













































 chemistry
chemistrySimilar presentations:










Обмен липидов
1.
Кафедра общей ибиологической химии
Доцент кафедры общей и
биологической химии, к.б.н.
Романова Л. В.
2. Цель лекции
Выработать умения использовать знания ометаболизме холестерола, кетоновых тел в
практической деятельности врача
3. План лекции
1. Холестерол. Синтез холестерола.Регуляция.
2. Пути превращения и выведения
холестерола.
3. Гиперхолестеренемия. Атеросклероз.
Желчекаменная болезнь.
4. Метаболизм кетоновых тел.
5. Сфинголипидозы.
4.
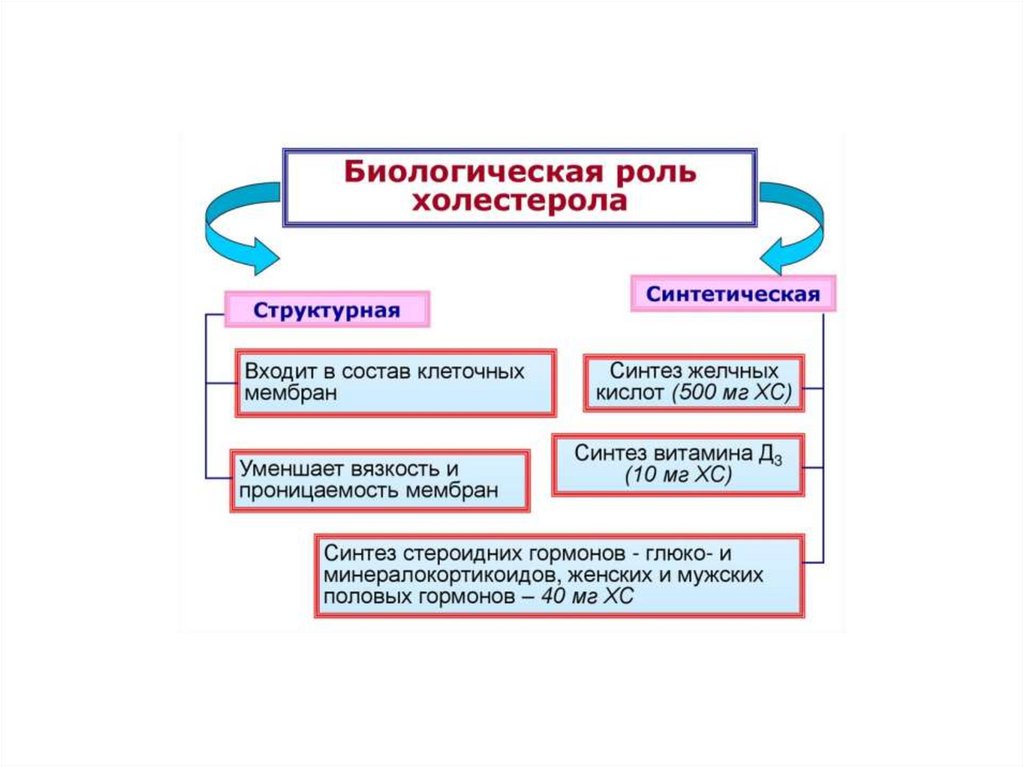
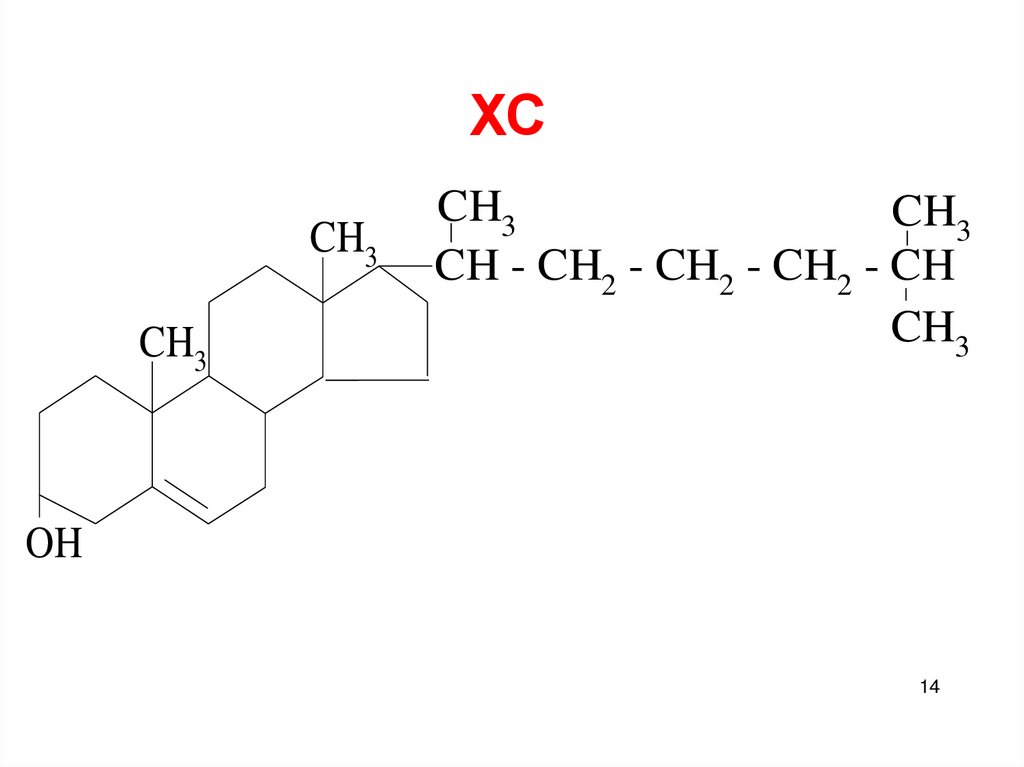
ХолестеролХС – стероид, характерный только для
животных организмов. 50% синтезируется в
печени, 15-20% в тонком кишечнике, остальной – в коже, коре надпочечников, половых
железах. В сутки синтезируется 1г ХС. С пищей поступает 300-500мг.
N в крови = 3,9-5,2 ммоль/л
У новорожденных ХС – 1,3-2,6 ммоль/л
В 12-14 лет – достигает нормы.
5.
6.
ХСХС
CH3
CH3
OH
CH3
CH3
CH - CH2 - CH2 - CH2 - CH
CH3
N – 3,9 – 5,2 ммоль/л
14
7.
