


























































































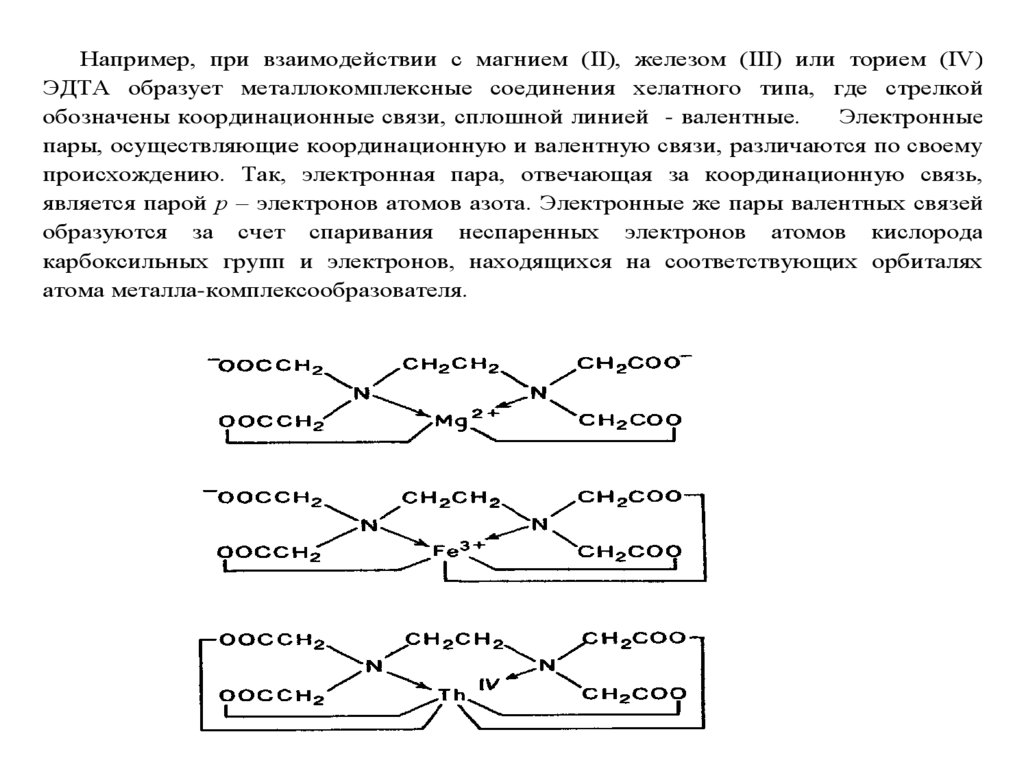








 chemistry
chemistrySimilar presentations:










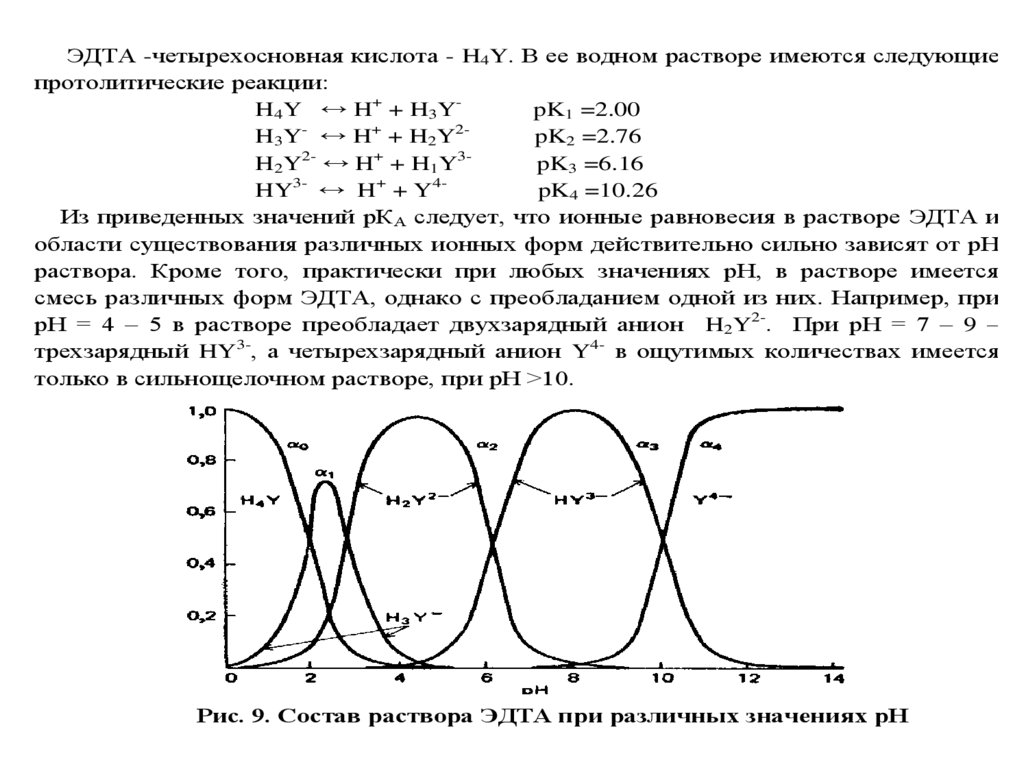
Титриметрический метод анализа
1.
