















 medicine
medicineSimilar presentations:










Мышечные дистонии, спастическая параплегия Штрюмпеля: классификация, этиология, патогенез, клиническая картина, диагностика
1. Мышечные дистонии, спастическая параплегия Штрюмпеля: классификация, этиология, патогенез, клиническая картина, диагностика,
диф.диагностика, медикаментозные инемедикаментозные методы терапии.
Выполнила: студентка 4 курса группы
№ 3 Забияка А.Р.
Преподаватель: Шевченко В.В.
2.
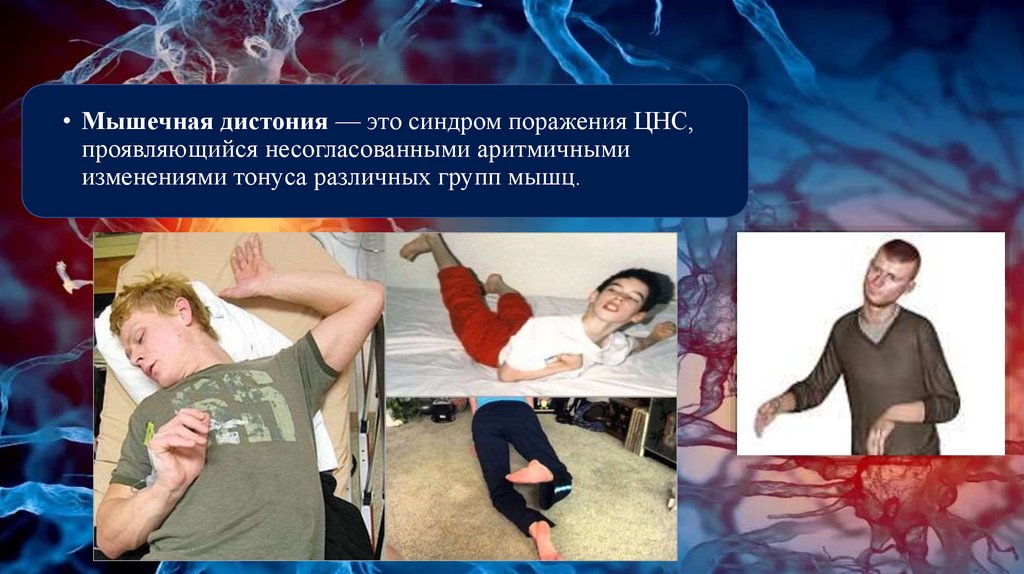
• Мышечная дистония — это синдром поражения ЦНС,проявляющийся несогласованными аритмичными
изменениями тонуса различных групп мышц.
3.
ЭТИОЛОГИЯ• Генетические изменения. Генные мутации обуславливают изменения медиаторной или рецепторной части нервных
синапсов, участвующих в передаче регулирующих тонус воздействий. К наследственным формам МД
относятся идиопатический блефароспазм, миоклоническая дистония, первичная торсионная дистония.
• Черепно-мозговая травма. Повреждение подкорковых структур при ЧМТ, проводящих трактов экстрапирамидной
системы обуславливает расстройство центральной регуляции тонуса мышц. При неполном восстановлении мышечная
дистония сохраняется в посттравматическом периоде.
• Опухоль головного мозга. Прорастая церебральные ткани, агрессивные внутримозговые неоплазии разрушают нейроны
и проводящие пути. При неинвазивном росте по мере увеличения размеров опухоли возникает компрессия окружающих
образований. МД возникает при вовлечении в патологический процесс экстрапирамидных структур.
• Энцефалит. Воспалительное поражение различных уровней экстрапирамидной системы при энцефалите влечёт
расстройство её регулирующего воздействия на мышечный тонус. МД может проявляться на фоне стихания острого
воспалительного процесса и регресса более грубой неврологической симптоматики.
• Медикаментозное воздействие. Спровоцировать дистонические расстройства способны атипаркинсонические
фармпрепараты, нейролептики, антидепрессанты, антипсихотики. Факторами риска возникновения побочных эффектов
терапии являются большая длительность приёма и высокие дозировки.