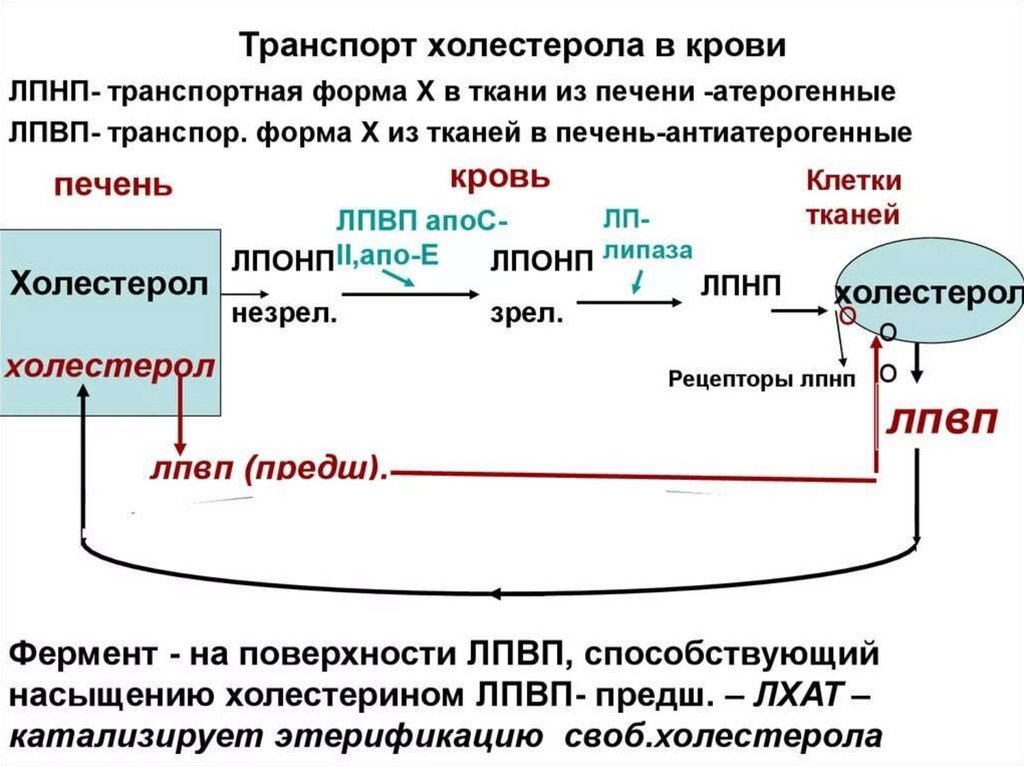
8. МЕТАБОЛИЗМ ХОЛЕСТЕРОЛА
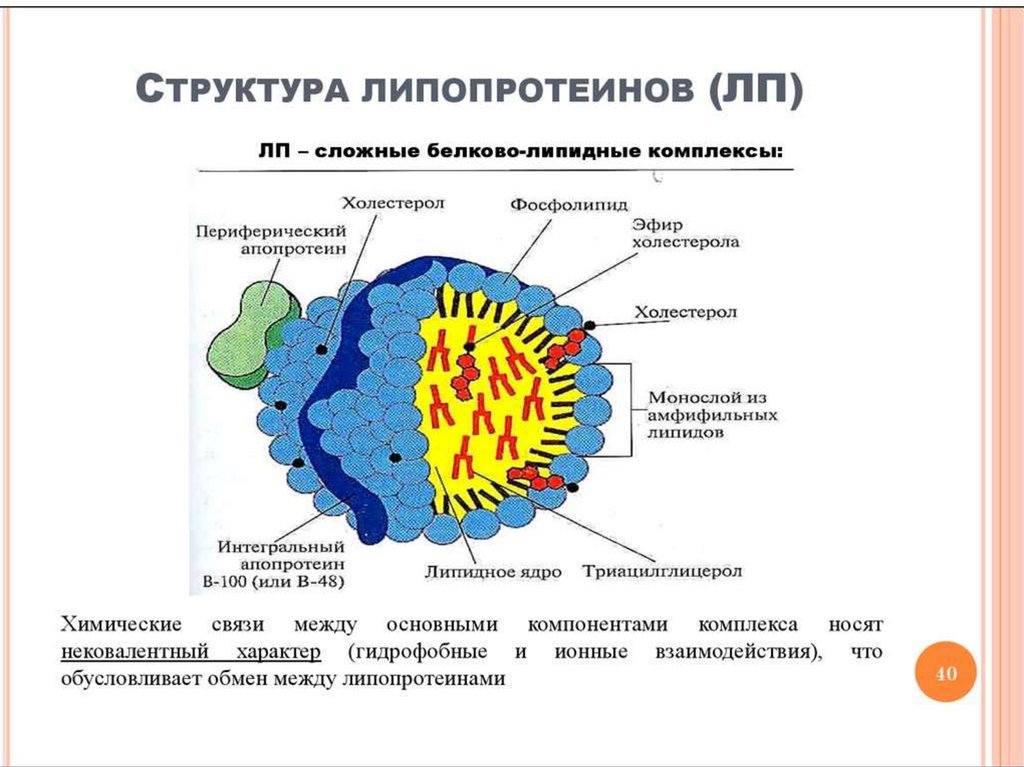
Транспорт холестерола и его эфировосуществляется липопротеинами
низкой(ЛПНП) и высокой плотности (ЛПВП).
9.
10.
Липопротеины высокой плотности. образуются в печени de novo, в плазме крови
при распаде
хиломикронов, некоторое количество в
стенке кишечника,
• в составе частицы примерно половину
занимают белки, еще четверть фосфолипиды,
остальное холестерин и ТАГ (50% белка, 25%
ФЛ, 13% эфиров ХС и 5% свободного ХС, 7%
ТАГ),
• структурным апобелком является апо А1,
также содержат апоЕ и апоСII.
11.
Функция• Транспорт свободного ХС от тканей к печени.
• Фосфолипиды ЛПВП являются источником
полиеновых кислот для синтеза клеточных
фосфолипидов и эйкозаноидов.
12.
Метаболизм1. Синтезированные в печени частицы (первичные ЛПВП)
содержат в основном фосфолипиды и апобелки. Остальные
липидные компоненты накапливаются в ЛПВП по мере
метаболизма в плазме крови.
2. Новосинтезированные ЛПВП выглядят на электронных
микрофотографиях как двухслойные диски из двух монослоев
фосфолипидов (таблеткообразная форма). Синтез апоЕ и апоС
происходит главным образом в печени, в отличие от апо А-I,
образующегося как в печени, так и в тонком кишечнике.
3. В плазме крови насцентный ЛПВП сначала превращается в
ЛПВП3 (условно его можно назвать "зрелый").
13.
В этом превращении главным является то, чтоЛПВП
• забирает от клеточных мембран свободный
холестерин при непосредственном контакте или при
участии специфических транспортных белков,
• взаимодействуя с мембранами клеток, отдает им
часть фосфолипидов из своей оболочки, доставляя
таким образом полиеновые жирные кислоты в
клетки,
• тесно взаимодействует с ЛПНП и ЛПОНП, получая
от них свободный холестерин. В обмен
ЛПВП3 отдают эфиры ХС, образованные благодаря
переносу жирной кислоты от фосфатидилхолина
(ФХ) на холестерин
14.
4. Внутри ЛПВП активно протекает реакцияпри участии лецитин:холестеролацилтрансферазы (ЛХАТ-реакция). В этой
реакции остаток полиненасыщенной жирной
кислоты переносится от 2-го
положения фосфатидилхолина (из оболочки
самого ЛПВП) на получаемый
свободный холестерин с образованием
лизофосфатидилхолина (лизоФХ) и эфиров ХС.
ЛизоФХ остается внутри ЛПВП, эфир
холестерина отправляется в ЛПНП.
15.
5. В результате первичный ЛПВП постепенно,через зрелую форму ЛПВП3, преобразуется в
ЛПВП2 (остаточный, ремнантный). При этом
происходят и дополнительные события:
• взаимодействуя с разными формами ЛПОНП
и ХМ, ЛПВП получают ацил-глицеролы
(МАГ, ДАГ, ТАГ), и обмениваются
холестерином и его эфирами,
• ЛПВП отдают апоЕ- и апоСII-белки на
первичные формы ЛПОНП и ХМ, и потом
забирают обратно апоСII-белки от
остаточных форм.
16.
Таким образом, при метаболизме ЛПВП в немпроисходит накопление свободного ХС, МАГ,
ДАГ, ТАГ, лизоФХ и утрата фосфолипидной
оболочки. Функциональные способности
ЛПВП снижаются.