ТитриметрияТитриметрический метод анализа был
предложен Ж.Л.Гей-Люссаком в XVIII веке и
благодаря простоте выполнения, высокой
скорости и точности, а также возможности
использования реакций самого разного типа
получил очень широкое распространение
для определения неорганических и
органических веществ, как в водных, так и
неводных растворах.
2.
Титриметрическим методом количественного химическогоанализа называют метод, основанный на измерении
необходимого эквивалентного количества реагента А,
затраченного на реакцию с эквивалентным количеством
определяемого компонента В, с использованием аналитической
химической реакции.
NAVA = NBVB
Закон эквивалентности. Химические элементы или их
соединения вступают в химические реакции друг с другом в
строго определенных весовых количествах,
соответствующих их химическим эквивалентам (мольэквивалентам). Другими словами, количество мольэквивалентов одного вещества реагирует с точно таким же
количеством моль-эквивалентов другого вещества. Этим
правилом руководствуются при расчете результатов анализа.
3.
Для титриметрических операций можно использовать реакцииразличных типов, но все они должны удовлетворять следующим
требованиям.
Реакция должна протекать по строго определенному
стехиометрическому уравнению, побочные реакции должны
быть исключены.
Реакция должна протекать количественно. Это значит, что
константа равновесия реакции должна быть достаточно
большой и, следовательно, погрешность из-за незавершенности
реакции будет минимальна – степень полноты протекания
реакции в момент эквивалентности должна быть не менее 99.9%
Реакция должна протекать быстро, чтобы в любой момент
титрования состояние равновесия наступило практически
мгновенно.
4.
Указанным выше требованиям удовлетворяют следующие реакции.Реакции кислотно-основного взаимодействия, т.е. реакции, сопровождающиеся
переносом протонов. Для частного случая в водном растворе: Н+ + ОН- = Н2О
Метод, основанный на реакциях этого типа, называют методом кислотно-основного
титрования.
Реакции окисления-восстановления, т.е. реакции, сопровождающиеся изменением
степеней окисления:
aAок.+ bBвос. = aAвос. + bBoк.
Метод, основанный на реакциях этого типа, называют методом окислительновосстановительного титрования (редоксометрия).
Реакция комплексообразования, т.е. реакция образования малодиссоциирующих
комплексных соединений: Mm+ + nLp- = [MLn]m-np
Метод, основанный на реакциях этого типа, называют методом комплексометрического
титрования.
Реакция осаждения, т.е. реакция образования малорастворимого электролита:
nKtm+ + mAnn- = ↓ KtnAnm
Метод, основанный на реакциях этого типа, называют методом осадительного
титрования (седиментация).
В каждый момент титрования концентрацию компонентов системы определяют
константой равновесия реакции и количеством добавленного титранта.
Различают 4 этапа титрования, сопровождающиеся определением концентраций
компонентов системы:
1. До начала титрования.
3. В момент эквивалентности.
2. До момента эквивалентности 4. После момента эквивалентности.
5.
СПОСОБЫ ТИТРОВАНИЯ1. Прямое титрование – это титрование определяемого вещества В
непосредственно раствором титранта А. Его применяют в том случае,
когда реакция между А и В протекает быстро. Содержание компонента В
при прямом титровании титрантом А рассчитывают на основе равенства
NAVA = NBVB .
В качестве примеров определения веществ способом прямого титрования
можно рассмотреть реакции:
1. CH3COOH + NaOH = CH3COONa + H2O
υ (CH3COOH) = υ (NaOH)
2. 10FeSO4 + 2KMnO4 + 8H2SO4 = 5Fe2(SO4)3 + 2MnSO4 + K2SO4 + 8H2O
υ (FeSO4) = υ (1/5KMnO4)
6.
2. Обратное титрование заключается в добавлении к определяемомувеществу В избытка точно известного количества стандартного раствора А
и в последующем титровании оставшегося количества вещества А
раствором титраната С. Этот способ применяют в тех случаях, когда
реакция между А и В протекает недостаточно быстро, либо нет
подходящего индикатора для фиксирования точки эквивалентности этой
реакции.
Количество молей эквивалента определяемого вещества В при
обратном титровании всегда равно разности между количеством молей
эквивалента вещества А и С.