4. ПАТОГЕНЕЗ
• Выделяют основные нейробиологические механизмы, потенциальноприводящие к развитию дистонии: нарушения синтеза или метаболизма
дофамина, наиболее значимые для развития ДОФА-чувствительной
дистонии; митохондриальная дисфункция; накопление металлов в головном
мозге; нарушения работы кальциевых каналов и гомеостаза кальция;
нарушения регуляции транскрипции генов; изменения эндоплазматического
ретикулума или ядерной оболочки.
5. КЛИНИЧЕСКАЯ КАРТИНА
• Клиническая картина характеризуется совокупностью моторных инемоторных проявлений. Моторные проявления, связанные с дистонией,
проявляются особой неестественной дистонической позой с дистальным
тремором или без него, специфическими избыточными движениями на пике
дистонических движений, зеркальными движениями на пораженной стороне
при выполнении движений контралатеральной стороной и наличием
корригирующих жестов (сенсорных или антагонистических движений) для
устранения/уменьшения патологической позы или
движения. Недвигательные (немоторные) симптомы дистонии - боль,
депрессия, тревожность, социальные фобии - характерные клинические
проявления, которые, наряду с моторными симптомами, составляют
клиническую картину заболевания, существенно влияют на качество жизни
пациентов
6. КЛАССИФИКАЦИЯ
Распространённость патологическогопроцесса на мышечные группы:
Степени выраженности МД:
• Фокальная. Патологический процесс охватывает не более
одной мышечной группы. К фокальным формам
относится писчий спазм, блефароспазм, спастическая
дисфония.
• I — изменения выражены минимально. Дистонический
синдром проявляется в ситуации повышенного
психического напряжения, при переутомлении.
• Сегментарная. Тоническое сокращение распространяется на
несколько рядом расположенных мышечных групп.
Примером может служить
комбинированная оромандибулярная МД.
• Мультифокальная. Фокальные дистонические феномены
наблюдаются в нескольких участках тела. Патологические
мышечные сокращения в них могут происходить
одновременно и независимо друг от друга.
• Генерализованная. Непроизвольное мышечное напряжение
распространяется практически на всю скелетную
мускулатуру. Начавшись с фокальных форм, МД способна
трансформироваться в генерализованную форму.
• II — мышечная дистония регулярно возникает при
физической активности. В покое дистонические
феномены отсутствуют.
• III — мышечная дистония носит постоянный характер,
усиливается при произвольных движениях. Отмечаются
затруднения при выполнении действий с участием
подверженных дистонии мышечных групп. Возникают
ограничения в профессиональной деятельности.
• IV — выраженная МД, лишающая пациента возможности
самостоятельного выполнения отдельных движений.
Инвалидизирует больного.
7. ДИАГНОСТИКА
• 1) клиническая оценка − выявление синдрома дистонии, и оценка егоклинических характеристик в соответствии с Классификацией 2013 г.;
• 2) документирование клинических проявлений: видео- и фоторегистрация, оценка
клинических проявлений по стандартным оценочным шкалам для каждой
клинической формы дистонии;
• 3) генетическое тестирование при подозрении на наследственный характер
первичной дистонии, при планировании глубокой стимуляции мозга пациентам с
сегментарной и генерализованной дистонией;
• 4) инструментальная диагностика для исключения повреждений мозга и других
заболеваний при подозрении на вторичный характер дистонии, для выявления
паттерна активации мышц и в некоторых специфических случаях;
• 5) диагностический тест с L-DOPA при подозрении на ДОФА-зависимые формы
дистонии.
8. ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
• Болезнь Фара (код МКБ: G23.8) ‒ это редкое нейродегенеративное заболевание, связанное с неатеросклеротическимобызвествлением коры полушарий, базальных ганглиев и зубчатых ядер мозжечка, вследствие отложения солей кальция и железа в
стенки мелких артерий и артериол, а также в вещество головного мозга на фоне патологии щитовидной и паращитовидных желез.
• Гемидистония (код МКБ: G23.8) всегда имеет симптоматическую вторичную природу и указывает на органическое поражение
контралатерального полушария.
• Дистония в результате медикаментозной интоксикации (код МКБ: G24.0) особенно часто возникает при применении фенитоина,
карбамазепина, фенотиазина, бутирофенона, бензамина, трициклических антидепрессантов, антигистаминных препаратов,
кетамина, лития и церукала.