6. Далее ЛПВП2 захватывается гепатоцитами
при помощи апоА-1-рецептора, происходит
эндоцитоз и частица разрушается.
17.
Липопротеины низкой плотности• образуются в гепатоцитах de novo и в сосудистой
системе печени под воздействием печеночной ТАГлипазы из ЛПОНП,
• в составе преобладают холестерол и его эфиры,
другую половину массы делят белки и фосфолипиды
(38% эфиров ХС, 8% свободного ХС, 25% белки,
22% фосфолипидов, 7% триацилглицеролов),
• основным апобелком является апоВ-100,
• нормальное содержание в крови 3,2-4,5 г/л,
• самые атерогенные
18.
Функция1. Транспорт холестерола в клетки,
использующих его
• для реакций синтеза половых
гормонов, глюкокортикоидов и минералокортикоидов ,
• для превращения в холекальциферол (кожа),
• для образования желчных кислот (печень),
• для выведения в составе желчи (печень).
19.
2. Транспорт полиеновых жирных кислот ввиде эфиров ХС в некоторые клетки рыхлой
соединительной ткани (фибробласты,
тромбоциты, эндотелий, гладкомышечные
клетки), в клетки костного мозга, в клетки
роговицы глаз, в нейроциты, в базофилы
аденогипофиза.
20.
Клетки рыхлой соединительной тканиактивно синтезируют эйкозаноиды. Поэтому им
необходим постоянный приток
полиненасыщенных жирных кислот (ПНЖК),
что осуществляется через апо-В-100-рецептор,
т.е. регулируемым поглощением ЛПНП,
которые несут ПНЖК в составе эфиров
холестерола.
Особенностью клеток, поглощающих ЛПНП,
является наличие лизосомальных кислых
гидролаз, расщепляющих эфиры ХС. У других
клеток таких ферментов нет.
21.
Метаболизм• 1. В крови первичные ЛПНП взаимодействуют с
ЛПВП, отдавая свободный ХС и получая
этерифицированный. В результате в них происходит
накопление эфиров ХС, увеличение гидрофобного
ядра и "выталкивание" белка апоВ-100 на
поверхность частицы. Таким образом, первичный
ЛПНП переходит в зрелый.
• 2. На всех клетках, использующих ЛПНП, имеется
высокоафинный рецептор, специфичный к ЛПНП –
апоВ-100-рецептор. Около 50% ЛПНП
взаимодействует с апоВ-100-рецепторами разных
тканей и примерно столько же поглощается
гепатоцитами.
22.
3. При взаимодействии ЛПНП с рецепторомпроисходит эндоцитоз липопротеина и его
лизосомальный распад на составные части –
фосфолипиды, белки (и далее до аминокислот),
глицерол, жирные кислоты, холестерол и его эфиры.
– ХС превращается в гормоны или включается в
состав мембран,
– излишки мембранного ХС удаляются с помощью
ЛПВП,
– принесенные с эфирами ХС ПНЖК используются
для синтеза эйкозаноидов или фосфолипидов.
– при невозможности удалить ХС часть
его этерифицируется с олеиновой или линолевой
кислотами ферментом ацил-SКоА:холестеролацилтрансферазой (АХАТ-реакция)
23.
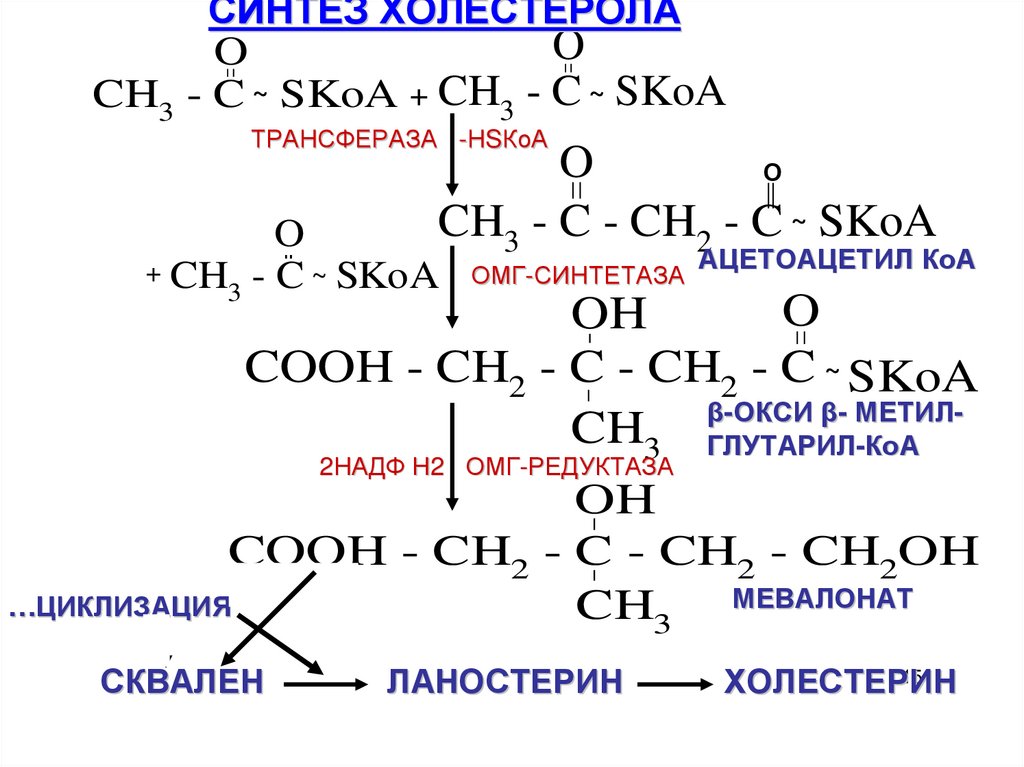
24. СИНТЕЗ ХОЛЕСТЕРОЛА
Синтез ХС состоит из 3-х этапов.1. Образование мевалоновой кислоты
из ацетил-КоА.
2. Образование ланостерина из мевалоновой кислоты.
3. Образование ХС из ланостерина.
25.
СИНТЕЗ ХОЛЕСТЕРОЛАO
O
CH3 - C ~ SKoA + CH3 - C ~ SKoA
ТРАНСФЕРАЗА -НSКоА
O
О
CH3 - C - CH2 - C ~ SKoA
O
+ CH - C ~ SKoA
3
ОМГ-СИНТЕТАЗА
АЦЕТОАЦЕТИЛ КоА
O
OH
COOH - CH2 - C - CH2 - C ~ SKoA
-ОКСИ β- МЕТИЛCH3 βГЛУТАРИЛ
-КоА
2НАДФ Н2 ОМГ-РЕДУКТАЗА
OH
COOH - CH2 - C - CH2 - CH2OH
…ЦИКЛИЗАЦИЯ
CH3 МЕВАЛОНАТ
СКВАЛЕН
ЛАНОСТЕРИН
15
ХОЛЕСТЕРИН
26.
27.
