральная
амилоидная ангиопатия
Прионовые
белки
Локально
Локально
Приобретенный
Наследственны
й
Болезнь Крейтцфельдта-Якоба
Герстманна-Straussler-Scheinker
болезнь
Фатальная семейная бессонница
ABri
ABri белок
Локально/
Системно
Наследственны
й
Британская семейная деменция
ACys
Цистатин С
Системно
Наследственны
й
Исландская наследственная
церебральная амилоидная
ангиопатия
Ab2M
Beta2микроглобули
н
Системно
Приобретенный
Пациенты на гемодиализе
AL
Легкие цепи
иммуноглобулина
Системно/
локально
Приобретенный
Первичный амилоидоз
Миеломная болезнь
Molecular Mechanisms of Amyloidosis
Giampaolo Merlini, M.D., and Vittorio Bellotti, M.D., Ph.D./ N Med 2003; 349: 583-96
37. 4. Болезнь Альцгеймера
БА имеет достаточно сложныйпатогенез, но по процессу образования
амилоидных включений данная патология
чрезвычайно близка к прионным
наследственным и инфекционным
болезням (De Strooper B. et.al.,1998)
38. БА – медленно и длительно текущее заболевание, имеющее 3 основные стадии
Образование и накопление Аβ – пептидов –продуктов протеолиза АРР
(предшественника амилоидного белка)
Интеграция Аβ – пептидов в мембраны
нейронов и образование токсигенных ионных
каналов, вызывающих гибель нейронов и
одновременную активацию микроглиальных
элементов, и, следовательно, воспалительную
реакцию в очагах поражения
Образование амилоидных включений
39. Патогенез
Синтез АРР• Образование Аβ – пептидов и формирование
токсигенных ионных каналов в нейронах
• Активация микроглии Аβ - пептидами
воспаление
• Освобождение цитокинов
• Синтез патологических шаперонов (Апо Е и др)
Гибель
нейронов
• Активация процесса образования амилоидных
бляшек
• Нейрофибрилярные образования погибших
нейронов
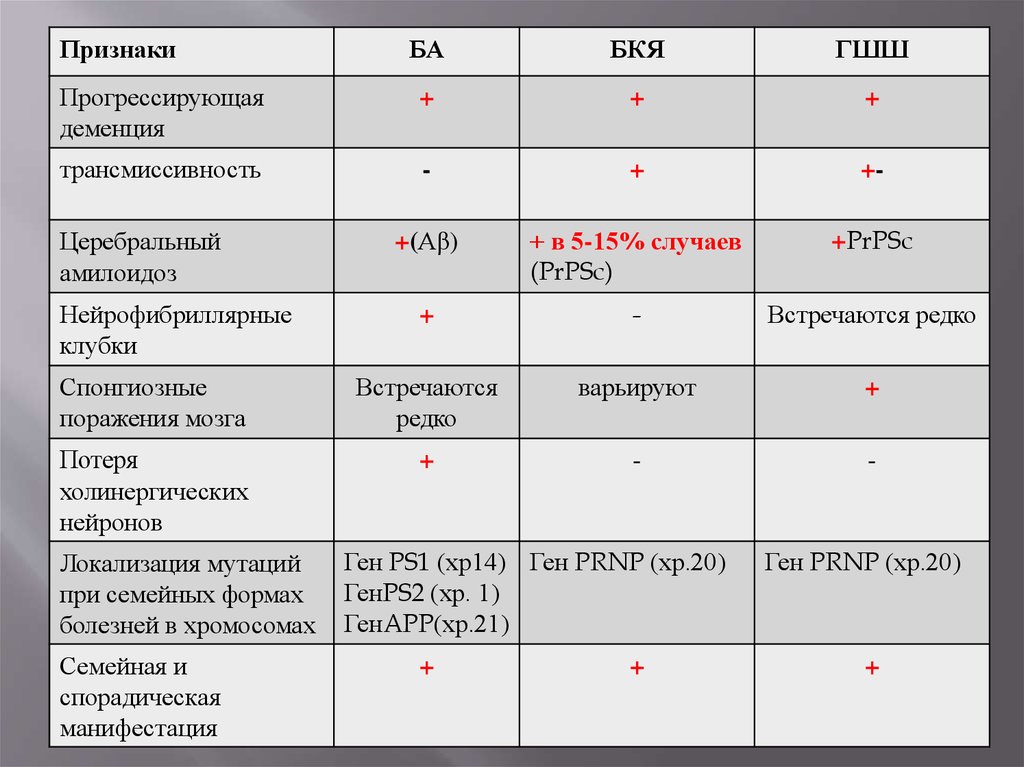
40.
ПризнакиБА
БКЯ
ГШШ
Прогрессирующая
деменция
+
+
+
трансмиссивность
-
+
+-
+(Аβ)
+ в 5-15% случаев
(PrPSc)
+PrPSc
+
-
Встречаются редко
Спонгиозные
поражения мозга
Встречаются
редко
варьируют
+
Потеря
холинергических
нейронов
+
-
-
Церебральный
амилоидоз
Нейрофибриллярные
клубки
Локализация мутаций
при семейных формах
болезней в хромосомах
Семейная и
спорадическая
манифестация
Ген PS1 (хр14) Ген PRNP (хр.20)
ГенPS2 (хр. 1)
ГенAPP(хр.21)
+
+
Ген PRNP (хр.20)
+
41.
По мнению D.Westaway (1998г) сходствосимптоматики этих болезней отражает
близость фундаментальных элементов
патогенеза. Эти болезни могут быть
объединены в 4 группы, что облегчает их
анализ и, возможно, поможет понять генез
этих болезней:
Семейные прионные болезни, напоминающие
БА
Семейная БА с признаками прионных болезней
Спорадическая БА с признаками прионных
болезней
Миозит телец включения, Aβ – PrP - типа
42.
СПАСИБО ЗА ВНИМАНИЕ43. Список, используемой литературы
Патоморфология головного мозга при прионныхболезнях (В.Я. Кармышева, Т.С.Гулевская, В.В.
Погодина, В.М. Ройхель, И.А. Завалишин )
Прионы и прионные болезни (В.И. Покровский,
О.И. Киселев, Б.Л. Черкасский
Основы клинической неврологии (С.В. Котов)