(υ = υA - υC)
Примерами обратного титрования могут служить следующие реакции:
СаСO3 + 2НСl(избыток) = CaCl2 + CO2 + H2O + (HCl(остаток))
HCl(остаток) + NaOH = NaCl + H2O
υ (1/2CaCO3) = υ (HCl) (избыток) – υ (NaOH)
MnO2 + H2C2O4(избыток) + H2SO4 = MnSO4 + 2CO2 + 2H2O + (H2C2O4 (остаток)).
5H2C2O4 (остаток) + 2KMnO4 + H2SO4 = 2MnSO4 +10CO2 + K2SO4 +8H2O
υ (1/2MnO2) = υ (1/2H2C2O4) (избыток) – υ (1/5KMnO4).
7.
3. Титрование по заместителю заключается в титровании титрантом А неопределяемого вещества В , а эквивалентного ему количества заместителя
С, получающегося в результате предварительно проведенной реакции
между определяемым веществом В и каким-либо реагентом.
Титрование заместителя применяют обычно в тех случаях, когда
невозможно провести прямое титрование.
Количество молей эквивалента определяемого вещества при
титровании заместителя всегда равно количеству молей эквивалента
титранта:
(υ B= υ A= υ C).
(2.3)
В качестве примера можно рассмотреть реакцию
:
K2Cr2O7 + 6KI + 7H2SO4 = Cr2(SO4)3 + 3I2(заместитель) +4K2SO4 + 7H2O
I2(заместитель) + 2Na2S2O3 = 2NaI + Na2S4O6
υ (1/6K2Cr2O7) = υ (1/2I2) = υ (Na2S2O3)
8.
ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИЙ ИОНОВ ВОДОРОДА.В водных растворах сильных кислот и оснований. Концентрация
ионов водорода [H+] в предельно разбавленных водных растворах
сильных кислот и оснований (типа HCl, HNO3; NaOH, KOH )
практически равняется концентрации этих кислот и оснований
(CHan, CKtOH):
[H+] = CHАn ; рН = -lg[H+] ; pH = -lgCHAn
[OH-] = CKtOH ; pH = 14- рОН ; pH = 14 – (-lgCKtOH )
Пример. Вычислить pH в 0,05 М растворе HCl
Решение. [H+] = CHCl и, соответственно, [H+] = 0,05 = 5.10-2 моль/л,
откуда рН = lg5.10-2 = 2 – lg5 = 2 – 0.7 = 1.3
Пример. Вычислить рН в 0.01 М растворе гидроксида калия
Решение. [OH-] = CKOH, соответственно, [OH-]=10-2; рОН =-lg10-2;
рОН = 2
рН = 14 – 2 = 12
9.
В водных растворах слабых кислот. В данном случае концентрацию ионов водорода[H+] слабой кислоты нельзя приравнять к концентрации этой кислоты (CHАn). В водном
растворе слабая кислота диссоциирует согласно уравнению: HAn = H+ + AnПрименим закон действия масс:
[ H ] [ An ]
= KHAn
[ HAn]
Так как [H+] = [An-], то можно это уравнение записать как:
[H ]2
= KHAn или
[ HAn]
[H+] =
K HAn [HAn ]
(3.3)
Концентрация непродиссоциированной части молекул слабой кислоты [HAn] равна
разности первоначальной концентрации слабой кислоты С НAn и продиссоциированной
ее части [An-], или концентрации ионов водорода [H+], что одно и тоже, следовательно:
[HAn] = СНAn – [H+]
(3.4)
Подставляя равенство (3.4) в уравнение (3.3), получим:
[H+] = K HAn (C HAn [ H ])
(3.5)
Для слабо диссоциированной кислоты концентрация ионов водорода [H+] - величина
сравнительно малая по отношению к СНAn, и ею можно пренебречь. Уравнение при этом
примет следующий вид:
K HAn C HAn , откуда:
[H+] =
рН = ½ рК HAn - ½ lgСНAn
Пример. Вычислить концентрацию ионов водорода и рН 0.01 молярного раствора
уксусной кислоты, если КСН3СООН = 1.82 . 10-5
Решение. На основании уравнений (3.6) и (3.7) можно записать:
[H+] =
K HAn C HAn
[H+] =
K HAn (C HAn [ H ]) ; [H+]2 + KHAn [H+] - KHAn СНAn = 0 ,
5
2
10
; [H+] = 1.82 10
= 4.3 . 10-4 моль/л
рН = -lg[H+] = 4 – lg4.3 = 3.3
+
Если [H ] составляет более 5% от СНAn, то нельзя приравнивать [HAn] и СНAn . В таких
случаях пользуются более точным уравнением, которое можно вывести из уравнения
(1.5), проведя соответствующие преобразования:
откуда
[H+] = -1/2KHAn ±
2
1 / 4 K HAn
K HAn C HAn
10.