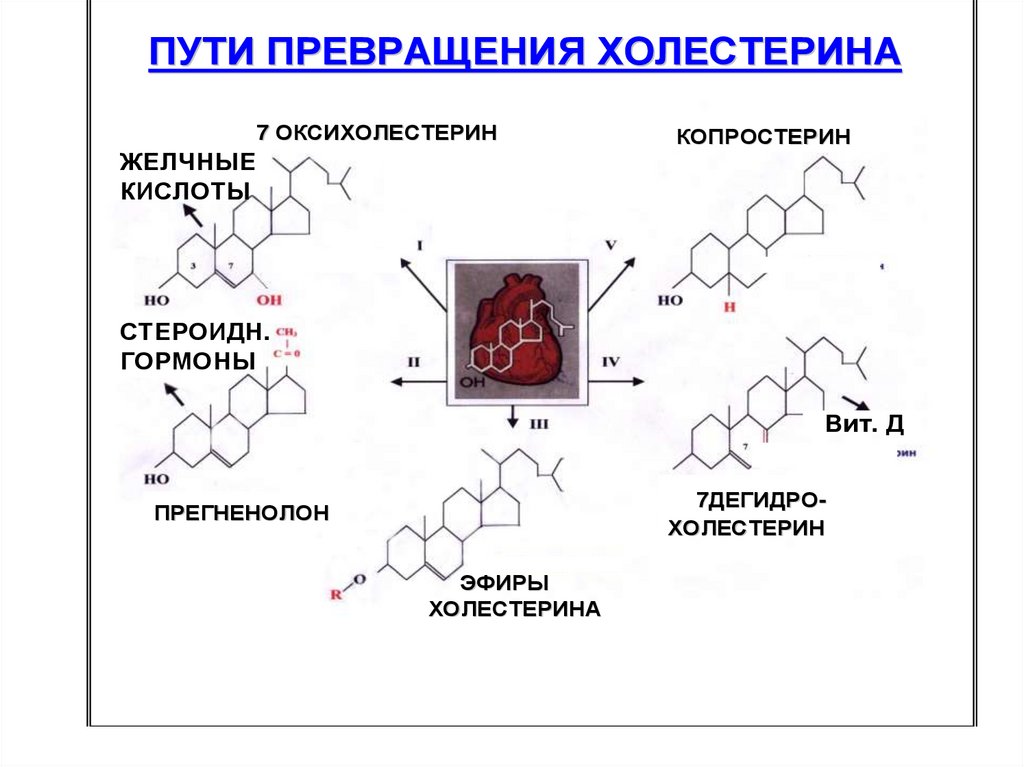
ПУТИ ПРЕВРАЩЕНИЯ ХОЛЕСТЕРИНА7 ОКСИХОЛЕСТЕРИН
ЖЕЛЧНЫЕ
КИСЛОТЫ
КОПРОСТЕРИН
СТЕРОИДН.
ГОРМОНЫ
Вит. Д
7ДЕГИДРОХОЛЕСТЕРИН
ПРЕГНЕНОЛОН
ЭФИРЫ
ХОЛЕСТЕРИНА
16
28.
Пути превращенияХС
1. Окисление. В печени ХС окисляется в желчные кислоты (80%), в стероидные гормоны (3%).
В результате - повышается растворимость ХС в
воде, что способствует его выведению из организма.
29.
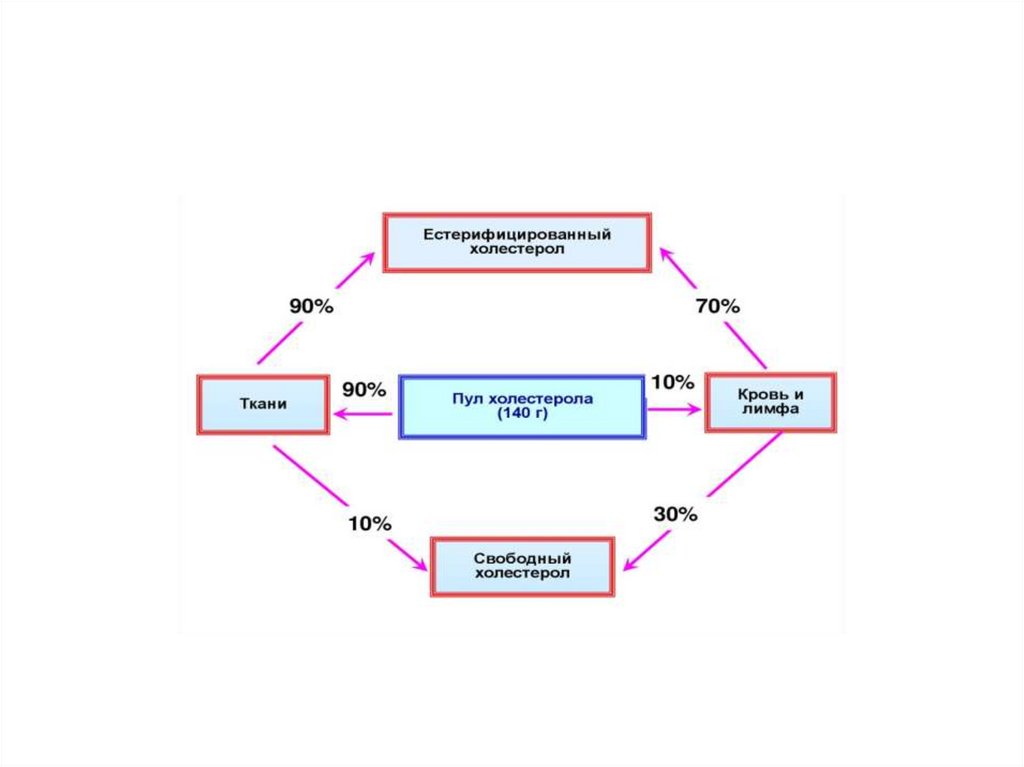
2. Этерификация. Образование эфиров ХС.ЭХС составляют около 10% от общего количества ХС в организме. При этом растворимость уменьшается и это приводит к его накоплению. У новорожденных относительно низкий
коэффициент этерификации по сравнению с
более старшим возрастом детей = 0,58-1
ммоль/л, что связано с дефицитом ПНЖК.
3. Дегидрирование. В положении 7, 8 образуются двойные связи, что приводит к образованию 7-дегидрохолестерола (предшественника витамина Д3).
30. Выведение холестерола
Чтобы поддерживать постоянный уровеньего в организме необходимо выводить 1,5г в
сутки. 1г в сутки окисляется в желчные кислоты, 200-300 мг в сутки с калом в виде копростанола, 100 мг со слущенным эпителием, 40
мг - на синтез стероидных гормонов, с мочой
1-2 мг в сутки.
