













































































 biology
biologySimilar presentations:










Импринтинг и наследственная патология у человека
1.
ЭПИГЕНЕТИКАЧасть 2
ИМПРИНТИНГ И НАСЛЕДСТВЕННАЯ
ПАТОЛОГИЯ У ЧЕЛОВЕКА
проф. Дмитрий Залетаев
zalnem@mail.ru
кафедра медицинской генетики ИВиДПО ФГБНУ «МГНЦ»
2.
Геномный импринтинг - эпигенетический механизм регуляцииэкспрессии гомологичных генов в процессе развития организма в
зависимости от родительского происхождения гена, хромосомы
или генома.
Эпигенотип (импринт) - совокупность модификаций, которые
по-разному маркируют родительские аллели и обеспечивают
моноаллельный характер экспрессии импринтированных генов на
хромосомах отцовского или материнского происхождения.
Импринтированный ген - ген, который дифференциально
экспрессируется в зависимости от материнского или отцовского
происхождения. Импринтированные гены в диплоидной клетке
млекопитающих обычно экспрессируются только с одного аллеля.
3.
Геномный импринтингЭпигенетический феномен, дифференцирующий
материнские и отцовские копии генов в геноме организма.
Подобная дифференцировка обусловливает моноаллельную
экспрессию импринтированных генов в зависимости от пола
родителя, их передавшего.
Helen Crouse
В 1960 году был предложен термин «хромосомный
импринтинг»
У мушки Sciara Coprophila зигота содержит два набора аутосом
и три половых хромосомы, две из которых отцовские. В
течение эмбриогенеза одна отцовская Х хромосома теряется
из эмбрионов обоих полов (у самки - AmApXmXp) и обе
отцовские Х хромосомы - из эмбрионов самца (AmApXm). Это
свидетельствует о том, что хромосомы каким-то образом
маркируются (или импринтируются) перед слиянием гамет в
соответствие со своим родительским происхождением.
4.
5.
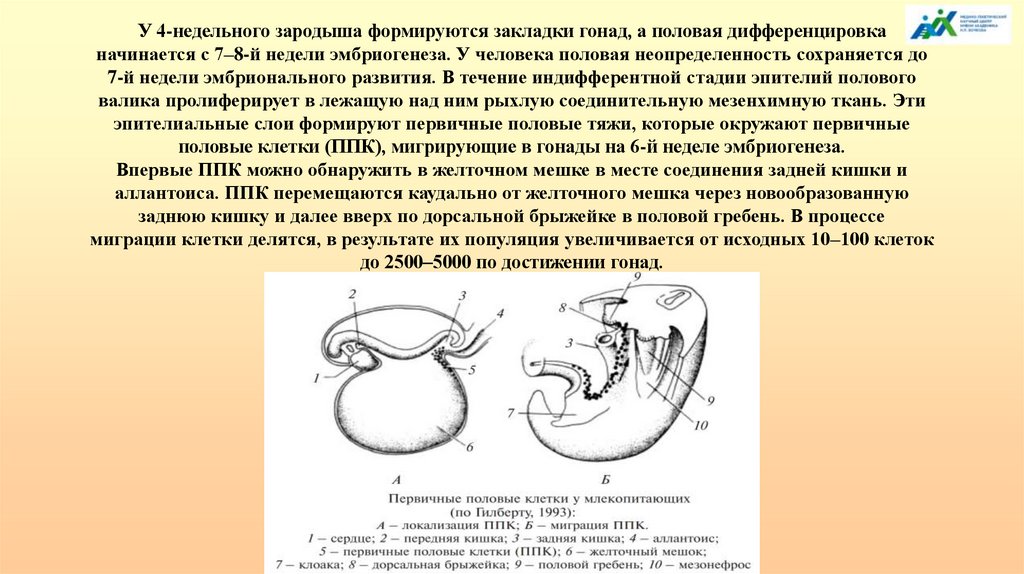
У 4-недельного зародыша формируются закладки гонад, а половая дифференцировканачинается с 7–8-й недели эмбриогенеза. У человека половая неопределенность сохраняется до
7-й недели эмбрионального развития. В течение индифферентной стадии эпителий полового
валика пролиферирует в лежащую над ним рыхлую соединительную мезенхимную ткань. Эти
эпителиальные слои формируют первичные половые тяжи, которые окружают первичные
половые клетки (ППК), мигрирующие в гонады на 6-й неделе эмбриогенеза.
Впервые ППК можно обнаружить в желточном мешке в месте соединения задней кишки и
аллантоиса. ППК перемещаются каудально от желточного мешка через новообразованную
заднюю кишку и далее вверх по дорсальной брыжейке в половой гребень. В процессе
миграции клетки делятся, в результате их популяция увеличивается от исходных 10–100 клеток
до 2500–5000 по достижении гонад.
6.
7.
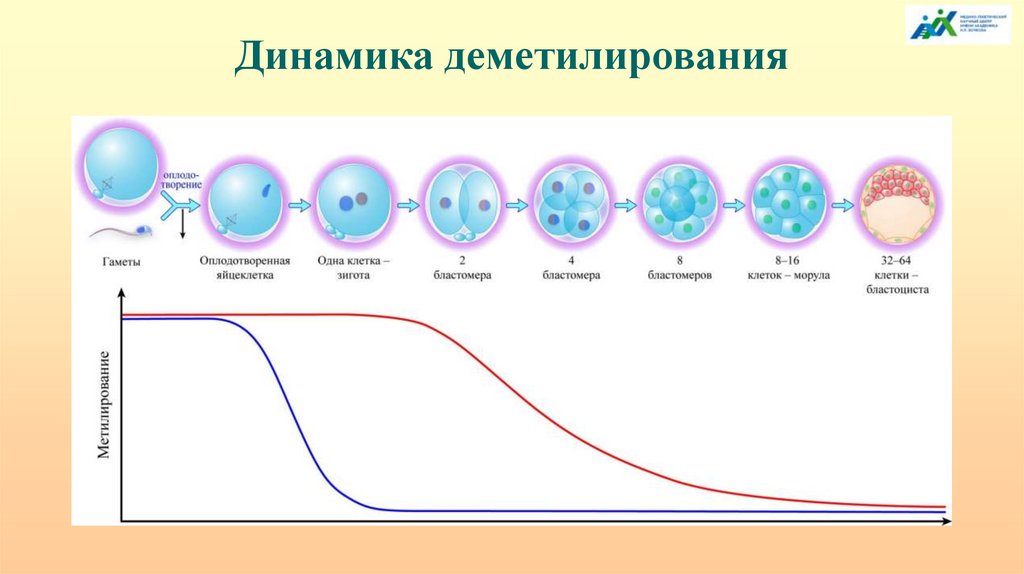
Динамика деметилирования8.
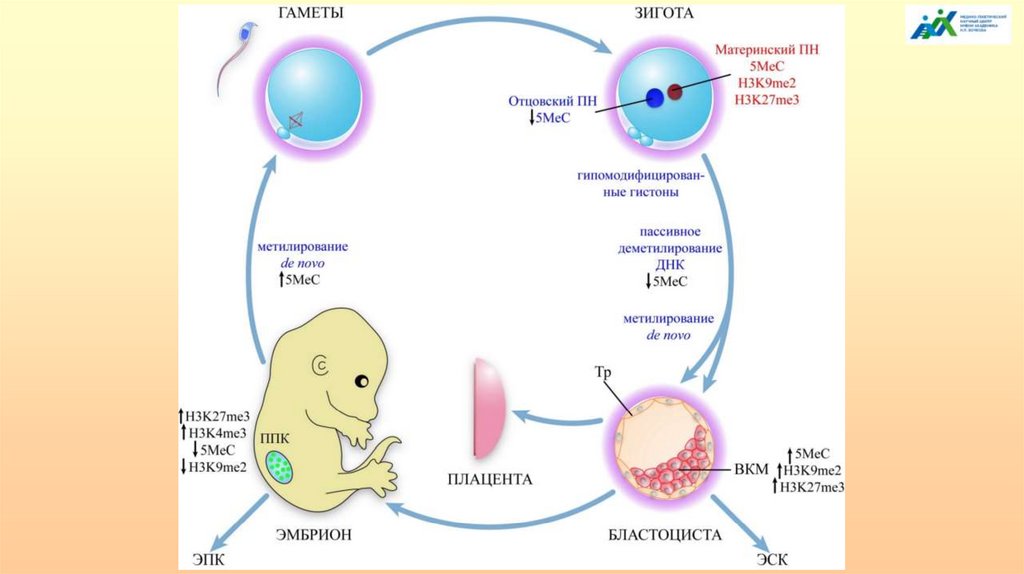
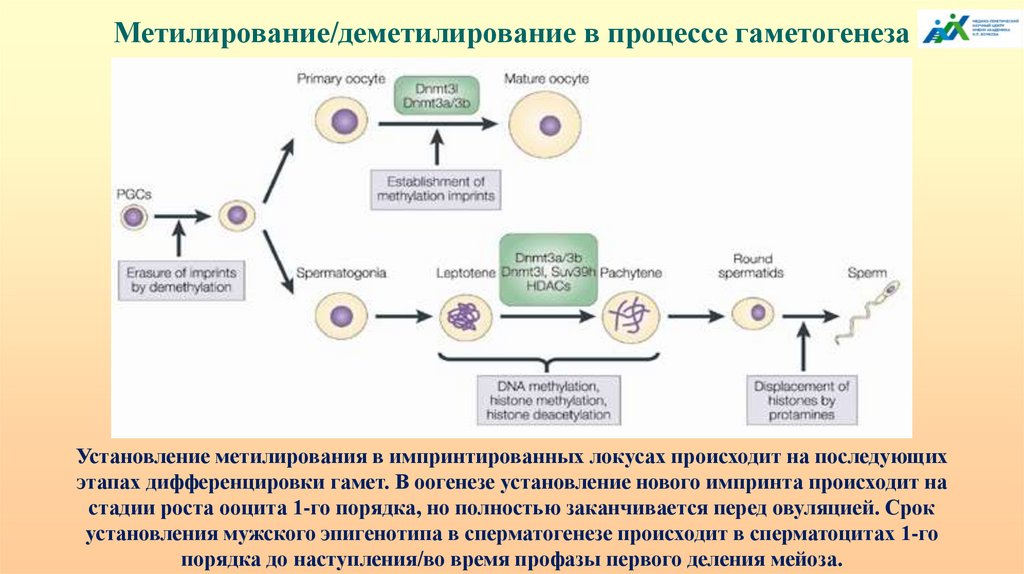
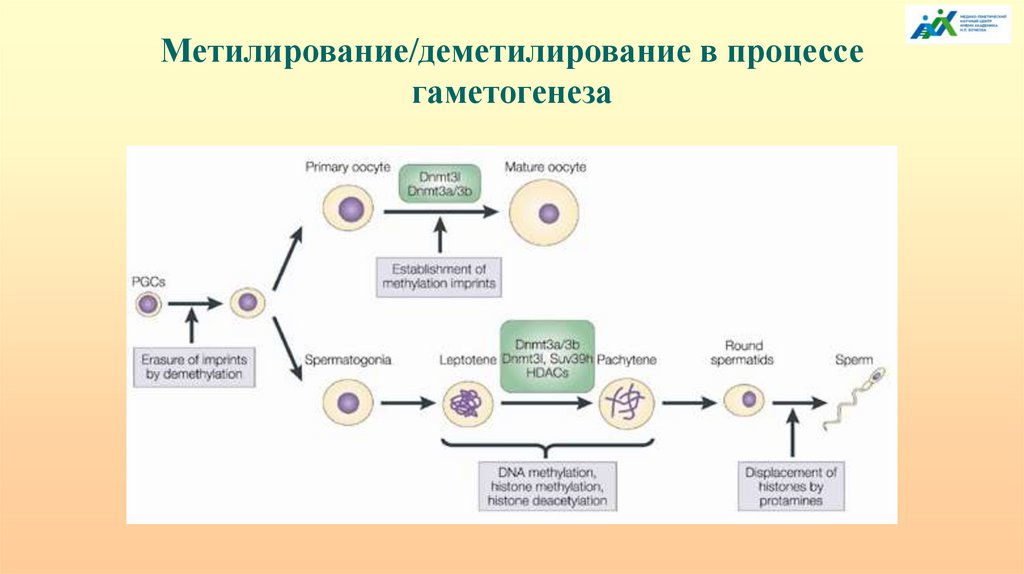
Метилирование/деметилирование в процессе гаметогенезаУстановление метилирования в импринтированных локусах происходит на последующих
этапах дифференцировки гамет. В оогенезе установление нового импринта происходит на
стадии роста ооцита 1-го порядка, но полностью заканчивается перед овуляцией. Срок
установления мужского эпигенотипа в сперматогенезе происходит в сперматоцитах 1-го
порядка до наступления/во время профазы первого деления мейоза.
9.
Механизмом распознавания в отцовском и материнском гаметогенезепоследовательностей, которые должны быть по-разному
метилированы, возможно, является существование различных
вариантов DNMT1. В результате альтернативного сплайсинга 5’экзонов гена DNMT1 образуются два варианта, один из которых
реализуется в оогенезе, а второй — в сперматогенезе.
Оказалось, что в сперматоцитах 1-го порядка на стадии профазы
первого деления мейоза содержится та сплайс-форма DNMT1 — мРНК,
которая не транслируется и не дает белкового продукта. Ооцитспецифический сплайсинг гена DNMT1 сопровождается продукцией
укороченных с N-конца молекул фермента, которые в большом
количестве присутствуют в растущем ооците 1-го порядка и
специфически метилируют будущие материнские аллели
импринтированных генов.
10.
11.
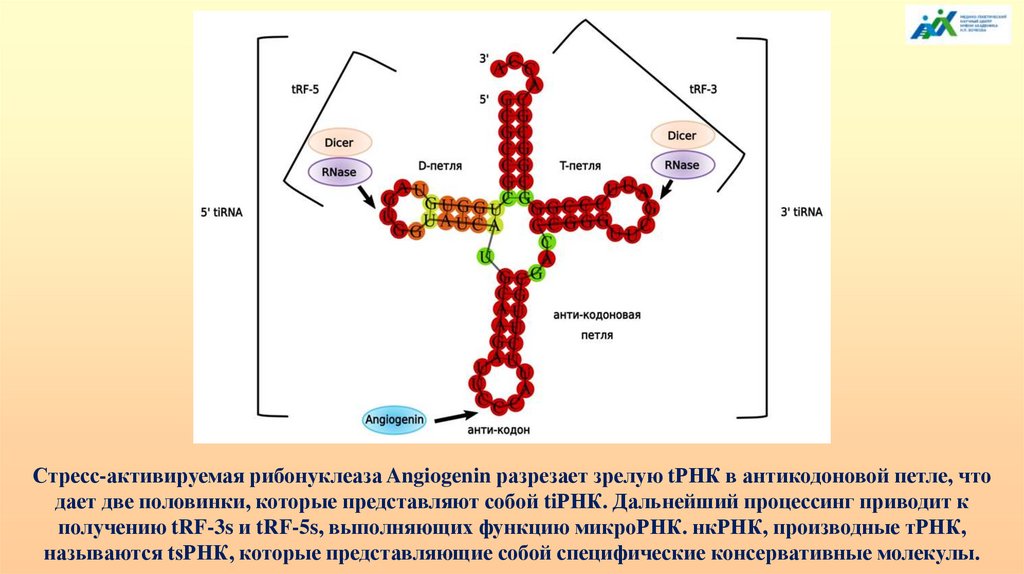
Стресс-активируемая рибонуклеаза Angiogenin разрезает зрелую tРНК в антикодоновой петле, чтодает две половинки, которые представляют собой tiРНК. Дальнейший процессинг приводит к
получению tRF-3s и tRF-5s, выполняющих функцию микроРНК. нкРНК, производные тРНК,
называются tsРНК, которые представляющие собой специфические консервативные молекулы.
12.
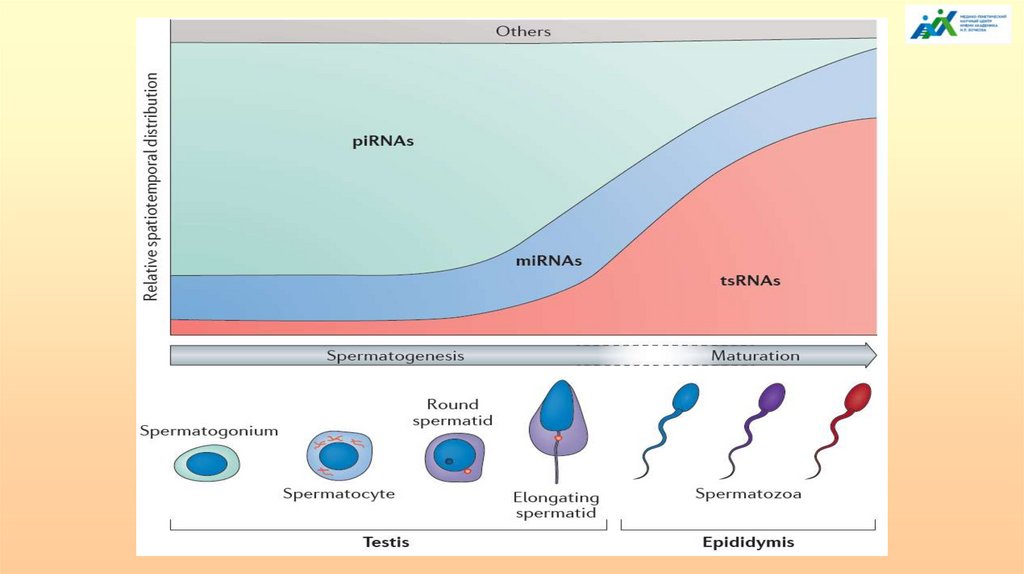
- Несколько сотен важных для развития малых РНК соматического происхождениядоставляются в сперматозоид специальным классом экстраклеточных везикул
эпидидимиса, называемых эпидидимосомами. Эпидидимис является ключевым
участником формирования эпигенома спермы, т.к. может включать РНК из экзосом
соматического происхождения.
- Состав РНК сперматозоидов отражает образ жизни и несет в себе «память»
отцовского опыта; эта «память» на основе РНК передается потомству как
приобретенные характеристики, способные повлиять на здоровье и общую
биологическую судьбу потомства.
- Недавние эксперименты показали потенциал РНК сперматозоидов в качестве
трансгенерационных модификаторов, свойства которых появились в ответ на
условия окружающей среды или стресса, включая диету, сигаретный дым,
чувствительность к запаху и когнитивные и поведенческие условия.
- В результате убедительных экспериментов показано, что нкРНК является
трансгенерационным модификатором - потомство из нормальных зигот, в которые
вводили РНК сперматозоидов, повторяет фенотипические черты животных-доноров
РНК.
- РНК, поставляемые сперматозоидами при оплодотворении выполняют
регуляторные функции и ремоделируют профиль экспрессии генов в ранних
эмбрионах.
13.
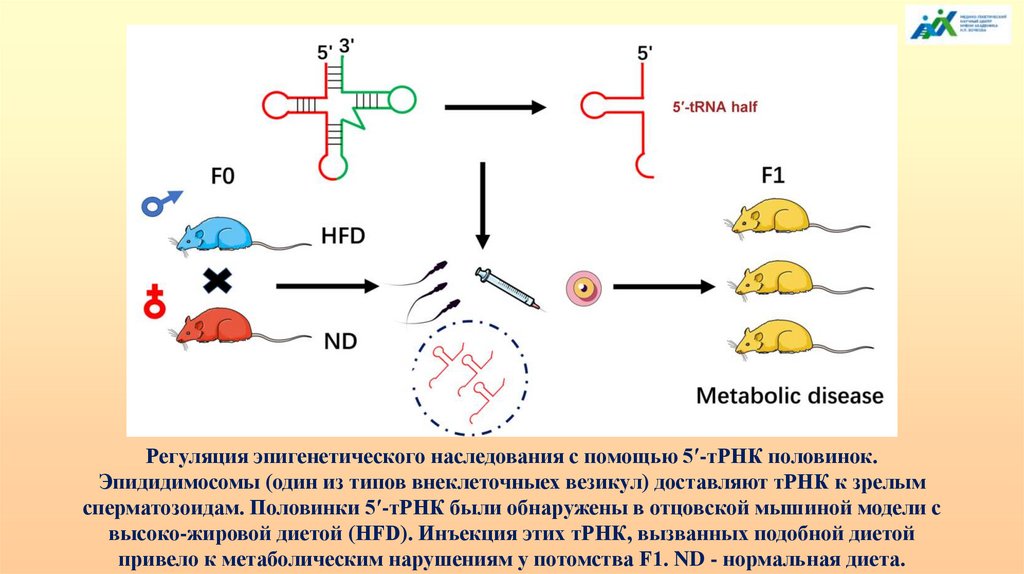
Регуляция эпигенетического наследования с помощью 5′-тРНК половинок.Эпидидимосомы (один из типов внеклеточныех везикул) доставляют тРНК к зрелым
сперматозоидам. Половинки 5′-тРНК были обнаружены в отцовской мышиной модели с
высоко-жировой диетой (HFD). Инъекция этих тРНК, вызванных подобной диетой
привело к метаболическим нарушениям у потомства F1. ND - нормальная диета.
14.
Было показано, что внеклеточные везикулы в эпидидимисе содержатбольшое количество tsРНК, подобно зрелым сперматозоидам, а
эксперименты in vitro показали, что эпидидимосомы могут сливаться и
переносить tsРНК в сперматозоиды. Этот сценарий предполагает
передачу tsРНК между эпидидимисом и сперматозоидом. Подобный
перенос может происходить в семенной жидкости, где внеклеточные
везикулы обогащены miРНК и tsРНК. Более того, обогащение miРНК
и tsРНК также обнаружено в сыворотке крови и в синаптических
везикулах нейронов, которые чувствительны к физиологическим
изменениям при таких состояниях организма, как воспаление,
старение или ограничение калорий.
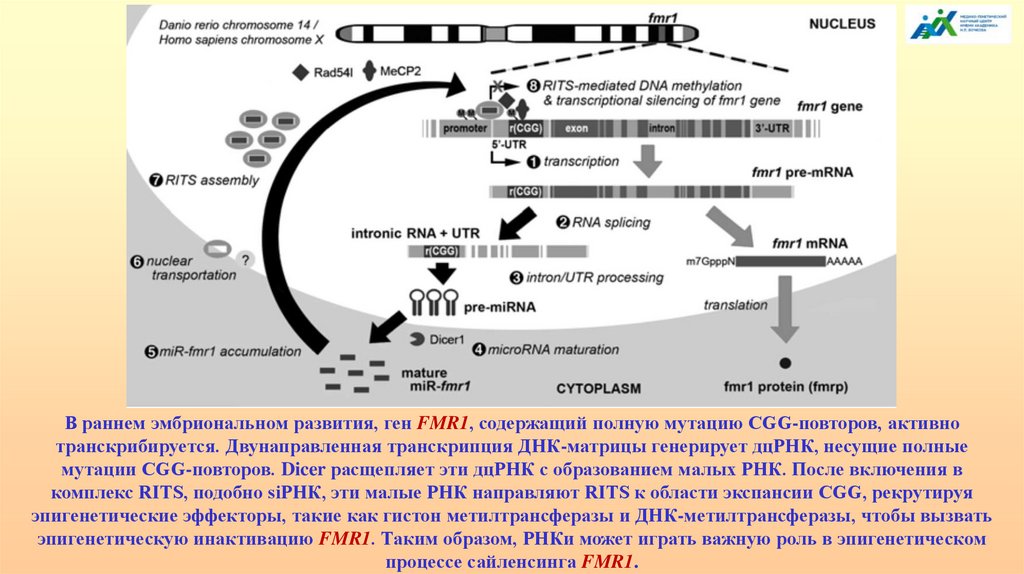
15.
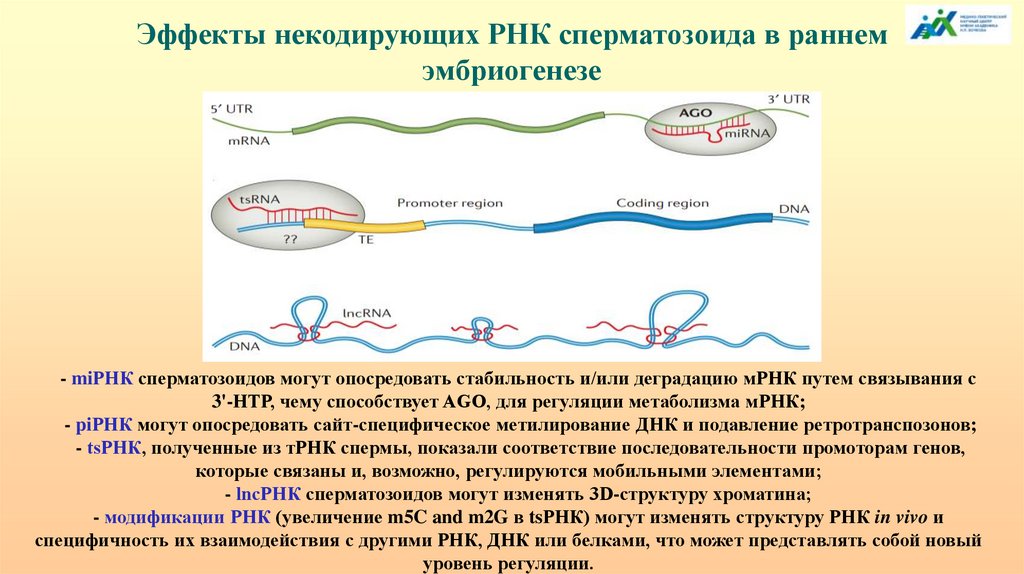
Эффекты некодирующих РНК сперматозоида в раннемэмбриогенезе
- miРНК сперматозоидов могут опосредовать стабильность и/или деградацию мРНК путем связывания с
3'-НТР, чему способствует AGO, для регуляции метаболизма мРНК;
- piРНК могут опосредовать сайт-специфическое метилирование ДНК и подавление ретротранспозонов;
- tsРНК, полученные из тРНК спермы, показали соответствие последовательности промоторам генов,
которые связаны и, возможно, регулируются мобильными элементами;
- lncРНК сперматозоидов могут изменять 3D-структуру хроматина;
- модификации РНК (увеличение m5C and m2G в tsРНК) могут изменять структуру РНК in vivo и
специфичность их взаимодействия с другими РНК, ДНК или белками, что может представлять собой новый
уровень регуляции.
16.
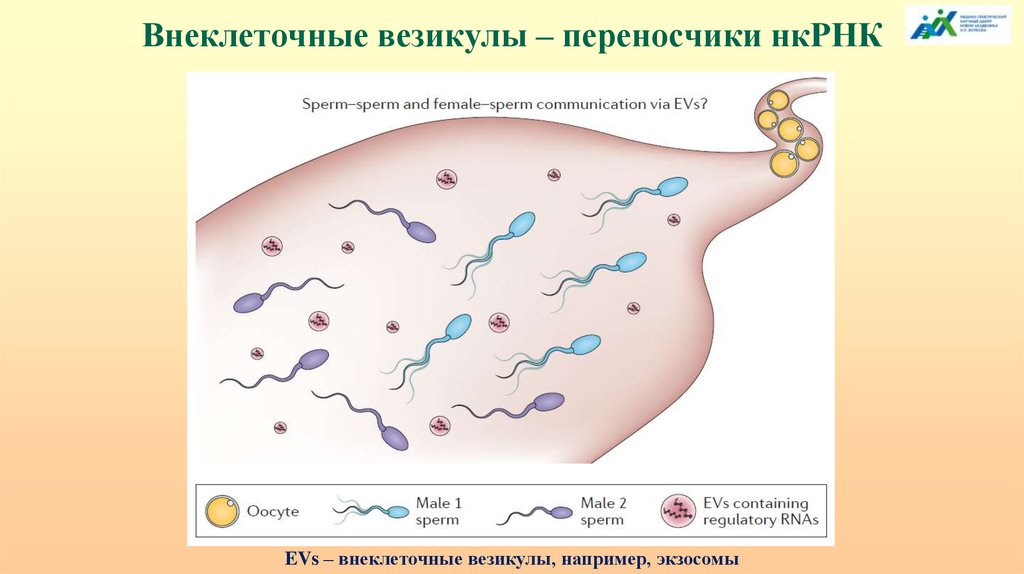
Внеклеточные везикулы – переносчики нкРНКEVs – внеклеточные везикулы, например, экзосомы
17.
Барьер Вейсмана – миф!«Клетки тела не могут передавать информацию половым клеткам. Ни
внешние воздействия, ни приобретенный опыт, ни упражнение органов не
могут приводить к адекватному или отчасти неслучайному изменению
наследственности», 1893 г.
18.
TелегонияВ XIX в. лорд Мортон, близкий друг Ч. Дарвина отважился на биологический
опыт: скрестил чистопородную кобылу с жеребцом-зебры. Потомства не
получилось, однако спустя два года, после скрещивания с самцом своей породы у
кобылы родились жеребята с едва заметными полосами на крупе. Мортон назвал
это явление телегонией. Дарвин считал это проявлением архаичного признака,
присущего предку рода лошадиных.
Tелегония (с древнегреческого τῆλε – «далекий» и γόνος – «рождение») - это
проявление признаков первого самца у потомства в животном мире, даже если при
спаривании в первый раз не наступала беременность. Вера в телегонию в основном
распространена у племенных заводчиков и селекционеров. Известные факты:
- чистопородные собаки и кошки при случке с беспородными, дают в последующем
«плохое» потомство, поэтому даже при единичной вязке, таких животных
«выбраковывают»;
- среди профессиональных голубятников существует жестокий обычай сворачивать
голову породистой самке голубя, если она имела контакт с сизарем - «диким»
представителем семейства голубиных.
19.
А так ли неправ был Ламарк?Эволюционная теория Ламарка больше соответствует определению
«примитивизм», но, в настоящее время, это уже неоламаркизм.
Передача фенотипически значимой эпигенетической информации через половые
клетки может обеспечить механизм для улучшения приспособленности потомства на
основе опыта отцов. Убедительный пример такой передачи выявлен при исследовании
интенсивного заживления ран, наблюдаемого у потомков самцов крыс, подвергшихся
хирургическим повреждениям печени.
20.
Трансгенерационное наследование приобретенных признаков.(Модифицировано из Р. Киплинга, "Дитя слона", 1912)
21.
Эксперимент Конрада Уоддингтона (1953). У взрослых особей дрозофилы, подвергшихсятепловому шоку на стадии куколки, наблюдалась повышенная склонность к развитию крыльев,
лишенных поперечных жилок. Результаты исследования показали, что приобретенный признак
может передаваться практически без изменений в течение 23 поколений.
22.
Гендерная справедливость - не только самцыХорошо известный пример влияния недоедания во время
беременности на метилирование ДНК получен в результате
исследования женщин, подвергшихся воздействию Голландской
Зимы (1944-1945)..
Сначала установили
снижение метилирования импринтированного гена
инсулиноподобного фактора роста IGF2. Затем выявили
аномальное метилирование других генов, кодирующих:
инсулин, гуанин-связывающий белок - GNAS, длинную
некодирующую РНК с материнской экспрессией - MEG3, лептин
– LEP и АТФ-связывающую кассету A1 - ABCA1.
Исследования показали, что эпигенетические нарушения в
ооцитах лежат в основе нездоровья потомства.
На моделях животных было показано, что материнское недоедание во время периконцепции и зачатия
изменяет метилирование ДНК, модификации гистонов и микроРНК плода: - белок-дефицитная диета
нарушает экспрессию DNMT, метилирование ДНК и модификации гистонов у потомства, что приводит
к внутриутробной задержке роста и аномальному метилированию ДНК в плаценте;
- дефицит фолатов во время беременности вызывает гипометилирование ДНК в плаценте, которое
сохраняется у плода;
- цинк-дефицитная диета в течение 3-5 дней до овуляции нарушает глобальное метилирование ДНК
и H3K4me3 в ооцитах.
23.
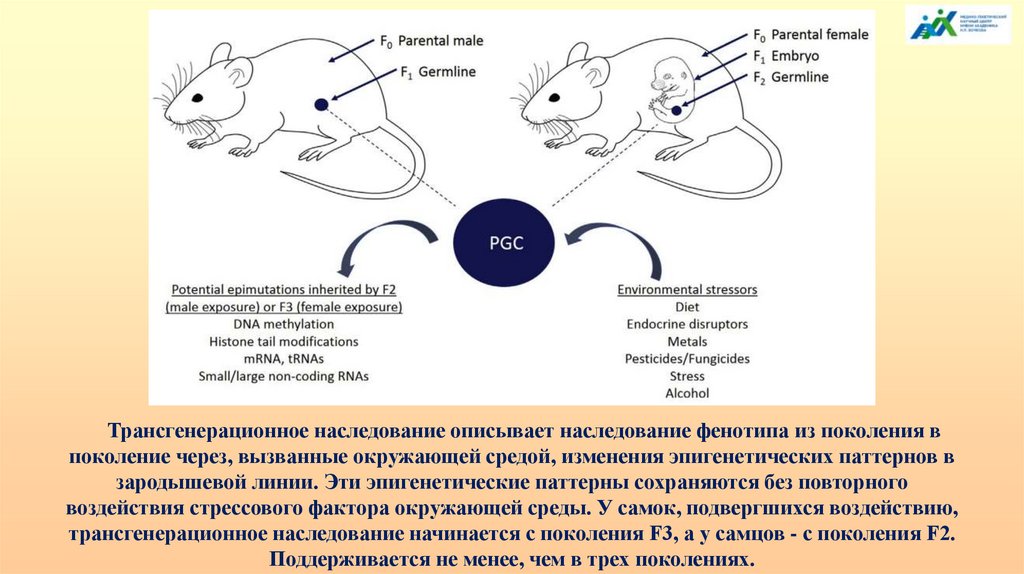
Трансгенерационное наследование описывает наследование фенотипа из поколения впоколение через, вызванные окружающей средой, изменения эпигенетических паттернов в
зародышевой линии. Эти эпигенетические паттерны сохраняются без повторного
воздействия стрессового фактора окружающей среды. У самок, подвергшихся воздействию,
трансгенерационное наследование начинается с поколения F3, а у самцов - с поколения F2.
Поддерживается не менее, чем в трех поколениях.
24.