В водных растворах слабых оснований.Концентрацию
ионов
гидроксила
([OH-])
слабого
основания
нельзя
приравнять к самой концентрации данного слабого основания (С KtOH). В
водном растворе слабое основание диссоциирует согласно уравнению:
KtOH = Kt
+
[ Kt ] [OH ]
Применим закон действия масс:
= KKtOH
[ KtOH ]
-
+ OH
Так как [Kt+] = [OH-], то
Концентрация
[OH ]2
= KKtOH или
[ KtOH ]
K KtOH [KtOH ] (3.9)
[OH-]=
непродиссоциированной
части
молекул
слабого
основания [KtOH] равна разности первоначальной концентрации слабого
основания СKtOH и продиссоциированной части данного основания [Kt+],
или соответствующей ей концентрации ионов гидроксила [ОH-] .
[KtОH] = СKtOH – [ОH-]
(3.10)
Подставляя равенство (3.10) в уравнение, (3.9) получим:
[OH-] =
Для
слабо
K KtOH (С KtOH [OH ])
диссоциированного
(3.11)
основания
концентрация
ионов
годроксила [OH-] величина сравнительно малая по отношению к С KtOH, и
ею можно пренебречь. Уравнение при этом примет следующий вид:
[OH-] =
но,
K KtOH С KtOH
(3.12); рOН = ½ рКKtOH –
½ lgСKtOH (3.14)
pH = pKW – pOH тогда: pH = 14 – ½ рКKtOH + ½ lgСKtOH
Пример. Вычислить концентрацию ионов гидроксила и рН
раствора гидроксида аммония, если КNH4OH = 1.76
.
10
0.01 молярного
-5
Решение. Подставим в уравнения (3.12) и (3.14) значения константы
концентрации гидроксида аммония:
[OH-] =
K KtOH С KtOH
=
1.76 10 5 10 2
= 4.2 . 10-4 моль/л
рН = 14 – рОН = 14 – lg 4.2 . 10-4 = 14 – (4 – lg 4.2) = 14 – 3.3 = 10.7
и
11.
БУФЕРНЫЕ РАСТВОРЫБуферным называется раствор, рН которого не изменяется при
разбавлении или при добавлении небольших количеств кислоты или
основания.
Буферные
растворы
представляют
собой
смесь
слабой
кислоты и ее соли, или смеси слабого основания и его соли, а также
кислую или основную соли.
Буферный
раствор
препятствует
резкому
изменению
рН,
которое
может происходить при добавлении к раствору сильной кислоты или
сильного основания.
Сильная кислота (НCl) будет подавляться ацетатом
натрия, который вступает в реакцию с сильной кислотой по уравнению:
CH3COOH
HCl +
= CH3COOH
CH3COO- Na+ = CH3COOH + NaCl
Или в ионной форме:
H+ + CH3COO- = CH3COOH
Влияние оснований будет подавляться слабой уксусной кислотой, которая
вступает в реакцию нейтрализации с сильным основанием:
NaОН +
CH3COONa
= CH3COONa
CH3COOH
= CH3COONa + H2O
В ионной форме: ОН- + CH3COOH = CH3COO- + H2O
При этом рН раствора при прибавлении сильной кислоты понизится с 4.76
до 4.67, а при прибавлении сильного основания повысится с 4.76 до 4.84.
Буферные растворы широко применяют при проведении химических и
биохимических реакций. Буферные растворы образуются и при титровании
слабой кислоты сильным основанием и наоборот.
12.
Расчет рН буферных смесей.из
уравнений
константы
Значение рН буферной смеси рассчитывают
ионизации
слабых
кислот
или
оснований,
входящих в состав буферной смеси.
Рассчитаем рН в смеси слабой кислоты СН3СООН и ее соли СН3СООNa.
Из уравнения ионизации кислоты рассчитывают концентрацию ионов [Н+]
СН3СООН = СН3СОО- + Н+,
K CH 3COOH
[CH 3 COO ] [ H ]
,
[CH 3 COOH ]
откуда
K CH 3COOH [CH 3 COOH ]
[H+] =
(4.1)
[CH 3 COO ]
Но CH3COOH – кислота слабая и, как уже говорилось ранее, в растворе
находится
в
основном
в
виде
неионизированных
молекул,
поэтому
концентрацию неионизированной части кислоты принимают равной общей
концентрации ее в растворе, т.е. [CH3COOH] = Скислоты. С другой стороны,
соль СН3СООNa ионизирована в растворе полностью, а CH3COOH – очень
мало,
поэтому
почти
все
имеющиеся
в
растворе
CH3COO-
анионы
образуются за счет ионизации соли. Поскольку из каждой молекулы соли
при
ионизации
Ссоли.
[H+] =
образуется
Следовательно,
K КИС . С КИС .
С СОЛИ
один
ион
CH3COO-, очевидно, [CH3COO-] =
уравнение
(4.1)
принимает
(4.2)
Логарифмируя уравнение (4.2) и меняя знак на обратное, получим:
-lg[H+] = - lgKкисл – lg
рН = рКкисл – lg
C КИСЛ .
С СОЛИ
C КИСЛ .
С СОЛИ
(4.3)
вид:
13.
Для смеси слабого основания и его соли, например, NH4OH и NH4Сl:-
[OH ] =
K NH 4OH [ NH 4 OH ]
NH 4 Cl
К ОСН . С ОСН .