Злокачественный нейролептический синдром (код МКБ: G21.0) − неврологическое расстройство, которое возникает после
назначения нейролептиков, блокирующих дофаминовые рецепторы. Для этого синдрома характерна орофациальная дистония.
• Болезнь Вильсона–Коновалова (гепато-лентикулярная дегенерация, код МКБ: Е83.0) характеризуется аутосомно-рецессивным
типом наследования.
• Ювенильный паркинсонизм (код МКБ G21) является аутосомнорецессивной патологией, обусловленной мутациями гена PRKN.
• Атетоз (код МКБ: G25.8, G80.3) является экстрапирамидным гиперкинезом и в качестве варианта дистонии рассматривается не
всеми авторами, хотя клинически представляет собой медленный дистонический гиперкинез, для которого характерны
непроизвольные стереотипные, ритмические, червеобразные, вычурные движения небольшого объема.
• Дистония в результате травмы периферической нервной системы (код МКБ: G23.8) может развиться только у детей старшего
возраста и взрослых. Механизмы возникновения данной дистонии не ясны.
• Дистония в результате перинатального поражения головного мозга (код МКБ: G23.8) появляется у детей с патологией
перинатального периода до трех лет жизни, по мере развития у ребенка гиперкинетической формы ДЦП.
9. ЛЕЧЕНИЕ
• Ботулинотерапия стала основным методом лечения мышечной дистониинезависимо от этиологии.
• Ботулинотерапия рекомендована при цервикальной дистонии (Класс
рекомендации А), блефароспазме (Класс рекомендации В), гемифациальном
спазме (Класс рекомендации С), фокальной дистонии верхних конечностей
(Класс рекомендации В), cпастической аддукторной дисфонии (Класс
рекомендации В).
• Существует 7 серотипов ботулотоксина: А, В, C1, C2, D, E, F и G, которые
вырабатываются различными штаммами Clostridium botulinum. Все они
являются нейротоксичными, за исключением С2, у которого доказано
вазодилятационное действие. Наиболее мощным является серотип А.
• В нашей стране имеется несколько препаратов ботулинического токсина
типа А: 1) онаботулотоксин А (Ботокс, 1994); 2) абоботулотоксин А
(Диспорт, 1999); 3) инкоботулотоксин А (Ксеомин, 2008); 4) лантокс (2008);
5) релатокс (2012).
10.
• Хирургические методы лечения. В тех случаях, когда у пациентавыявляется резистентность к ботулинотерапии, требуется проведение
хирургического вмешательства, которое включает глубокую
стимуляцию мозга (DBS). В основном этот метод лечения предлагается
для генерализованной и сегментарной дистонии.
• Интратекальное введение баклофена применяется при
неэффективности от проводимой терапии у пациентов с тяжелыми
генерализованными дистониями, преимущественно вторичного
характера (гиперкинетическая форма детского церебрального
паралича).
• Медикаментозная коррекция мышечных дистоний до сих пор остается
недостаточно эффективной и после внедрения в широкую врачебную
практику ботулинотерапии уходит в прошлое. Тем не менее, на
сегодняшний день холинолитики, баклофен, бензодиазепины и
леводопа продолжают применяться в лечебных схемах.
11. НЕМЕДИКАМЕНТОЗНЫЕ МЕТОДЫ ТЕРАПИИ
• Неинвазивная анодная стимуляции мозжечка и первичной моторной коры;• Специализированные физические упражнения, направленные на релаксацию и растяжение
мышц шеи;
• БОС-терапия - метод лечения и реабилитации, основанный на развитии у пациента навыков
самоконтроля и саморегуляции различных функций организма. Цель - помочь осознать и
скорректировать физиологические реакции, которые обычно не поддаются сознательному
контролю.
• Так, было показано положительное влияние комбинированной терапии (сочетание
ботулинотерапии с немедикаментозными методами) в виде уменьшения болевого синдрома,
увеличения периода действия ботулинотерапии, уменьшение последующей разовой дозы
ботулинотерапии, увеличение повседневной активности.
• Для коррекции астении, тревожно-депрессивных, диссомнических нарушений
целесообразно рассматривать когнитивно-поведенческую терапию, что также требует
проведения дополнительных исследований.
12.