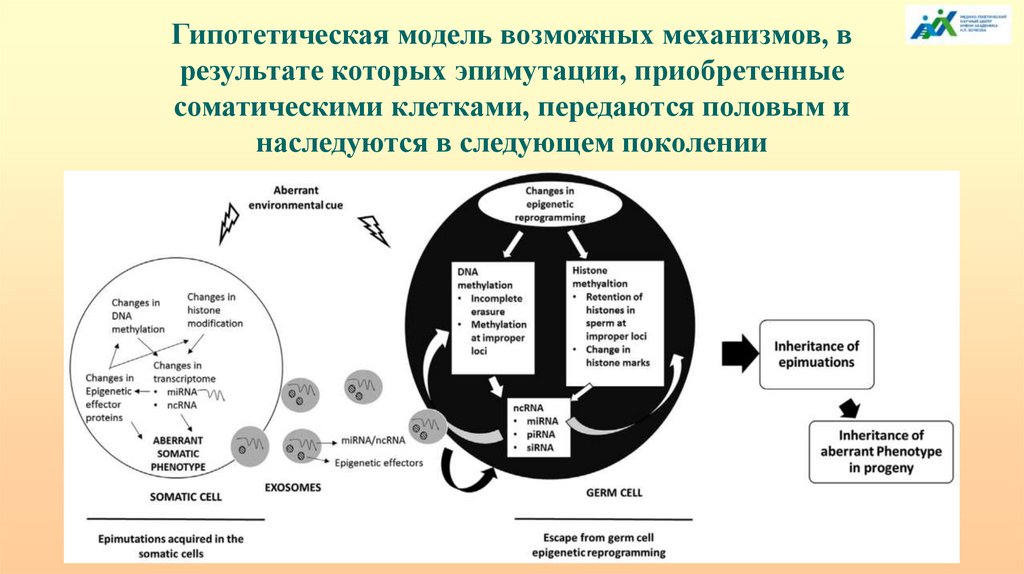
Гипотетическая модель возможных механизмов, врезультате которых эпимутации, приобретенные
соматическими клетками, передаются половым и
наследуются в следующем поколении
25.
Для нормального развития необходим равный вклад обоихродителей
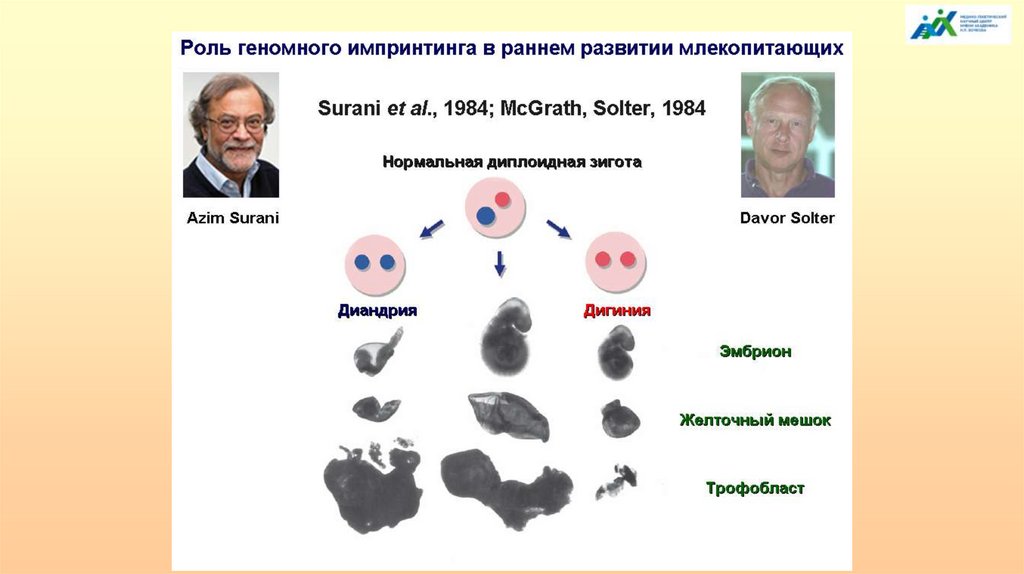
Трансплантация пронуклеусов.
Андрогенетические зиготы - нормальное развитие зародышевых мембран и
плаценты, практически нет развития эмбриональных структур.
Гиногенетические зиготы - нормальное развитие эмбриональных структур и
плохое - зародышевых мембран и плаценты.
Патология у человека.
Пузырный занос - гидатиформный моль. Нет эмбриональных структур - два
набора отцовских хромосом (22+Х)
Тератома - эмбриональная опухоль, включающая все три эмбриональных слоя
и отсутствие плацентарной ткани - два набора материнских хромосом.
Триплоидия.
2n - отец + n - мать -> андроид: большая кистозная плацента, у плода: большая
голова, маленькое веретенообразное тело, синдактилия, отставание в росте и
развитии. Если плод рождается, то, как правило, есть мозаицизм.
2n - мать + n - отец -> гиноид: недоразвитая плацента, клеточная масса,
эмбрион и плод не развивается.
26.
27.
Наши основоположникиА.П. Дыбан, 1922-2002
В.С. Баранов, 1940-2022
А.П. Дыбан и В.С.Баранов внесли значительный вклад в экспериментальную цитогенетику
развития млекопитающих, создали новую технику хромосомного анализа ранних зародышей
млекопитающих, что позволило детально проанализировать влияние числовых и структурных
хромосомных аберраций на ключевые звенья эмбриогенеза. На основании полученных данных
удалось сформулировать новые представления о роли различных хромосом в раннем развитии и о
сочетанном участии генов и эпигеномных факторов в контролирующих механизмах раннего
эмбриогенеза млекопитающих.
28.
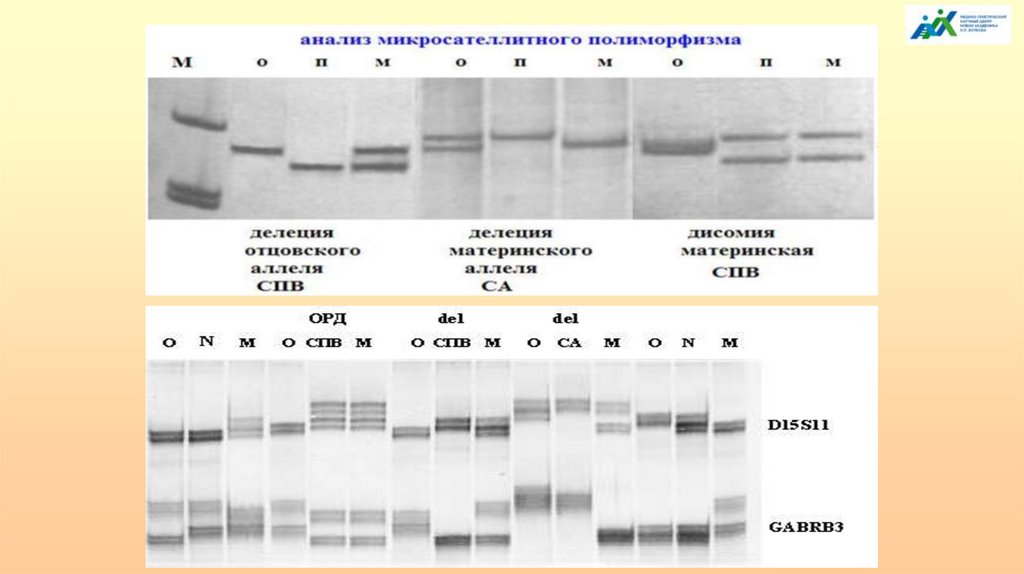
Однородительская дисомияЭрик Энжел, 1980 г.
На мышиных транслокационных гибридах, несущих отдельные хромосомные участки,
имеющие как отцовское, так и материнское происхождение, показано, что, либо потомство
отличается по альтернативным признакам (хромосомы 2 и 11 - гиперактивныгипоактивны, маленькие-большие 70%-130% ), либо не жизнеспособно. У мышей
отцовская дупликация и материнская делеция проксимальной части короткого плеча
хромосомы 6 фенотипически не проявляется, а материнская дупликация и отцовская
делеция летальны на ранних сроках эмбрионального развития. То же с хромосомами 2р и
7р.
В двух случаях муковисцидоза, сопровождавшегося задержкой умственного и
физического развития было показано, что обе хромосомы 7 имели материнское
происхождение.
Частичная три- и тетрасомия по проксимальной части длинного плеча хромосомы 15
приводят, либо к фенотипу с-ма Прадера-Вилли, либо к необычному аномальному
фенотипу.
Частичная трисомия хромосомы 11р , унаследованная от отца приводит к
фенотипическим проявлениям синдрома Видеманна-Беквита, а от матери – к синдрому
Сильвера-Рассела.
29.
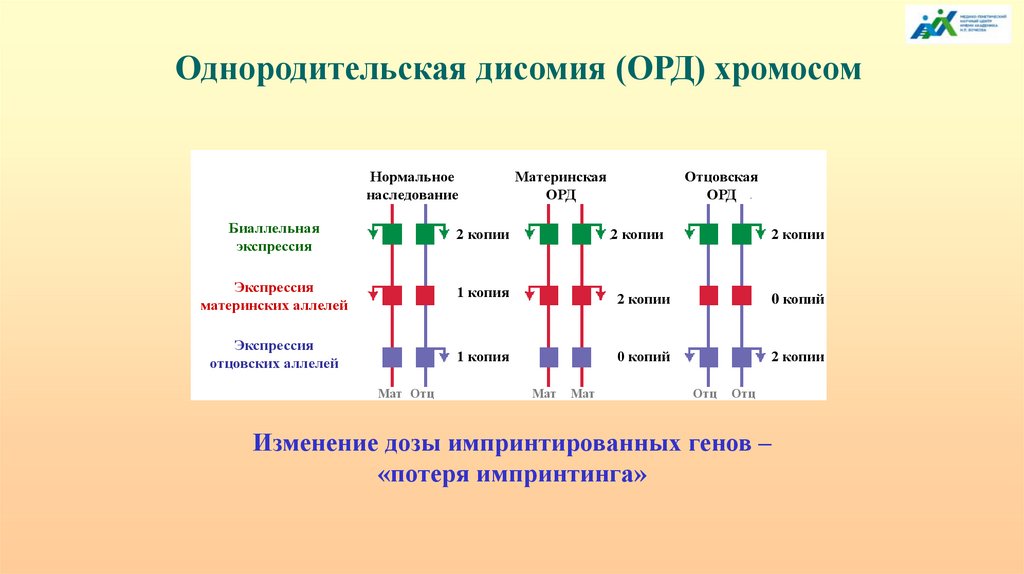
Однородительская дисомия (ОРД) хромосомНормальное
наследование
Материнская
ОРД
Отцовская
ОРД
Биаллельная
экспрессия
2 копии
2 копии
2 копии
Экспрессия
материнских аллелей
1 копия
2 копии
0 копий
Экспрессия
отцовских аллелей
1 копия
0 копий
2 копии
Мат Отц
Мат
Мат
Отц
Отц
Изменение дозы импринтированных генов –
«потеря импринтинга»
30.
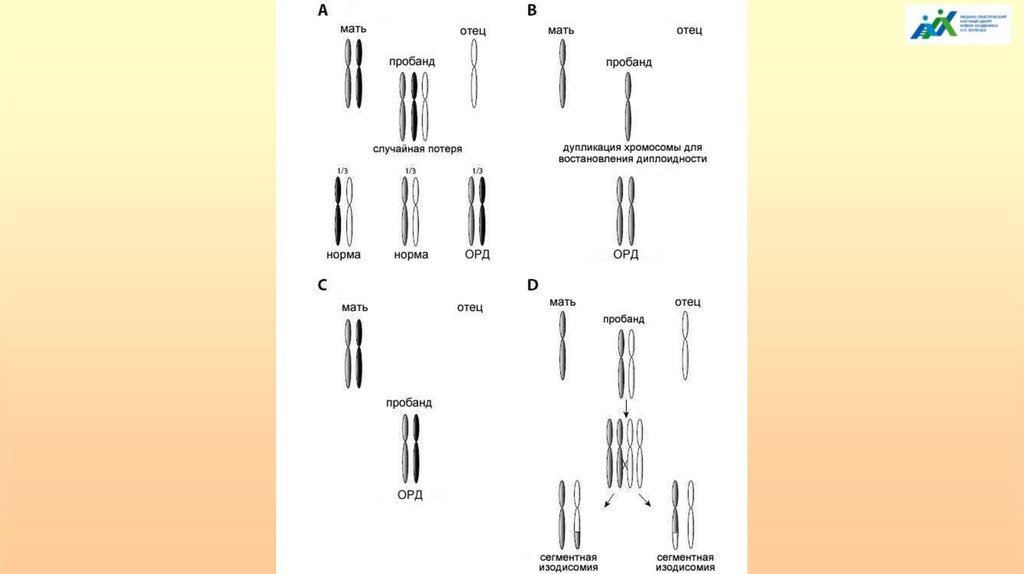
Механизмы формирования ОРД у человека1) Комплементация гамет – дополнение нуллисомной по определенной
хромосоме набора одной гаметы дисомной по этой же хромосоме другой гаметы
(1:3000 гамет).
2) Коррекция трисомии до дисомии, т.е. слияние одной дисомной и одной
нормальной гамет и формирование трисомной зиготы с элиминацией в
последующих делениях дробления той хромосомы, которая содержалась в
нормальной (моносомной) гамете.
3) Коррекция моносомии до дисомии возможна при слиянии нормальной и
нуллисомной гамет с образованием моносомной зиготы и последующей
дупликацией моносомной хромосомы.
4) Соматическая рекомбинация - обмен между хроматидами гомологичных
хромосом в соматических клетках. Это ведет к ОРД по отдельным хромосомным
районам.
Эти механизмы обеспечивают возникновение ОРД двух типов:
1) гетеродисомию - наследование двух разных гомологичных хромосом от
одного из родителей (возникает по механизмам 1 и 2);
2) изодисомию - наследование от одного из родителей двух копий одной и той
же хромосомы. Этот тип ОРД возникает путем коррекции моносомии до дисомии.
31.
32.
ОРД по целым хромосомам или их фрагментам выявленные прианализе наследственной патологии у человека
ОРД по хромосомам 1pat, 2pat/mat, 6mat, 9pat, 10mat, 16pat/mat, 22pat/mat =>
признаки дисэмбриогенеза, отставание в умственном и физическом развитии, без
синдромального фенотипа;
отцовская ОРД по длинному плечу хромосомы 6(q23 - q24) => транзиторный неонатальный диабет;
материнская ОРД по хромосоме 7 => синдром Сильвера – Рассела;
отцовская сегментная ОРД по хромосоме 11 => синдром Видеманна-Беквита;
материнская ОРД по хромосоме 14 => гипотония, черепно-лицевые
аномалии, акромикрия, сколиоз, задержка физического, моторного и умственного развития синдром Темпл;
отцовская ОРД по хромосоме 14 => сильная умственная отсталость и скелетно-мышечные
аномалии - синдром Кагами-Огата;
материнская ОРД по хромосоме 15 => синдром Прадера-Вилли;
отцовская ОРД по хромосоме 15 => синдром Ангельмана;
отцовская ОРД по длинному плечу хромосомы 20 (GNAS1) => псевдогипопаратироидизм 1В;
материнская ОРД по хромосоме 20 => cиндром Мулчандани–Божж–Конлин.
33.
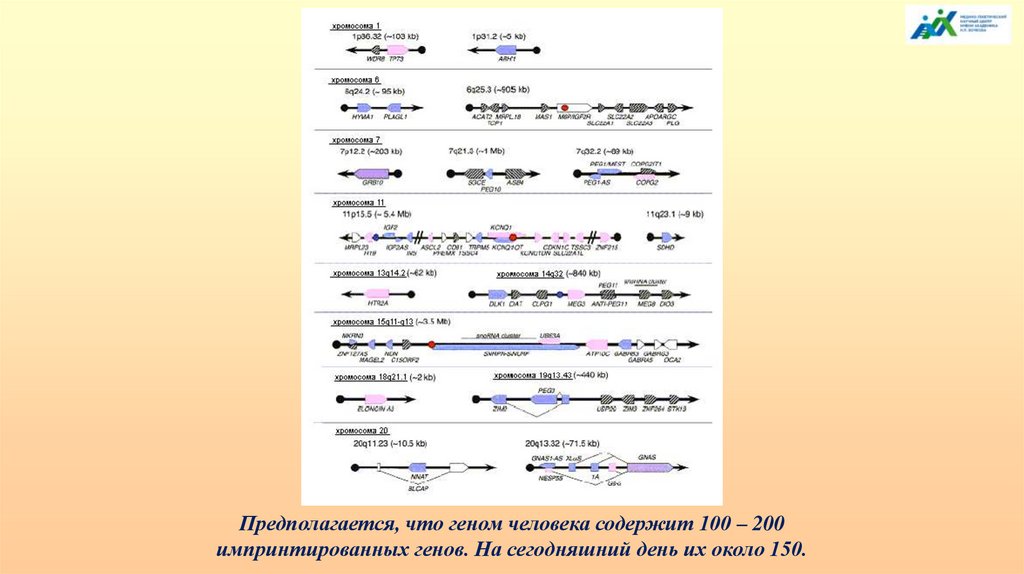
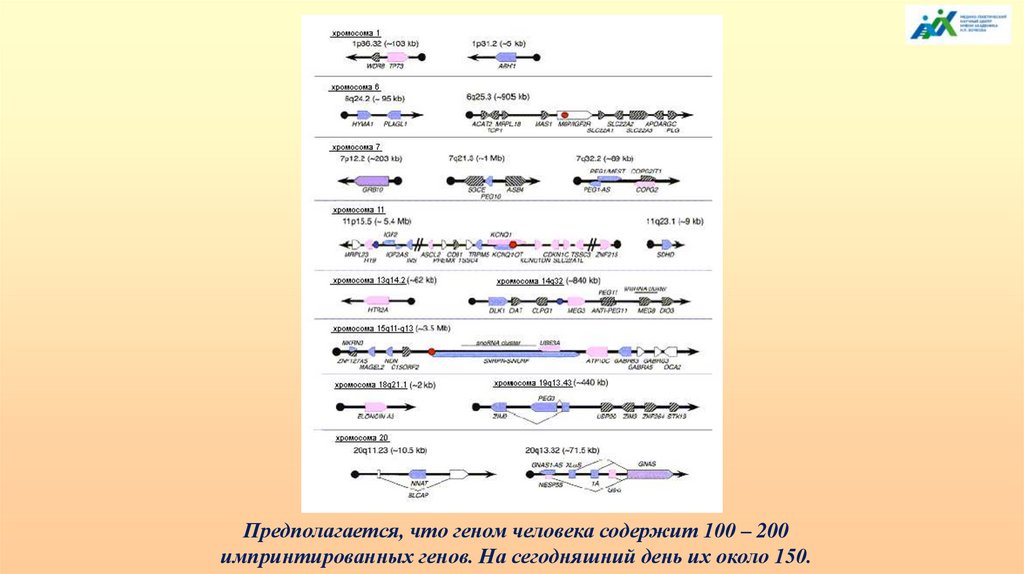
Предполагается, что геном человека содержит 100 – 200импринтированных генов. На сегодняшний день их около 150.
34.
Болезни импринтингаТранзиторный неонатальный диабет - хромосома 6q24;
Синдром Бирка–Барела - хромосома 8q24.3 (материнские миссенс-мутации KCNK9/TASK3;
Синдром Видеманна-Беквита - хромосома 11р15.5;
Синдром Сильвера-Рассела – хромосомы 7 и 11p15.5;
Синдром IMAGe - хромосома 11р15.5 (материнские миссенс-мутации CDKN1C);
Ретинобластома - хромосома 13q14.3, 2-ой интрон RB1;
Синдромы Темпл и Кагами-Огата - ОРД (материнская/отцовская) по хромосоме 14;
Синдромы Прадера-Вилли и Ангельмана – хромосома 15(q11.2-q13);
Синдром Шаафа–Янга – отцовские нонсенс- и миссенс-мутации MAGEL2;
Синдром центрального преждевременного полового созревания 2-го типа - отцовские мутации
MKRN3;
Псевдогипопаратироидизм 1В типа и родственные заболевания – хромосома 20q13;
Множественные аномалии метилирования импринтированных регуляторных районов – MLID;
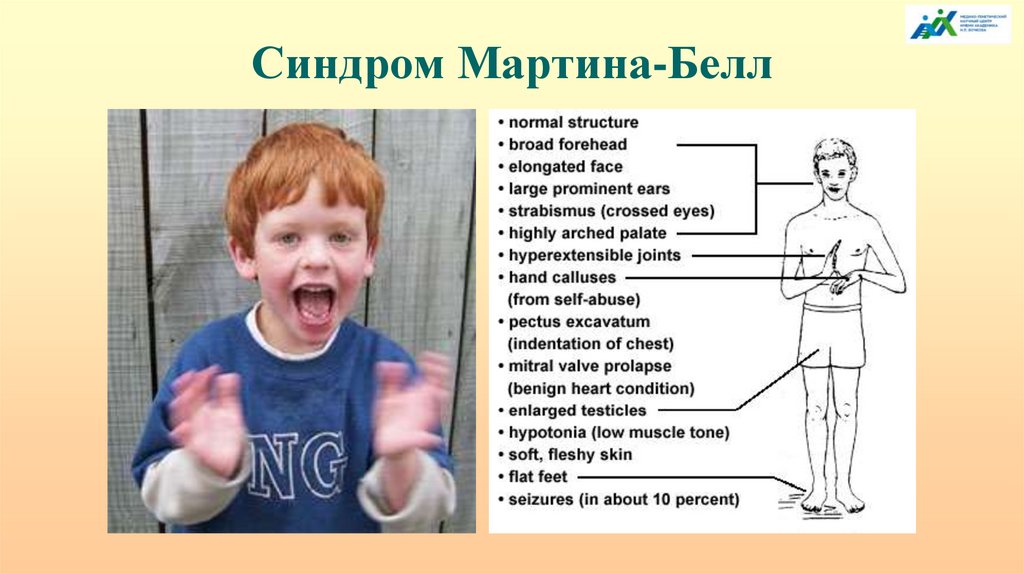
Синдром Мартина-Белл – хромосома Xq27.3 (условно).
35.
36.
В музее Прадо в Мадриде есть пара картин придворного художника XVII столетия ХуанаКарреньо де Миранда с названиями «La Monstrua vestida» и «La Monstrua desnuda» («Одетый
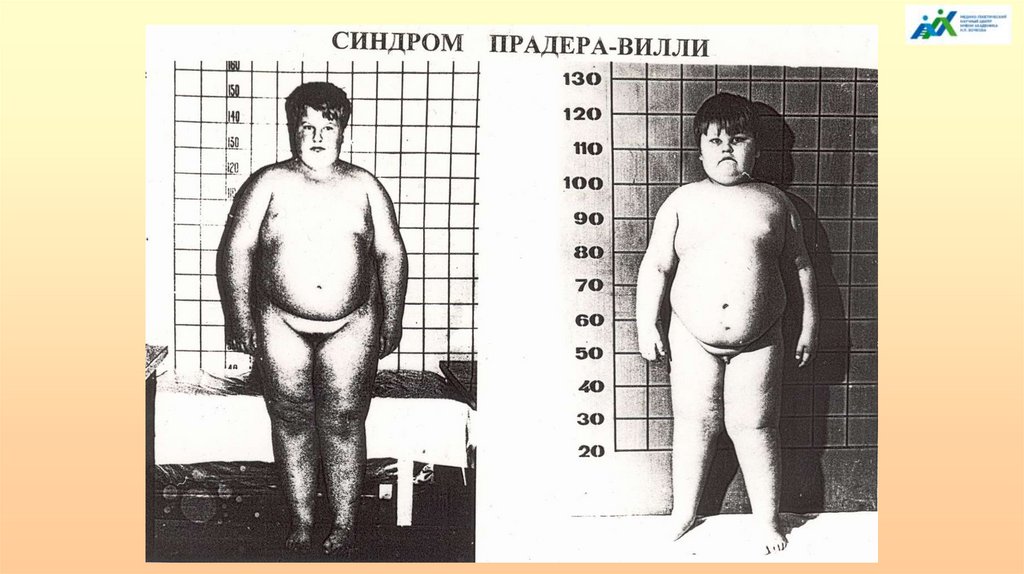
монстр» и «Раздетый монстр»). На картинах изображена очень толстая пятилетняя девочка
Евгения Мартинес Валлехо с синдромом Прадера-Вилли.
37.
38.
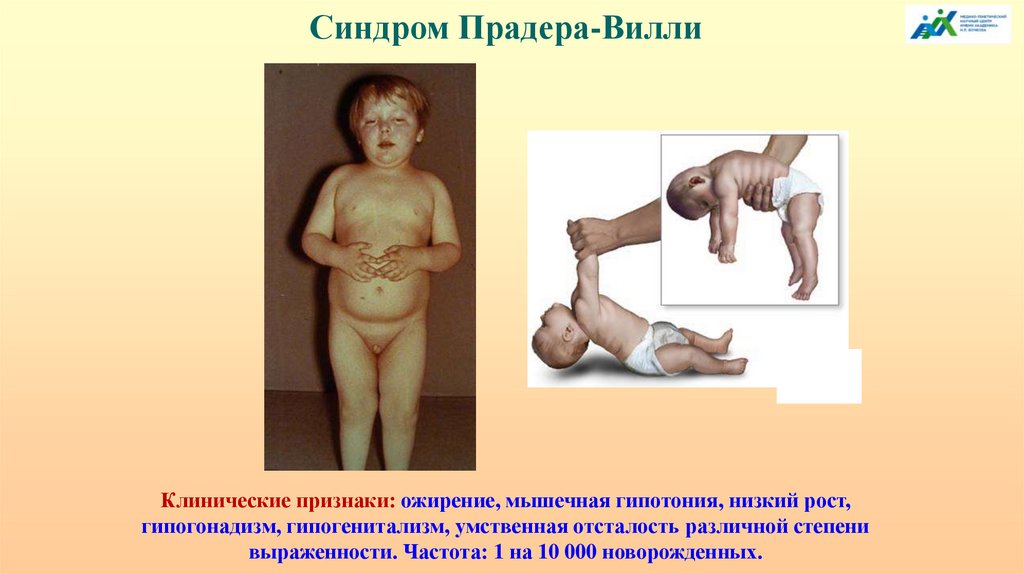
Синдром Прадера-ВиллиКлинические признаки: ожирение, мышечная гипотония, низкий рост,
гипогонадизм, гипогенитализм, умственная отсталость различной степени
выраженности. Частота: 1 на 10 000 новорожденных.
39.
Синдром АнгельманаМикробрахицефалия, большая нижняя челюсть, приоткрытый рот,
макростомия, редко растущие зубы, гипопигментация. Умственная
отсталость, атаксия, судорожная готовность, гиперрефлексия,
гиперкинезия (приступы неконтролируемого смеха, хлопанье в ладоши и
специфическое выражение лица). Частота 1:12000–20000.
40.
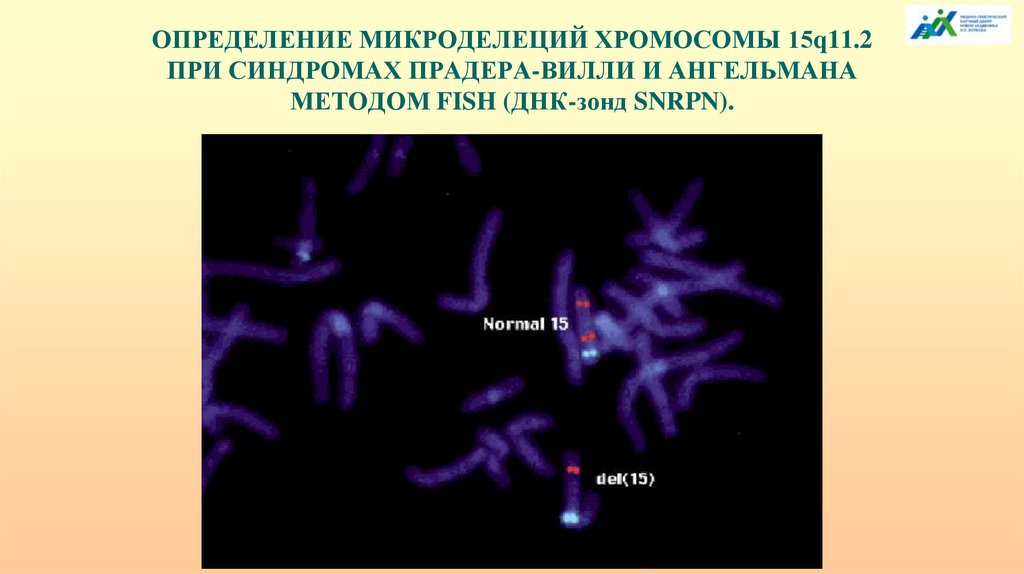
ОПРЕДЕЛЕНИЕ МИКРОДЕЛЕЦИЙ ХРОМОСОМЫ 15q11.2ПРИ СИНДРОМАХ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА
МЕТОДОМ FISH (ДНК-зонд SNRPN).
41.
42.
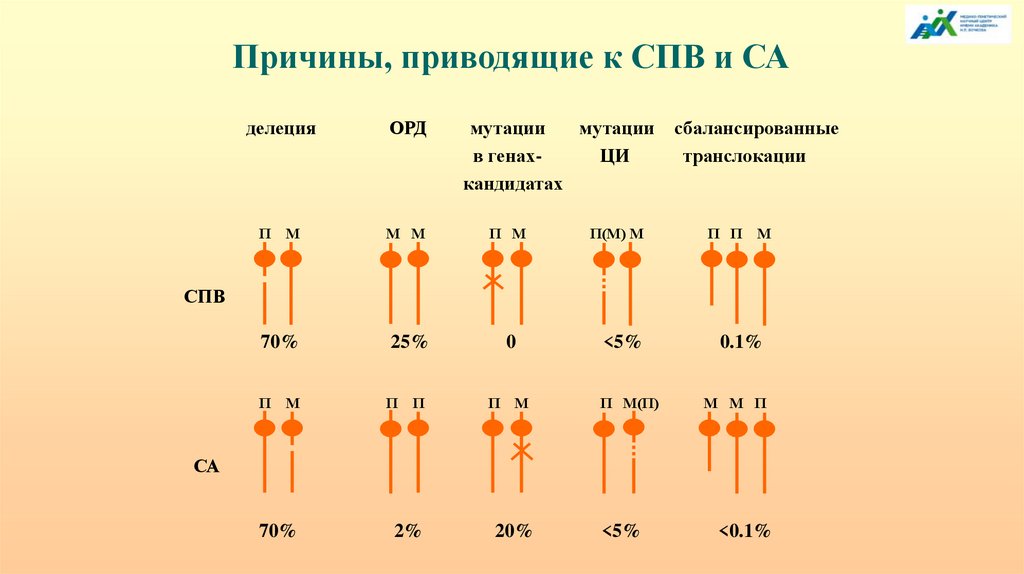
Причины, приводящие к СПВ и САделеция
ОРД
мутации
мутации
в генахЦИ
кандидатах
сбалансированные
транслокации
П М
М М
П М
П(М) М
П П М
70%
25%
0
<5%
0.1%
П
М
П П
П М
П М(П)
М М П
70%
2%
20%
<5%
<0.1%
СПВ
СА
43.
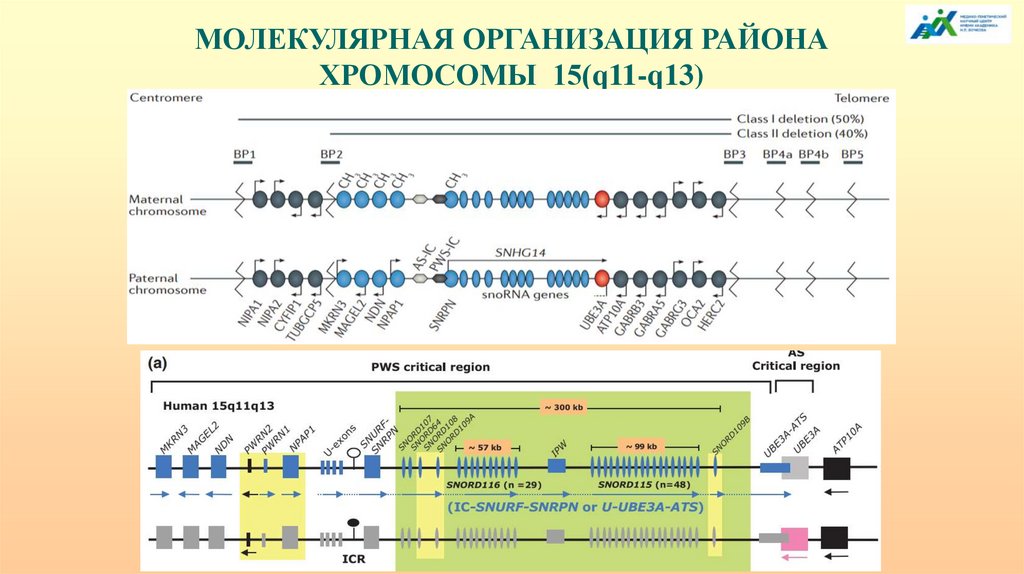
МОЛЕКУЛЯРНАЯ ОРГАНИЗАЦИЯ РАЙОНАХРОМОСОМЫ 15(q11-q13)
44.
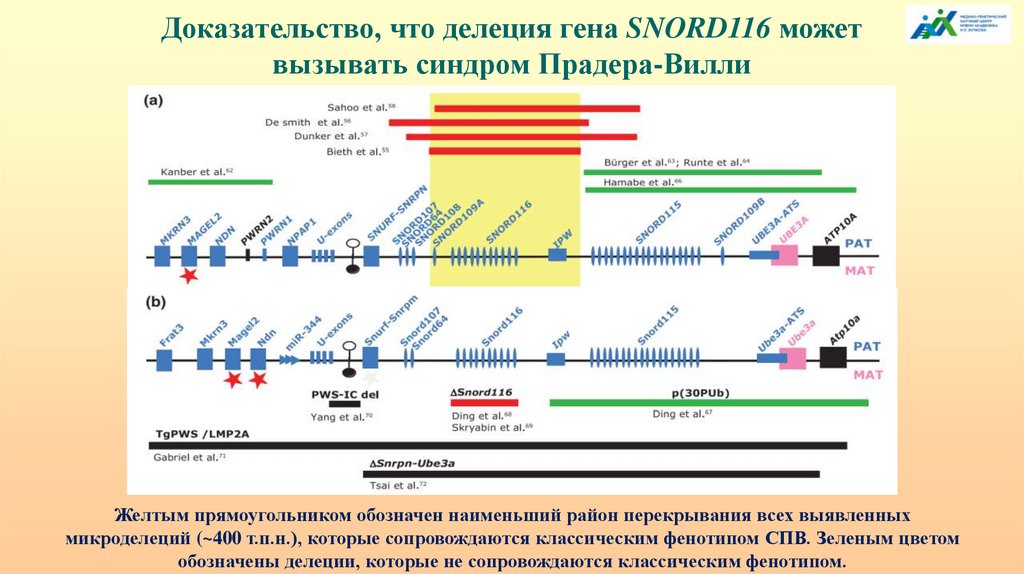
Доказательство, что делеция гена SNORD116 можетвызывать синдром Прадера-Вилли
Желтым прямоугольником обозначен наименьший район перекрывания всех выявленных
микроделеций (~400 т.п.н.), которые сопровождаются классическим фенотипом СПВ. Зеленым цветом
обозначены делеции, которые не сопровождаются классическим фенотипом.