С СОЛИ
Сделаем допущение, что [NH4OH] Сосн., а [NH4Cl] Cсоли
Тогда:
[OH-] =
К ОСН .СОСН .
ССОЛИ
СОСН .
ССОЛИ
(4.5)
СОСН .
рН = 14 – рКосн.+ lg
ССОЛИ
(4.6)
рН = 14 – рКосн. – lgСсоли. + lgСосн.
(4.7)
рОН = рКосн. – lg
но,
откуда
(4.4)
рН = 14 – рОН ,
Способность буферных смесей поддерживать, практически постоянное
значение рН в растворе не безгранична, предел ее зависит от концентрации
компонентов буферной смеси. Таким образом, буферная смесь обладает
определенной
буферной
емкостью.
Наибольшая
буферная
емкость
наблюдается у растворов, содержащих равные концентрации слабой
кислоты и ее соли или слабого основания и его соли.
14.
Например, добавим 1 мл 0,1 М раствора сильного основания к 100 млацетатной
буферной
смеси
(СН3СООН
+ СН3СООNa), в
которой
концентрации кислоты и соли равны между собой и, соответственно,
равны 10-1 моль/л. (Ккислоты = 1.8 10-5). Концентрация кислоты в результате
реакции
нейтрализации
(100 1) 0.1
0.099 моль / л ,
101
а
уменьшится
и
станет
равной
концентрация
соли
(ацетата
натрия)
(100 1) 0.1
0.1моль / л , Тогда концентрация
увеличится и станет равной
101
ионов водорода, согласно уравнению (4.2), будет равна:
[H+] = 1.8 . 10-5 . 0.099 : 0.1 = 1.75 . 10-5 моль/л.
Эта величина мало отличается от Ккислоты = 1.8 10-5.
Таким образом, в тех случаях, когда концентрации кислоты и соли в
буферной
смеси
близки,
значение
[H+]
численно
приближается
к
константе диссоциации слабой кислоты Ккислоты даже при прибавлении к
ней сильной кислоты или основания.
В равной мере это относится к концентрации ионов [ОН-] для буферной
смеси, состоящей из слабого основания и его соли.
15.
ГИДРОЛИЗ СОЛЕЙГидролизом называют взаимодействие ионов растворенной соли с ионами Н+
или ОН- воды, сопровождающееся нарушением равновесия электролитической
диссоциации воды и изменением рН раствора.
Различают три варианта гидролиза.
1). Гидролиз солей, образованных взаимодействием сильных оснований и
слабых кислот. Например, соль KCN в растворе полностью ионизирована:
K+ + CN- + H2O = HCN + K+ + OHCN- + H2O = HCN + OHСоль KCN гидролизуется за счет образования слабой кислоты HCN. В растворе
накапливаются ионы ОН- , среда щелочная.
2). Гидролиз солей, образованных взаимодействием слабых оснований и
сильных кислот. Например, гидролиз NH4Cl.
NH4+ + Cl- + H2O = NH4OH + H+ + ClNH4+ + H2O = NH4OH + H+
Соль NH4Cl гидролизуется за счет образования малоионизированного слабого
основания NH4OH. В растворе накапливаются свободные ионы [H+], и реакция
среды становится кислой.
3). Гидролиз солей, образованных взаимодействием слабых оснований и слабых
кислот. Например, CH3COONH4
CH3COO- + NH4+ + H2O = CH3COOH + NH4OH
Константы ионизации CH3COOH и NH4OH почти равны, поэтому ионы Н+ и ОНсвязываются с ионами соли практически в равной степени, и реакция раствора
остается почти нейтральной.
16.
Расчет концентрации ионов гидроксила и рН в водных растворахгидролизующихся бинарных солей, образованных слабой кислотой и сильным
основанием.
Гидролиз соли, образованной слабой кислотой и сильным основанием, как
говорилось ранее, идет по аниону, так как катион при гидролизе не образует
малоионизированного соединения.
Запишем уравнение гидролиза соли KtAn, где Kt – cильное основание, An – слабая
кислота.
KtAn + Н2О = KtOH + HAn
(5.1)
+
+
Полное ионное уравнение Kt + An + HOH = Kt + OH + HAn
(5.1а)
Краткое ионное уравнение An + HOH = OH + HAn
(5.1б)
Запишем константу гидролиза, исходя из закона действия масс:
[ HAn] [OH ]
K ГИД .
[ An ] [ H 2 O]
(5.2)
(При математических расчетах, связанных с применением констант равновесия,
взаимодействие и образование воды не принимают в расчет).
[ HAn] [OH ]
K ГИД .
[ An ]
(5.3)
Умножив и разделив правую часть уравнения (5.3) на [H+], получим выражение:
[ HAn] [OH ] [ H ]
K ГИД .
, где выражение [OH-] . [H+] - это ионное
[ An ] [ H ]
произведение воды КW,
а
[ HAn]
- это обратная величина константы диссоциации слабой кислоты
[ An ] [ H ]
( 1/KHAn), и тогда уравнение (5.3) примет вид:
K ГИД .