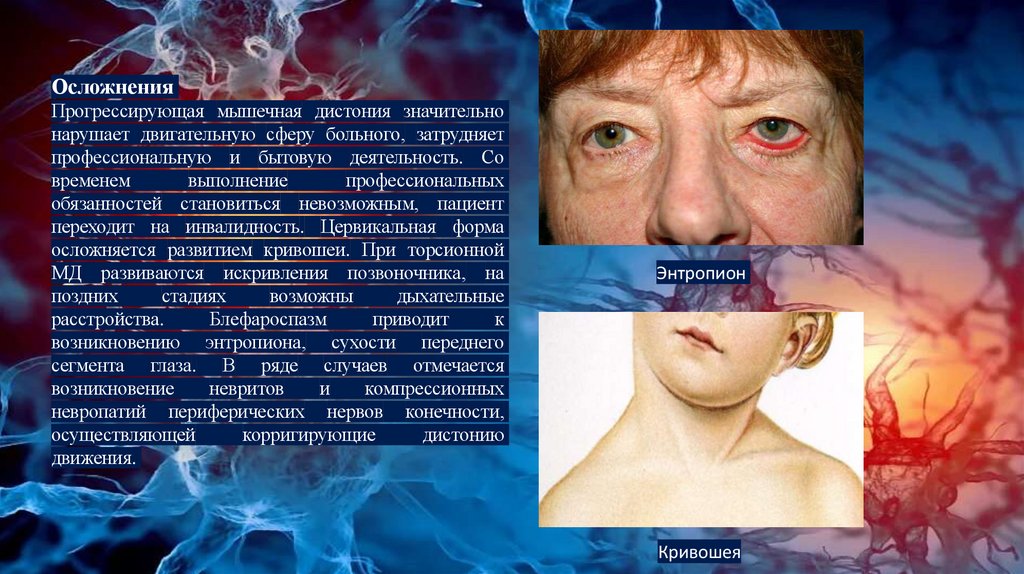
ОсложненияПрогрессирующая мышечная дистония значительно
нарушает двигательную сферу больного, затрудняет
профессиональную и бытовую деятельность. Со
временем
выполнение
профессиональных
обязанностей становиться невозможным, пациент
переходит на инвалидность. Цервикальная форма
осложняется развитием кривошеи. При торсионной
МД развиваются искривления позвоночника, на
поздних
стадиях
возможны
дыхательные
расстройства.
Блефароспазм
приводит
к
возникновению энтропиона, сухости переднего
сегмента глаза. В ряде случаев отмечается
возникновение
невритов
и
компрессионных
невропатий периферических нервов конечности,
осуществляющей
корригирующие
дистонию
движения.
Энтропион
Кривошея
13. Спастическая параплегия Штрюмпеля
• Дегенеративная наследственнаямиелопатия с двусторонним
поражением боковых и передних
спинномозговых столбов
преимущественно на поясничном
уровне.
14. ЭТИОЛОГИЯ
• Достоверно определить, что стало причиной развития поражениярефлексов и начала болезни Штрюмпеля практически
невозможно, так как активно развивающаяся генетика уже
обнаружила больше 50 вариантов хромосомных локусов, дефекты
в которых могут привести к развитию той или иной формы
болезни. При этом в некоторых случаях присутствует
доминантное наследование, в других рецессивное, а в самых
редких вариантах болезнь передаётся сцепленной только с Xхромосомой.
15. КЛАССИФИКАЦИЯ
В зависимости от варианта наследования патологиивыделяют:
По силе проявления симптомов:
•Аутосомно-доминантные. Если у одного из родителей
проявилась болезнь или есть мутация, необходимая для
запуска патологии, то с вероятностью 50% его ребёнок
также получит такой диагноз. [3]
•Аутосомно-рецессивные. Болезнь проявится, если оба
родителя будут болеть или являться носителями одного и
того типа мутации. Вероятность развития патологии – 25%.
•Х-сцепленные формы. В этом случае женщины являются
носительницами мутации в своих хромосомах, но не
заболевают. А вот у мужчин, чья мать была носительницей,
риск заболеть крайне велик.
•Неосложнённая форма. Единственным
заметным симптомом на протяжении всего
течения болезни является спастический нижний
парапарез.