45.
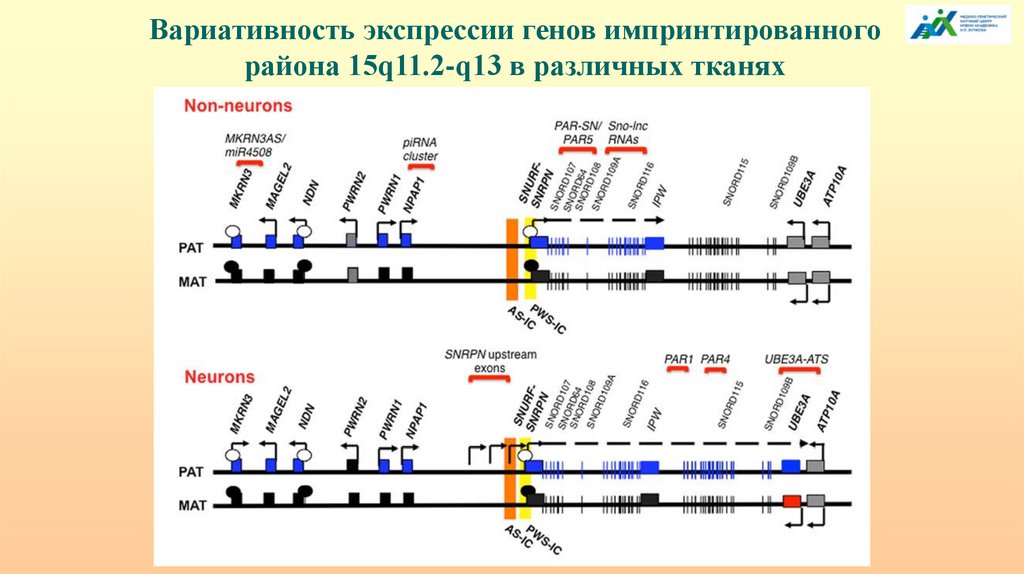
Вариативность экспрессии генов импринтированногорайона 15q11.2-q13 в различных тканях
46.
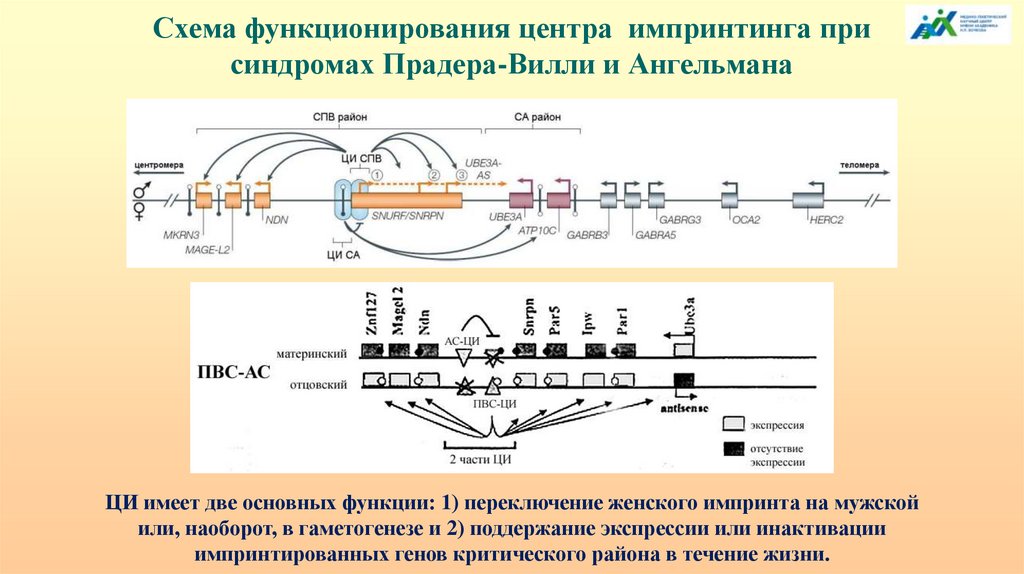
Схема функционирования центра импринтинга присиндромах Прадера-Вилли и Ангельмана
ЦИ имеет две основных функции: 1) переключение женского импринта на мужской
или, наоборот, в гаметогенезе и 2) поддержание экспрессии или инактивации
импринтированных генов критического района в течение жизни.
47.
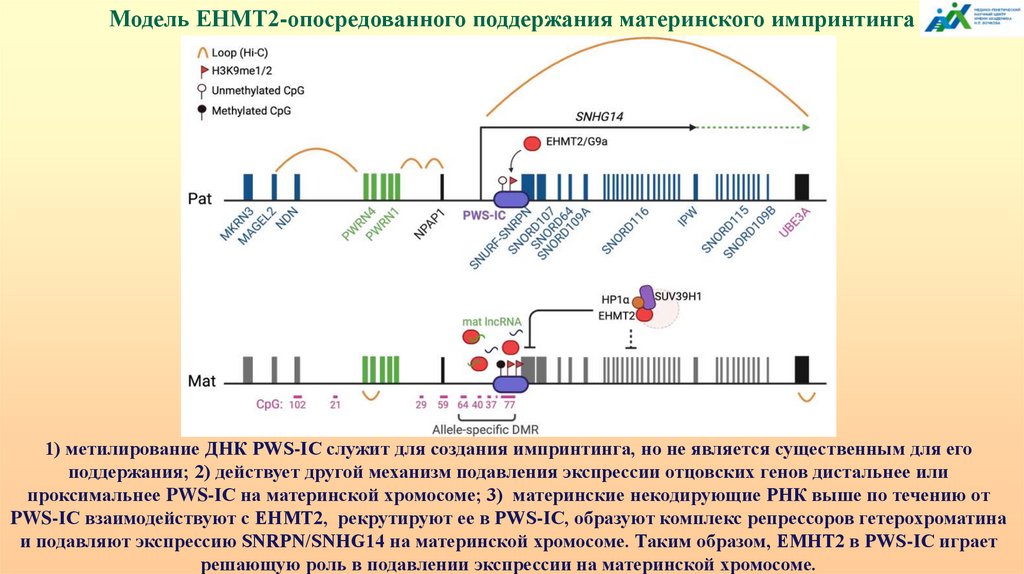
Модель EHMT2-опосредованного поддержания материнского импринтинга1) метилирование ДНК PWS-IC служит для создания импринтинга, но не является существенным для его
поддержания; 2) действует другой механизм подавления экспрессии отцовских генов дистальнее или
проксимальнее PWS-IC на материнской хромосоме; 3) материнские некодирующие РНК выше по течению от
PWS-IC взаимодействуют с EHMT2, рекрутируют ее в PWS-IC, образуют комплекс репрессоров гетерохроматина
и подавляют экспрессию SNRPN/SNHG14 на материнской хромосоме. Таким образом, EMHT2 в PWS-IC играет
решающую роль в подавлении экспрессии на материнской хромосоме.
48.
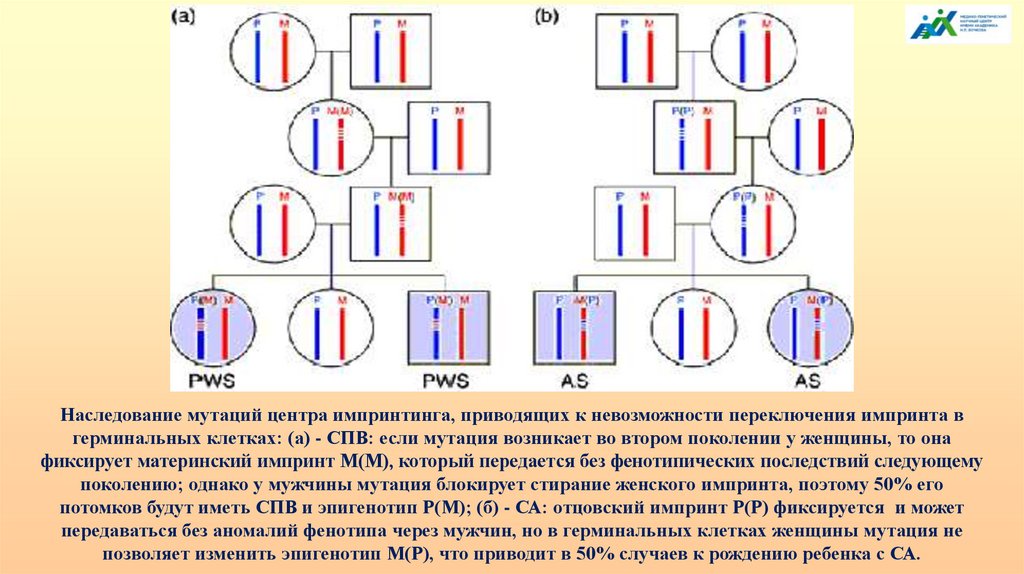
Наследование мутаций центра импринтинга, приводящих к невозможности переключения импринта вгерминальных клетках: (а) - СПВ: если мутация возникает во втором поколении у женщины, то она
фиксирует материнский импринт М(М), который передается без фенотипических последствий следующему
поколению; однако у мужчины мутация блокирует стирание женского импринта, поэтому 50% его
потомков будут иметь СПВ и эпигенотип Р(М); (б) - СА: отцовский импринт Р(Р) фиксируется и может
передаваться без аномалий фенотипа через мужчин, но в герминальных клетках женщины мутация не
позволяет изменить эпигенотип М(Р), что приводит в 50% случаев к рождению ребенка с СА.
49.
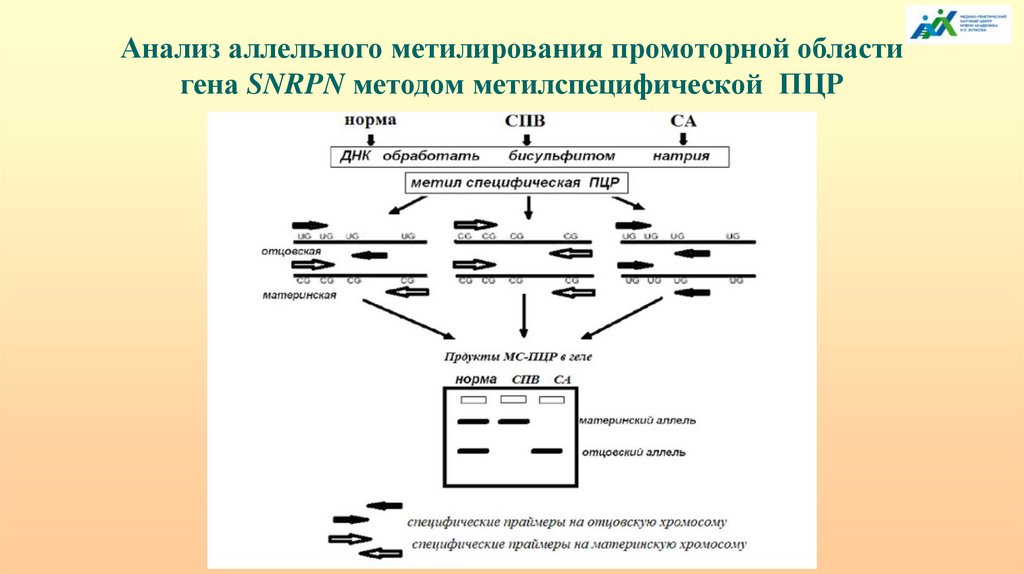
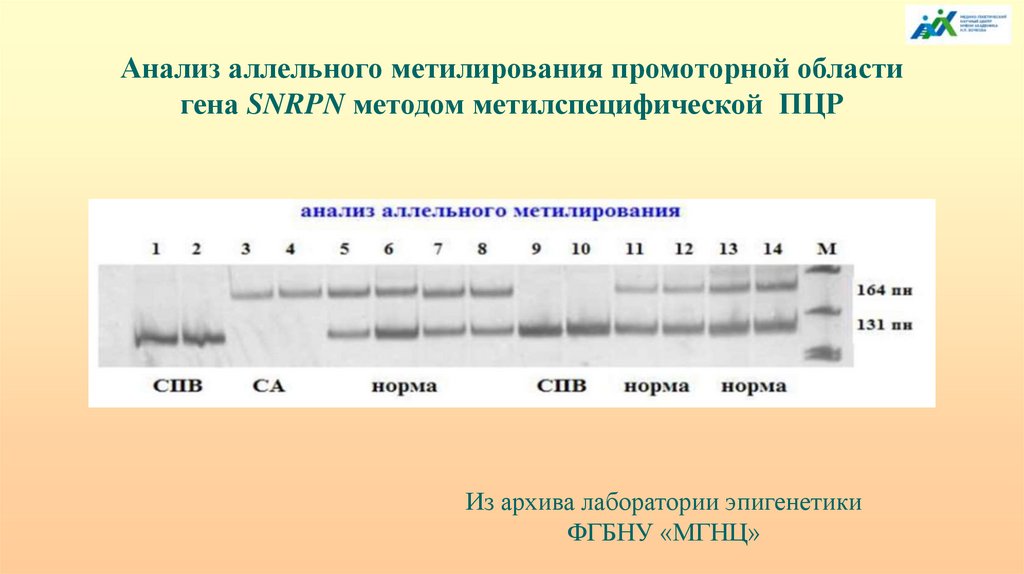
Анализ аллельного метилирования промоторной областигена SNRPN методом метилспецифической ПЦР
50.
Анализ аллельного метилирования промоторной областигена SNRPN методом метилспецифической ПЦР
Из архива лаборатории эпигенетики
ФГБНУ «МГНЦ»
51.
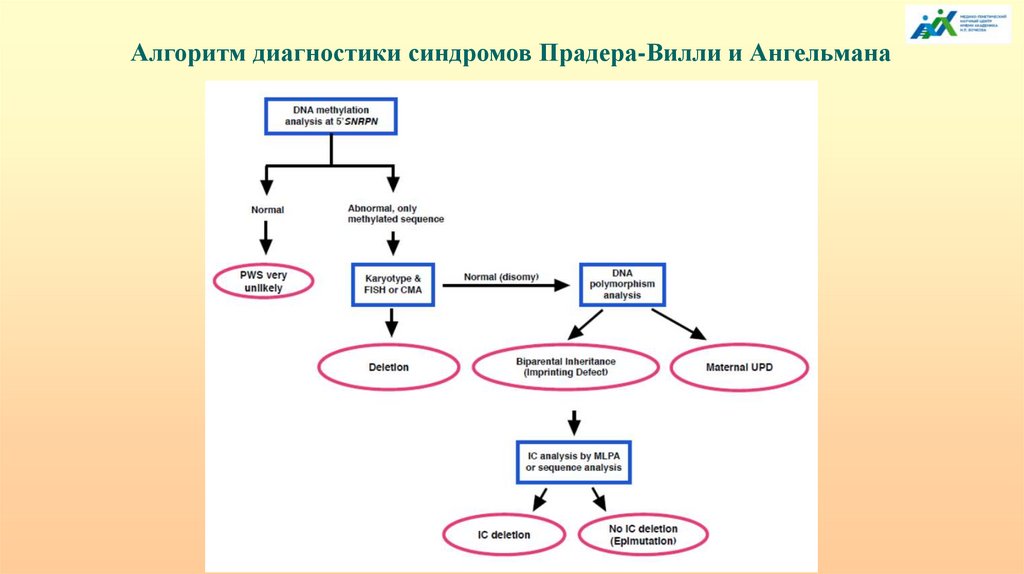
52.
Алгоритм диагностики синдромов Прадера-Вилли и Ангельмана53.
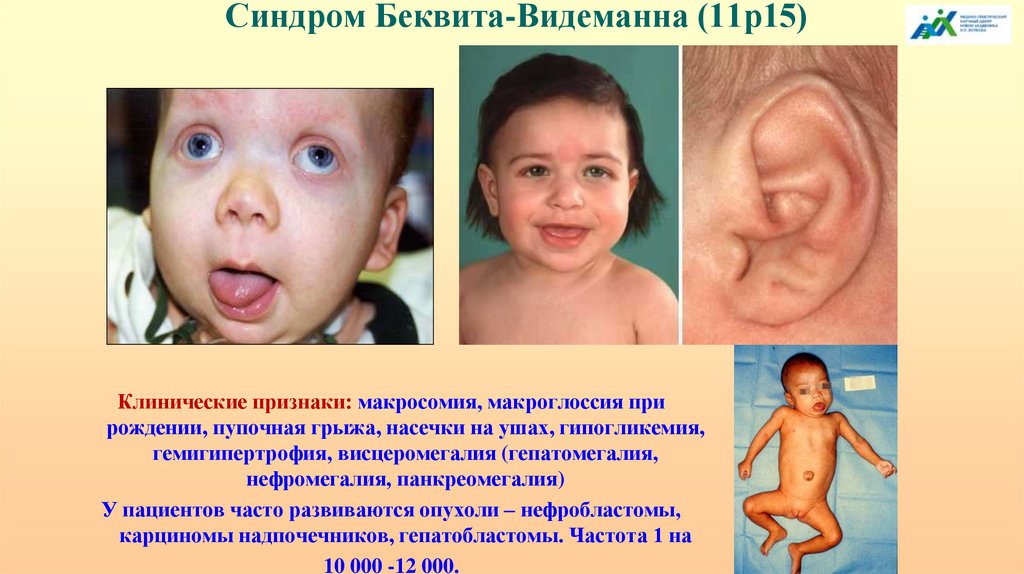
54.
Синдром Беквита-Видеманна (11р15)Клинические признаки: макросомия, макроглоссия при
рождении, пупочная грыжа, насечки на ушах, гипогликемия,
гемигипертрофия, висцеромегалия (гепатомегалия,
нефромегалия, панкреомегалия)
У пациентов часто развиваются опухоли – нефробластомы,
карциномы надпочечников, гепатобластомы. Частота 1 на
10 000 -12 000.
55.
56.
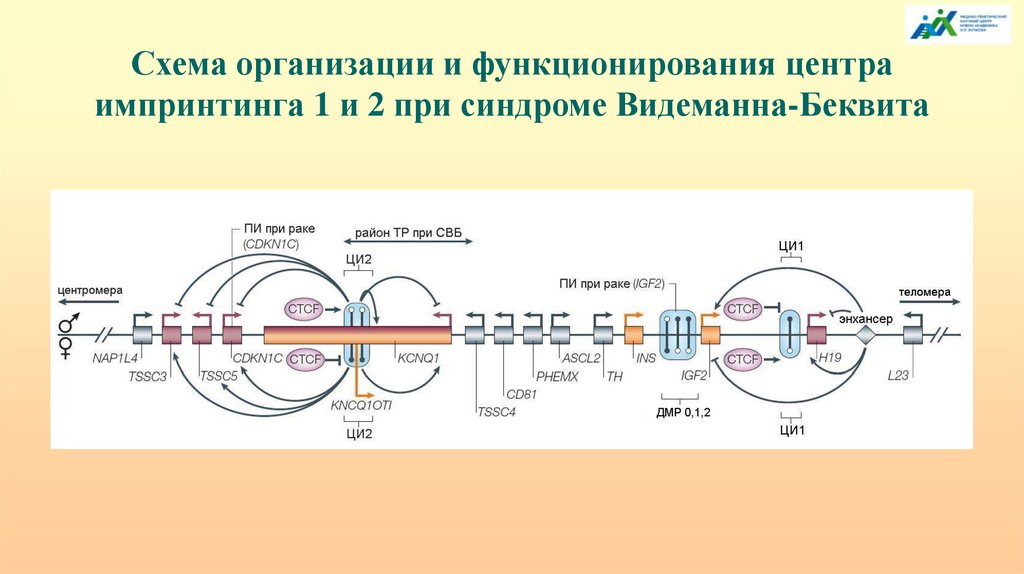
Схема организации и функционирования центраимпринтинга 1 и 2 при синдроме Видеманна-Беквита
57.
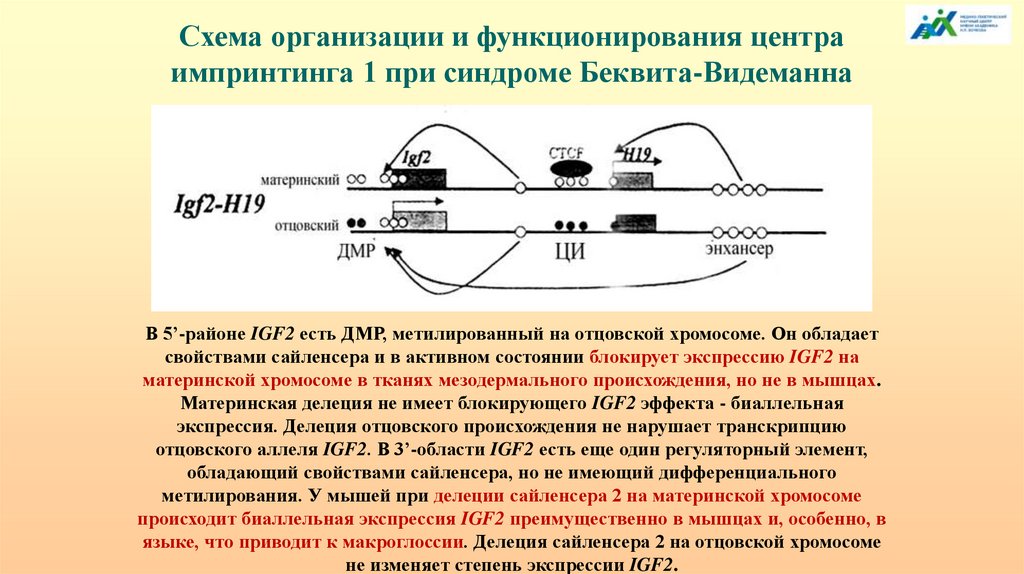
Схема организации и функционирования центраимпринтинга 1 при синдроме Беквита-Видеманна
В 5’-районе IGF2 есть ДМР, метилированный на отцовской хромосоме. Он обладает
свойствами сайленсера и в активном состоянии блокирует экспрессию IGF2 на
материнской хромосоме в тканях мезодермального происхождения, но не в мышцах.
Материнская делеция не имеет блокирующего IGF2 эффекта - биаллельная
экспрессия. Делеция отцовского происхождения не нарушает транскрипцию
отцовского аллеля IGF2. В 3’-области IGF2 есть еще один регуляторный элемент,
обладающий свойствами сайленсера, но не имеющий дифференциального
метилирования. У мышей при делеции сайленсера 2 на материнской хромосоме
происходит биаллельная экспрессия IGF2 преимущественно в мышцах и, особенно, в
языке, что приводит к макроглоссии. Делеция сайленсера 2 на отцовской хромосоме
не изменяет степень экспрессии IGF2.
58.
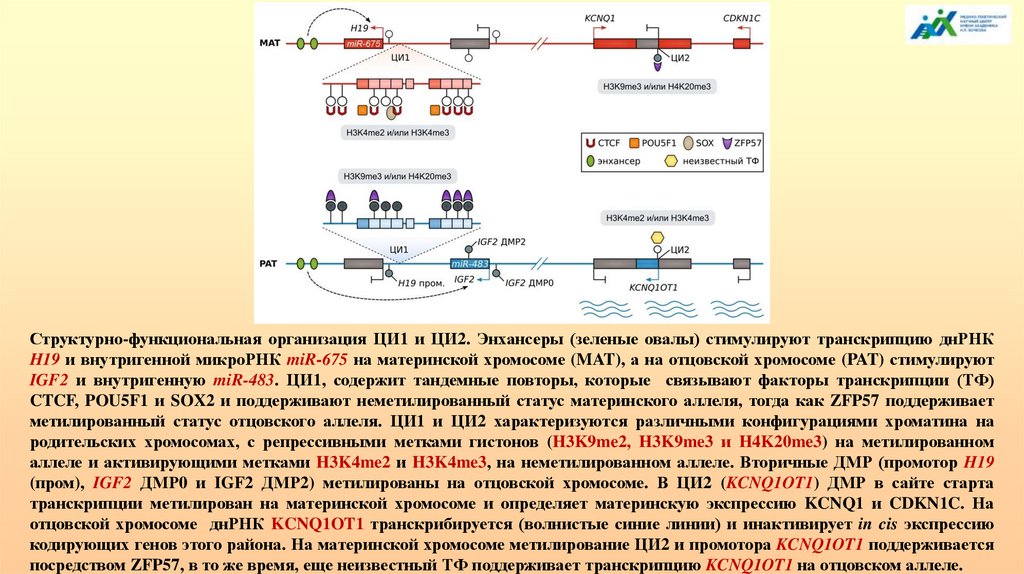
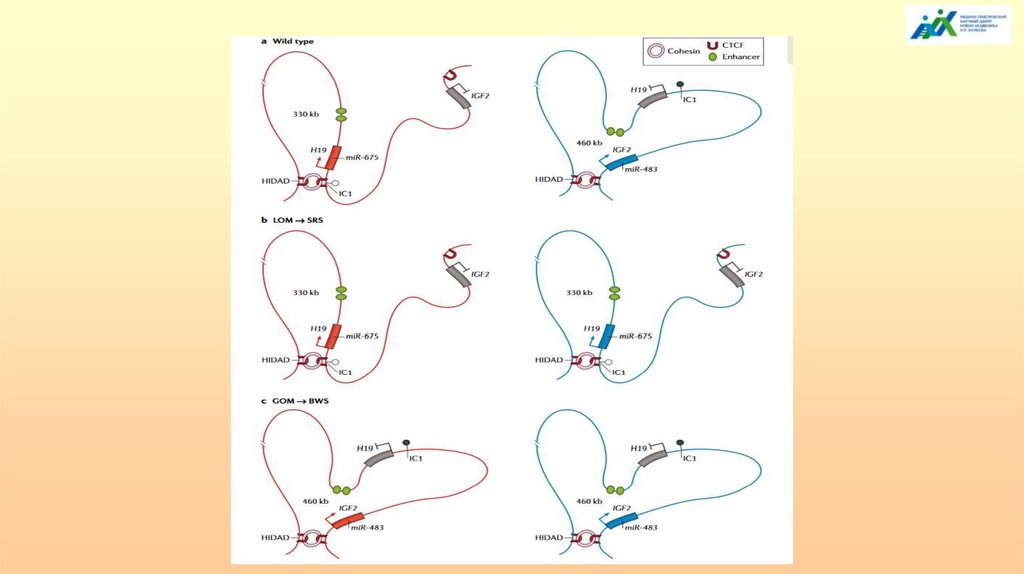
Структурно-функциональная организация ЦИ1 и ЦИ2. Энхансеры (зеленые овалы) стимулируют транскрипцию днРНКH19 и внутригенной микроРНК miR-675 на материнской хромосоме (MAT), а на отцовской хромосоме (PAT) стимулируют
IGF2 и внутригенную miR-483. ЦИ1, содержит тандемные повторы, которые связывают факторы транскрипции (TФ)
CTCF, POU5F1 и SOX2 и поддерживают неметилированный статус материнского аллеля, тогда как ZFP57 поддерживает
метилированный статус отцовского аллеля. ЦИ1 и ЦИ2 характеризуются различными конфигурациями хроматина на
родительских хромосомах, с репрессивными метками гистонов (H3K9me2, H3K9me3 и H4K20me3) на метилированном
аллеле и активирующими метками H3K4me2 и H3K4me3, на неметилированном аллеле. Вторичные ДМР (промотор H19
(пром), IGF2 ДМР0 и IGF2 ДМР2) метилированы на отцовской хромосоме. В ЦИ2 (KCNQ1OT1) ДМР в сайте старта
транскрипции метилирован на материнской хромосоме и определяет материнскую экспрессию KCNQ1 и CDKN1C. На
отцовской хромосоме днРНК KCNQ1OT1 транскрибируется (волнистые синие линии) и инактивирует in cis экспрессию
кодирующих генов этого района. На материнской хромосоме метилирование ЦИ2 и промотора KCNQ1OT1 поддерживается
посредством ZFP57, в то же время, еще неизвестный ТФ поддерживает транскрипцию KCNQ1OT1 на отцовском аллеле.
59.
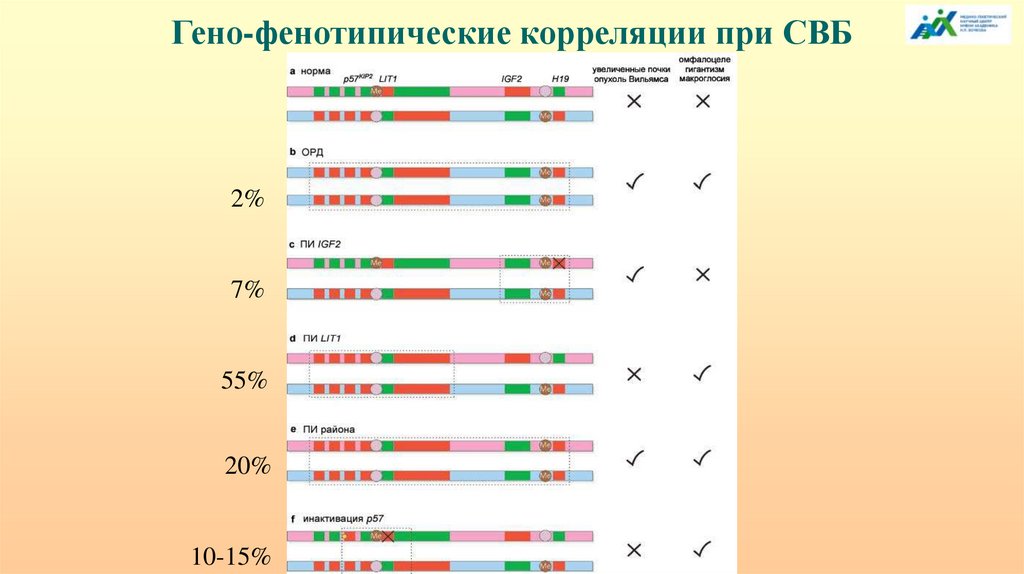
Гено-фенотипические корреляции при СВБ2%
7%
55%
20%
10-15%
60.
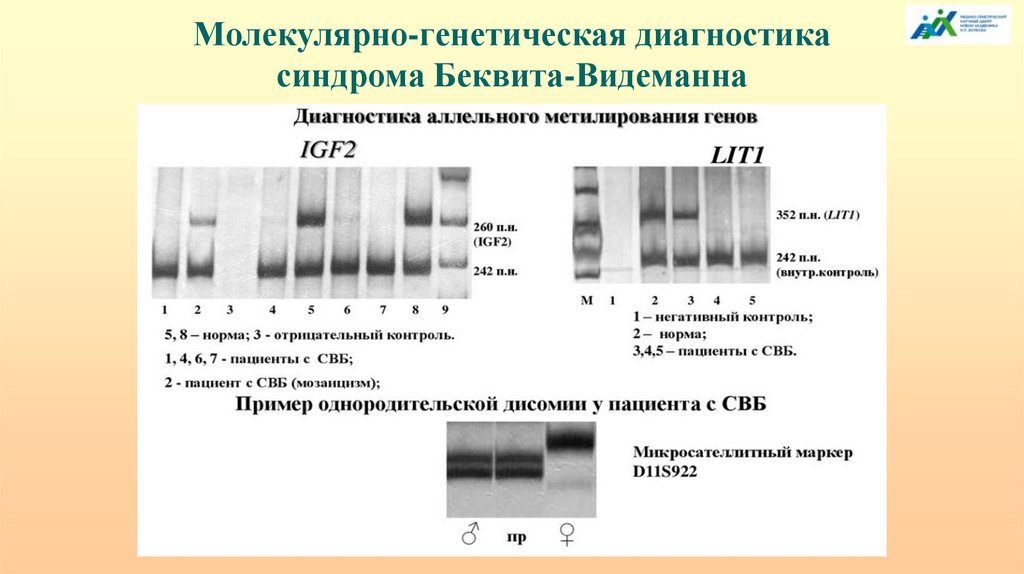
Молекулярно-генетическая диагностикасиндрома Беквита-Видеманна
61.
Структура молекулярно-генетической иэпигенетической патологии при СВБ
1) Потеря метилирования в ЦИ2 (KCNQ1OT1) – 50%;
2) Отцовская мозаичная сегментная ОРД хромосомы 11 – 20%;
3) Нарушение метилирования ЦИ1, приводящее к
метилированию Н19 на материнской хромосоме и
биаллельной экспрессии IGF2 – 5–10%;
4) Материнские мутации, инактивирующие CDKN1C – 5–10%;
5) Хромосомные перестройки (дупликации, транслокации,
инверсии, делеции) 11p15.5 – 1%;
6) MLID – 25%
62.
63.
Синдром Рассела-СильвераПренатальная и
постнатальная задержка
роста; треугольное лицо с
выступающим лбом;
клинодактилия или
брахидактилия;
макроцефалия;
скелетная асимметрия;
мышечная гипотрофия;
гипотония.
Хромосомные перестройки, затрагивающие хромосомы
трис.1q42, 7, 8, 11p15, 15q26.3, 17q24 и 18
Мат ОРД 7 (5-15% случаев), тандемные дупликации 7p11.2-p13.
1) 7p11.2-p13 (GRB10 - ингибитор роста); 2) 7q31-qter (MEST);
3) 7q21.3 - PEG10
В 30-65% случаев обнаруживается гипометилирование H19 на
отцовской хромосоме 11.
64.
7p11.2-p13. У человека отцовская экспрессия GRB10 установлена вголовном и спинном мозге, материнская – в скелетных мышцах, в остальных
тканях ген экспрессируется биаллельно. Мышиный ген импринтирован,
экспрессируется с материнской хромосомы во всех тканях кроме мозга, где
экспрессируется отцовский аллель. Делеции материнского аллеля гена приводят к
увеличению роста у потомства, свидетельствуя о его функции, как негативного
регулятора роста.
7q32.2 содержит 5 импринтированных генов, включая гены MEST,
COPG2IT1, MESTIT1, которые экспрессируются с отцовской хромосомы, а CPA4 и
KLF14 – с материнской. MEST имеет две изоформы, одна из которых
экспрессируется с отцовского аллеля, а вторая (использующая альтернативный
первый экзон) экспрессируется биаллельно во всех тканях, кроме плаценты. Нокаут
гена у мышей приводит к малому размеру потомства.
7q21.3 содержит гены PEG10 и SGCE, имеющих отцовскую экспрессию,
PPP1R9A экспрессируется с материнского аллеля в эмбриональных скелетных
мышцах и экстраэмбриональных тканях, а ген TFP12 экспрессируется с
материнского аллеля в плаценте. Делеции PEG10 у мышей приводят к ранней
эмбриональной гибели.
65.
Схема эпигенетической патологии при СРС(гипометилирование H19 на отцовской хромосоме 11)
66.