KW
K HAn
(5.4)
17.
Согласно ионному уравнению гидролиза соли KtAn (5.1б) на каждыйобразующийся ион ОН- образуется одна молекула HAn, т.е. [OH-] = [HAn].
Кроме того, так как ионизация слабой кислоты HAn очень мала, то концентрацией
анионов [An-], получаемых за счет ионизации данной кислоты, можно пренебречь.
Концентрацию анионов [An-] принимают равной общей концентрации соли СKtAn,
ионизирующей в растворе нацело. Принимая это допущение, уравнение (5.3) можно
записать в виде:
[OH ] 2
К ГИД .
C KtAn
откуда
[OH] =
К ГИД .
(5.5)
С KtAn
Подставив значение Кгид из уравнения(5.4) в уравнение (5.5), получим выражение:
[OH ]
K W C KtAn
K HAn
(5.6)
Уравнение (5.6 ) показывает, что концентрация ионов гидроксила в водном
растворе бинарных солей, образованных катионами сильных оснований и анионами
слабых кислот, прямо пропорциональна корню квадратному из произведения
константы ионизации воды на молярную концентрацию соли и обратно
пропорциональна константе ионизации слабой кислоты.
Логарифмируя это уравнение, получим:
-lg[OH-] = – ½ lgKW + ½ lg KHAn – ½ lg С KtAn .
Отсюда
рOH = 7 – ½ рKHAn – ½ lg С KtAn
Так как рН = 14 – рОН,
рН = 7 + ½ pKHAn + ½ lg С KtAn
(5.7)
18.
Расчет концентрации ионов водорода и рН в водных растворах гидролизующихсябинарных солей, образованных сильной кислотой и слабым основанием.
Гидролиз соли, образованной сильной кислотой и слабым основанием, идет по
катиону, так как анион при гидролизе не образует малоионизированного соединения.
Запишем уравнение гидролиза соли KtAn, где Kt – слабое основание,
An – слабая кислота.
KtAn + Н2О = KtOH + HAn
(5.8)
+
+
Полное ионное уравнение Kt + An + HOH = KtOH + H + An
(5.8а)
+
+
Краткое ионное уравнение Kt + HOH =KtOH + H
(5.8б)
Запишем константу гидролиза, исходя из закона действия масс:
[ KtOH ] [ H ]
К ГИД
[ Kt ]
(5.9)
Умножив и разделив левую часть уравнения (5.9) на [OH-], получим выражение:
[ KtOH ] [OH ] [ H ]
К ГИД
,где выражение [OH-] . [H+] - это ионное произведение
[ Kt ] [OH ]
[ KtOH ]
воды КW, а
- это обратная величина константы диссоциации слабого
[ Kt ] [OH ]
основания (1/K KtOH), и тогда уравнение (5.9 ) примет вид:
KW
К ГИД
K KtOH
(5.10 )
19.
Согласно ионному уравнению гидролиза соли KtAn (5.8б) на каждый образующийсяион Н+ приходится одна молекула слабого основания KtOH, т.е. [H+] = [KtOH]. Так как
ионизация слабого основания KtOH очень мала, то концентрацией катионов,
получаемых за счет ионизации данного основания, можно пренебречь. Концентрацию
катионов [Kt+] принимают равной общей концентрации соли СKtAn, ионизирующей в
растворе нацело. Принимая это допущение, уравнение (5.9) можно записать в виде:
[ H ]2
К ГИД
C KtAn
откуда [H+] =
K ГИД .
C KtAn
(5.11)
Подставив значение Кгид из уравнения(5.10) в уравнение (5.11), получим выражение:
[H ]
K W C KtAn
K KtOH
(5.12)
Уравнение (5.12) показывает, что концентрация ионов водорода в водном растворе
бинарных солей, образованных катионами слабых оснований и анионами сильных
кислот, прямо пропорциональна корню квадратному из произведения константы
диссоциации воды на молярную концентрацию соли и обратно пропорциональна
константе ионизации слабого основания.
Логарифмируя это уравнение, получим:
-lg[H+] = – ½ lgKW – ½ lg KKtOH + ½ lg С KtAn .
Отсюда
рH = 7 – ½ рKKtOH – ½ lg СKtAn
(5.13)
20.
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ6.1. Титрование сильной кислоты сильным основанием.
Вычисление рН раствора в различные моменты титрования.
Предположим, что для титрования взято 100 мл 0,1 М раствора НСl,
который титруется 0,1 М раствором NaOH. Пользуясь формулой (3.1),
можно вычислить рН раствора в любой точке процесса нейтрализации
сильной кислоты сильным основанием.
До начала титрования. Концентрация ионов водорода и значение рН
определяются по формуле:
[H+] = CHCl
рH = -lg[H+] = -lgCHCl= -lg10-1 = 1
До точки эквивалентности. Рассмотрим, что произойдет со 100 мл
0,1 М раствором HCl, если прилить к нему 50; 90; 99; 99,9 мл 0,1 М
раствора NaOH
Для вычисления концентрации непрореагировавшей в
процессе титрования свободной кислоты СHCl (с учетом разбвления) можно
использовать формулу:
С HCl
V HCl V NaOH
M HCl
V HCl V NaOH
(6.1)
Далее рассчитаем значения рН в процессе всего титрования.