31. Патология обмена ХС
Гиперхолестеролемия – повышение уровняХС в крови. Факторы риска:
- стресс;
- гиперкалорийное питание (избыточное поступление углеводов, жиров);
- гиподинамия;
- курение;
- сопутствующие заболевания (гипертоническая болезнь, СД, гипотиреоз, ожирение)
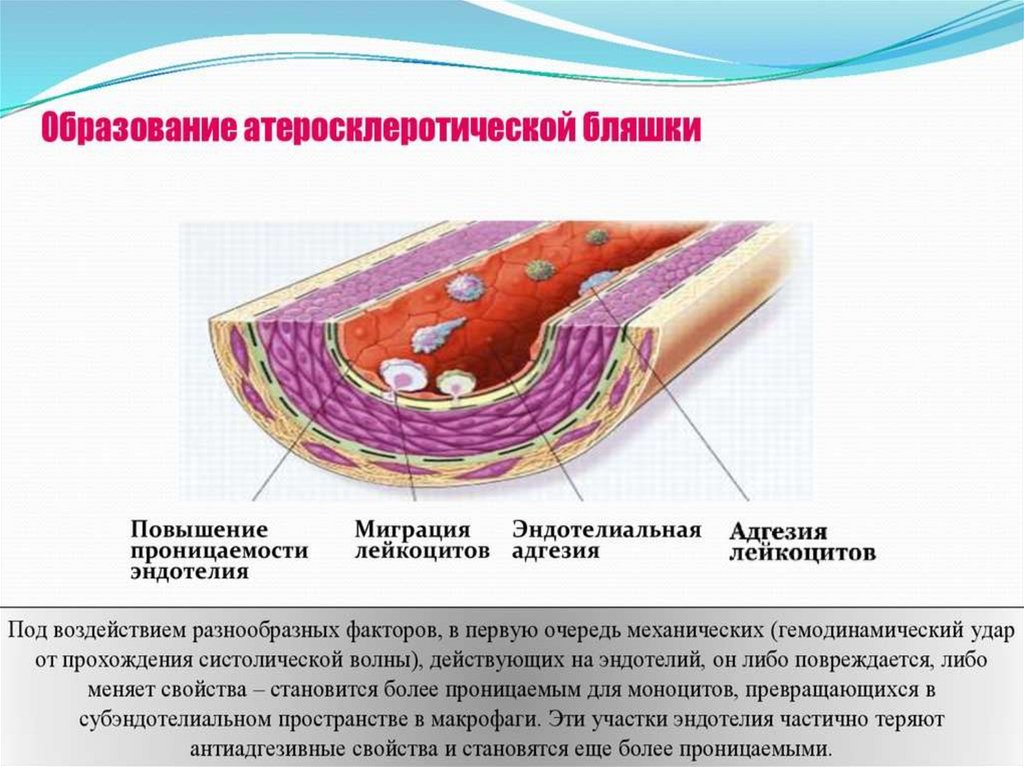
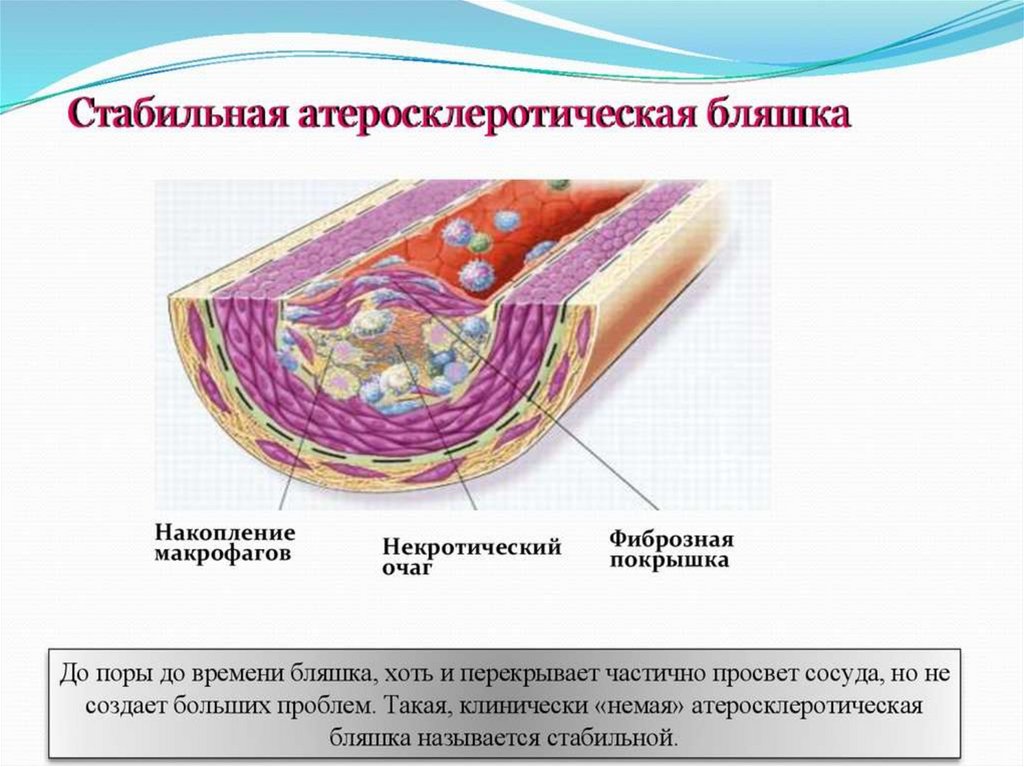
32. Атеросклероз
Заболевание, связанное с накоплениемхолестерола и его эфиров в ЛПНП, в поврежденном эндотелии сосудов.
В области бляшки часто образуются тромбы, суживающие просвет сосуда, что приводит к острому нарушению кровообращения и
развитию инфаркта миокарда, инсульта.
33.
34.
Непосредственой биохимической причинойатеросклероза является модификация ЛП в
зоне воспаления, в основном окислением, гликозилированием. Окисление ЛПНП нарастает
при недостаточной активности антиоксидантных систем –авитаминозах Е и С, нехватке металлов (железо, селен, медь, цинк), входящих в
состав антиоксидантных ферментов (каталазы,
пероксидазы, супероксиддисмутазы).
Гликозилирование белков ЛПНП увеличивается при СД. Такие модифицированные ЛП
теряют способность связываться с апоВ-100рецептором и проникать в клетки-мишени и, в
результате,накапливаются в крови и в интиме
сосудов.
35.
1736.
37.
38.
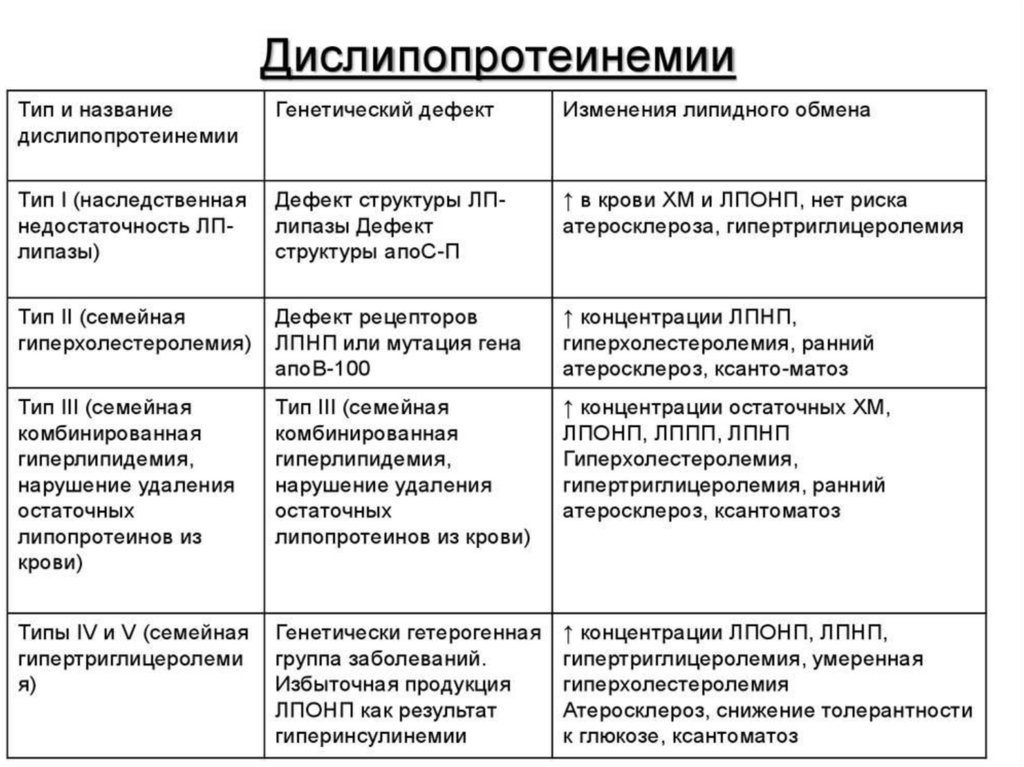
39. Дислипопротеинемии – нарушения обмена ЛП.