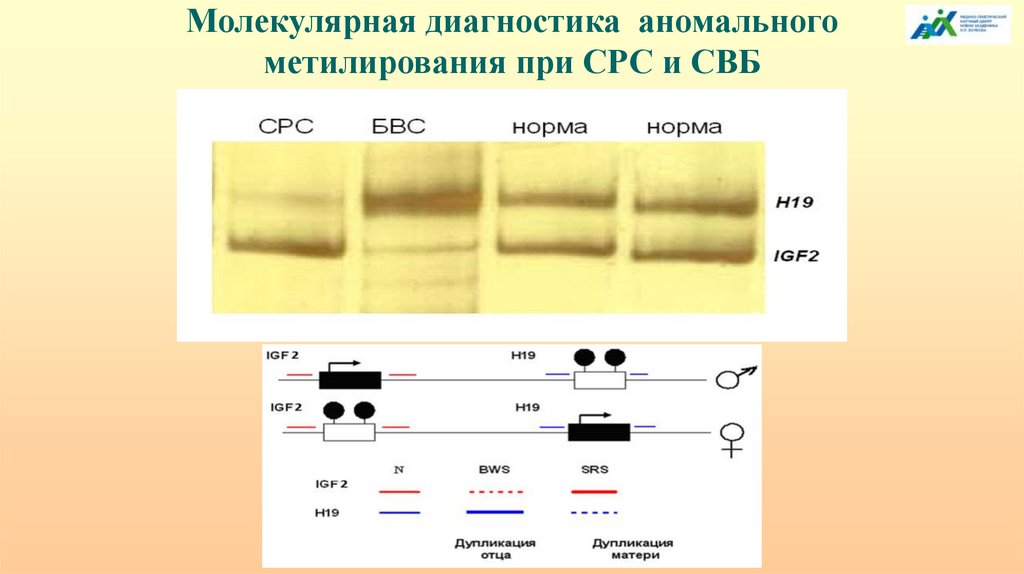
Молекулярная диагностика аномальногометилирования при СРС и СВБ
67.
Структура молекулярно-генетической иэпигенетической патологии при СРС
1) Потеря метилирования ЦИ1 на отцовской хромосоме, что приводит к
биаллельной экспрессии Н19 – 40–60%;
2) Материнская ОРД 11p15 – редко;
3) Точковые мутации в генах CDKN1C, IGF2, HMGA2, PLAG1 –
единичные случаи;
4) Метилирования ЦИ2 на отцовской хромосоме – редко;
5) MLID – 7–10%;
6) Изодисомия/материнская дупликация и гетеродисомия материнского
происхождения хромосомы 7 – 5–10%.
68.
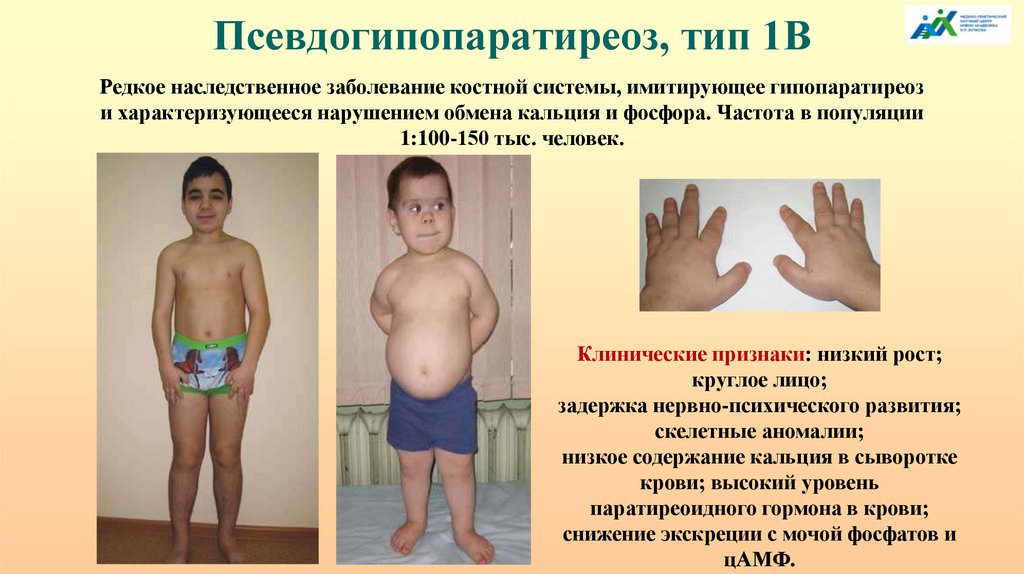
Псевдогипопаратиреоз, тип 1ВРедкое наследственное заболевание костной системы, имитирующее гипопаратиреоз
и характеризующееся нарушением обмена кальция и фосфора. Частота в популяции
1:100-150 тыс. человек.
Клинические признаки: низкий рост;
круглое лицо;
задержка нервно-психического развития;
скелетные аномалии;
низкое содержание кальция в сыворотке
крови; высокий уровень
паратиреоидного гормона в крови;
снижение экскреции с мочой фосфатов и
цАМФ.
69.
Псевдогипопаратиреоз 1В проявляется гипокальцемией игиперфосфатемией в результате резистентности к ПТГ. Описаны как
спорадические, так и семейные случаи заболевания, причем последние
наследуются аутосомно-доминантно с неполной пенетрантностью. Анализ
больших семей показал, что резистентность к ПТГ развивается только в
том случае, если дефект наследуется по материнской линии. У пациентов с
ПГП 1А, как правило, выявляются материнские мутации в GNAS
(кодирует α-субъединицу белка, связывающего гуанин), а у пациентов с
ПГП 1В таковых не обнаружено. В то же время у последних
обнаруживается потеря метилирования ДМР локуса GNAS (20q13.2), что
приводит к биаллельной экспрессии альтернативных транскриптов.
70.
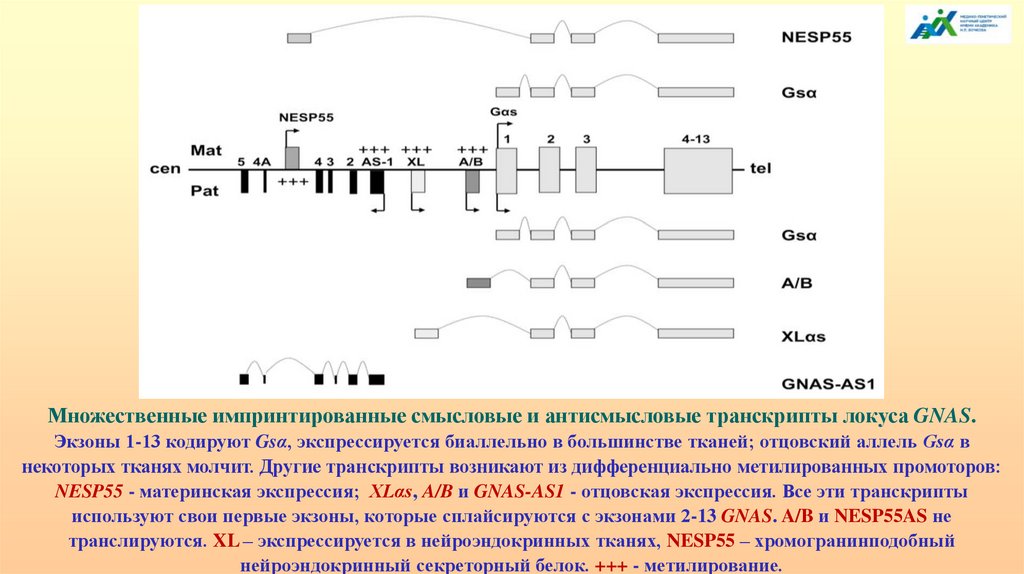
Множественные импринтированные смысловые и антисмысловые транскрипты локуса GNAS.Экзоны 1-13 кодируют Gsα, экспрессируется биаллельно в большинстве тканей; отцовский аллель Gsα в
некоторых тканях молчит. Другие транскрипты возникают из дифференциально метилированных промоторов:
NESP55 - материнская экспрессия; XLαs, A/B и GNAS-AS1 - отцовская экспрессия. Все эти транскрипты
используют свои первые экзоны, которые сплайсируются с экзонами 2-13 GNAS. A/B и NESP55AS не
транслируются. XL – экспрессируется в нейроэндокринных тканях, NESP55 – хромогранинподобный
нейроэндокринный секреторный белок. +++ - метилирование.
71.
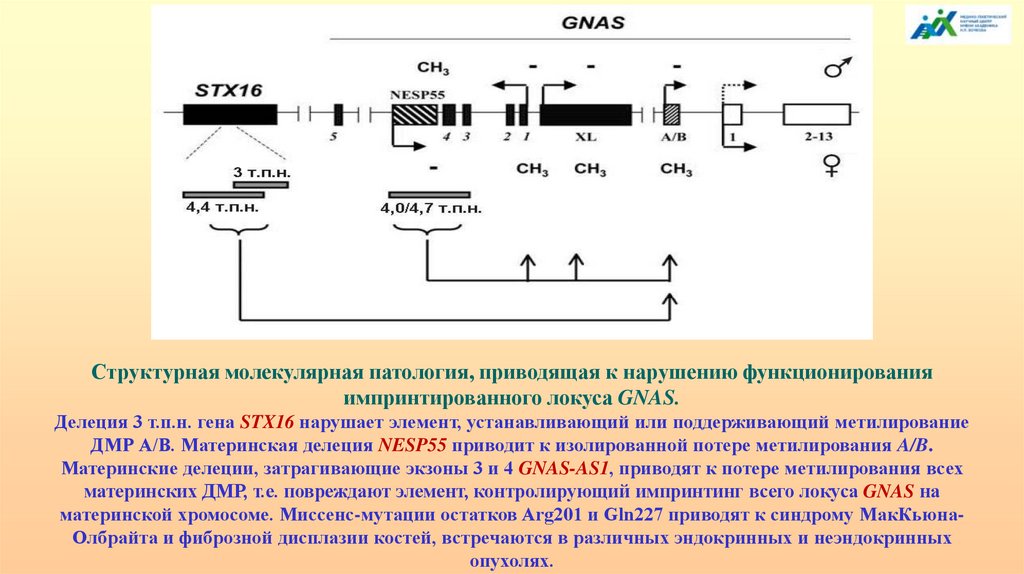
Структурная молекулярная патология, приводящая к нарушению функционированияимпринтированного локуса GNAS.
Делеция 3 т.п.н. гена STX16 нарушает элемент, устанавливающий или поддерживающий метилирование
ДМР А/В. Материнская делеция NESP55 приводит к изолированной потере метилирования A/B.
Материнские делеции, затрагивающие экзоны 3 и 4 GNAS-AS1, приводят к потере метилирования всех
материнских ДМР, т.е. повреждают элемент, контролирующий импринтинг всего локуса GNAS на
материнской хромосоме. Миссенс-мутации остатков Arg201 и Gln227 приводят к синдрому МакКьюнаОлбрайта и фиброзной дисплазии костей, встречаются в различных эндокринных и неэндокринных
опухолях.
72.
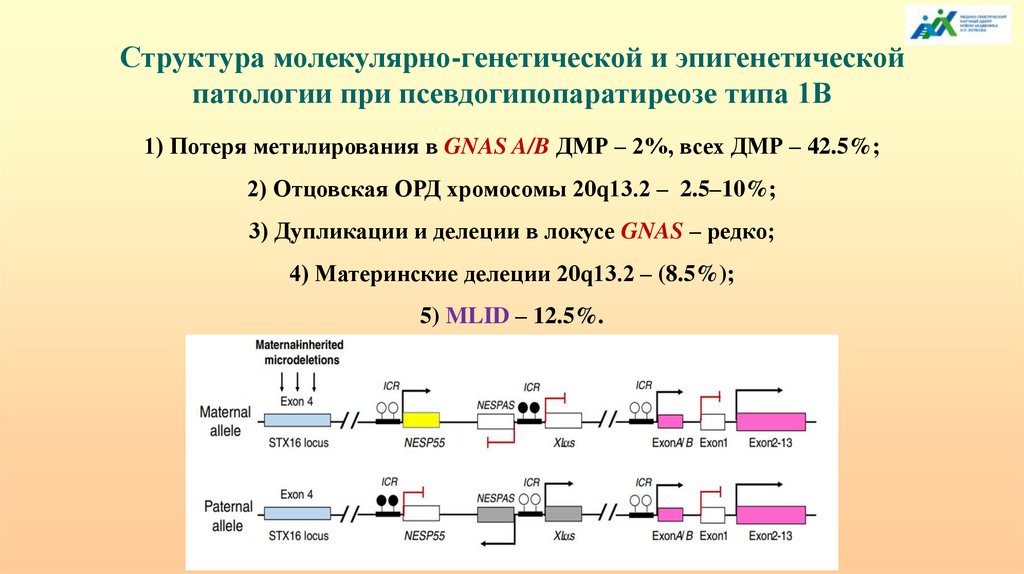
Структура молекулярно-генетической и эпигенетическойпатологии при псевдогипопаратиреозе типа 1В
1) Потеря метилирования в GNAS A/B ДМР – 2%, всех ДМР – 42.5%;
2) Отцовская ОРД хромосомы 20q13.2 – 2.5–10%;
3) Дупликации и делеции в локусе GNAS – редко;
4) Материнские делеции 20q13.2 – (8.5%);
5) MLID – 12.5%.
73.
Родственные заболевания, связанные с геном GNASПсевдогипопаратиреоз типа 1A.
Генерализованная гормонорезистентность различной степени, умственная отсталость, ожирение,
связанное со снижением энергетических затрат в состоянии покоя, наследственная остеодистрофия
Олбрайта, проявляющаяся низкорослостью, округлым лицом, подкожными оссификатами,
брахидактилией и другими скелетными аномалиями. Частота неизвестна. Мутации с потерей
функции в материнском аллеле гена GNAS – 100%.
Псевдопсевдогипопаратиреоз.
Фенотип остеодистрофии Олбрайта без сопутствующих эндокринных нарушений. Частота
неизвестна. Мутации с потерей функции в отцовском аллеле гена GNAS – 100%.
Прогрессирующая костная гетероплазия.
Подкожная оссификация, появляющаяся в детском возрасте и прогрессирующая с возрастом, с
вовлечением подкожных и более глубоких соединительных тканей. Остеодистрофия Олбрайта или
гормонорезистентность отсутствуют. Частота неизвестна. Мутации с потерей функции в отцовском
аллеле гена GNAS – 100%.
Синдром Мулчандани–Божж–Конлин.
Пренатальная и постнатальная задержка роста, экстремальные сложности вскармливания,
вызванные невозможностью сосания в первые годы жизни, микроцефалия, признаки
дисморфогенеза, напоминающие синдром Рассела–Сильвера, задержка умственного развития.
Частота неизвестна. Материнская ОРД по хромосоме 20 – 100%.
74.
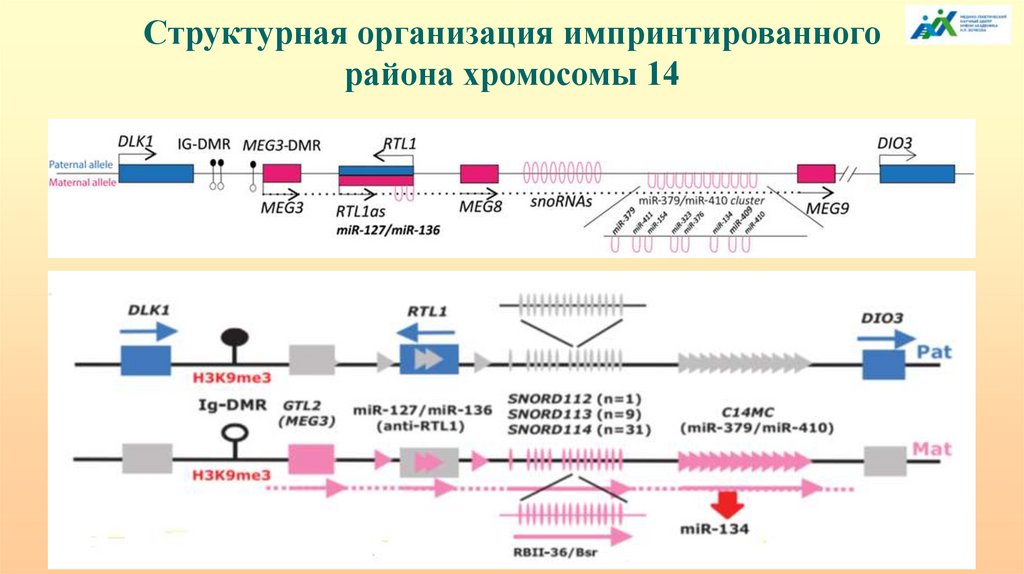
Структурная организация импринтированногорайона хромосомы 14
75.
Структурная организация импринтированногорайона хромосомы 14
Район хромосомы 14q32.2 содержит кластер импринтированных генов: часть которых
экспрессируется с отцовской хромосомы – DLK1, RTL1 и DIO3, а другие – гены днРНК
MEG3/GTL2, MEG8, RTL1as, мяРНК и miРНК – c материнской.
DLK1- регулирует дифференцировку преадипоцитов, экспрессируется в нейроэндокринных
тканях, особенно в корковом слое надпочечников. RTL1- ретротранспозон-подобный ген,
экспрессирующийся в плаценте и позднем фетальном периоде. DIO3- йодтиронин
дейодиназа 3 типа имеет несколько транскриптов: один, молекулярной массой 2,1 т.н.
экспрессируется в плаценте, фетальной печени и матке, другой – 3,2 т.н. – в яичках,
мочевом пузыре и матке, третий – 4,8 т.н. – в сердце и скелетных мышцах. MEG3 кодирует
днРНК, которая координирует широкий спектр различных клеточных процессов,
требующих эпигенетической регуляции генов и взаимодействия с ключевыми
сигнальными белками. Его интронный ДМР содержит два сайта связывания белка CTCF,
что предопределяет регуляторные функции. Функции RTL1as и MEG8, помимо
регуляторных, неизвестны. Паттерны экспрессии генов, зависящие от родительского
происхождения, регулируются межгенными ДМР, имеющими герминальное происхождение
(MEG3/DLK1 ДМР), и ДМР, функционирующим уже после оплодотворения (MEG3-ДМР).
Оба ДМР метилированы на отцовской хромосоме.
76.
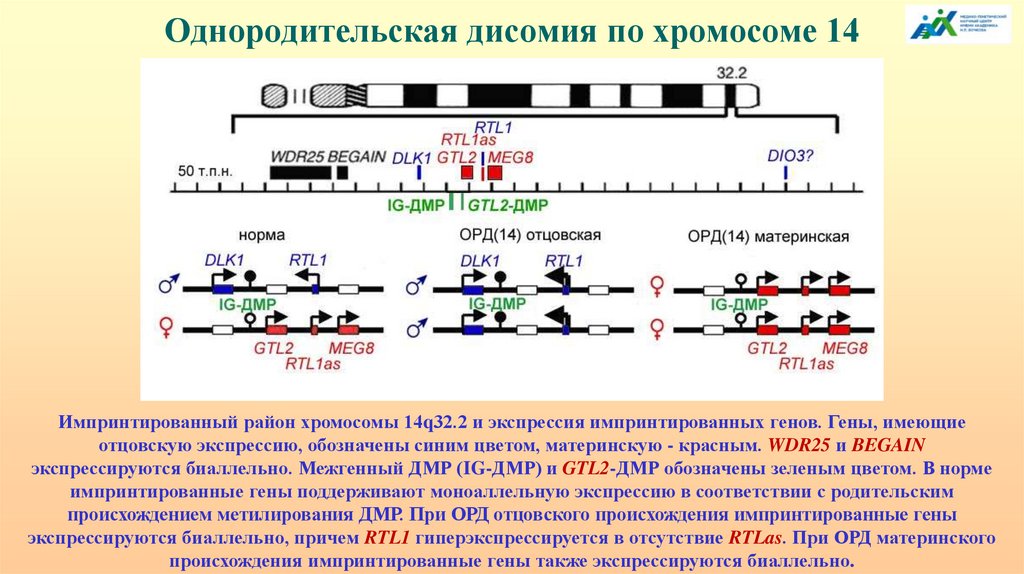
Однородительская дисомия по хромосоме 14Импринтированный район хромосомы 14q32.2 и экспрессия импринтированных генов. Гены, имеющие
отцовскую экспрессию, обозначены синим цветом, материнскую - красным. WDR25 и BEGAIN
экспрессируются биаллельно. Межгенный ДМР (IG-ДМР) и GTL2-ДМР обозначены зеленым цветом. В норме
импринтированные гены поддерживают моноаллельную экспрессию в соответствии с родительским
происхождением метилирования ДМР. При ОРД отцовского происхождения импринтированные гены
экспрессируются биаллельно, причем RTL1 гиперэкспрессируется в отсутствие RTLas. При ОРД материнского
происхождения импринтированные гены также экспрессируются биаллельно.
77.
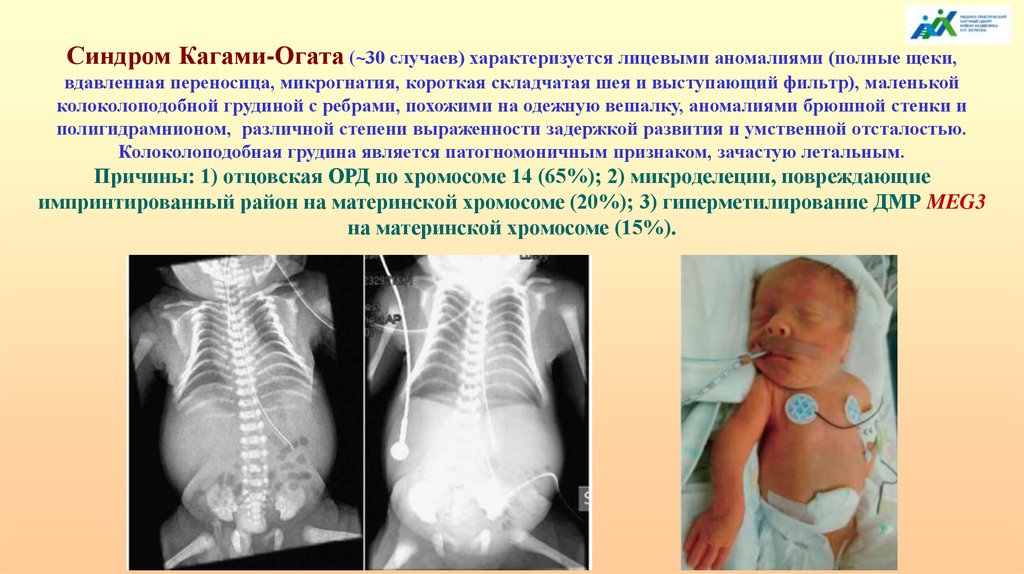
Синдром Кагами-Огата (~30 случаев) характеризуется лицевыми аномалиями (полные щеки,вдавленная переносица, микрогнатия, короткая складчатая шея и выступающий фильтр), маленькой
колоколоподобной грудиной с ребрами, похожими на одежную вешалку, аномалиями брюшной стенки и
полигидрамнионом, различной степени выраженности задержкой развития и умственной отсталостью.
Колоколоподобная грудина является патогномоничным признаком, зачастую летальным.
Причины: 1) отцовская ОРД по хромосоме 14 (65%); 2) микроделеции, повреждающие
импринтированный район на материнской хромосоме (20%); 3) гиперметилирование ДМР MEG3
на материнской хромосоме (15%).
78.
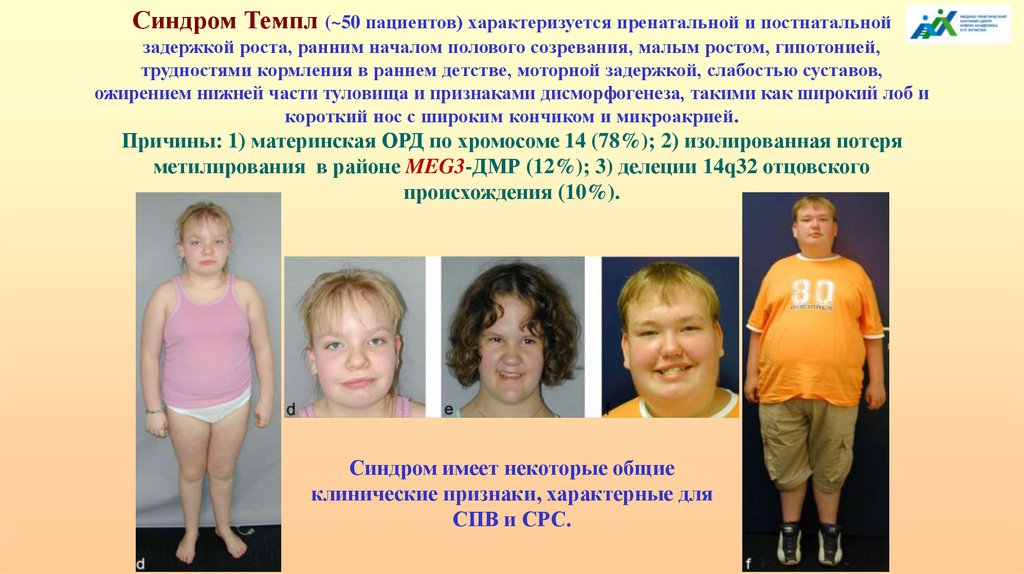
Синдром Темпл (~50 пациентов) характеризуется пренатальной и постнатальнойзадержкой роста, ранним началом полового созревания, малым ростом, гипотонией,
трудностями кормления в раннем детстве, моторной задержкой, слабостью суставов,
ожирением нижней части туловища и признаками дисморфогенеза, такими как широкий лоб и
короткий нос с широким кончиком и микроакрией.
Причины: 1) материнская ОРД по хромосоме 14 (78%); 2) изолированная потеря
метилирования в районе MEG3-ДМР (12%); 3) делеции 14q32 отцовского
происхождения (10%).
Синдром имеет некоторые общие
клинические признаки, характерные для
СПВ и СРС.
79.
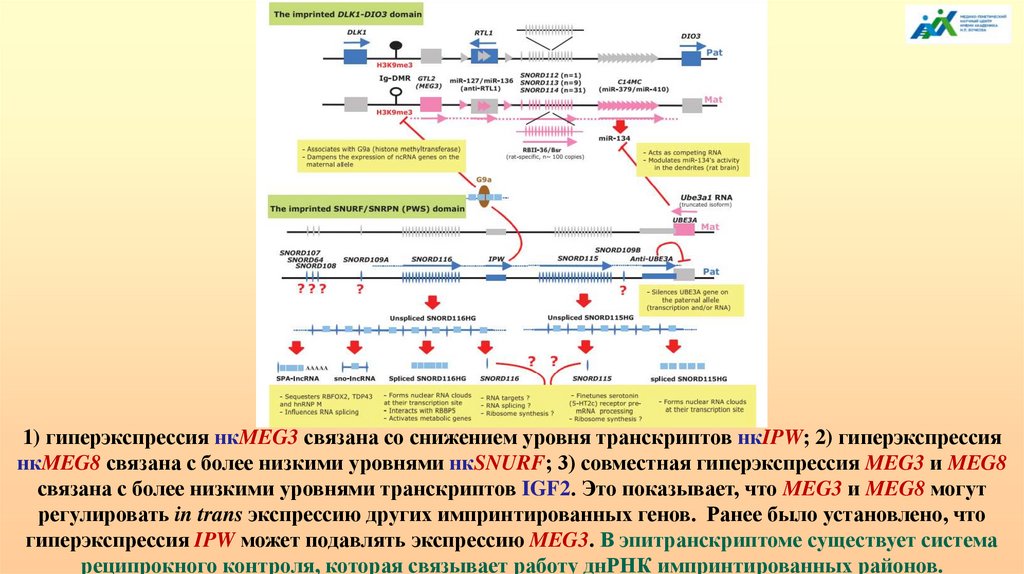
1) гиперэкспрессия нкMEG3 связана со снижением уровня транскриптов нкIPW; 2) гиперэкспрессиянкMEG8 связана с более низкими уровнями нкSNURF; 3) совместная гиперэкспрессия MEG3 и MEG8
связана с более низкими уровнями транскриптов IGF2. Это показывает, что MEG3 и MEG8 могут
регулировать in trans экспрессию других импринтированных генов. Ранее было установлено, что
гиперэкспрессия IPW может подавлять экспрессию MEG3. В эпитранскриптоме существует система
реципрокного контроля, которая связывает работу днРНК импринтированных районов.
80.
Гены SNORD в импринтированном домене SNURF-SNRPN генерирует многоразных некодирующих транскриптов. Предсказанные молекулярные функции,
связанные со SNORD, включая полностью процессированные SNORD,
сплайсированные Host-гены (HG), частично процессированные SNORDсодержащие РНК (sno-lncRNAs и SPA-lncRNAs), IPW lncRNA и анти-UBE3A
транскрипты, обобщены в желтых квадратах.
Импринтированный домен DLK1-DIO3 на 14q32 также содержит многочисленные
материнские гены SNORD (SNORD112, SNORD113 и SNORD114), чья организация
тандемно повторяющихся генов напоминает таковую в домене PWS. В 14q32 также
расположено множество генов миРНК (головки стрелок), большинство из которых
организованы в два кластера: кластеры miR-127/miR-136 и miR-379/miR-410.
Недавно в нейронах крысы было обнаружено еще одно взаимодействие между
областями Dlk1-Dio3 и Snurf-Snrpn: альтернативный транскрипт Ube3a (Ube3a-1)
функционирует как конкурирующая эндогенная РНК для инактивации miR-134,
производимой кластером miR-379/miR-410. Это, в свою очередь, влияет на развитие
нейронов, зависящее от активности.
Отсутствие экспрессии miR-127 и miR-136 у человека сопровождается аномалиями
развития.
81.
Схема молекулярной организации импринтированногорайона 6q24.
Транзиторный неонатальный сахарный диабет редкое заболевание (частота 1:500000
новорожденных), которое проявляется гипергликемией, глюкозоурией, сильной
дегидратацией организма и задержкой роста. В небольшом количестве описанных семейных
случаев наследование было исключительно отцовское и ассоциировалось с дупликацией
района хромосомы 6q24. Два импринтированных гена — PLAGL1 и HYMAI, активно
экспрессируются в поджелудочной железе. PLAGL1 регулирует транскрипцию и является
геном-супрессором опухолевого роста, его гиперэкспрессия приводит к аномальной работе βклеток поджелудочной железы. PLAGL1 регулирует работу ряда импринтированных и
неимпринтированных генов - IGF2, H19, SLC2A4, CDKN1C и PPARγ1, вовлеченных в
процессы клеточного роста и метаболизма. HYMAI кодирует днРНК. ZAC-AS/PLAGL1-AS.
82.
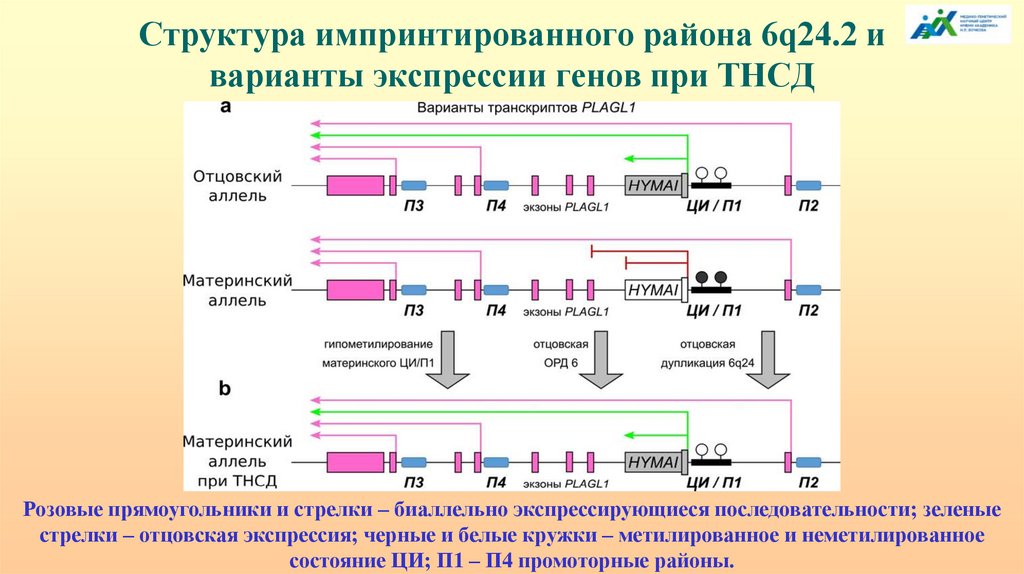
Структура импринтированного района 6q24.2 иварианты экспрессии генов при ТНСД
Розовые прямоугольники и стрелки – биаллельно экспрессирующиеся последовательности; зеленые
стрелки – отцовская экспрессия; черные и белые кружки – метилированное и неметилированное
состояние ЦИ; П1 – П4 промоторные районы.
83.
Структура молекулярно-генетической иэпигенетической патологии при транзиторном
неонатальном сахарном диабете
1) Отцовская ОРД хромосомы 6 – 35–40%;
2) Отцовская дупликация 6q24 – 30–40%;
3) Потеря метилирования ЦИ PLAGL1 материнского аллеля
– 20–30%.
4) Мутации ZFP57 и других генов, приводящих к MLID –
30–50%.
84.
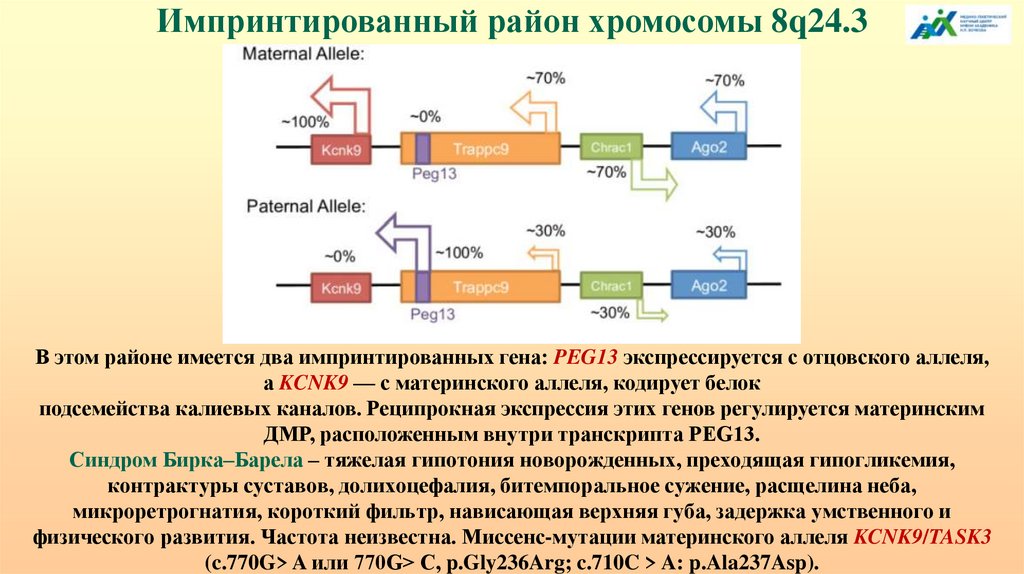
Импринтированный район хромосомы 8q24.3В этом районе имеется два импринтированных гена: PEG13 экспрессируется с отцовского аллеля,
а KCNK9 — с материнского аллеля, кодирует белок
подсемейства калиевых каналов. Реципрокная экспрессия этих генов регулируется материнским
ДМР, расположенным внутри транскрипта PEG13.