•Сложная форма. Сочетает проявление
непосредственно симптоматики патологии с
другими тяжёлыми заболеваниями, среди которых
эпилепсия, расстройство слуха, дизартрия,
мозжечковая атаксия, задержка психического
развития и другие.
16. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
• Спастический парапарез или параплегия, вначале может быть асимметричным, позднее – симметричный. Картина походки:круговые движения носков ступни, походка на пальцах ступни, повёрнутые внутрь колени, при быстрой ходьбе колени бьются друг
о друга.
• Положительный рефлекс Бабинского, клонус ахиллесового и пателлярного рефлекса. Обыкновенно нормальные брюшные
рефлексы.
• Спастические контрактуры сгибающей мускулатуры, pes cavus.
• Иногда (чаще в поздней стадии) повышенный уровень собственных мышечных рефлексов верхних конечностей.
• Нарушение двигательной функции руки.
• Редко: псевдобульбарный синдром
• Нарушение глубокой чувствительности, в особенности ощущения вибрации.
• Внезапный, непреодолимый посыл к мочеиспусканию, невозможность контролировать процесс опорожнения мочевого пузыря
• Дополнительные симптомы (в случае сложных наследственных форм)
• Повреждения вторичных двигательных нейронов и, как следствие, амиотрофия (чаще нижних конечностей), нейропатия
зрительного нерва, ретинопатия, экстрапирамидальные симптомы, умственная отсталость и деменция, целебеллярная атаксия,
нарушения слуха, эпилептические припадки (височной доли мозга).
• Нарушения распространения сердечного импульса, глаукома, гематологические нарушения, изменения кожи и скелета, малый
рост, рефлюксная болезнь, грыжа пищеводного отверстия диафрагмы.
• Течение заболевания
• Заболевание проявляется обычно в детском возрасте. Оно характеризуется медленным развитием, редко приводит к потери
способности движения. Длительность жизни – нормальная.
17. ДИАГНОСТИКА
• Диагностирование наследственной спастической параплегии осуществляетсяна основании характерной клинической картины с характерными
симптомами: спастическим симметричным парапарезом ног, повышенным
уровнем рефлексов, положительным рефлексом Бабинского, положительным
семейным анамнезом и нарушением вибрационной чувствительности. К
возможным дополнительным симптомам относятся: центральное нарушение
мочеиспускания, парез и повышенный уровень рефлексов в области рук.
• Лабораторные параметры: уровень витамина B12 (метилмалоновая кислота,
гомоцистеин), жирные кислоты с длинной цепью, антитела против HTLV-1,
аквапорин 4 и MOG.
• В рамках дифференциальной диагностики проводятся: сканирование в
магнитном томографе, исследование спинномозговой жидкости (ликвора).
• Консультация у специалиста по генетике.
18. ЛЕЧЕНИЕ
• Основу терапии составляют миорелаксанты (баклофен, толперизон) и транквилизаторы(тазепам, диазепам), которые также оказывают расслабляющий мышцы эффект. Лечение
стартует с минимальной дозировки препарата, которая постепенно увеличивается. При
достижении эффекта в виде существенного ослабления спастики, дозу препарата
прекращают наращивать. При возникновении побочных эффектов останавливают
увеличение дозы, если это не помогает — производят ее постепенное снижение. Резкая
отмена препарата опасна синдромом отмены, т. е. быстрым нарастанием спастики до
степени, превышающей первоначальные проявления. В случаях, когда пероральный прием
не дает желаемого эффекта, препараты вводят внутримышечно. Возможно эндолюмбальное
локальное введение. При грубой спастике прибегают к установке помпы для
постоянной интратекальной инфузии баклофена. Указанное лечение является
симптоматическим, оно не позволяет полностью излечить болезнь Штрюмпеля, а лишь дает
возможность уменьшить скованность в ногах и, таким образом, улучшить их подвижность.
• Альтернативным методом уменьшения спастики является введение ботулотоксина в задние
мышцы бедер и икроножные мышцы. Наряду с медикаментозным лечением применяется
специальный комплекс ЛФК, физиопроцедуры (парафинолечение, точечный
массаж, лечебные ванны). Показана консультация и наблюдение ортопеда, при
необходимости — использование ортезов. По показаниям возможно хирургическое
ортопедическое лечение возникших контрактур.