Причины: изменение активности ферментов обмена ЛП, снижение рецепции ЛП наклетках, нарушения синтеза апобелков.
Тип I. Гиперхиломикронемия
Обусловлена генетической недостаточностью липопротеинлипазы.
Проявляется в раннем возрасте ксантоматозом и гепатоспленомегалией в результате
отложения липидов в коже, печени и селезенке.
40.
Тип II. Гипер-β-липопротеинемияПодтип IIа (семейная гиперхолестеролемия).
Причины: мутации,приводящие к нарушению синтеза ЛПНП, апо-В-100, рецепторов.
Уровень ХС-12-20 ммоль/л.
Проявляется атеросклеротическими нарушениями и заканчивается летальным исходом
от инфаркта миокарда в молодом возрасте.
41.
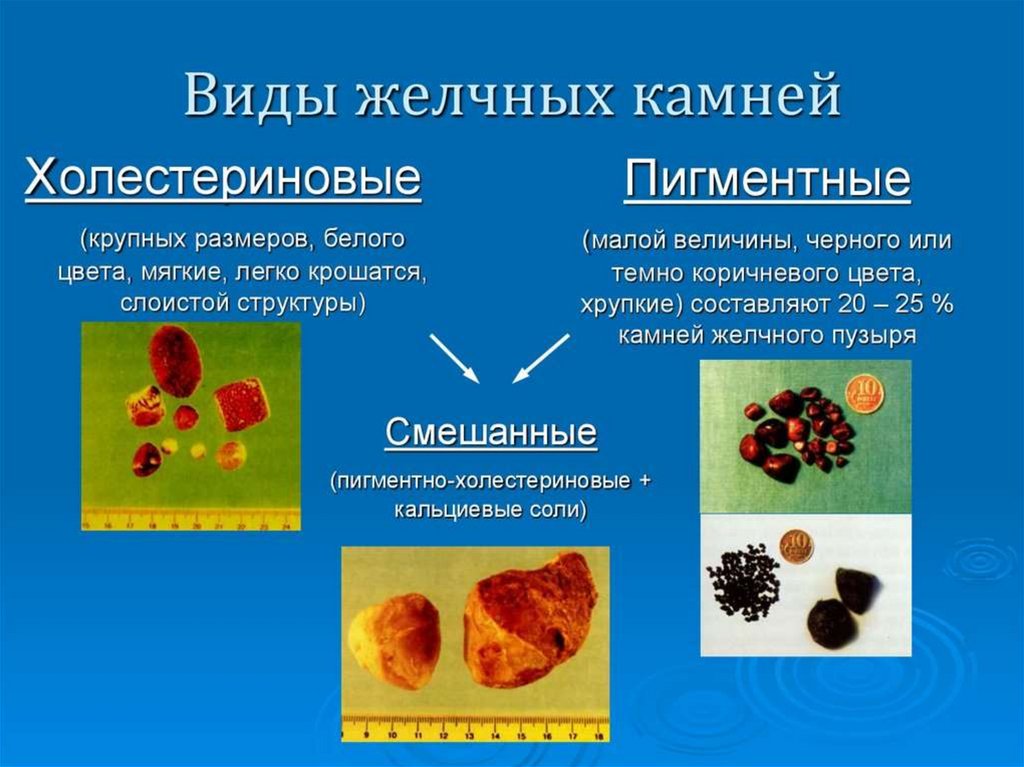
42. Желчнокаменная болезнь
Заболевание, при котором в желчном пузыреобразуются камни, основу которых составляет
ХС. Выделение ХС в желчь сопровождается
пропорциональным выделением желчных кислот и ФЛ, удерживающих гидрофобные молекулы ХС в желчи в мицеллярном состоянии.
Если эти пропорции нарушены, то ХС начинает
осаждаться в желчном пузыре, образуя вязкий