Синдром Бирка–Барела – тяжелая гипотония новорожденных, преходящая гипогликемия,
контрактуры суставов, долихоцефалия, битемпоральное сужение, расщелина неба,
микроретрогнатия, короткий фильтр, нависающая верхняя губа, задержка умственного и
физического развития. Частота неизвестна. Миссенс-мутации материнского аллеля KCNK9/TASK3
(c.770G> A или 770G> С, p.Gly236Arg; c.710C > A: p.Ala237Asp).
85.
Структура 5’- района гена RB1 и локализация в нем CpG-островковCpG 85 является промотором альтернативного транскрипта RB1, который экспрессируется только с
неметилированной отцовской хромосомы. Несмотря на ожидаемо более высокий общий уровень
экспрессии мРНК транскриптов, уровень экспрессии с отцовского аллея ниже в 2-3 раза за счет
интерференции транскрипции, вызванной совместной экспрессией регулярного и альтернативного
транскриптов. Транскрипционный комплекс альтернативного транскрипта гена RB1, который
связывается с неметилированным островком CpG 85 на отцовской хромосоме, выступает в качестве
блока для прохождения транскрипционного комплекса основного транскрипта на том же аллеле.
Низкопенетрантные герминальные мутации RB1, унаследованные от отца, приводят к развитию РБ
чаще и в более тяжелой форме по сравнению с таковыми, унаследованными от матери. В случае
наследования низкопенетрантной мутации по материнской линии, опухолевой супрессорной
активности pRB достаточно для предотвращения развития РБ. Напротив, при передаче мутантного
аллеля от отца, низкая остаточная активность белка за счет более низкой экспрессии будет
имитировать мутацию, приводящую к отсутствию белка, что приведет к развитию заболевания.
86.
Редкие синдромы, связанные с патологиейимпринтированных генов
Импринтированный район хромосомы 11p15.5.
Синдром IMAGe – задержка внутриутробного развития, метафизарная дисплазия, врожденная
гипоплазии надпочечников и аномалии половых органов. Частота неизвестна. Миссенс-мутации
материнского аллеля гена CDKN1C (p.Asp274Asn, p.Lys278Glu, p.Arg279Leu).
Импринтированный район хромосомы 15q11.2–q13.
Синдром Шаафа–Янга – гипотония новорожденных, гипогонадизм, контрактуры суставов, задержка
умственного и физического развития, аутизм. Частота неизвестна. Нонсенс- и миссенс-мутации
отцовского аллеля гена MAGEL2.
Синдром центрального преждевременного полового созревания типа 2 – гонадотропин-зависимое
преждевременное половое созревание (до 6-летнего возраста у девочек и 8- летнего – у мальчиков).
Частота неизвестна. Гетерозиготные инактивирующие мутации отцовского аллеля гена MKRN3/ZFP127.
Дупликации импринтированного района хромосомы 15q11.2-q13.1 – материнские дупликации
манифестируют гипотонией, задержкой моторного, физического и умственного развития, судорогами,
аутизмом, поведенческими аномалиями; отцовские дупликации фенотипически не проявляются.
ОРД хромосомы 16mat. Импринтированный ген ZNF597 экспрессируется только с материнской
хромосомы. Множественные аномалии и задержка внутриутробного развития без явного специфического
фенотипа. ОРД хромосомы 16pat крайне редки и сопровождаются аномалиями фенотипа.
87.
88.
Множественные аномалии метилированияимпринтированных регуляторных районов (MLID)
Описано более 10 пациентов с СВБ, у которых, помимо материнского
гипометилирования СВБ-ЦИ2 обнаружена потеря метилирования по другим
локусам. У 6 пациентов с ТНД, кроме материнского гипометилирования ДМР на
хромосоме 6q24, выявлены и другие локусы гипометилирования. В 2006 г. была
описана семья (близкородственный брак), в которой две дочери имели
фенотипические проявления ТНД с некоторыми признаками СВБ. При
исследовании статуса метилирования импринтированных районов установлено, что
потеря метилирования произошла не только в импринтированном районе PLAGL1
(6q24), но и в районах KCNQ1OT1 (11p15.5), GRB10 (7p11.2–р12), PEG3 (19q13),
PEG1/MEST (7q32) и NESPAS (20q13). Предполагалось, что в семье имеет место
некий аутосомно-рецессивный дефект, повреждающий механизмы метилирования у
потомства, или нарушен процесс установления импринта в ооцитах.
89.
В 2008 г. было установлено, что причиной такого многолокусного гипометилированияРДМ могут быть мутации в гене ZFP57, расположенном в 6р22.1. Ген является
транскрипционным репрессором, который формирует комплекс с белком корепрессором KAP1. KAP1 рекрутирует другие белки, такие, как метилтрансфераза
лизина 9 гистона Н3 (SETDB1), ядерный белок NP95, который, в свою очередь,
привлекает ДНК-метилтрансферазы. Таким образом, этот белковый комплекс играет
огромную роль в регуляции и поддержания метилирования ДНК в различных ЦИ.
ZFP57 связывается с метилированными ЦИ в предимплантационном развитии,
защищая их от деметилирования и сохраняя родительскую идентичность. Сайт
связывания ZFP57 обнаружен в 17 из 31 импринтированного ДМР.
Подобно ZFP57, ZFP445 может связываться с ЦИ, привлекать KAP1 и запускать
метилирование H3K9me3. Эти гены экспрессируются не одновременно – сначала
ZNF445, а затем ZFP57, поэтому они рекрутируют KAP1 по очереди. ZNF445
связывается с 13 импринтированными ДМР.
ZNF202, связывает только четыре импринтированных ДМР.
ZFP42, являющийся маркером стволовых клеток и активно экспрессирующийся в
преимплантационном эмбрионе, выполняет функцию защиты от метилирования
обычно неметилированных аллелей импринтированных ДМР.
90.
Множественные аномалии метилированияимпринтированных регуляторных районов (MLID)
Мутации в “материнских генах”, кодирующих белки субкортикального комплекса ооцита,
могут вызывать репродуктивные проблемы на эпигенетическом уровне. Этот комплекс играет
важную роль на раннем этапе эмбрионального развития и содержит не менее семи белков
(NLRP2, NLRP5, NLRP7, PADI6, KHDC3L, TLE6 и OOEP). Гены этих белки экспрессируются
исключительно материнским геномом в ооцитах и на ранних этапах развития эмбриона, а
затем инактивируются, когда геном эмбриона начинает функционировать самостоятельно.
Гены NLRP2, NLRP5, NLRP7, кодирующие небольшое подсемейство цитоплазматических
белков, содержащих пирин-домены, активно экспрессируются в растущих ооцитах и
необходимы для созревания ооцита, регуляции метилирования на ранних стадиях эмбриогенеза
и поддержания плоидности в раннем эмбрионе.
Мутации в генах NLRP2, NLRP5, NLRP7 сопровождаются фенотипами СБВ, СРС и ТНСД, в
генах: OOEP – ТНСД, ZAR1 – СБВ.
Мутации в гене PADI6 вызывают потерю метилирования в импринтированных ДМР:
KCNQ1OT1, PLAGL1, GRB10, MEST, H19/IGF2, GNAS-AS, GNAS-XL, MEG3, SNURF и могут
приводить к формированию фенотипов синдромов Рассела–Сильвера, Беквита–Видеманна и
Темпл.
91.
Частоты вклада MLID в фенотипическиепроявления болезней импринтинга
Транзиторный неонатальный сахарный диабет с потерей метилирования PLAGL1-DMR
в результате MLID по разным оценкам составляет 30–50%;
Синдром Рассела–Сильвера с потерей метилирования в H19-DMR может быть
обусловлен MLID в 7–30%;
Синдром Беквита–Видеманна с потерей метилирования в KCNQ1OT1 или биаллельным
метилированием в H19-DMR в результате MLID может составлять 20–50%.
Псевдогипопаратиреоз типа 1В, вызванный потерей метилирования в GNAS-A/B DMR в
результате MLID - 6.3–12.5% случаев.
При синдромах Прадера–Вилли и Ангельмана практически не наблюдаются нарушения
метилирования, обусловленные MLID.
У пациентов с MLID выявлены проявления специфического “классического” фенотипа
определенной болезни импринтинга, однако у некоторых из них развивается комплекс
симптомов, характерных для разных синдромов, обусловленных аномалиями
импринтинга. MLID часто бывают мозаичными, так как патология возникает в
ограниченном количестве клеток на самых ранних этапах развития эмбриона. Поэтому
корреляции между эпи-/генотипом и фенотипом у пациентов с MLID определить крайне
сложно.
92.
Изучение структурной организации, специфического аллельногометилирования и определенной аллельной структуры хроматина в
регуляторных районах, цис- и транс-взаимодействия днРНК с
первичными/герминальными (PLAGL1, H19/IGF2, PEG13, IGF2-DMR0,
KCNQ1OT1, RB1, MEG3/DLK1, SNURF, GNAS-AS1, GNAS-XL, GNAS A/B) и
вторичными/ соматическими (IGF2-DMR2, DLK1, GTL2, MEG8, MAGEL2,
NDN, SNRPN, SNORD116, GNAS/NESP) регуляторными районами (DMR) на
различных этапах онтогенеза позволяет понять, как функционирует
эпигеном в норме и при патологии. Нарушения концерта этих структурных и
функциональных составляющих генома могут приводить к патологическим
состояниям, таким как болезни импринтинга, не синдромальные формы
нарушений умственного и физического развития, многофакторные
заболевания, в том числе онкологические, аутоиммунные и другие.
93.
Импринтинги вспомогательные репродуктивные технологии
Наиболее распространенная патология:
С-м Ангельмана – 20 случаев
С-м- Прадера-Вилли – 13 случаев
С-м Видеманна-Беквита – 60 случаев
Синдром Сильвера-Рассела – 5
Вероятность рождения ребенка с СБВ после ВРТ примерно в 7 - 10 раз выше по
сравнению с естественным зачатием. В подавляющем большинстве (83,3-100%)
пациенты имели гипометилирование материнского аллеля в районе СБВ-ЦИ2,
регулирующим экспрессию генов СDKN1C и KCNQ1 (потеря импринтинга по LIT1).
Для других синдромов частота не превышает популяционную.
94.
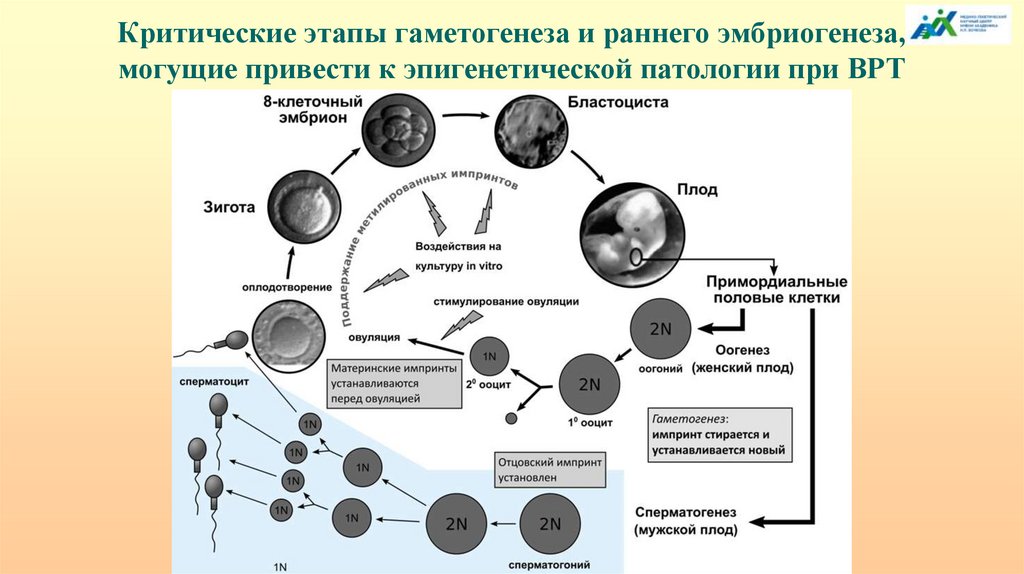
Целый ряд причин может приводить к эпигенетическим аномалиям: 1)бесплодие само по себе; 2) процесс стимуляции овуляции; 3) физические
манипуляции с эмбрионом в процессе оплодотворения in vitro (IVF), введение
сперматозоида в яйцеклетку (ICSI), непосредственно перенос эмбриона; 4)
особенности культивирование эмбриона in vitro. Эти критические
манипуляции совпадают с очень тонкими эпигенетическими процессами
стирания, установления и поддержания метилирования на ранних этапах
формирования гамет, оплодотворения и раннего эмбриогенеза. Большинство
случаев связано с нарушением метилирования материнского аллеля.
Материнский геном может быть более подвержен дефектам импринтинга и
метилирования в течение преимплантационного периода, когда эмбрион
полностью зависит от условий культивирования in vitro.
В ряде случаев установлена материнская ОРД. Этот факт имеет логическое
обоснование: нерасхождение хромосом характерно для возрастных женщин,
которые составляют значительную группу, прибегающую к ВРТ.
95.
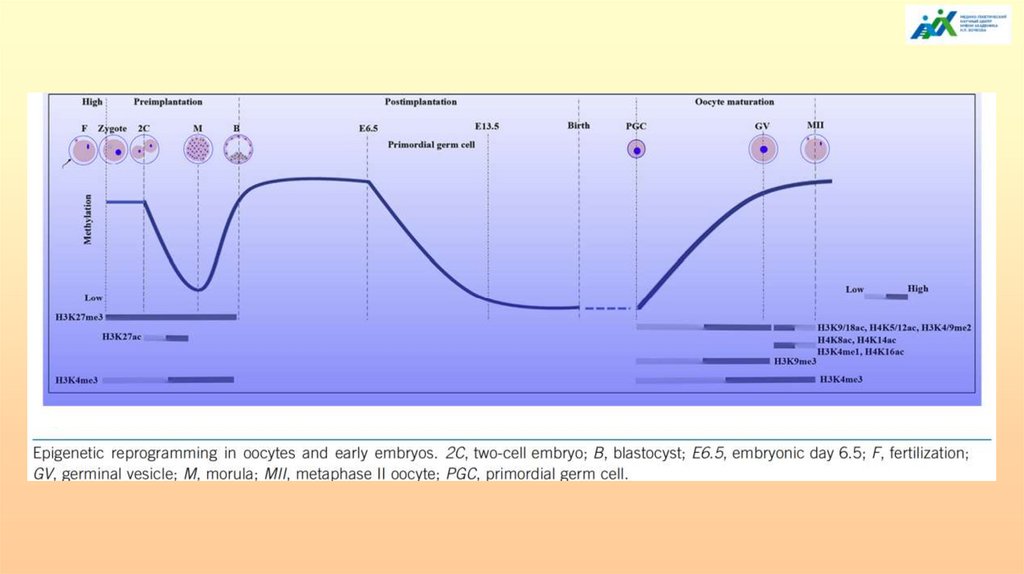
Метилирование/деметилирование в процессегаметогенеза
96.
Критические этапы гаметогенеза и раннего эмбриогенеза,могущие привести к эпигенетической патологии при ВРТ
97.
Но…Недавнее клиническое исследование, в котором анализировалась сперма фертильных
и суб-фертильных/бесплодных мужчин, показало, что изменения гистонов спермы были
связаны со значительными изменениями в метилировании ДНК и изменением миРНК
сперматозоидов. В бластоцистах измененные миРНК сперматозоидов хорошо
коррелировали с изменениями в уровнях транскриптов соответствующих мРНКмишеней. Эти наблюдения приводят к мысли о том, что оценки эпигенетических сигнатур
в сперме могут и должны использоваться в качестве прогностических факторов
успешного исхода вспомогательных репродуктивных технологий.
Denomme MM, et al. Alterations in the sperm histone retained epigenome are associated with
unexplained male factor infertility and poor blastocyst development in donor oocyte IVF cycles.
Hum Reprod 2017;32:2443e55.
Olszewska M, et al. Global 5mC and 5hmC DNA Levels in Human Sperm Subpopulations with
Differentially Protaminated Chromatin in Normo- and Oligoasthenozoospermic Males. Int J Mol
Sci. 2022;23(9):4516.
Становится все более очевидным, что эпигеном сперматозоидов чувствителен к
различным видам воздействия окружающей среды в течение жизненного цикла мужских
половых клеток, и что эпигенетическая сигнатура сперматозоида/спермы конкретного
мужчины изменчива и будет отражать текущие обстоятельства жизни.
98.
Синдром Мартина-БеллЧастота СМБ - 1: 3000-4000 мужчин и 1:8000 женщин
99.
Синдром Мартина-Белл100.
Ломкость Х-хромосомы101.
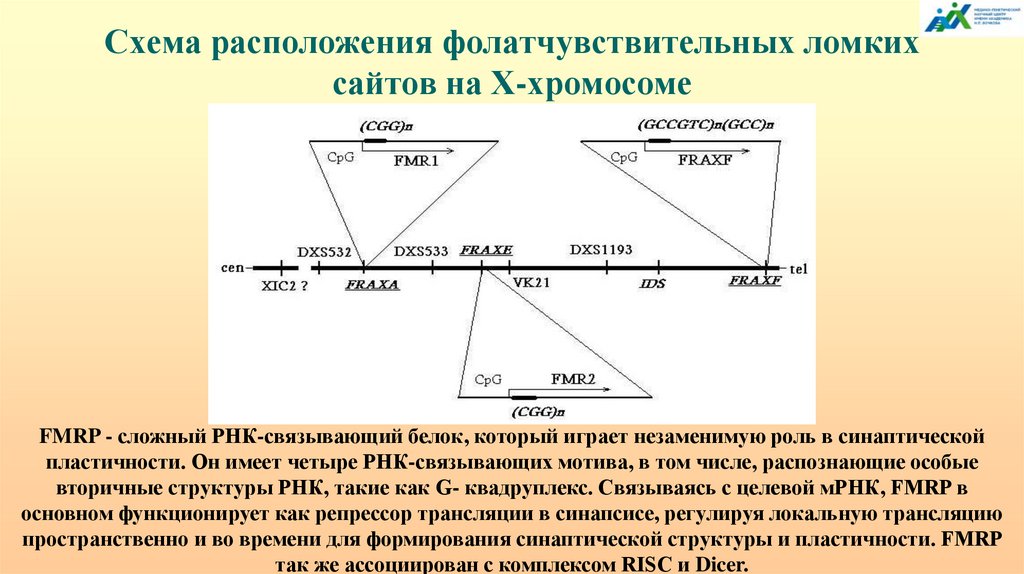
Схема расположения фолатчувствительных ломкихсайтов на Х-хромосоме
FMRP - сложный РНК-связывающий белок, который играет незаменимую роль в синаптической
пластичности. Он имеет четыре РНК-связывающих мотива, в том числе, распознающие особые
вторичные структуры РНК, такие как G- квадруплекс. Связываясь с целевой мРНК, FMRP в
основном функционирует как репрессор трансляции в синапсисе, регулируя локальную трансляцию
пространственно и во времени для формирования синаптической структуры и пластичности. FMRP
так же ассоциирован с комплексом RISC и Dicer.
102.
Синдром Мартина-БеллЧисло
триплетов
CGG
Метилирование Фенотип
промотора гена
СМБ
FMR1
норма
2-54
-
-
премутация
55-230
-
-
полная
мутация
230
+
+
Аллели в состоянии премутации выявляются у всех нормальных трансмиттеров и, по
крайней мере, у 80% бессимптомных носительниц. Нормальные трансмиттеры всегда
передают своим дочерям премутацию в неизменном виде. В то же время, премутации,
передаваемые женщинами, в 80% случаев трансформируются в полные мутации, причём
вероятность такой трансформации прямо зависит от размера премутации. Полная мутация
выявляется практически у всех больных обоего пола и у 20% бессимптомных носительниц.
103.
Наследование СМБ носит необычный характер:передача заболевания происходит через фенотипически
нормальных мужчин (нормальные трансмиттеры); дочери
нормальных трансмиттеров никогда не бывают умственно
отсталыми и никогда или почти никогда не имеют ломкого
сайта на Х-хромосоме, однако в следующем поколении треть
женщин умственно субнормальны, а их сыновья, в свою
очередь, как правило, оказываются больными. Братья
клинически нормальных мужчин-носителей маркерной
хромосомы имеют низкий риск заболевания СМБ, в то время
как для их внуков и правнуков риск значительно выше. Такой
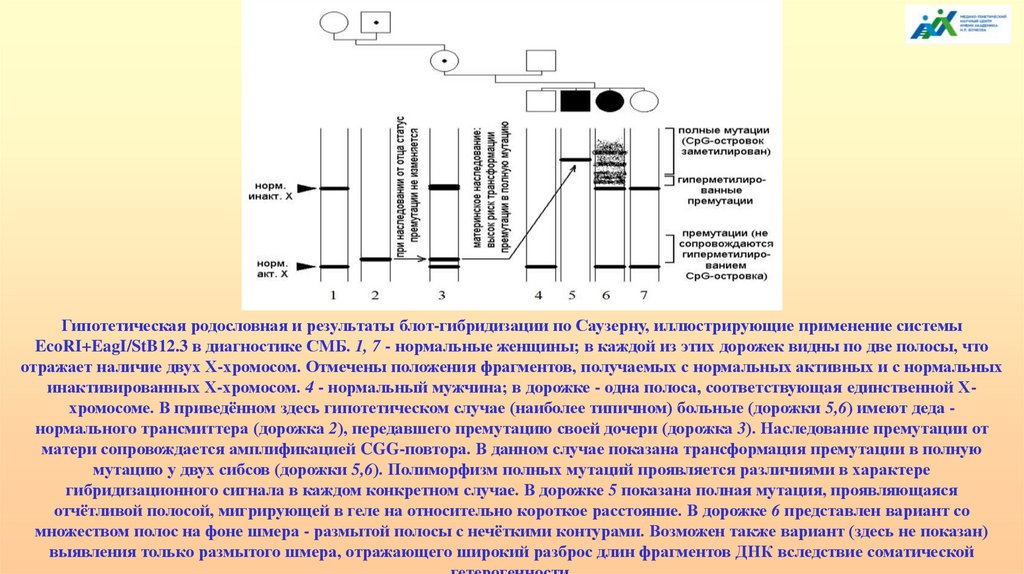
необычный характер наследования СМБ получил название
парадокса Шерман.
104.
Гипотетическая родословная и результаты блот-гибридизации по Саузерну, иллюстрирующие применение системыEcoRI+EagI/StB12.3 в диагностике СМБ. 1, 7 - нормальные женщины; в каждой из этих дорожек видны по две полосы, что
отражает наличие двух Х-хромосом. Отмечены положения фрагментов, получаемых с нормальных активных и с нормальных
инактивированных Х-хромосом. 4 - нормальный мужчина; в дорожке - одна полоса, соответствующая единственной Ххромосоме. В приведённом здесь гипотетическом случае (наиболее типичном) больные (дорожки 5,6) имеют деда нормального трансмиттера (дорожка 2), передавшего премутацию своей дочери (дорожка 3). Наследование премутации от
матери сопровождается амплификацией CGG-повтора. В данном случае показана трансформация премутации в полную
мутацию у двух сибсов (дорожки 5,6). Полиморфизм полных мутаций проявляется различиями в характере
гибридизационного сигнала в каждом конкретном случае. В дорожке 5 показана полная мутация, проявляющаяся
отчётливой полосой, мигрирующей в геле на относительно короткое расстояние. В дорожке 6 представлен вариант со
множеством полос на фоне шмера - размытой полосы с нечёткими контурами. Возможен также вариант (здесь не показан)
выявления только размытого шмера, отражающего широкий разброс длин фрагментов ДНК вследствие соматической
105.
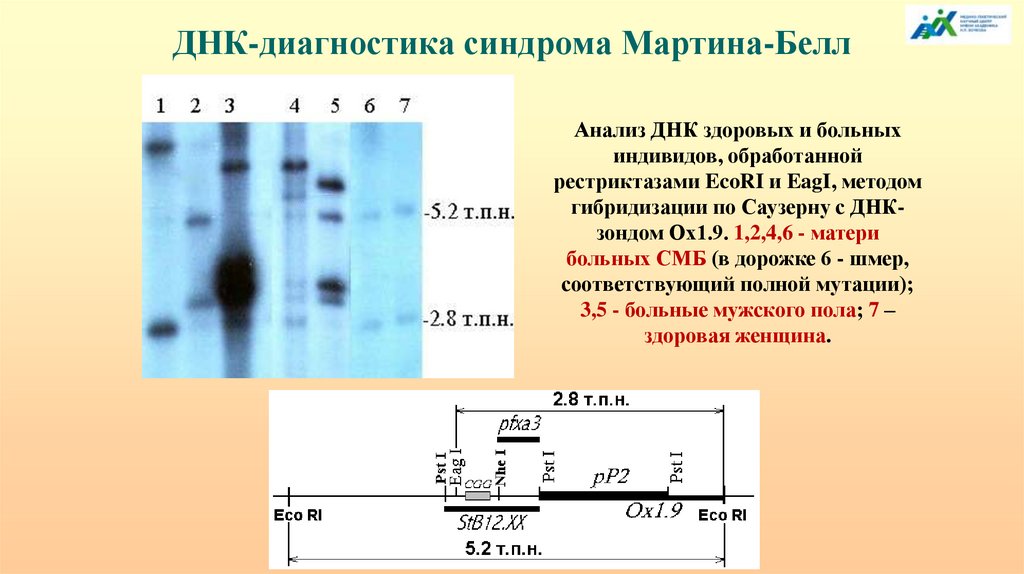
ДНК-диагностика синдрома Мартина-БеллАнализ ДНК здоровых и больных
индивидов, обработанной
рестриктазами EcoRI и EagI, методом
гибридизации по Саузерну с ДНКзондом Ох1.9. 1,2,4,6 - матери
больных СМБ (в дорожке 6 - шмер,
соответствующий полной мутации);
3,5 - больные мужского пола; 7 –
здоровая женщина.
106.
107.
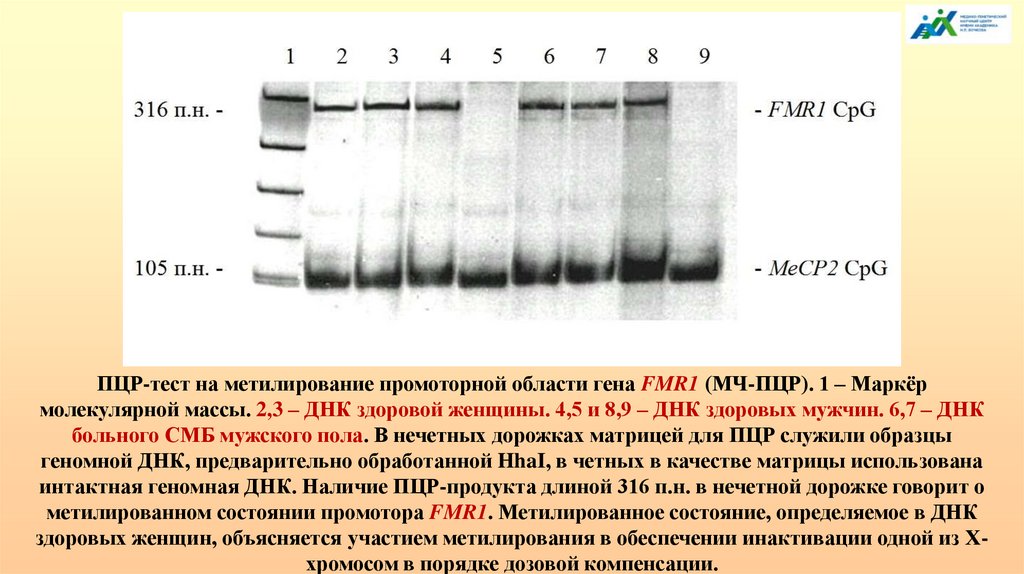
ПЦР-тест на метилирование промоторной области гена FMR1 (МЧ-ПЦР). 1 – Маркёрмолекулярной массы. 2,3 – ДНК здоровой женщины. 4,5 и 8,9 – ДНК здоровых мужчин. 6,7 – ДНК
больного СМБ мужского пола. В нечетных дорожках матрицей для ПЦР служили образцы
геномной ДНК, предварительно обработанной HhaI, в четных в качестве матрицы использована
интактная геномная ДНК. Наличие ПЦР-продукта длиной 316 п.н. в нечетной дорожке говорит о
метилированном состоянии промотора FMR1. Метилированное состояние, определяемое в ДНК
здоровых женщин, объясняется участием метилирования в обеспечении инактивации одной из Ххромосом в порядке дозовой компенсации.
108.
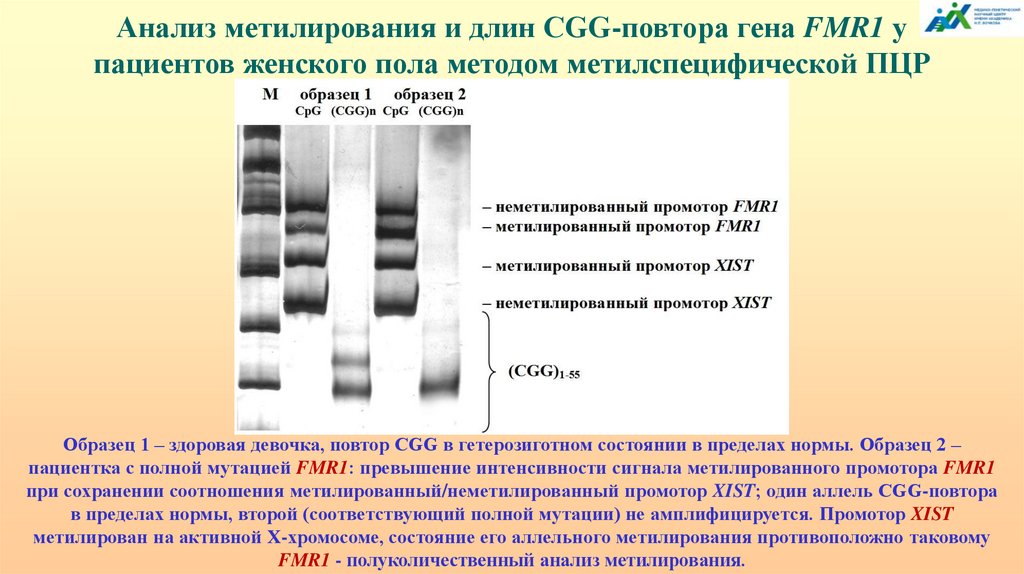
Анализ метилирования и длин CGG-повтора гена FMR1 упациентов женского пола методом метилспецифической ПЦР
Образец 1 – здоровая девочка, повтор CGG в гетерозиготном состоянии в пределах нормы. Образец 2 –
пациентка с полной мутацией FMR1: превышение интенсивности сигнала метилированного промотора FMR1
при сохранении соотношения метилированный/неметилированный промотор XIST; один аллель CGG-повтора
в пределах нормы, второй (соответствующий полной мутации) не амплифицируется. Промотор XIST
метилирован на активной Х-хромосоме, состояние его аллельного метилирования противоположно таковому
FMR1 - полуколичественный анализ метилирования.
109.
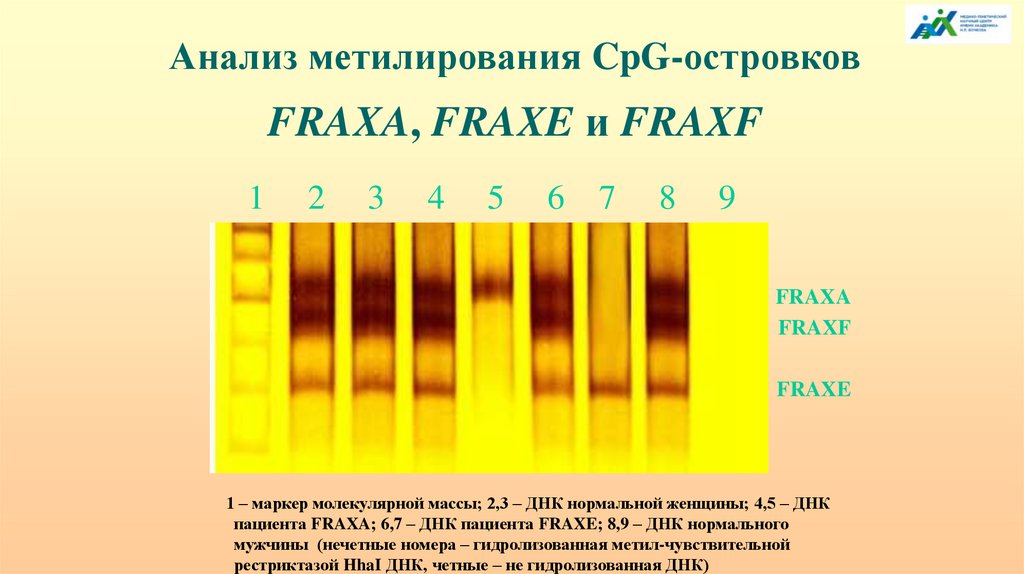
Анализ метилирования CpG-островковFRAXA, FRAXE и FRAXF
1
2
3
4
5
6
7
8
9
FRAXA
FRAXF
FRAXE
1 – маркер молекулярной массы; 2,3 – ДНК нормальной женщины; 4,5 – ДНК
пациента FRAXA; 6,7 – ДНК пациента FRAXE; 8,9 – ДНК нормального
мужчины (нечетные номера – гидролизованная метил-чувствительной
рестриктазой HhaI ДНК, четные – не гидролизованная ДНК)
110.