1. Прилито 50 мл р-ра NaOH
СHCl
100 50
0.1 3.3 . 10-2 моль/л
100 50
рН = - lgCHCl = - lg[H+] = 2- lg3.3 = 1.5
2. Прилито 90 мл р-ра NaOH
СHCl
100 90
0.1 5.3 . 10-3 моль/л
100 90
рН = - lgCHCl = - lg[H+] = 3 - lg5.3 = 2.3
3. Прилито 99 мл р-ра NaOH
СHCl
100 99
0.1 5.0 . 10-4 моль/л
100 99
рН = - lgCHCl = - lg[H+] = 4 - lg5 = 3.3
4. Прилито 99.9 мл р-ра NaOH
СHCl
100 99.9
0.1 5.5 . 10-5 моль/л
100 99.9
рН = - lgCHCl = - lg[H+] = 5 - lg5 = 4.3
В точке эквивалентности. Если прилить к 100 мл 0.1 М раствора HCl
100 мл 0.1 М раствора NaOH , то достигается полная нейтрализация
кислоты и в точке эквивалентности [H+] = [OH-], откуда:
рН = рОН =7
21.
После точки эквивалентности. Если прилить к 100 мл 0.1 М раствораHCl 100.1; 101; 110 мл 0.1 М раствора NaOH, то раствор станет щелочным.
Концентрация свободных ионов гидроксила [OH-] в этом случае будет
равна концентрации избытка гидроксила натрия, и определяться по
формуле:
С NaOH
VNaOH VHCl
M NaOH
VHCl V NaOH
(6.2)
Значения рН рассчитываются следующим образом:
Прилито 100.1 мл р-ра NaOH
СNaOH 100.1 100 0.1 5.0 . 10-5 моль/л
100 100.1
pOH = -lgCNaOH = 5- lg5 = 4.3
рН = 14 – pOH = 14 – 4.3 = 9.7
6. Прилито 101 мл р-ра NaOH
СNaOH
101 100
0.1 5.0 . 10-4 моль/л
100 101
pOH = -lg CNaOH = 4 – lg 5 = 3.3
рН = 14 – pOH = 14 – 3.3 = 10.7
7. Прилито 110 мл р-ра NaOH
СNaOH
110 100
0.1 4.8 . 10-3 моль/л
100 110
pOH = -lg CNaOH =3 – lg 4.8 = 2.3
рН = 14 – pOH = 14 – 2.3 = 11.7
Заметим, что для перехода от рН = 4.3 к рН = 7 (т.е. для увеличения рН
на 2.7 единиц) потребовалось всего 0.1 мл 0.1 М раствора NaOH. В то
время как в начале титрования для возрастания рН от 1 до рН = 3.3
(т.е. на 2.3 единицы рН) потребовалось 99.0 мл 0.1 М раствора NaOH, т.е в
1000 раз больше. Особо резкое изменение рН наблюдается в интервале,
когда осталось 0,1% неоттитрованной HCl или прилито 0,1% избытка
щелочи. В этом интервале рН быстро изменяется от 4.3 до 9.7 (ΔрН = 5.4).
Резкое изменение рН раствора, наблюдающееся вблизи точки
эквивалентности, т.е. в конце титрования называют скачком рН, или
скачком титрования. Чем больше скачек рН, тем точнее можно
оттитровать определяемое вещество.
22.