осадок, который постепенно становится твердым. Камни могут состоять не только из ХС
(белого цвета), но и из смеси ХС, билирубина,
белков, кальция – смешанные (коричневого
цвета).
43.
Причины: гиперкалорийное питание, избыток ХС в пище, застой желчи, нарушениекишечно-печеночной циркуляции желчных
кислот, а также их синтеза, инфекции желчного пузыря.
44.
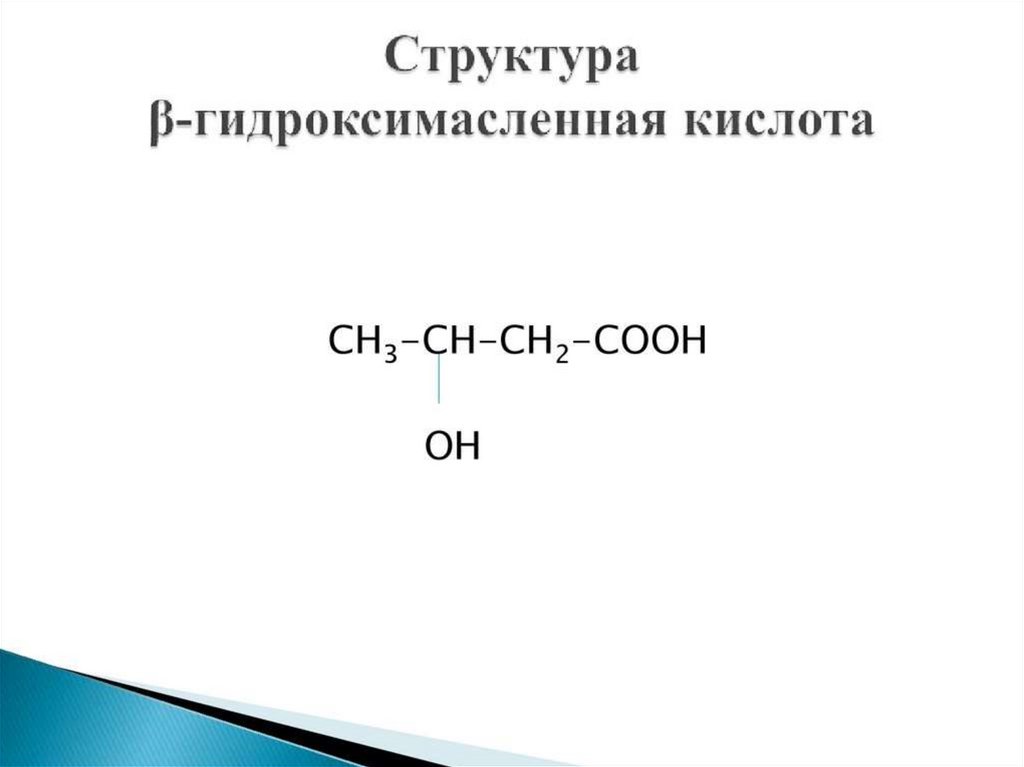
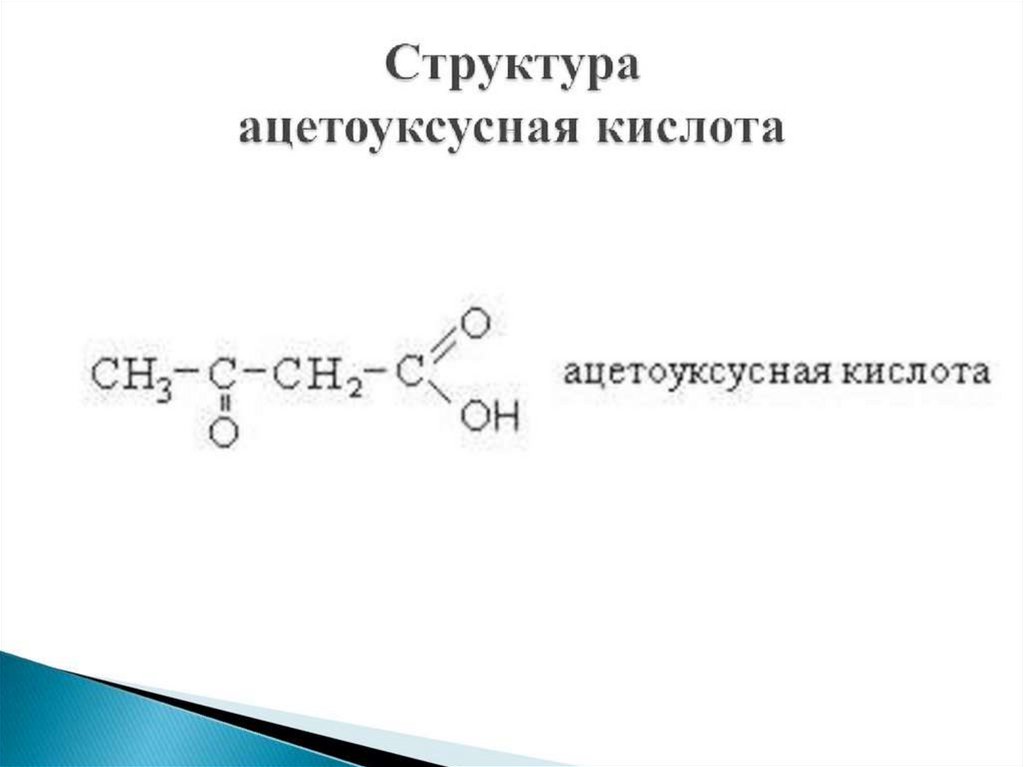
45. Метаболизм кетоновых тел
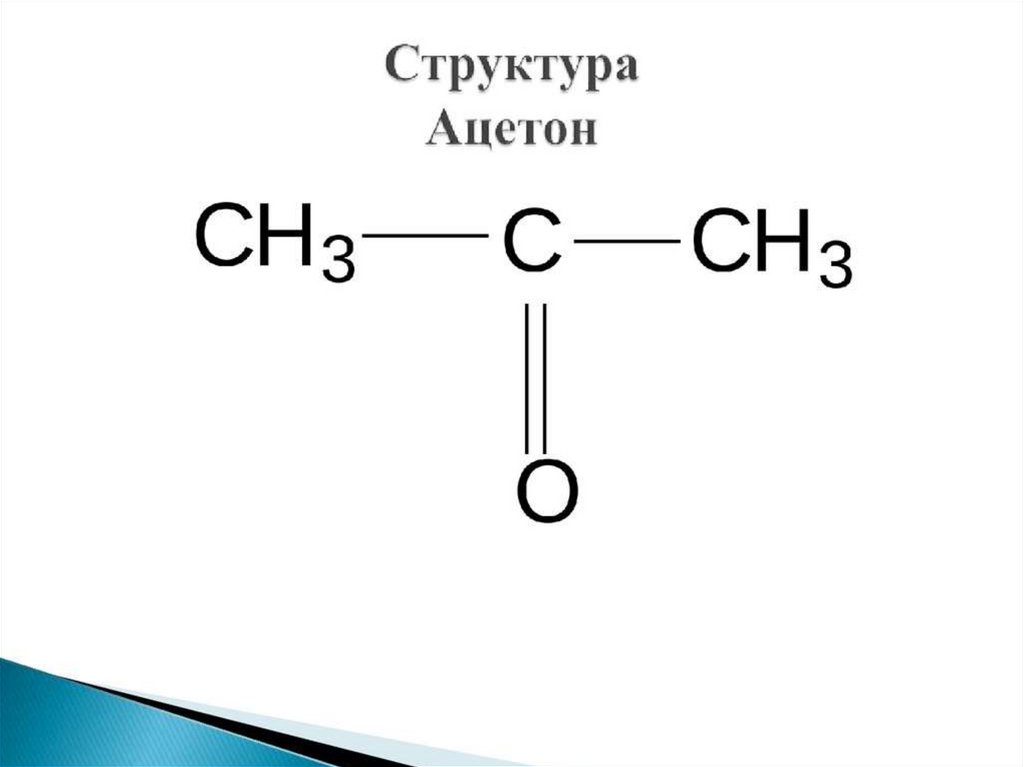
Кетоновые тела:• Ацетоуксусная кислота
• β оксимасляная (β оксибутират)
• Ацетон
В норме 0,9-1,7 ммоль/л
46.
47.
48.
49.
50.
51.
52.
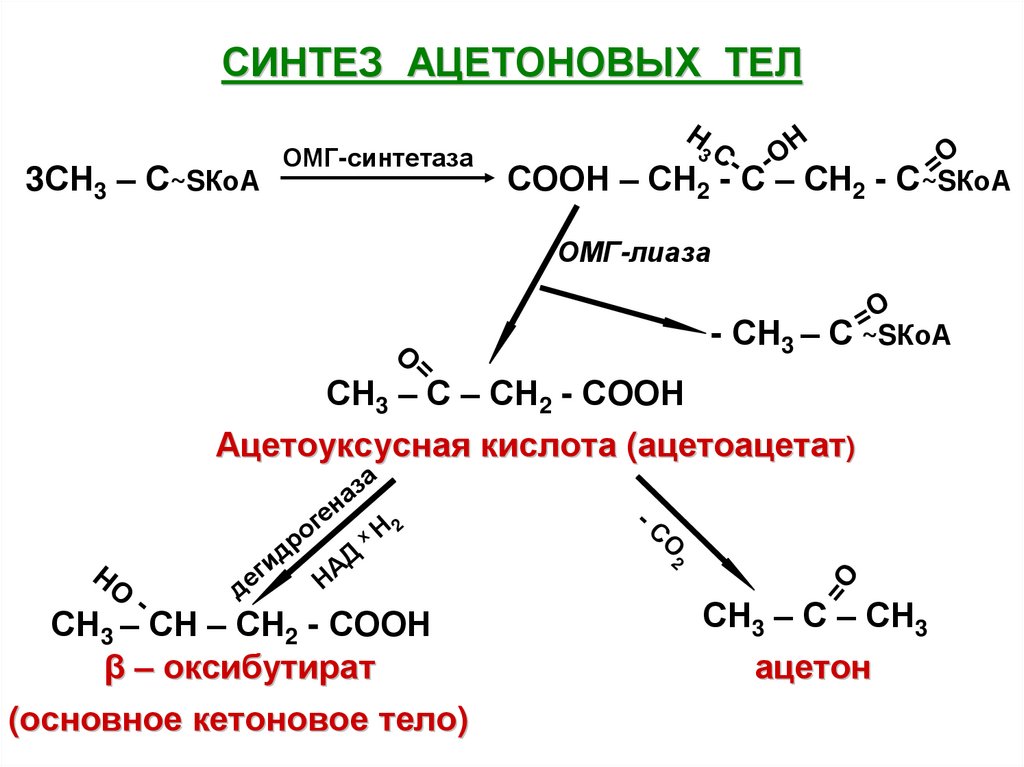
СИНТЕЗ АЦЕТОНОВЫХ ТЕЛ3СН3 – С~SКоА
ОМГ-синтетаза
Н
3С
Н
О
-
=О
СООН – СН2 - С – СН2 - С~SКоА
-
ОМГ-лиаза
=О
- СН3 – С ~SКоА
=О
СН3 – С – СН2 - СООН
Ацетоуксусная кислота (ацетоацетат)
О
Н
СН3 – СН – СН2 - СООН
β – оксибутират
(основное кетоновое тело)
-С
О
2
=О
за
а
ен
г
2
о
х Н
р
Д
ид
А
г
Н
де
СН3 – С – СН3
ацетон
53.