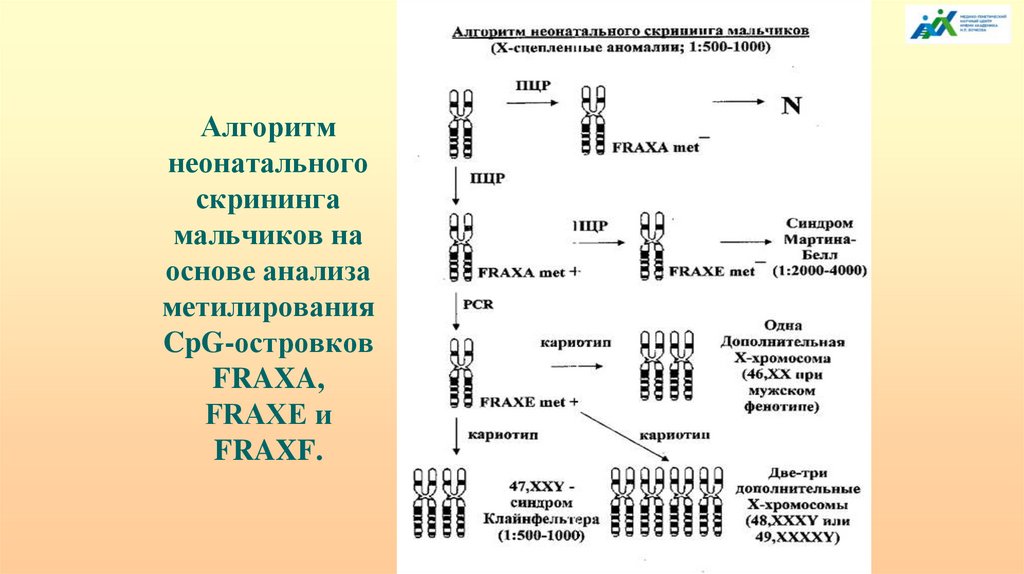
Алгоритмнеонатального
скрининга
мальчиков на
основе анализа
метилирования
CpG-островков
FRAXA,
FRAXE и
FRAXF.
111.
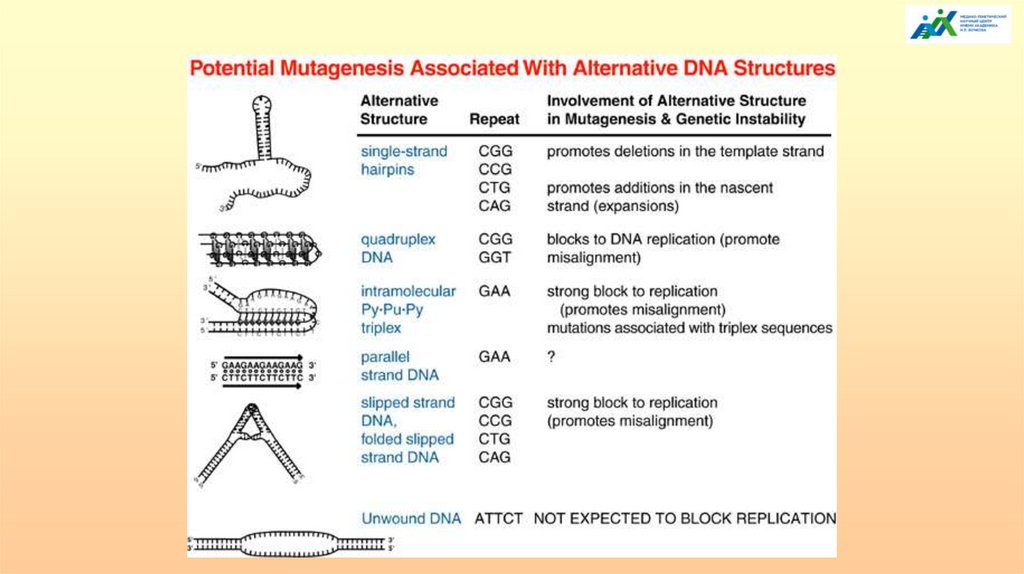
К настоящему времени предложено несколько моделейэпигенетической инактивации FMR1
Первая модель ДНК-ориентированная. Вторичные структуры,
образованные CGG-повторами служат субстратами для ДНКметилтрансфераз, инициирующих de novo метилирования ДНК, или в
качестве мишеней для белков, связывающих повторы, которые, затем,
привлекают репрессорные комплексы.
Вторая модель РНК-ориентированная. Шпилечные структуры в мРНК,
образованные CGG-повторами, превышающие определенную величину,
запускают процесс РНКи для депонирования репрессивных эпигенетических
маркеров.
Третья модель - смешанная, в которой действует гибрид ДНК:РНК. Во время
транскрипции гибридизация вновь синтезированной РНК с ее
распакованной ДНК образуют особую R-петлю, которая может играть роль
структурного блока или аналога нуклеосомы, чтобы вызвать
эпигенетическую инактивацию.
112.
В раннем эмбриональном развития, ген FMR1, содержащий полную мутацию CGG-повторов, активнотранскрибируется. Двунаправленная транскрипция ДНК-матрицы генерирует дцРНК, несущие полные
мутации CGG-повторов. Dicer расщепляет эти дцРНК с образованием малых РНК. После включения в
комплекс RITS, подобно siРНК, эти малые РНК направляют RITS к области экспансии CGG, рекрутируя
эпигенетические эффекторы, такие как гистон метилтрансферазы и ДНК-метилтрансферазы, чтобы вызвать
эпигенетическую инактивацию FMR1. Таким образом, РНКи может играть важную роль в эпигенетическом
процессе сайленсинга FMR1.
113.
Механизм патогенеза FXS, опосредованного миРНКЧрезмерная экспансия r(CGG) в гене FMR1, кодирующего FMRP, лежит в основе FXSассоциированных расстройств, таких как умственная отсталость и аутизм. Экспансия
повторов более чем 200 копий (полная мутация) приводит к полной потере экспрессии FMR1 и
отсутствию FMRP. Патологический процесс, приводящий к отсутствию этого важного белка
включает восемь этапов: (1) Транскрипция гена FMR1 начинается после стадии бластоцисты
(на 7-10 день эмбрионального развития у человека. (2) РНК-сплайсинг и дальнейший
процессинг разрезают амплифицированный повтор 5′-НТР r(CGG) мРНК FMR1 на
фрагменты. (3) Вырезанные r(CGG) фрагменты далее обрабатываются определенными
рибонуклеазами до различных предшественников миРНК, связанных с повторами. (4) Эти
предшественники миРНК экспортируются в цитоплазму, а затем щепятся до зрелых miRfmr1s РНКазой Дайсером. (5) Избыточное количество miR-fmr1s накапливается в цитоплазме
вокруг клеточного ядра. (6) miR-fmr1s с сигналом NIS попадают в клеточное ядро. (7)
Накопление miR-fmr1s в ядре приводит к образование комплексов РНК-индуцированного
транскрипционного сайленсинга – RITS (RISC-подобный гетерохроматин-таргетирующий
комплекс) с Rad54l и MeCP2 вблизи промотора гена FMR1. (8) Комплексы RITS вызывают
гиперметилирование ДНК и, следовательно, приводят к транскрипционной инактивации
FMR1, что наблюдается почти у всех пациентов с FXS.
114.
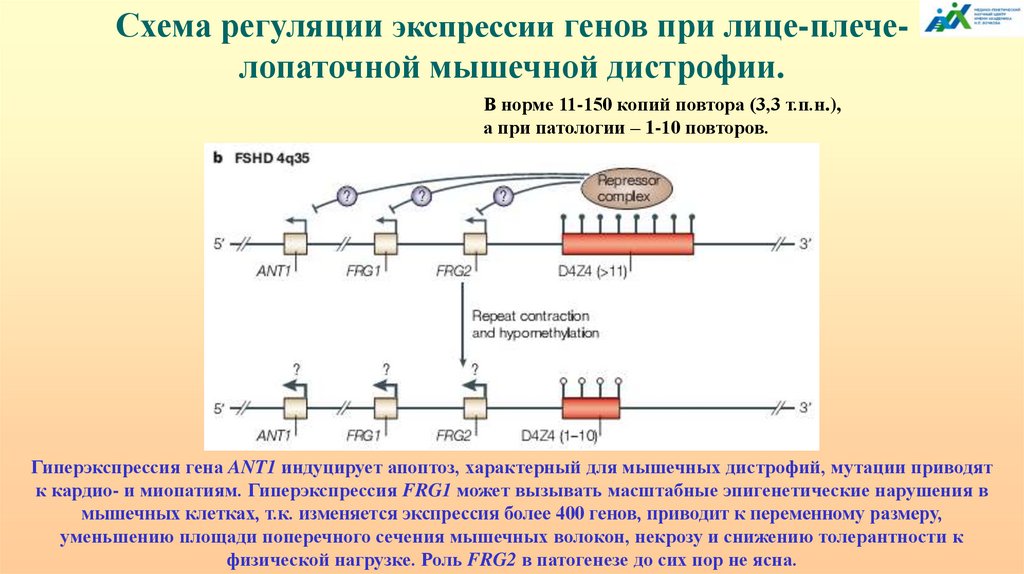
Схема регуляции экспрессии генов при лице-плечелопаточной мышечной дистрофии.В норме 11-150 копий повтора (3,3 т.п.н.),
а при патологии – 1-10 повторов.
Гиперэкспрессия гена ANT1 индуцирует апоптоз, характерный для мышечных дистрофий, мутации приводят
к кардио- и миопатиям. Гиперэкспрессия FRG1 может вызывать масштабные эпигенетические нарушения в
мышечных клетках, т.к. изменяется экспрессия более 400 генов, приводит к переменному размеру,
уменьшению площади поперечного сечения мышечных волокон, некрозу и снижению толерантности к
физической нагрузке. Роль FRG2 в патогенезе до сих пор не ясна.
115.
Ген DUX4 расположен внутри каждого D4Z4 и кодирует транскрипционный фактор,участвующий в активации генома зиготы на самой ранней стадии развития организма. Было
установлено, что его экспрессия во взрослых дифференцированных мышечных клетках
вызывает нарушение экспрессии генов, что приводит к апоптозу, индукции окислительного
стресса и воспалительных путей. Снижение экспрессии FAT1 приводит к нарушению миграции
и гибели миобластов, изменению формы мышц, включая лицевые и лопаточно-плечевые.
Аномальная экспрессия DUX4c приводит к снижению миогенной дифференцировки,
уменьшению слияния, снижению пролиферации сателлитных клеток, нарушению регуляции
миогенных факторов MYOD, PAX7 и MYF5.
116.
Предполагается, что геном человека содержит 100 – 200импринтированных генов. На сегодняшний день их около 150.
117.
Характерные черты импринтированных генов1. Кластеризация.
Импринтированные гены распределены не случайным образом в геноме, а встречаются
группами, причем в кластерах имеются гены, экспрессирующиеся, как с материнской, так и с
отцовской хромосомы.
Кластер в коротком плече хромосомы 11p15.5 (2 м.п.н.) содержит 6 генов имеющих
материнскую экспрессию: CDKN1C, KCNQ1, HASH2, ORCTL2, TSSC3, H19 и два - отцовскую:
KCNQ1OT1/LIT1 и IGF2.
Кластер в проксимальном районе длинного плеча хромосомы 15q11.2 (5 м.п.н.) содержит
пять белок-кодирующих генов (MKRN3, MAGEL2, NECDIN, NPAP1/C15orf2, SNURF-SNRPN),
гены нетранслируемых РНК (MKRN3-AS, IPW, UBE3A-AS/SNHG14) и гены малых
ядрышковых РНК (SNORD116, SNORD115, SNORD64, SNORD107, SNORD108, SNORD109A и
SNORD109B), имеющих отцовскую экспрессию. Материнская экспрессия - UBE3A.
Общие черты кластеров:
1) гены распределены на достаточно большом расстоянии;
2) наличие в кластере генов, экспрессирующихся только с
отцовской или материнской хромосомы;
3) наличие генов, которые продуцируют не кодирующую РНК;
4) наличие повторяющихся последовательностей.
118.
2. Консервативность импринтинга.Характер импринтинга генов H19, IGF2, p57KIP и SNRPN идентичен у
человека и мыши.
3. Асинхронность репликации ДНК
импринтированных генов.
Импринтированные гены имеют асинхронную репликацию, показанную
в кластерах импринтированных генов с использованием гибридизации in
situ. Но временной характер репликации может варьировать в различных
клетках, подобно мозаичному эффекту положения.
119.
4. Онтогенетическая и тканевая регуляцияимпринтинга.
INS2 импринтирован только в экстраэмбриональных тканях мышиного
эмбриона, но экспрессируется биаллельно в клетках поджелудочной железы;
KCNQ1 экспрессируется с материнского аллеля во всех тканях кроме
сердца;
UBE3A - экспрессируется биаллельно во всех тканях, а в мозге - только
с материнского аллеля;
IGF2R и MASH2 биаллельно экспрессируются на ранних стадиях
эмбриогенеза у мыши, а на поздних стадиях развития активным остается
только материнский аллель.
IGF2 имеет отцовскую экспрессию в большинстве тканей, но оба аллеля
экспрессируются в choroid plexus и lepthomenenges в течение развития мозга
и в зрелом состоянии. Кроме того, IGF2 в процессе развития экспрессируется
с трех различных промоторов.
Отдельные клетки трофобласта плаценты содержат не
импринтированный H19, но на более поздних стадиях развития экспрессия
становится полностью моноаллельной.
120.
5. Импринтированные гены кодируют как белки, так и толькоРНК.
Некоторые импринтированные гены не кодируют белков, но кодируют
консервативную РНК.
H19 кодирует РНК, аккумулирующуюся в больших количествах в
течение развития фетальных тканей мезодермального и эндодермального
происхождения.
XIST. Транскрипция гена с инактивированной Х-хромосомы в
экстраэмбриональных тканях заставляет предполагать регуляторную роль
импринтированной РНК.
MKRN3-AS, IPW, UBE3A-AS/SNHG14 экспрессируются с отцовской
хромосомы и дают только РНК.
Общие черты РНК-кодирующих генов:
1) большие первый и последний экзоны и маленькие внутренние экзоны;
2) консервативны по нуклеотидной последовательности;
3) предсказанная вторичная структура РНК - стебель-петля.
Все это свидетельствует в пользу того, что эти РНК имеют
биологическую функцию, необходимую в эволюции.
121.
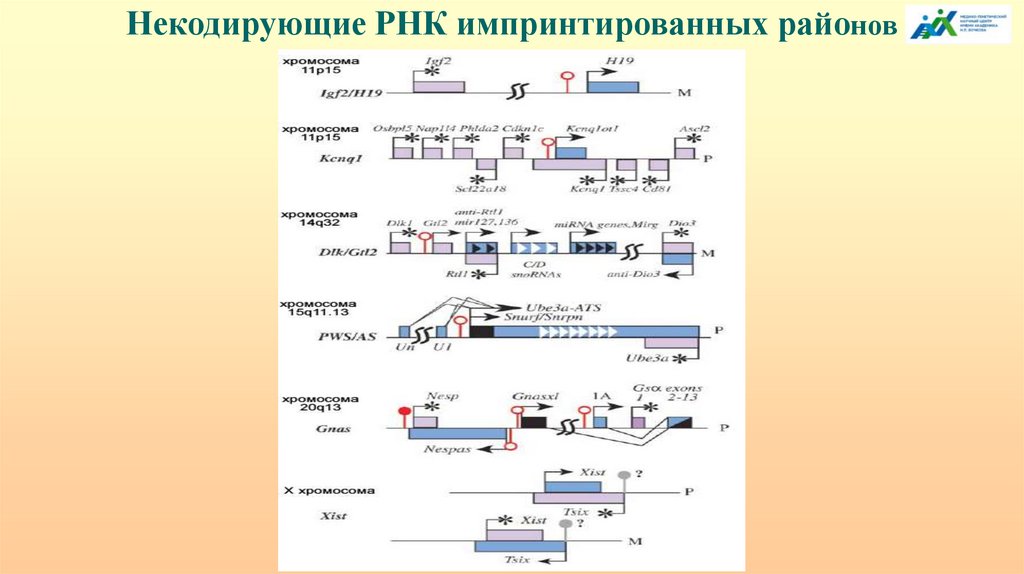
Некодирующие РНК импринтированных районов122.
По крайней мере одна днРНК (волнистая линия) присутствует в импринтированном домене(пунктирная волнистая линия представляет собой потенциальное распространение
транскрипта днРНК). Регуляция импринтинга в импринтированном домене осуществляется
через цис-действующий механизм, который контролируется ЦИ и связанным с ним геном
днРНК. Некоторые импринтированные гены в локусе проявляют тканеспецифическую
импринтированную экспрессию. Метилированное состояние ЦИ показано большими черными
кругами, а неметилированное - белыми. Меньшие кружки обозначают метилирование
вторичных, соматических ДМР. Эндогенные интерферирующие РНК изображены в виде
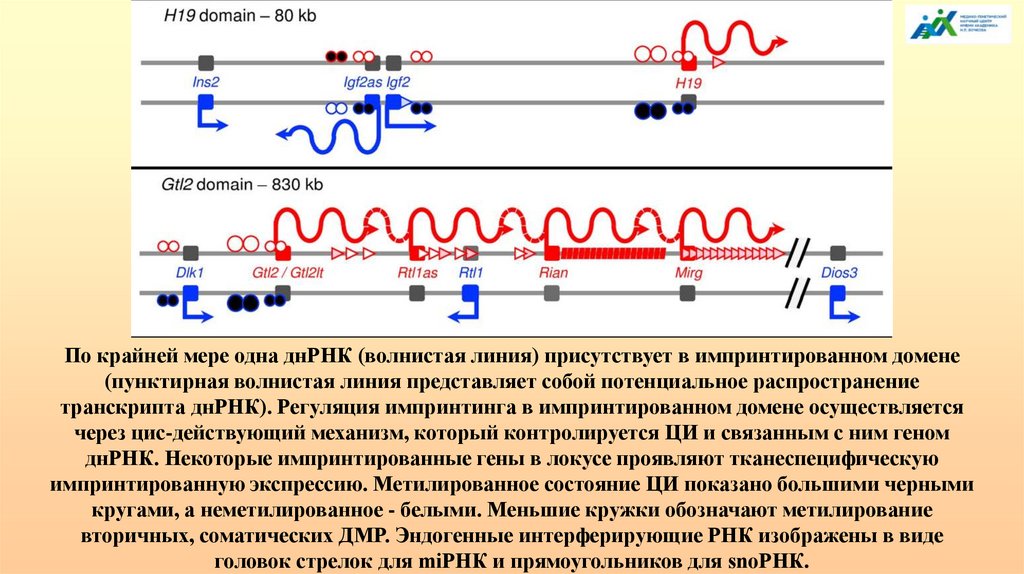
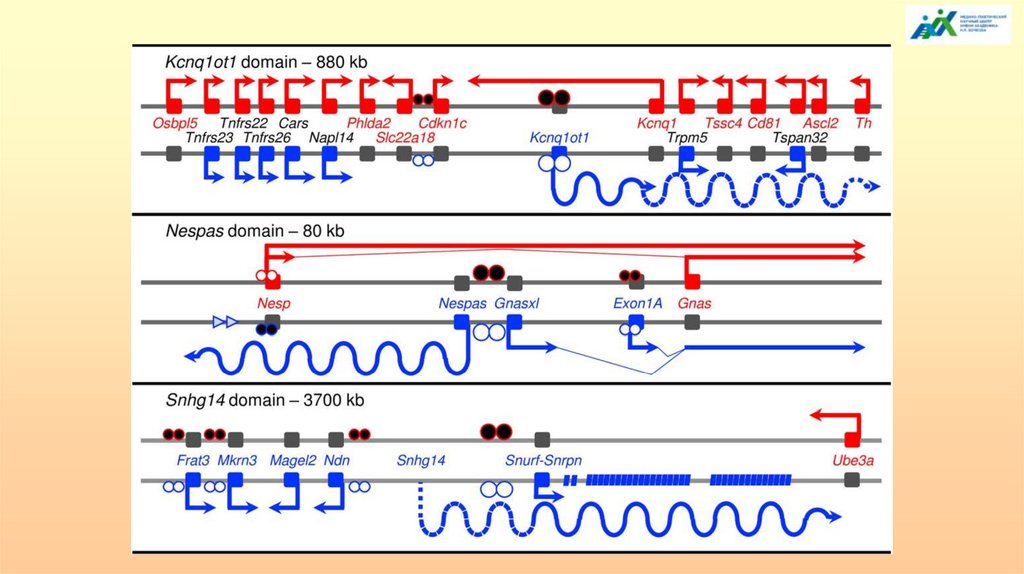
головок стрелок для miРНК и прямоугольников для snoРНК.
123.
124.
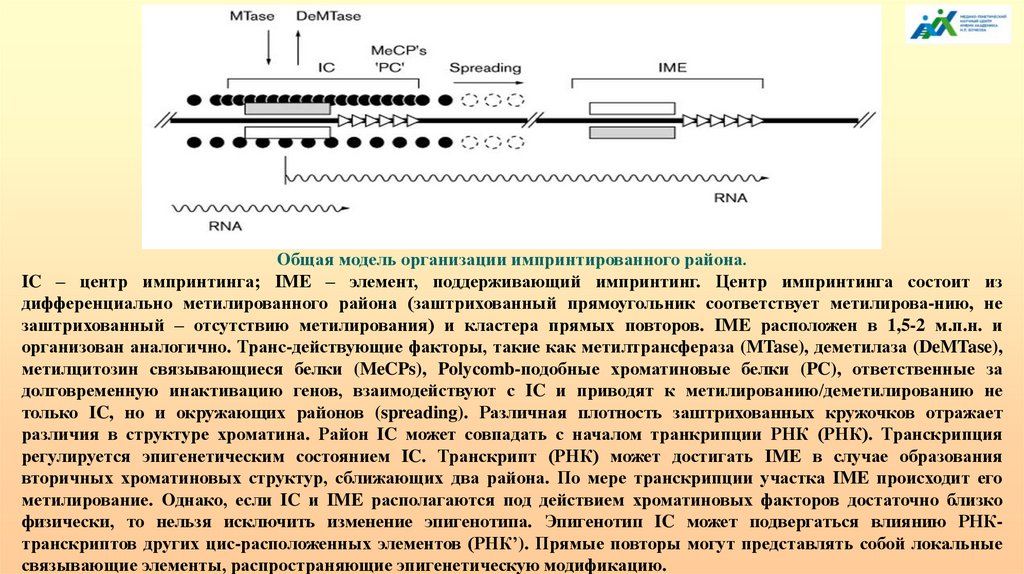
Общая модель организации импринтированного района.IC – центр импринтинга; IME – элемент, поддерживающий импринтинг. Центр импринтинга состоит из
дифференциально метилированного района (заштрихованный прямоугольник соответствует метилирова-нию, не
заштрихованный – отсутствию метилирования) и кластера прямых повторов. IME расположен в 1,5-2 м.п.н. и
организован аналогично. Транс-действующие факторы, такие как метилтрансфераза (MTase), деметилаза (DeMTase),
метилцитозин связывающиеся белки (MeCPs), Polycomb-подобные хроматиновые белки (PC), ответственные за
долговременную инактивацию генов, взаимодействуют с IC и приводят к метилированию/деметилированию не
только IC, но и окружающих районов (spreading). Различная плотность заштрихованных кружочков отражает
различия в структуре хроматина. Район IC может совпадать с началом транкрипции РНК (РНК). Транскрипция
регулируется эпигенетическим состоянием IC. Транскрипт (РНК) может достигать IME в случае образования
вторичных хроматиновых структур, сближающих два района. По мере транскрипции участка IME происходит его
метилирование. Однако, если IC и IME располагаются под действием хроматиновых факторов достаточно близко
физически, то нельзя исключить изменение эпигенотипа. Эпигенотип IC может подвергаться влиянию РНКтранскриптов других цис-расположенных элементов (РНК’). Прямые повторы могут представлять собой локальные
связывающие элементы, распространяющие эпигенетическую модификацию.
125.
Практически все импринтированные гены содержат повторы, в частности, первый интронгена SNRPN содержит структурно консервативные G-обогащенные повторы, а ген MAGEL2
содержит прямые 21- нуклеотидные повторы, расположенные в промоторной области. Повидимому, повторяющиеся последовательности могут быть вовлечены в установку
импринтинга и/или метилирования конкретного гена. Они могут служить мишенью для
маркирования определенного аллеля за счет организации вторичной структуры ДНК,
уникальной для одного из аллелей. Показано, что повторяющиеся последовательности
создают некие свернутые структуры, узнаваемые гетерохроматин-специфическими
белками.
Выявлены некоторые характерные особенности таких повторов. Во-первых, между ними
нет гомологии, во-вторых, длина единицы повтора может быть различной и, в-третьих, возможно любое расположение повторяющихся последовательностей по отношению к гену (рядом с геном в 5’-районе, в 3’-нетранслируемой области, внутри интрона или в кодирующей
части). Дифференциально метилированные районы в некоторых случаях перекрываются с
районами, с которых транскрибируются некодирующие и антисмысловые РНК, причем
транскрипты включают блоки тандемных повторов. Роль таких РНК определена - они
осуществляют регуляторные функции в импринтированных районах.
126.
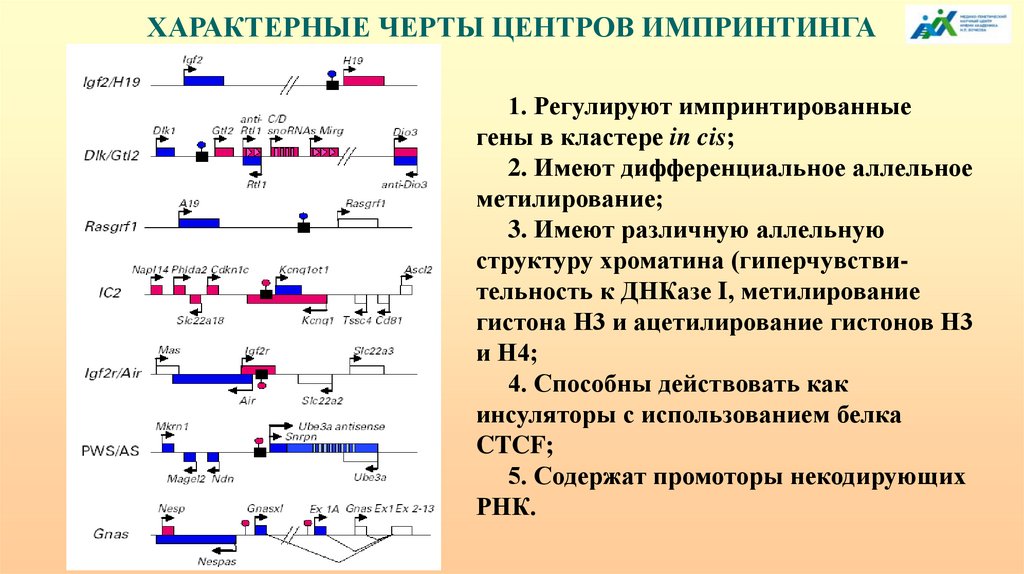
ХАРАКТЕРНЫЕ ЧЕРТЫ ЦЕНТРОВ ИМПРИНТИНГА1. Регулируют импринтированные
гены в кластере in cis;
2. Имеют дифференциальное аллельное
метилирование;
3. Имеют различную аллельную
структуру хроматина (гиперчувствительность к ДНКазе I, метилирование
гистона Н3 и ацетилирование гистонов Н3
и Н4;
4. Способны действовать как
инсуляторы с использованием белка
CTCF;
5. Содержат промоторы некодирующих
РНК.
127.
128.
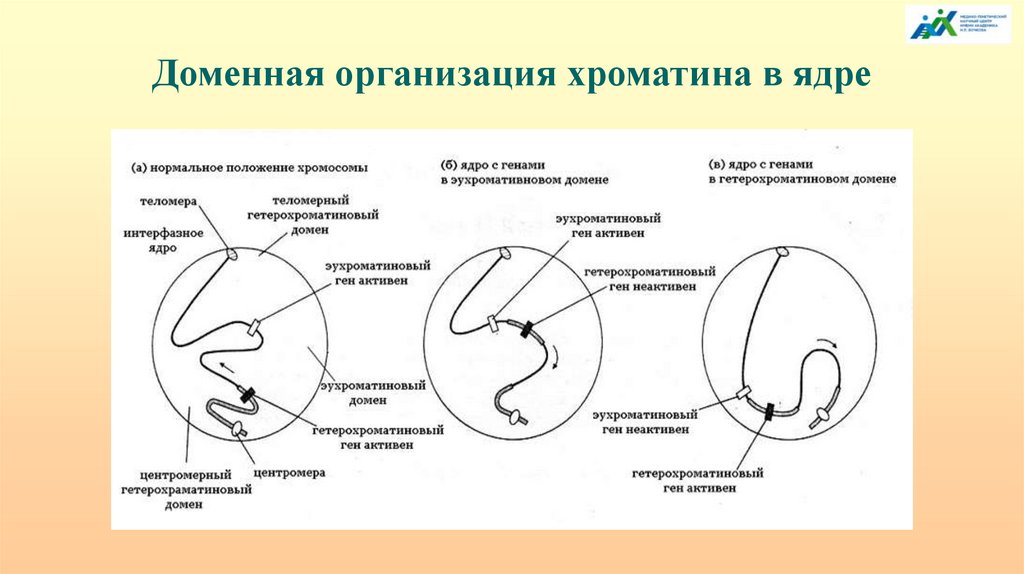
Доменная организация хроматина в ядре129.
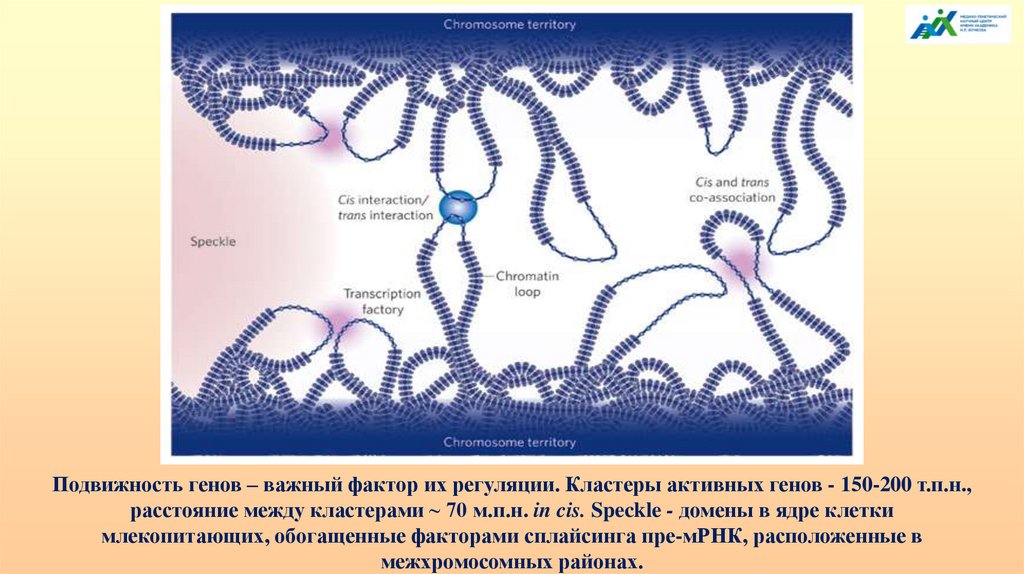
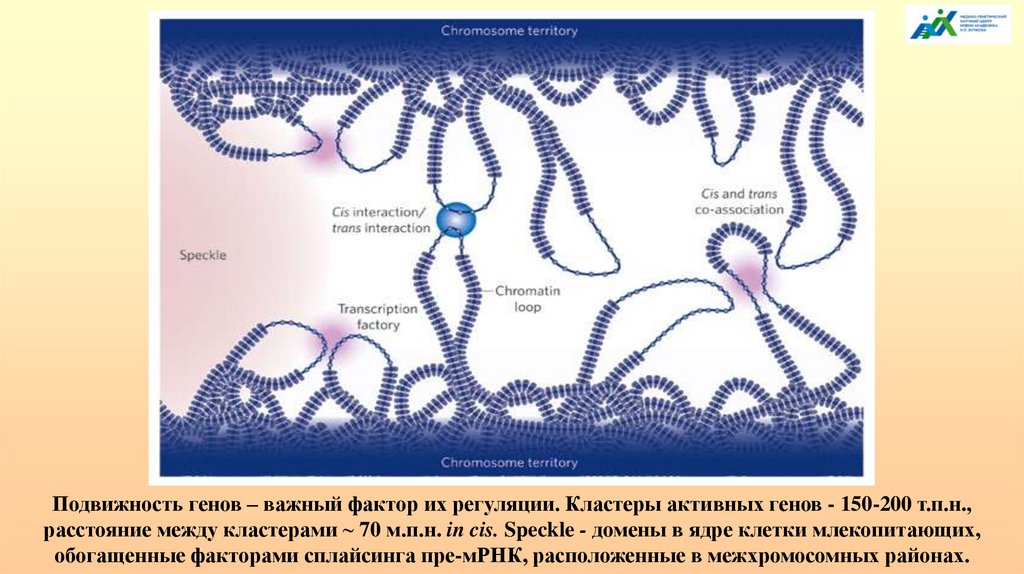
Подвижность генов – важный фактор их регуляции. Кластеры активных генов - 150-200 т.п.н.,расстояние между кластерами ~ 70 м.п.н. in cis. Speckle - домены в ядре клетки
млекопитающих, обогащенные факторами сплайсинга пре-мРНК, расположенные в
межхромосомных районах.
130.
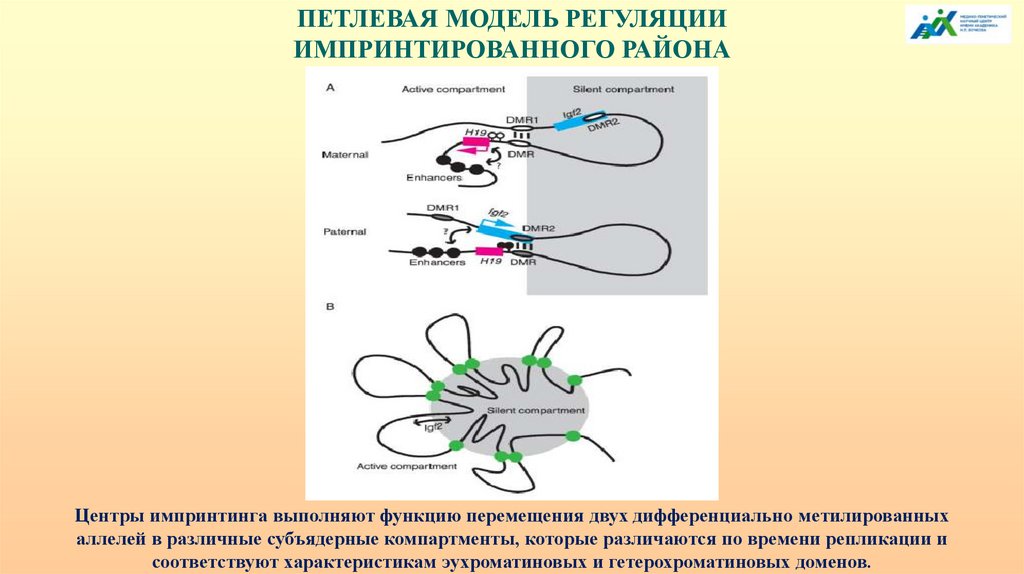
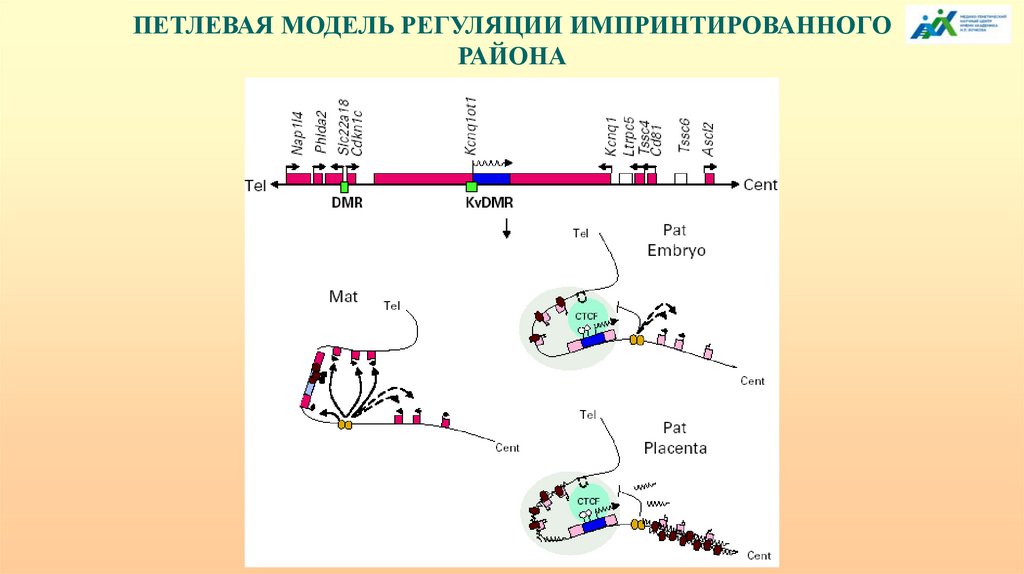
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИИМПРИНТИРОВАННОГО РАЙОНА
Центры импринтинга выполняют функцию перемещения двух дифференциально метилированных
аллелей в различные субъядерные компартменты, которые различаются по времени репликации и
соответствуют характеристикам эухроматиновых и гетерохроматиновых доменов.
131.
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГОРАЙОНА
132.
Показаны потенциальные структуры хроматина более высокого порядка всочетании с сайленсингом за счет некодирующей РНК и метилирования гистонов
на материнском и отцовских аллелях. На материнской хромосоме кластер ЦИ2
(синий) находится в доступной конформации, как в эмбрионе, так и в плаценте.
Энхансеры (желтые кружки) способны взаимодействовать с промоторами
«теломерных» генов, способствуя экспрессии в большинстве тканей. Те же или
другие энхансеры могут иметь доступ и к «центромерным» генам, часть из
которых экспрессируется только в плаценте.
На отцовской хромосоме неметилированный ЦИ связывается с белком CTCF
(большой голубой круг) в эмбриональных и плацентарных тканях. Это
взаимодействие либо устанавливает, либо поддерживает «теломерные» гены в
инактивированном хроматиновом домене (серый фон), в результате
взаимодействия с вышележащим элементом ДНК (заштрихованный
прямоугольник), где происходит метилирование гистонов (темно-красные
шестиугольники). Энхансеры лишены доступа к промоторам. Кроме того, в тканях
плаценты днРНК Kcnq1ot1 (волнистые линии) может покрывать хромосому,
способствуя метилированию гистонов по обе стороны ЦИ в качестве
дополнительного механизма репрессии.
133.
134.
135.
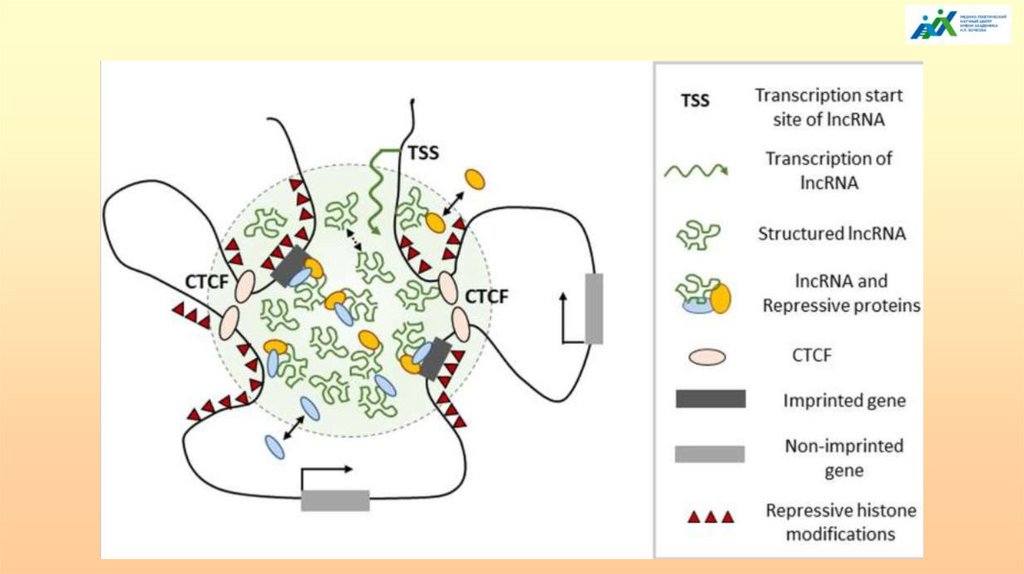
Модель, в которой импринтированная днРНК накапливается вблизи своегосайта транскрипции и взаимодействует с различными хроматиновыми и
комплексами репрессии (PRC1/2, EHMT2, другие). Это приводит к
образованию относительно стабильных РНК-белковых агрегатов (зеленая
штриховка), возможно частично, через механизмы жидкостно-жидкостного
фазового разделения (LLPS). Хромосомные/генные локусы, находящиеся в
непосредственной близости от агрегата, приобретают репрессивные
модификации гистонов и инактивируются. Этот процесс может быть
облегчен положительным влиянием нкРНК на связывание CTCF, тем
самым обеспечивая необходимые дальние хроматиновые взаимодействия,
которые сближают конкретный ген(ы) в непосредственную близость к
агрегату нкРНК-белок. Эта модель также может объяснить транс-эффекты
днРНК на гены, расположенные на других хромосомах, в случае, если они
расположены близко к таким комплексам, хотя бы временно.
136.
137.
Не основные эпигенетическиемеханизмы регуляции экспрессии генов
РЕГУЛЯТОРНЫЕ МОТИВЫ пре- и мРНК
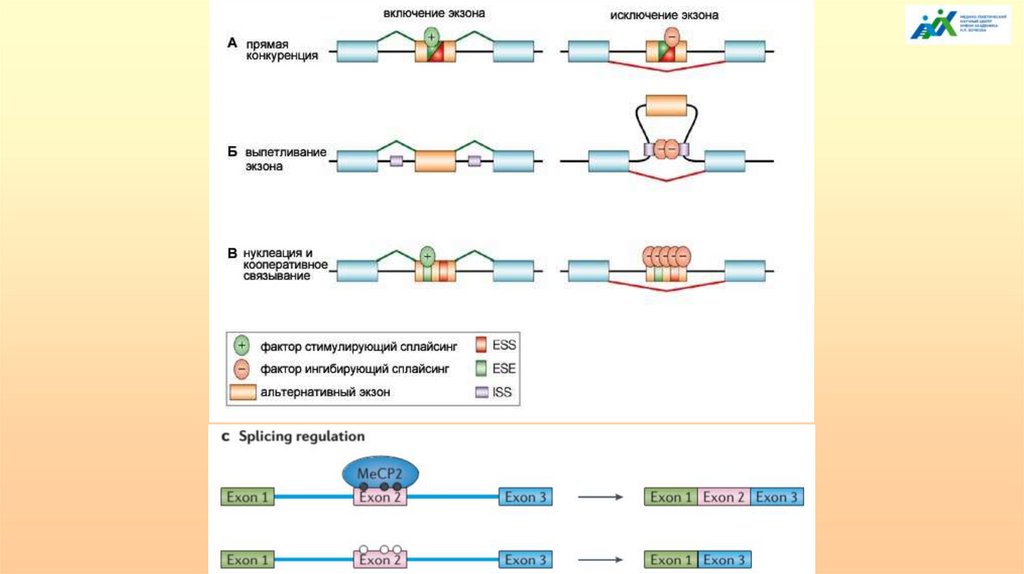
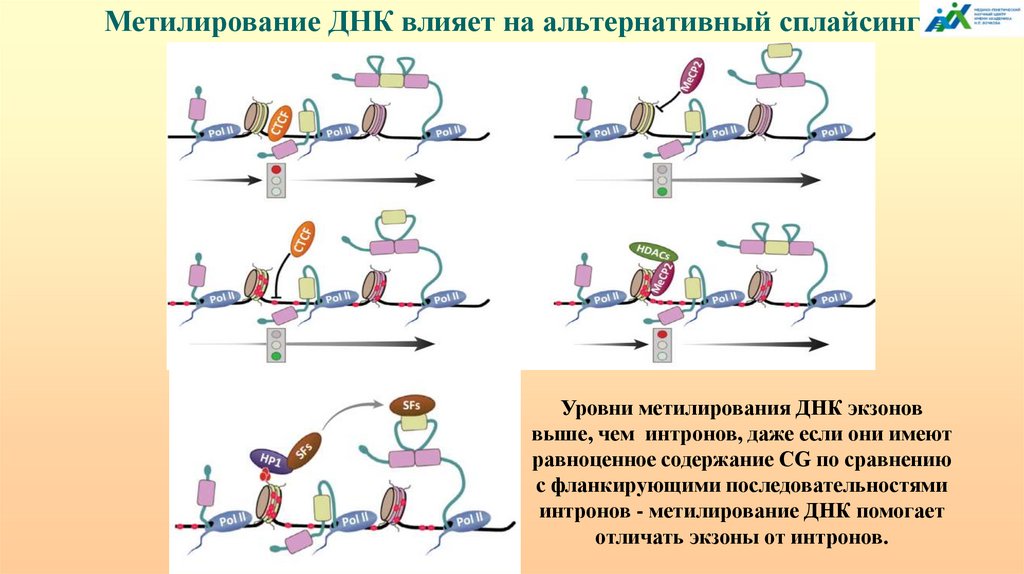
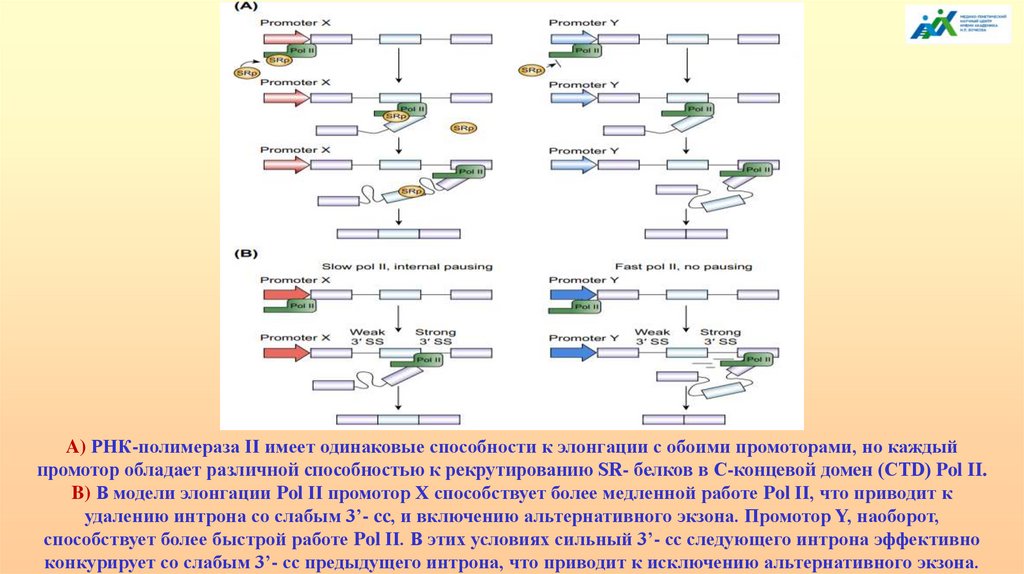
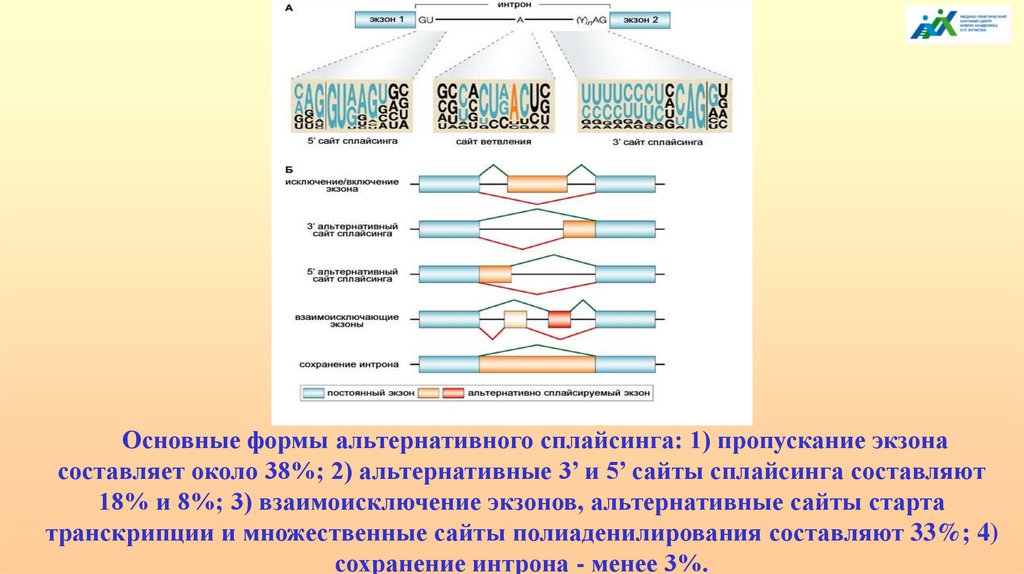
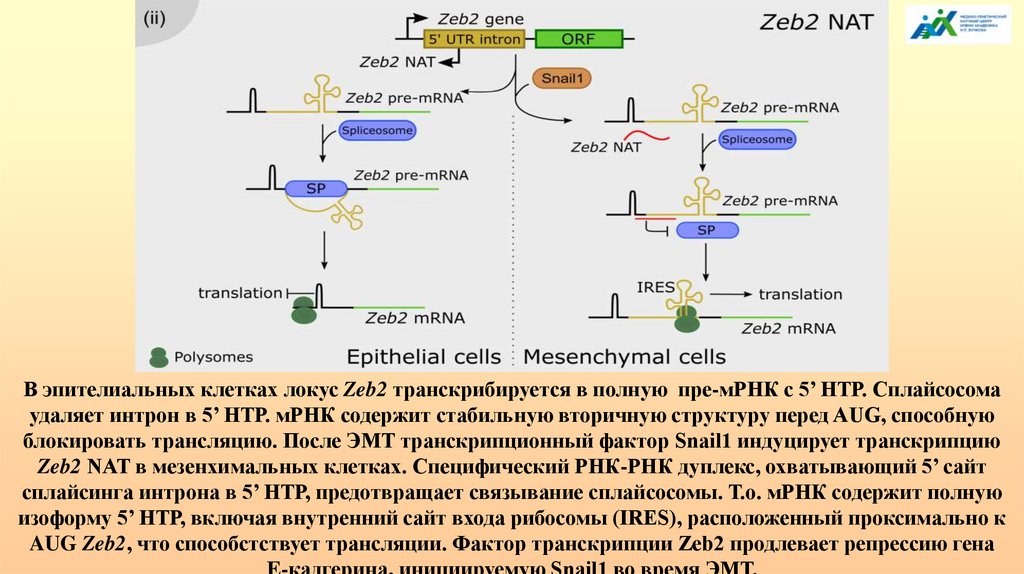
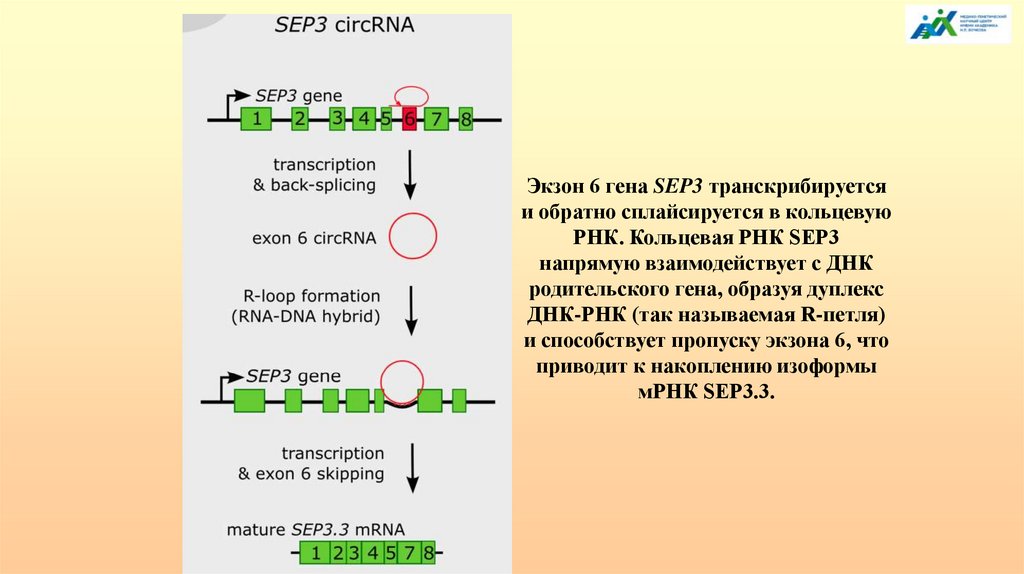
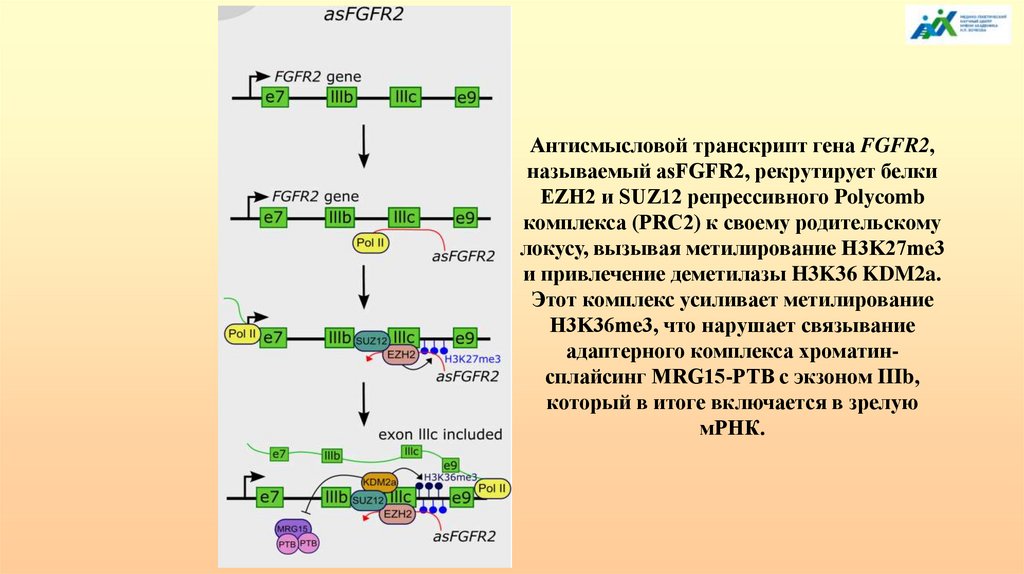
АЛЬТЕРНАТИВНЫЙ СПЛАЙСИНГ
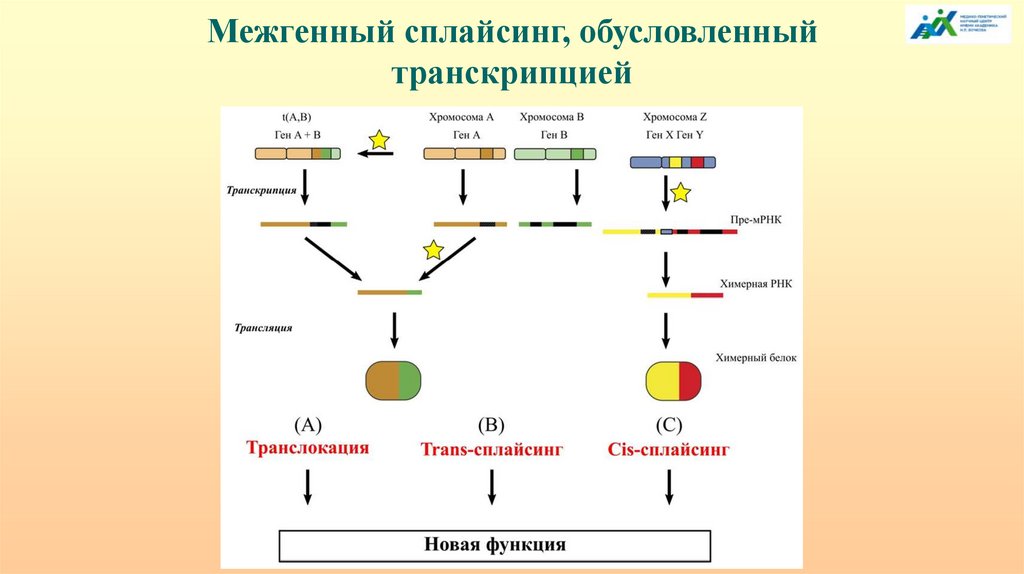
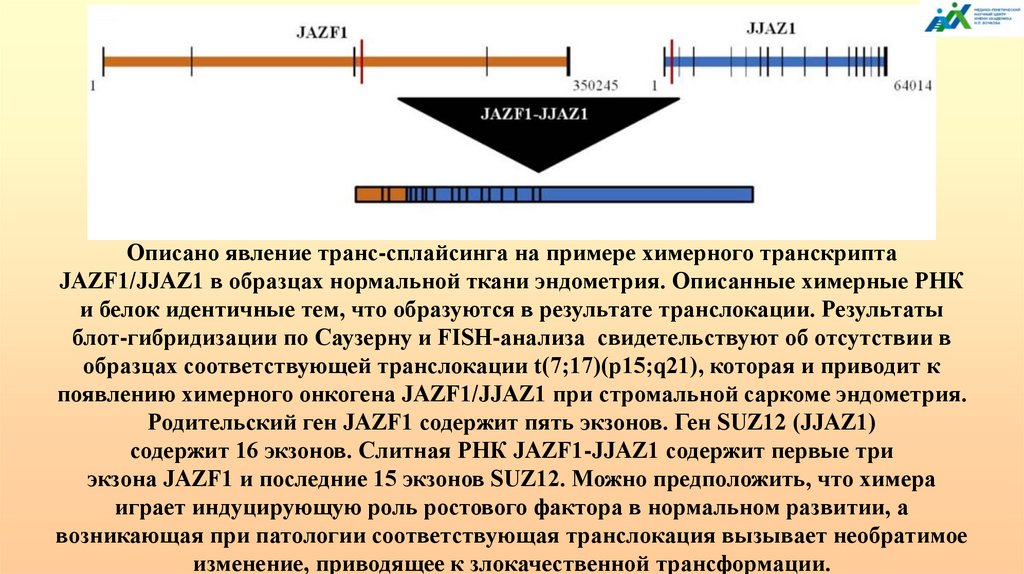
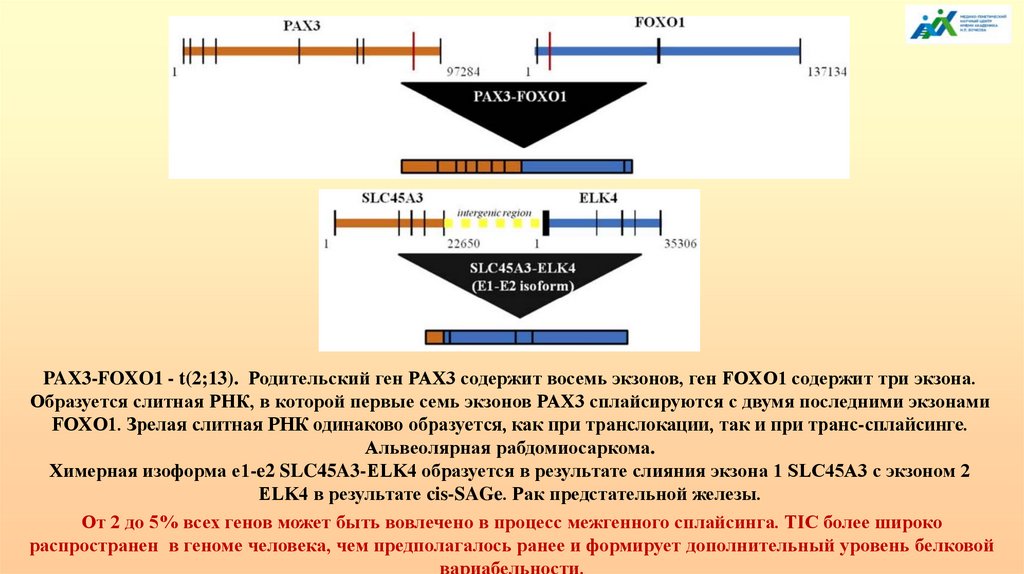
ТРАНСКРИПЦИОННО ИНДУЦИРОВАННЫЙ
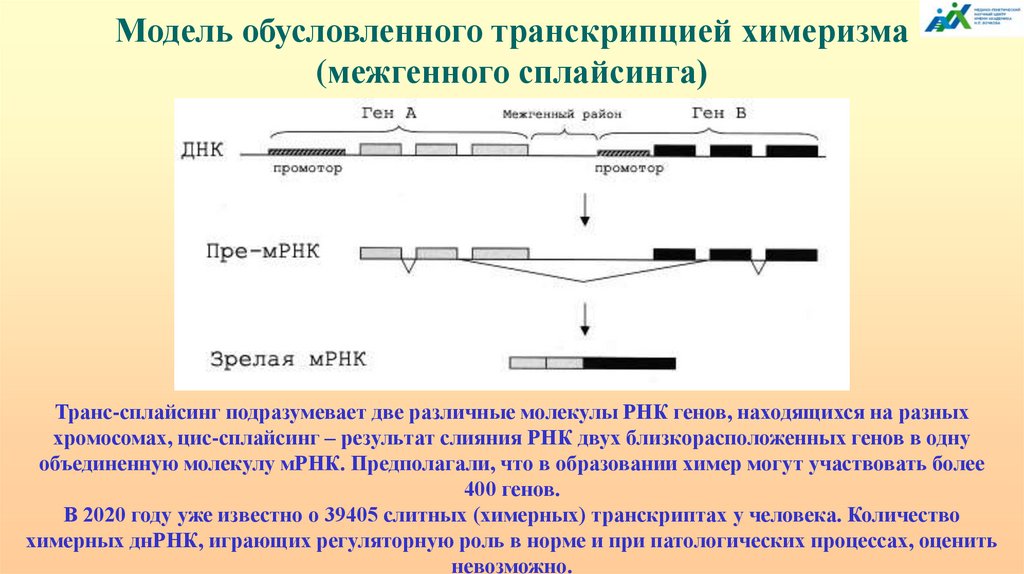
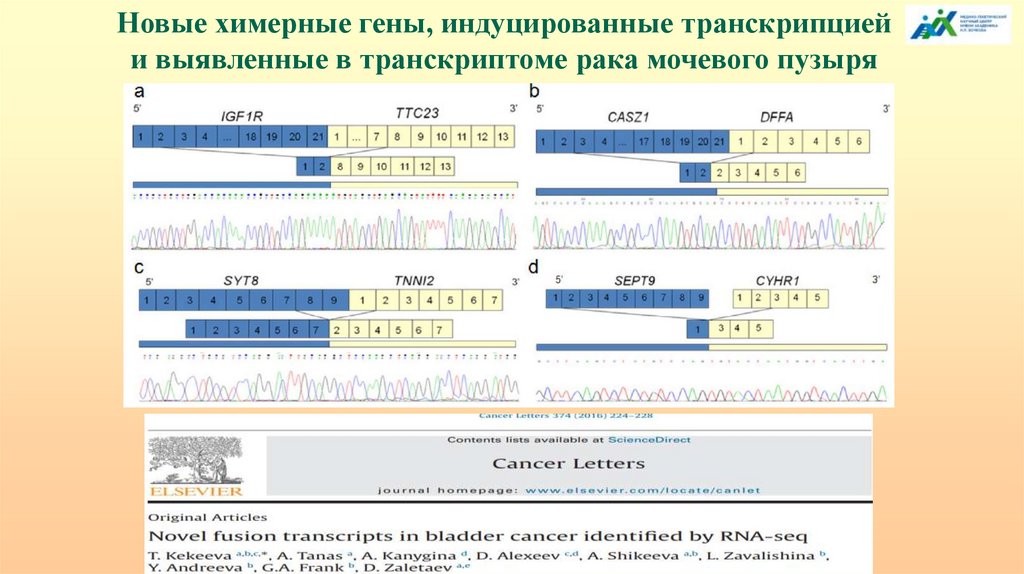
ХИМЕРИЗМ
138.
Уровни эпигенетической регуляцииМетилирование CpG. Метилирование CpH, где Н
ДНК (геном)
- любой нуклеотид, кроме G; 5hmC, 5fC, 5caC;
мутации отдаленных регуляторных элементов и
транспозиции генетического материала,
повторяющиеся последовательности
(«динамические» мутации).
РНК-интерференция. miРНК, siРНК, shРНК и
РНК
др., длинные некодирующие, антисмысловые и
(эпитранскриптом) кольцевые РНК; регуляторные мотивы мРНК;
альтернативный сплайсинг; цис- и транссплайсинг, индуцированный транскрипцией;
метилирование 6mA (ApH); редакция РНК.
Белки (протеом)
Гистоновый код.
Метилирование/деметилирование лизина 4, 9, 27,
36 и 79 гистона Н3 и лизина 20 гистона Н4;
ацетилирование/деацетилирование гистонов Н3
и Н4; белковые комплексы, ремоделирующие
хроматин; рибонуклеопротеиновые комплексы.
139.
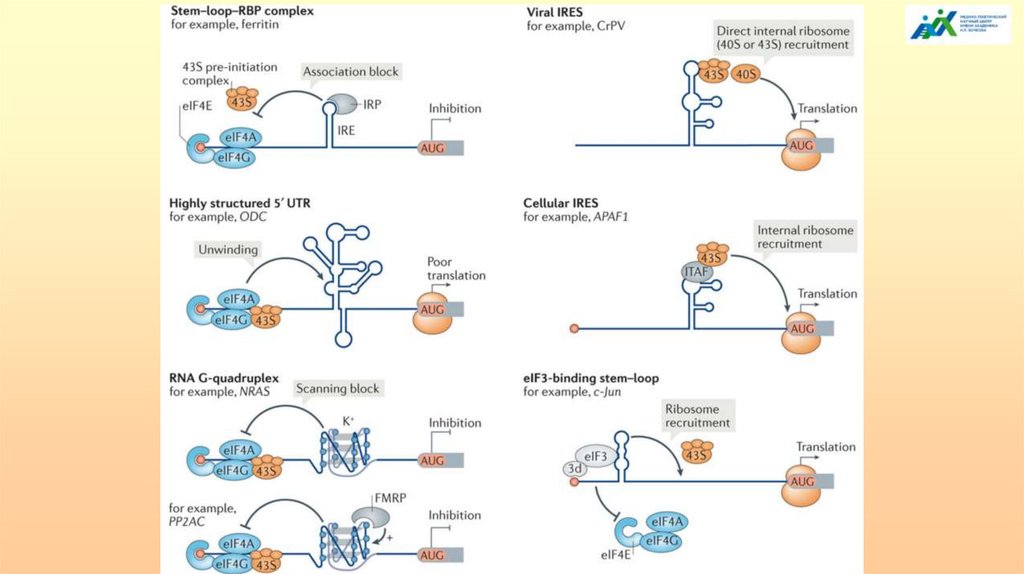
Роль 5`-нетранслируемой области в инициации трансляцииДлина в геноме человека варьирует от нескольких до более тысячи нуклеотидов (в среднем ~218
нуклеотидов). Кэп (красный кружок) из 7 метилгуанозина (m7G) на 5′ конце мРНК и поли(А) хвост
(An) на 3′ конце стабилизируют мРНК и стимулируют трансляцию. Нетранслируемая область 5′-UTR
содержит вторичные, третичные структуры и другие элементы последовательности. Такие структуры
РНК, как псевдоузлы, шпильки и G-квадруплексы (RG4s), а также, расположенные выше, короткие
открытые рамки считывания (uORF) и стартовые кодоны (uAUG), в основном ингибируют
трансляцию. Внутренние сайты входа рибосомы (IRESs) опосредуют инициацию трансляции
независимо от кэпа. Модификации РНК или РНК-связывающие белки (RBP) и длинные
некодирующие РНК (lncRNA), которые взаимодействуют с сайтами связывания РНК или образуют
рибонуклеопротеиновые (RNP) комплексы, а также последовательность Козак (AUG-кодон в
оптимальном контексте), вокруг стартового кодона могут дополнительно регулировать инициацию
трансляции.
140.
141.