В качестве источника энергии кетоновые тела используются клетками всех тканей, кромепечени и эритроцитов. Активно их потребляет
миокард и кора надпочечников.
В цитозоле β-оксибутират окисляется, образующийся ацетоацетат проникает в митохондрии и превращается в ацетил-SКоА, который
сгорает в ЦТК и ЦПЭ, что приводит к образованию энергии.
54.
Стимулом для повышенного образованиякетоновых тел является поступление большого количества ВЖК в печень, образующихся в
результате липолиза, который активируется
при:
- длительном голодании;
- СД l типа;
- интенсивных физических нагрузках;
- алкогольном отравлении;
- употреблении жирной пищи.
55.
56.
57.
У детей до 7 лет под влиянием различныхфакторов (краткосрочное голодание, инфекции, эмоциональное возбуждение) повышается синтез кетоновых тел и может легко
возникать кетоацидоз, сопровождающийся
неукротимой рвотой ("ацетонемическая
рвота"). Причиной этому служит неустойчивость углеводного обмена и малые запасы
гликогена у детей, что усиливает липолиз в
адипоцитах, накопление ВЖК в крови и, следовательно, кетогенез в печени.
58. Сфинголипидозы
Это генетические наследственные заболевания, когда из-за отсутствия ферментов происходит неполное расщепление сфинголипидов.59.
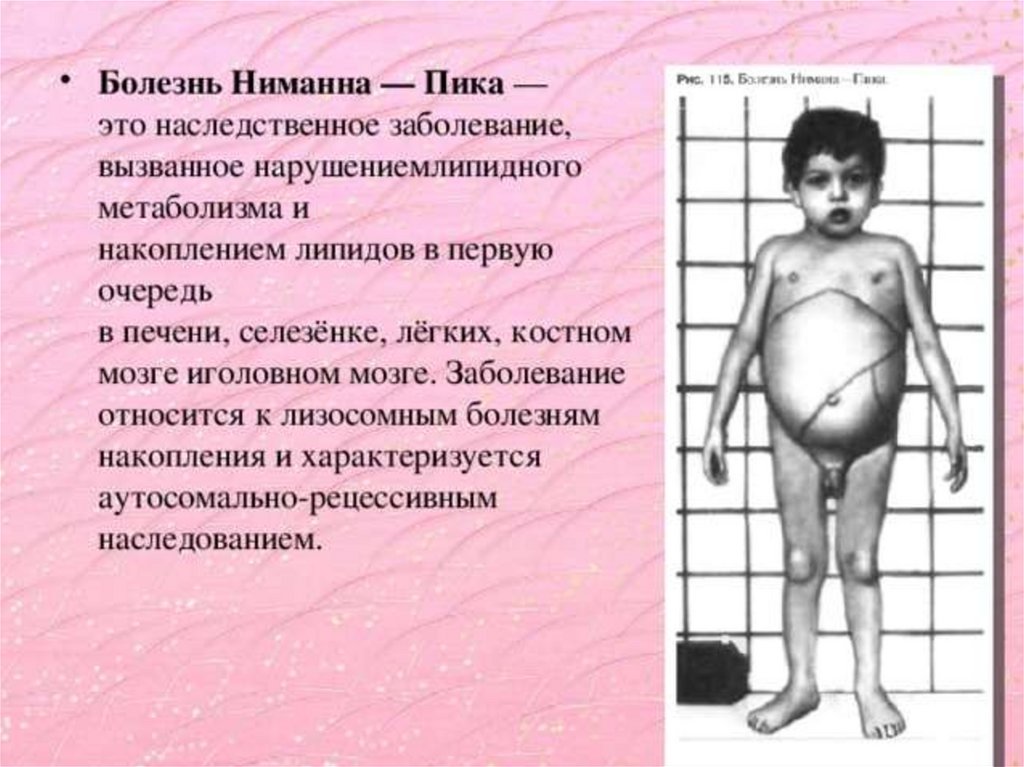
Болезнь Нимана – Пика.Причина: отсутствие фермента сфингомиелиназы.
Проявления: задержка роста, умственного развития, гепатоспленомегалия, частые респираторные инфекции и слепота (вишнево красное
пятно на сетчатке глаз в 50% случаев). Смерть
в возрас-те 3-4х лет.
Болезнь Тея-Сакса.
Причина: отсутствие фермента ганглиозидазы.
Проявления: мышечная гипотрофия, неврологические изменения, полная слепота, а после
года – макроцефалия.
60.
61.
62.
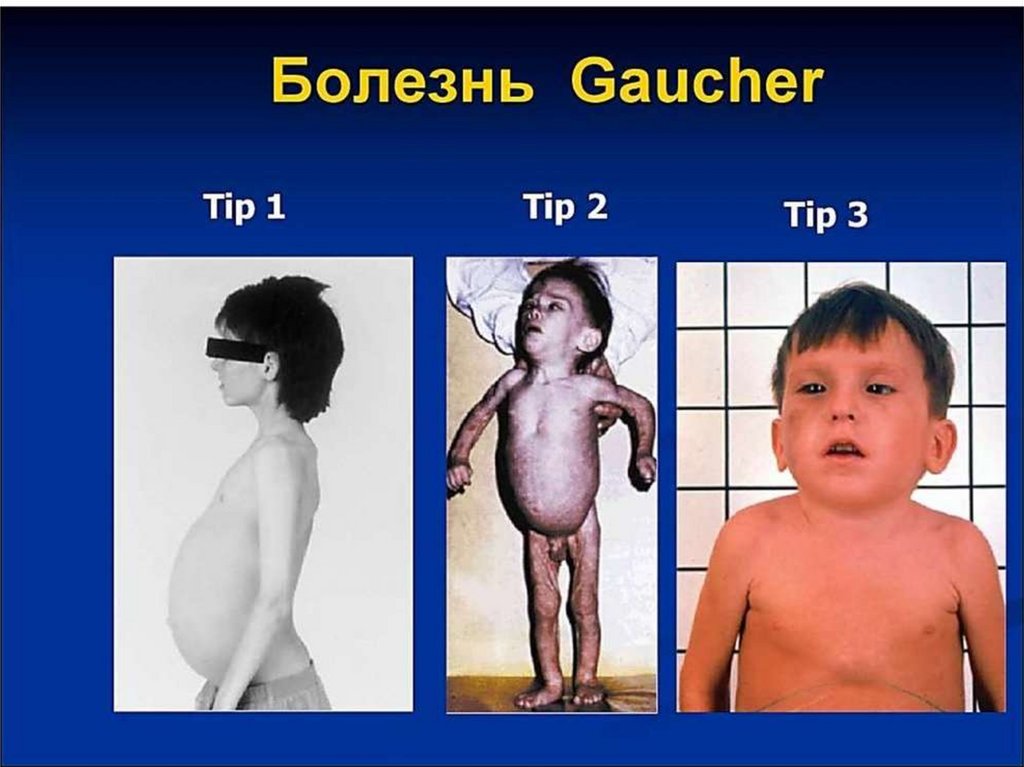
Болезнь Гоше.Причина: отсутствие фермента цереброзидазы.
Существует несколько форм. У детей, как
правило сопровождается задержкой роста,
умственного развития, неврологическими
расстройствами, заканчивающиеся летальным исходом.
У взрослых – отложением во внутренних
органах и костях цереброзидов.