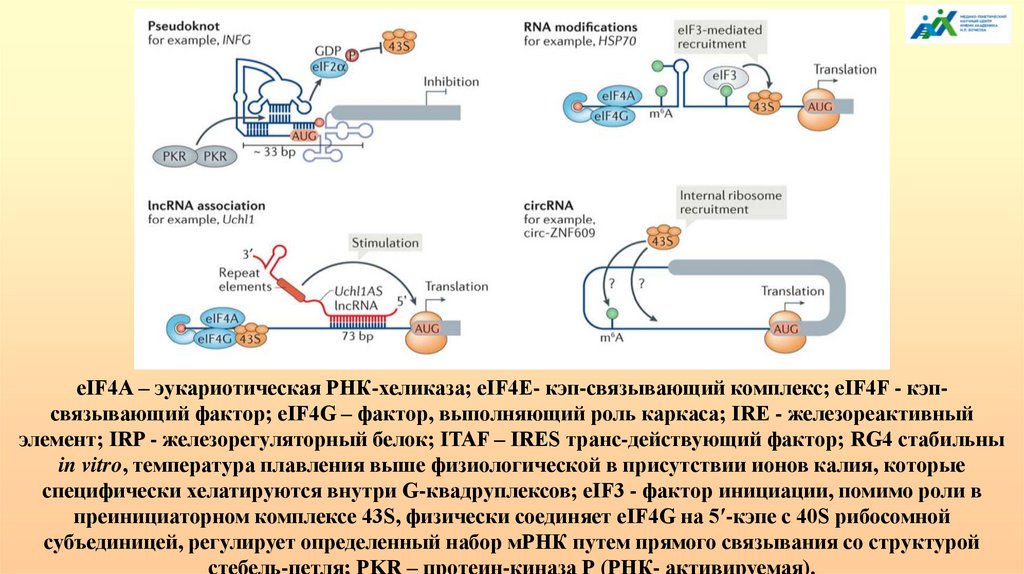
eIF4А – эукариотическая РНК-хеликаза; eIF4Е- кэп-связывающий комплекс; eIF4F - кэпсвязывающий фактор; eIF4G – фактор, выполняющий роль каркаса; IRE - железореактивныйэлемент; IRP - железорегуляторный белок; ITAF – IRES транс-действующий фактор; RG4 стабильны
in vitro, температура плавления выше физиологической в присутствии ионов калия, которые
специфически хелатируются внутри G-квадруплексов; eIF3 - фактор инициации, помимо роли в
преинициаторном комплексе 43S, физически соединяет eIF4G на 5′-кэпе с 40S рибосомной
субъединицей, регулирует определенный набор мРНК путем прямого связывания со структурой
стебель-петля; PKR – протеин-киназа Р (РНК- активируемая).
142.
Вспомните, пожалуйста, лекциюАлександры Юрьевны Филатовой
Около половины генов человека имеют uORF. Их можно разделить на две группы: (1) uORFs, которые не
перекрываются с основной белок-кодирующей частью (CDS) гена, т.е. их старт- и стоп-кодоны находятся в 5`UTR;
(2) перекрывающиеся uORFs, которые начинаются в 5`UTR, а заканчиваются в белок-кодирующей части гена,
таким образом их трансляция перекрывается с CDS.
Экспрессия многих uORF специфична для конкретных тканей и стадий развития – примерно половина всех
выявленных uORF транслировалась только в одном из исследуемых типов тканей, менее 20% транслировались
более, чем в 3-х тканях. Суммарно - около 77% всех uORF экспрессировались либо только в эмбриональном, либо
только во взрослом состоянии, что показывает их роль в регуляции экспрессии генов в онтогенезе.
143.
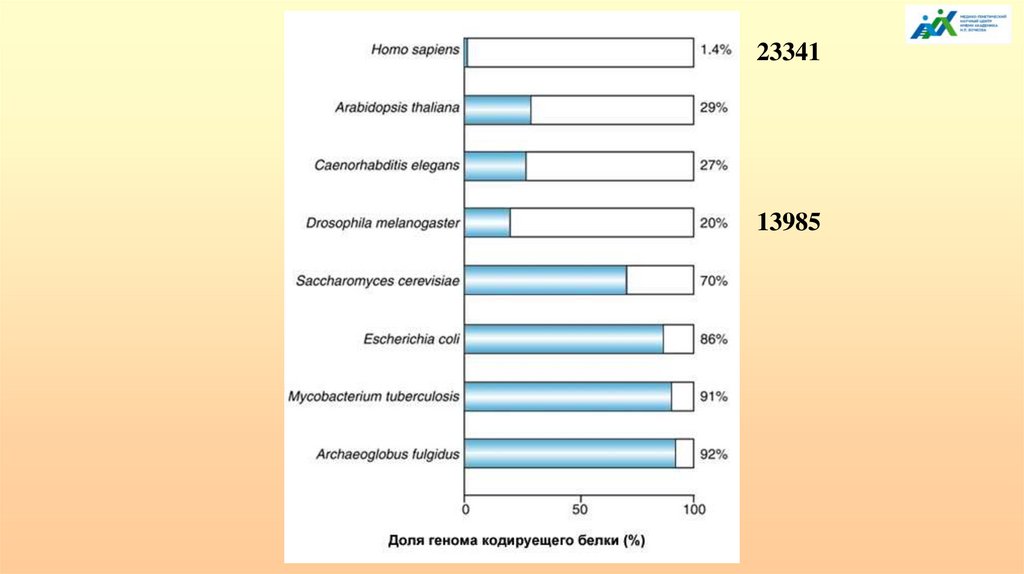
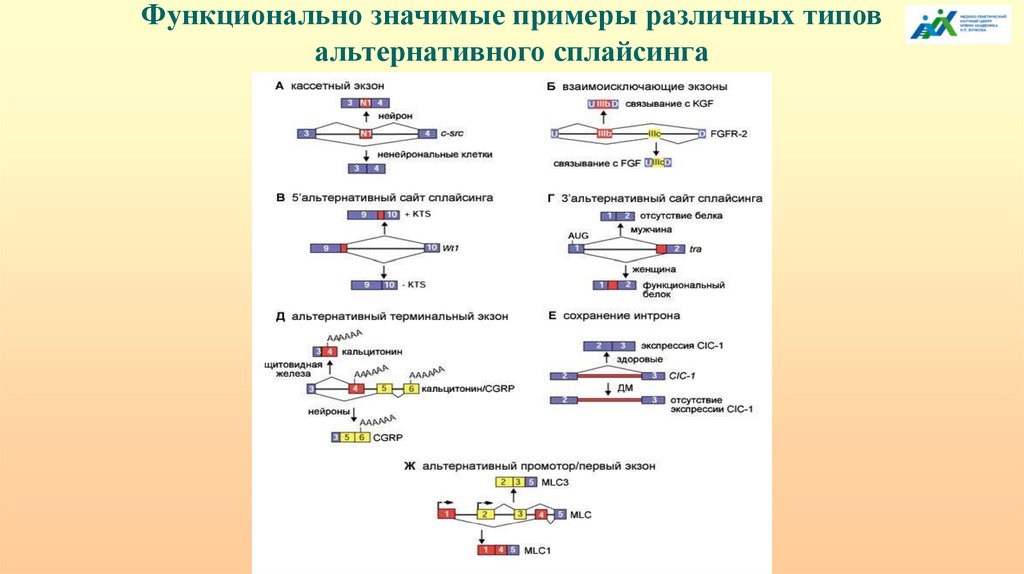
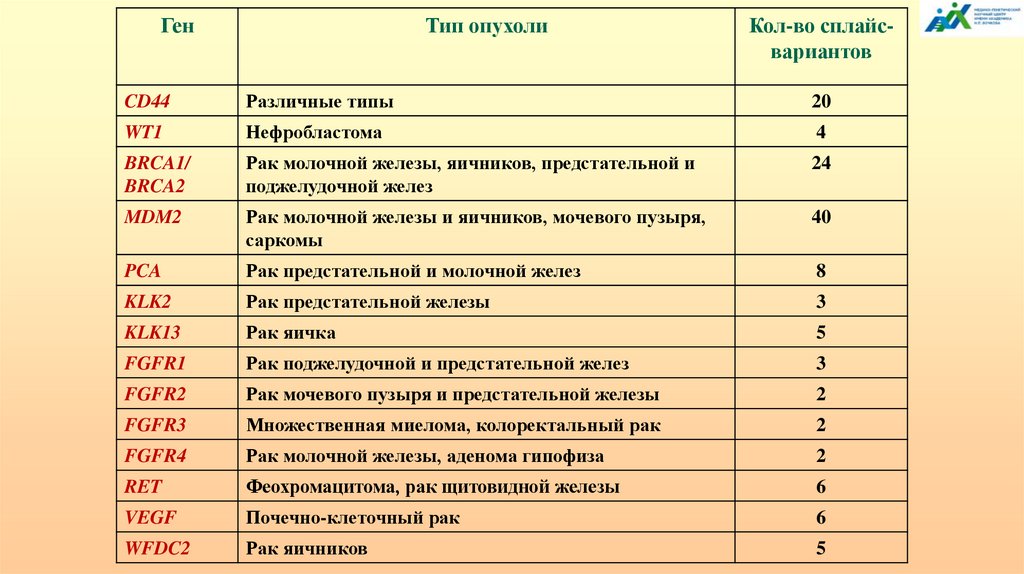
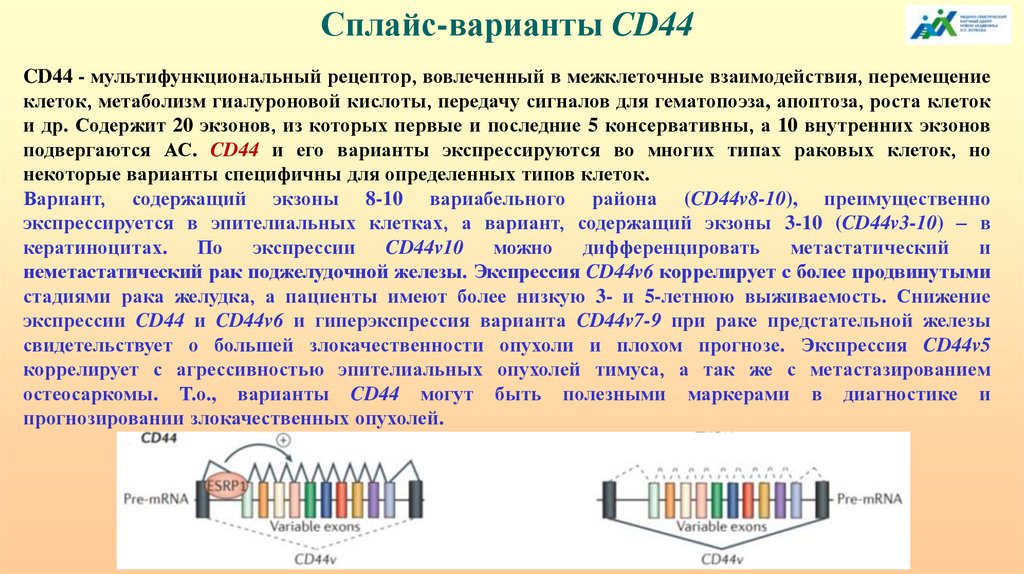
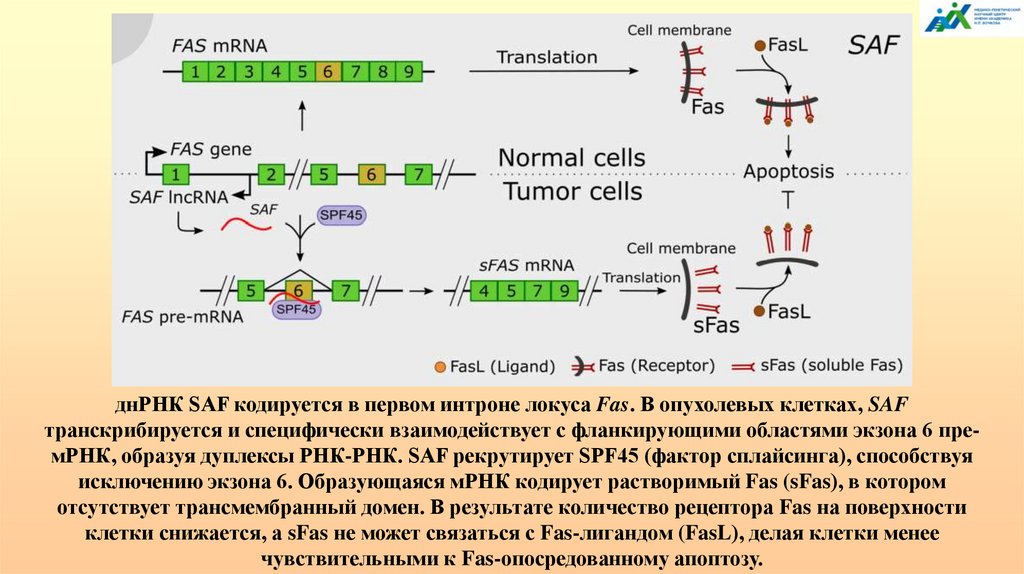
2334113985
144.
Около 95% генов человека могут подвергатьсяальтернативному сплайсингу, что вносит значительный
вклад в разнообразие протеома и объясняет несоответствие
между количеством генов (около 23341) и количеством
белков человека (~100000). Первичный транскрипт может
иметь несколько районов альтернативного сплайсинга и в
результате различных комбинаций ген может кодировать от
десятков до сотен различных изоформ.
145.
А. – консенсусные последовательности донорного сайта сплайсинга,акцепторного сайта сплайсинга и branch-сайта в интронах эукариот;
В. – механизм сплайсинга РНК с участием branch-сайта.
146.
Малые ядерные РНК (U1, U2, U4, U5 и U6), PRPF8 – фактор сплайсинга, взаимодействующий смяРНК и другими белками сплайсосомы.
147.
Механизм АС требует тонкой настройки активности многочисленных цисдействующих элементов и транс-действующих факторов, доступность и активностькоторых может варьировать на разных стадиях развития и в разных тканях.
Сплайсосома, большой рибонуклеопротеиновый комплекс, образованный более чем
170 белками и малыми ядерными РНК (U1, U2, U4, U5 и U6), составляет ядро
машины сплайсинга, которая собирается около 5’- и 3’- сс и удаляет интроны. У
позвоночных, из-за коротких экзонов и длинных интронов, сплайсосома часто
формируется поперек экзонов, что позволяет точно определить экзоны.
Распознавание сайтов сплайсинга сплайсосомой определяется комбинацией
множества факторов, включая силу и структурный контекст 5’- и 3’- сс,
полипиримидиновый тракт, «бранч-сайт» и вспомогательных регуляторных
сигналов цис-действующих экзоннных и интронных сайленсеров и энхансеров
сплайсинга. Последние, преимущественно, взаимодействуют с двумя широко
представленными семействами факторов, координирующих процессинг РНК,
включая регулярный сплайсинг и АС: серин-аргинин богатые (SR) белки, которые
усиливают включение альтернативных экзонов и гетерогенные ядерные
рибонуклеопротеины (hnRNPs), которые противодействуют им. В то же время, SR
белки могут выступать в качестве сайленсеров, а hnRNPs - в качестве энхансеров АС.
148.
Регуляторные элементы сплайсинга в пре-мРНКСхема определения альтернативного экзона компонентами сплайсосомы (выделены серым
цветом), вспомогательными цис-действующими элементами (ESS, ESE, ISE, ESS, A, YYYYY) и
транс-действующими белковыми факторами (SR, hnRNP, MBNL). Альтернативный экзон
выделен оранжевым цветом; SR - белки, богатые серином/аргинином; hnRNP - гетерогенный
ядерный рибонуклеопротеин; MBNL - Muscleblind-like protein; ESE и ESS - экзонные энхансер и
сайленсер сплайсинга; ISE и ISS - интронные энхансер и сайленсер сплайсинга; A - сайт
ветвления; YYYYY – полипиримидиновый тракт; зеленые стрелки - положительная регуляция
сплайсинга; красные стрелки - отрицательная регуляция сплайсинга; черные стрелки –
взаимосвязанная регуляция.
149.
Цис-действующие сайленсеры и энхансеры представляют собой платформу длявзаимодействия вспомогательных транс-действующих факторов, распознающих строго
определенные или вырожденные мотивы и представленные различными семействами
РНК-связывающих белков, экспрессия которых регулируется пространственно и в
процессе развития. Примером этого является белок MBNL1, который на более поздних
стадиях развития способствует генерации сотен альтернативных транскриптов в
результате взаимодействия с мотивом 5’ -YGCY- 3’ (где Y - пиримидин) в пре-мРНК.
Экспрессия MBNL1 низка на пренатальных стадиях и увеличивается в процессе
развития, становясь важнейшим фактором альтернативного сплайсинга, главным
образом, в мышцах, мозге и сердце взрослого организма. Кроме того, MBNL1 может
функционировать для подавления/активации сплайсинга в зависимости от
местоположения. Его ассоциация с интронными цис-сайтами, расположенными после
экзона, способствует включению альтернативного экзона, в то время как потеря экзона
происходит в результате связывания MBNL1 внутри альтернативного экзона и/или
интрона перед экзоном. Подобная позиционно-зависимая активность характерна и для
других факторов сплайсинга, таких, как РНК-связывающий гомолог FOX-1 (RBFOX1)
и белок, связывающий полипиримидиновый тракт, 1 (PTBP1).
150.
151.
Метилирование ДНК влияет на альтернативный сплайсингУровни метилирования ДНК экзонов
выше, чем интронов, даже если они имеют
равноценное содержание CG по сравнению
с фланкирующими последовательностями
интронов - метилирование ДНК помогает
отличать экзоны от интронов.
152.
Три белка передают информацию, закодированную в метилированой ДНК, механизму сплайсинга.Связывание CTCF с ДНК снижает скорость считывания Pol II и способствует включению экзона. Не
менее 100 альтернативных экзонов, содержащих сайты связывания CTCF, регулируются снижением
скорости транскрипции Pol II, что приводит к включению в мРНК. Если ДНК метилирована,
связывание CTCF невозможно, что приводит к исключению экзона. MeCP2 связывается с
метилированной ДНК и снижает скорость транскрипции, что приводит к включению экзона. Еще
один механизм - метилирование ДНК индуцирует метилирование H3K9me3. HP1 связывается с
H3K9me3 и несколькими факторами сплайсинга. HP1 накапливается в районах альтернативных
экзонов, а факторы сплайсинга, которые связываются с HP1, влияют на включение/исключение
этих экзонов.
Если CTCF и MeCP2 модулируют скорость транскрипции Pol II и регулируют АС экзонов,
расположенных вблизи их сайтов связывания, то HP1 влияет на связывание факторов сплайсинга с
хроматином; они могут связываться с пре-мРНК и, таким образом, регулировать АС.
Метилирование ДНК специфично для определенной ткани или стадии развития, а
дифференциально метилированные локусы обнаружены во многих регуляторных областях генома.
Метилирование ДНК влияет на включение около 22% альтернативно сплайсирующихся экзонов в
эмбриональных стволовых клетках мыши. Нокдаун MeCP2 затрагивает сплайсинг нескольких
тысяч альтернативных экзонов. Около 20% регуляции АС экзонов, подверженных метилированию,
можно объяснить механизмом, обусловленным HP1. В районах ряда альтернативных экзонов в 5’ и
3’ сайтах сплайсинга располагаются CpG, которые подвергаются метилированию, именно они могут
быть важны в определении точек альтернативного сплайсинга.
153.
A) РНК-полимераза II имеет одинаковые способности к элонгации с обоими промоторами, но каждыйпромотор обладает различной способностью к рекрутированию SR- белков в С-концевой домен (CTD) Pol II.
B) В модели элонгации Pol II промотор X способствует более медленной работе Pol II, что приводит к
удалению интрона со слабым 3’- cc, и включению альтернативного экзона. Промотор Y, наоборот,
способствует более быстрой работе Pol II. В этих условиях сильный 3’- сс следующего интрона эффективно
конкурирует со слабым 3’- сс предыдущего интрона, что приводит к исключению альтернативного экзона.
154.
Основные формы альтернативного сплайсинга: 1) пропускание экзонасоставляет около 38%; 2) альтернативные 3’ и 5’ сайты сплайсинга составляют
18% и 8%; 3) взаимоисключение экзонов, альтернативные сайты старта
транскрипции и множественные сайты полиаденилирования составляют 33%; 4)
сохранение интрона - менее 3%.
155.
Функционально значимые примеры различных типовальтернативного сплайсинга
156.
А) Альтернативное включение кассетного экзона встречается очень часто. Нейрон-специфическое включениеэкзона N1 в протоонкоген c-src приводит к образованию вставки в домене белок-белкового взаимодействия SH3,
что изменяет его связывание с другими белками. (Б) Альтернативные экзоны могут быть
взаимоисключающимися, например, экзоны IIIb и IIIc в гене рецептора фактора роста фибробластов 2 (FGFR2). Включение IIIb приводит к образованию рецептора с высоким сродством к фактору роста кератиноцитов
(KGF), тогда как включение IIIc приводит к образованию высокоаффинного рецептора к FGF. Потерю
изоформы IIIb обнаруживают при раке простаты. (В) Выбор альтернативного 5’ сайта сплайсинга в генесупрессоре опухоли Вильмса Wt1 приводит к вставке трех аминокислот - лизина, треонина и серина (KTS).
Формы +KTS и - KTS играют разные роли в формировании почек и гонад. Cдвиг баланса в сторону формы KTS связан с синдромом Фразье. (Г) В гене трансформер (tra) у дрозофилы, выбор специфичного для самок 3’
альтернативного сайта сплайсинга приводит к образованию одной длинной открытой рамки считывания,
которая дает начало белку, контролирующему соматическую половую дифференцировку по женскому типу. У
самцов мРНК гена tra не имеет длинной открытой рамки считывания и белок не образуется. (Д)
Альтернативные терминальные экзоны в гене, кодирующем кальцитонин и кальцитонин-связанный пептид
(CGRP), дают начало гормону, участвующему в гомеостазе кальция в щитовидной железе, или нейропептиду,
участвующему в вазодилатации в нервной системе. (Е) Прочтение интрона - редкая форм альтернативного
сплайсинга у человека. Сохранение интрона 2 в специфическом для мышц человека хлоридном канале 1 (ClC-1)
у пациентов с миотонической дистрофией (МД) приводит к преждевременному стоп-кодону и снижению
экспрессии ClC-1, способствуя возникновению проблем с расслаблением мышц (миотония).
(Ж) Альтернативное использование промотора в гене легкой цепи миозина (MLC) приводит к тому, что к
разным первым экзонам, которые объединяются с взаимоисключающимися последующими экзонами, дает
начало разным изоформам белка, а именно MLC1 и MLC3. Этот тип альтернативного сплайсинга возникает в
результате транскрипционной регуляции, а не в результате регуляции выбора места сплайсинга, как такового.
157.
ГенТип опухоли
Кол-во сплайсвариантов
CD44
Различные типы
20
WT1
Нефробластома
4
BRCA1/
BRCA2
Рак молочной железы, яичников, предстательной и
поджелудочной желез
24
MDM2
Рак молочной железы и яичников, мочевого пузыря,
саркомы
40
PCA
Рак предстательной и молочной желез
8
KLK2
Рак предстательной железы
3
KLK13
Рак яичка
5
FGFR1
Рак поджелудочной и предстательной желез
3
FGFR2
Рак мочевого пузыря и предстательной железы
2
FGFR3
Множественная миелома, колоректальный рак
2
FGFR4
Рак молочной железы, аденома гипофиза
2
RET
Феохромацитома, рак щитовидной железы
6
VEGF
Почечно-клеточный рак
6
WFDC2
Рак яичников
5
158.
Сплайс-варианты CD44CD44 - мультифункциональный рецептор, вовлеченный в межклеточные взаимодействия, перемещение
клеток, метаболизм гиалуроновой кислоты, передачу сигналов для гематопоэза, апоптоза, роста клеток
и др. Содержит 20 экзонов, из которых первые и последние 5 консервативны, а 10 внутренних экзонов
подвергаются АС. CD44 и его варианты экспрессируются во многих типах раковых клеток, но
некоторые варианты специфичны для определенных типов клеток.
Вариант, содержащий экзоны 8-10 вариабельного района (CD44v8-10), преимущественно
экспрессируется в эпителиальных клетках, а вариант, содержащий экзоны 3-10 (CD44v3-10) – в
кератиноцитах. По экспрессии CD44v10 можно дифференцировать метастатический и
неметастатический рак поджелудочной железы. Экспрессия CD44v6 коррелирует с более продвинутыми
стадиями рака желудка, а пациенты имеют более низкую 3- и 5-летнюю выживаемость. Снижение
экспрессии CD44 и CD44v6 и гиперэкспрессия варианта CD44v7-9 при раке предстательной железы
свидетельствует о большей злокачественности опухоли и плохом прогнозе. Экспрессия CD44v5
коррелирует с агрессивностью эпителиальных опухолей тимуса, а так же с метастазированием
остеосаркомы. Т.о., варианты CD44 могут быть полезными маркерами в диагностике и
прогнозировании злокачественных опухолей.
159.
нкРНК могут регулировать альтернативный сплайсинг. Их возможное участие согласуется сданными о высокой консервативности последовательностей, примыкающих к альтернативным
сайтам сплайсинга, у различных видов. Было показано, что расположение сайтов сплайсинга можно
легко изменить, как в культуре клеток, так и in vivo, введением антисмысловых малых РНК, что
оказалось многообещающим подходом к генотерапии миодистрофии и некоторых других
наследственных болезней человека, вызванных мутациями сайтов сплайсинга. Предполагается, что
малые РНК и в норме контролируют выбор сайтов сплайсинга.
Роль днРНК в процессах АС, таких H19, XIST, SRA1, EVF2 и MALAT1 впервые была выявлена в
2004 г. Они влияют на сплайсинг в результате взаимодействий с факторами сплайсинга, выступают
в качестве молекулярных каркасов и/или губок, или активируют/инактивируют другие РНК,
вовлеченные в этот процесс.
днРНК, такие, как MALAT1, PVT1, XIST и PANDAR, могут взаимодействовать со
специфическими факторами сплайсинга - белками SR или гяРНП комплексами. Например,
MALAT1 способна взаимодействовать с SR-белками, изменяя уровень их фосфорилирования и/или
изменяя 3D-структуру генома для индукции процессов, связанных с AС, что приводит к изменению
всего профиля AС. В случае повышенной экспрессии днРНК MIAT может связываться с
несколькими факторами сплайсинга - SF1, SRSF1, QKI и др., нарушая их обычную функцию и
подавлять АС.
Еще два механизма АС: рекрутирование РНК- связывающих белков (РСБ) в регулируемые
сайты сплайсинга и связывание с рибосомными и/или сплайсинговыми комплексами; днРНК могут
образовывать РНК-РНК дуплексы с молекулами пре-РНК, чтобы влиять на AС.
160.
днРНК SAF кодируется в первом интроне локуса Fas. В опухолевых клетках, SAFтранскрибируется и специфически взаимодействует с фланкирующими областями экзона 6 премРНК, образуя дуплексы РНК-РНК. SAF рекрутирует SPF45 (фактор сплайсинга), способствуя
исключению экзона 6. Образующаяся мРНК кодирует растворимый Fas (sFas), в котором
отсутствует трансмембранный домен. В результате количество рецептора Fas на поверхности
клетки снижается, а sFas не может связаться с Fas-лигандом (FasL), делая клетки менее
чувствительными к Fas-опосредованному апоптозу.
161.
В эпителиальных клетках локус Zeb2 транскрибируется в полную пре-мРНК с 5’ НТР. Сплайсосомаудаляет интрон в 5’ НТР. мРНК содержит стабильную вторичную структуру перед AUG, способную
блокировать трансляцию. После ЭMT транскрипционный фактор Snail1 индуцирует транскрипцию
Zeb2 NAT в мезенхимальных клетках. Специфический РНК-РНК дуплекс, охватывающий 5’ сайт
сплайсинга интрона в 5’ НТР, предотвращает связывание сплайсосомы. Т.о. мРНК содержит полную
изоформу 5’ НТР, включая внутренний сайт входа рибосомы (IRES), расположенный проксимально к
AUG Zeb2, что способстствует трансляции. Фактор транскрипции Zeb2 продлевает репрессию гена
E-кадгерина, инициируемую Snail1 во время ЭМТ.
162.
Экзон 6 гена SEP3 транскрибируетсяи обратно сплайсируется в кольцевую
РНК. Кольцевая РНК SEP3
напрямую взаимодействует с ДНК
родительского гена, образуя дуплекс
ДНК-РНК (так называемая R-петля)
и способствует пропуску экзона 6, что
приводит к накоплению изоформы
мРНК SEP3.3.
163.
Канонический сплайсинг пре-мРНК может конкурировать сциркуляризацией экзонов и этот механизм достаточно консервативен.
Родительский ген может регулировать биогенез его кРНК: второй экзон
фактора сплайсинга MBL/MBNL1 закольцован и у Дрозофила, и у
человека. кРНК, происходящая из пре-мРНК MBNL1 может
препятствовать связыванию РСБ с другими транскриптами или
конкурировать с каноническим сплайсингом пре-мРНК, что приводит к
изменению состава зрелых мРНК, в том числе и MBNL1. Изменение уровня
MBL значительно влияет на образование кMbl, эффект, зависящий от
сайтов связывания MBL. Кроме того, обратный сплайсинг может
генерировать кРНК и соответствующую линейную РНК, в то же время,
образование кРНК может нарушать целостность зрелой линейной РНК, и
не допускать ее трансляции – явление, известное как «ловушка мРНК».
164.
Антисмысловой транскрипт гена FGFR2,называемый asFGFR2, рекрутирует белки
EZH2 и SUZ12 репрессивного Polycomb
комплекса (PRC2) к своему родительскому
локусу, вызывая метилирование H3K27me3
и привлечение деметилазы H3K36 KDM2a.
Этот комплекс усиливает метилирование
H3K36me3, что нарушает связывание
адаптерного комплекса хроматинсплайсинг MRG15-PTB с экзоном IIIb,
который в итоге включается в зрелую
мРНК.
165.
днРНК asFGFR2 происходит из локуса FGFR2 человека и вызываетэпителиально-специфический альтернативный сплайсинг FGFR2 в результате
модификации хроматина именно в локусе FGFR2. В эпителиальных клетках
обнаружено, что asFGFR2 привлекает модификаторы хроматина, такие как белки
группы Polycomb и H3K36 деметилазу KDM2a к локусу FGFR2. В результате
создается такая упаковка хроматина, которая препятствует связыванию
ингибирующих регуляторов сплайсинга и способствует включению экзона IIIb.
Белки, связанные с Polycomb и KDM2a по-разному располагаются вдоль FGFR2 в
разных типах клеток, что соответствует результатам сплайсинга FGFR2. EZH2 не
может вызвать включение экзона IIIb в отсутствие KDM2a, а PRC2 в комплексе с
KDM2a способствует включению этого экзона в результате поддержания низкого
уровня H3K36me2/3.
Такой эпигенетический ландшафт нарушает связывание адаптерного
комплекса хроматин-сплайсинг MRG15-PTB, который обычно ингибирует
включение экзона IIIb. Антагонистический эффект H3K36me3 и H3K27me3 на
сплайсинг FGFR2 показывает роль днРНК asFGFR2 в перекрестном
взаимодействии между этими модификациями гистонов.
166.
Модель обусловленного транскрипцией химеризма(межгенного сплайсинга)
Транс-сплайсинг подразумевает две различные молекулы РНК генов, находящихся на разных
хромосомах, цис-сплайсинг – результат слияния РНК двух близкорасположенных генов в одну
объединенную молекулу мРНК. Предполагали, что в образовании химер могут участвовать более
400 генов.
В 2020 году уже известно о 39405 слитных (химерных) транскриптах у человека. Количество
химерных днРНК, играющих регуляторную роль в норме и при патологических процессах, оценить
невозможно.
167.
В 1998 году появилось сообщение о первой человеческой химерной РНК,GALT-IL11Ra, полученной в результате межгенного сплайсинга двух соседних
генов. Родительские гены, галактоза-1-фосфат уридилилтрансфераза (GALT) и
α-цепь интерлейкина-11-рецептора (IL-11Ra), расположены в районе
хромосомы 9p13. Этот транскрипт представляет собой результат cisсплайсинга. Анализ экспрессии показал высокие уровни в клеточных линиях
LT5, LT6 и костном мозге плода человека, что подтвердило наличие GALTIL11Ra в нормальных клетках человека. Транскрипт кодирует белок с
несколькими доменами, он расположен на клеточной мембране, а его
структура содержит часть GALT, присоединенную к аминокислотной
последовательности белка IL-11Ra. Функция слитного белка неизвестна и,
видимо, отличается от родительских белков, его экспрессия обнаружена в
норме в толстой кишке, адипоцитах, яичнике и яичке.
168.
Подвижность генов – важный фактор их регуляции. Кластеры активных генов - 150-200 т.п.н.,расстояние между кластерами ~ 70 м.п.н. in cis. Speckle - домены в ядре клетки млекопитающих,
обогащенные факторами сплайсинга пре-мРНК, расположенные в межхромосомных районах.
169.
Межгенный сплайсинг, обусловленныйтранскрипцией
170.
Описано явление транс-сплайсинга на примере химерного транскриптаJAZF1/JJAZ1 в образцах нормальной ткани эндометрия. Описанные химерные РНК
и белок идентичные тем, что образуются в результате транслокации. Результаты
блот-гибридизации по Саузерну и FISH-анализа свидетельствуют об отсутствии в
образцах соответствующей транслокации t(7;17)(p15;q21), которая и приводит к
появлению химерного онкогена JAZF1/JJAZ1 при стромальной саркоме эндометрия.
Родительский ген JAZF1 содержит пять экзонов. Ген SUZ12 (JJAZ1)
содержит 16 экзонов. Слитная РНК JAZF1-JJAZ1 содержит первые три
экзона JAZF1 и последние 15 экзонов SUZ12. Можно предположить, что химера
играет индуцирующую роль ростового фактора в нормальном развитии, а
возникающая при патологии соответствующая транслокация вызывает необратимое
изменение, приводящее к злокачественной трансформации.
171.
PAX3-FOXO1 - t(2;13). Родительский ген PAX3 содержит восемь экзонов, ген FOXO1 содержит три экзона.Образуется слитная РНК, в которой первые семь экзонов PAX3 сплайсируются с двумя последними экзонами
FOXO1. Зрелая слитная РНК одинаково образуется, как при транслокации, так и при транс-сплайсинге.
Альвеолярная рабдомиосаркома.
Химерная изоформа e1-e2 SLC45A3-ELK4 образуется в результате слияния экзона 1 SLC45A3 с экзоном 2
ELK4 в результате cis-SAGe. Рак предстательной железы.
От 2 до 5% всех генов может быть вовлечено в процесс межгенного сплайсинга. TIC более широко
распространен в геноме человека, чем предполагалось ранее и формирует дополнительный уровень белковой
вариабельности.
172.
Новые химерные гены, индуцированные транскрипциейи выявленные в транскриптоме рака мочевого пузыря
173.
Существуют химерные РНК в клетках человека, которые не связаны соструктурной перестройкой в геноме, такие как CCND1-Trop2, FAS-ERa,
CYP3A43-CYP3A4, CYP3A43-CYP3A5, Yq12-CDC2L2 и многие другие.
Химеры, обычно присутствующие во многих тканях, могут представлять собой
набор функциональных единиц, участвующих в фундаментальных клеточных
механизмах общих для всех клеток. Например, подвижность клеток и их
пролиферативная жизнеспособность страдают в отсутствие химерных
транскриптов C15orf57-CBX3 или ADCK-NUMBL. Нокдаун ADCK4-NUMBL с
использованием siРНК приводит к полной инактивации химерного
транскрипта в клеточной культуре RWPE-1. В результате - значительное
снижение скорости пролиферации и подвижности клеток. В культуре клеток
астроцитов снижалась подвижность, но уровень пролиферации не изменился.
Химерные РНК обеспечивают дополнительные функциональные возможности
генома без соответствующего увеличения числа генов.