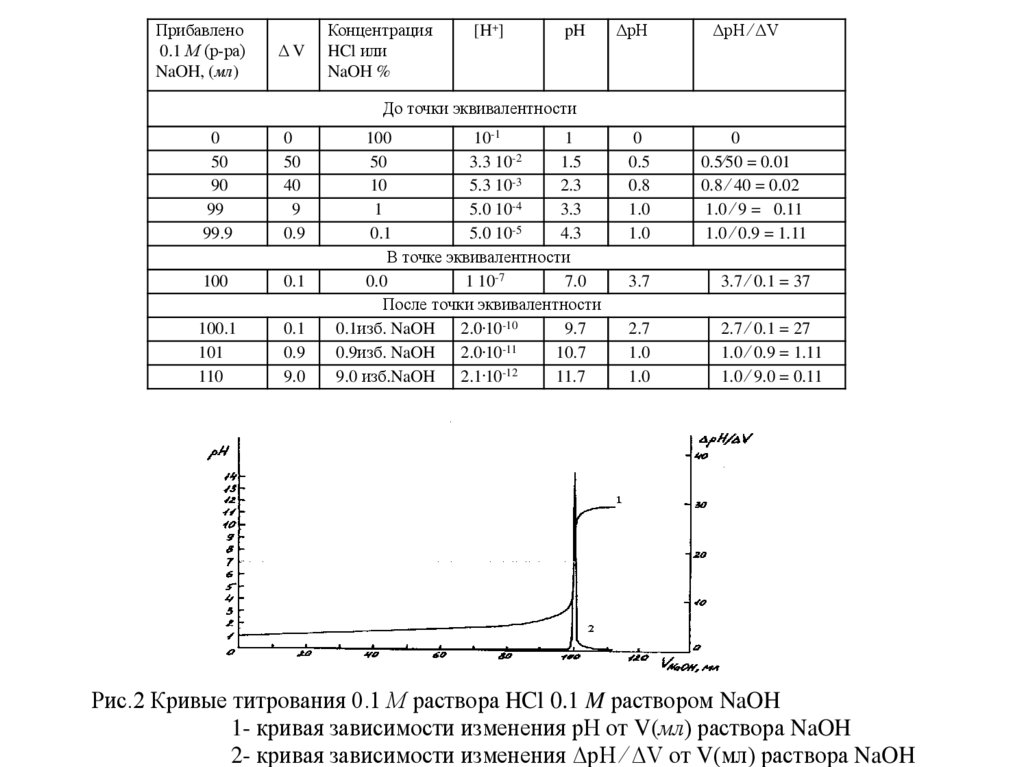
Прибавлено0.1 М (р-ра)
NaOH, (мл)
ΔV
Концентрация
HCl или
NaOH %
[H+]
pH
ΔpH
ΔpH ⁄ ΔV
0
0.5
0.8
1.0
1.0
0
0.5⁄50 = 0.01
0.8 ⁄ 40 = 0.02
1.0 ⁄ 9 = 0.11
1.0 ⁄ 0.9 = 1.11
3.7
3.7 ⁄ 0.1 = 37
2.7
1.0
1.0
2.7 ⁄ 0.1 = 27
1.0 ⁄ 0.9 = 1.11
1.0 ⁄ 9.0 = 0.11
До точки эквивалентности
0
50
90
99
99.9
0
50
40
9
0.9
100
0.1
100.1
101
110
0.1
0.9
9.0
100
10-1
1
-2
50
3.3 10
1.5
-3
10
5.3 10
2.3
1
5.0 10-4
3.3
0.1
5.0 10-5
4.3
В точке эквивалентности
0.0
1 10-7
7.0
После точки эквивалентности
0.1изб. NaOH
2.0.10-10
9.7
0.9изб. NaOH
2.0.10-11
10.7
.
-12
9.0 изб.NaOH 2.1 10
11.7
Рис.2 Кривые титрования 0.1 М раствора HCl 0.1 M раствором NaOH
1- кривая зависимости изменения рН от V(мл) раствора NaOH
2- кривая зависимости изменения ΔpH ⁄ ΔV от V(мл) раствора NaOH
23.
6.3 Титрование слабой кислоты сильным основаниемВычисление рН растворов в различные моменты титрования. Предположим, что
для титрования взято 100 мл 0.1 М раствора СН3СООН, титруемого 0.1М
раствором NaOH.
До начала титрования. В данном случае концентрацию ионов водорода слабой
кислоты [H+] нельзя приравнять к концентрации уксусной кислоты (CСН3СООН). В
водном растворе слабая кислота диссоциирует согласно уравнению:
СН3СООН = СН3СОО- + Н+
(6.3)
Применим закон действия масс:
[ H ] [CH 3 COO ]
= KCH3COOH
[CH 3 COOH ]
(6.4)
Так как [H+] = [CH3COO-], то уравнение (6.4) можно записать в виде:
[ H ]2
= KCH3COOH
(6.5)
[CH 3 COOH ]
или
[H+] =
[CH 3 COOH ] K CH 3COOH
(6.6)
Концентрация непродиссоциированной части молекул слабой кислоты равна:
[CH3COOH] = ССН3СООН – [H+]
(6.7)
Подставляя значение равенства (6.7) в уравнение (6.6), получим:
[H+] =
K CH 3COOH (СCH 3COOH [ H ]
(6.8)
Для слабо диссоциированной уксусной кислты кислоты концентрация ионов водорода
[H+] - величина сравнительно малая по отношению к С СН3СООН, и ею можно пренебречь.
Уравнение (6.8) при этом примет вид:
[H+] =
K CH 3COOH С CH 3COOH
(6.9)
Логарифмируя данное уравнение, получим:
рН = ½ рКCH3COOH – ½ lgССН3СООН
(6.10)
Подставляя в уравнение (6.10) значение логарифма константы (KCH3COOH = 1.82 . 10-5);
рКCH3COOH = -lgКCH3COOH = 5 – lg1.82 = 5- 0.26 = 4.74) и концентрации уксусной кислоты
(ССН3СООН = 0.1 мол/л), можно рассчить рН раствора до начала титрования.
рН = ½ 4.74 – ½lg 10-1 = 2.37 + 0.50 = 2.87
24.
До точки эквивалентности. Если прилить к титруемой уксусной кислоте 50; 90; 99;99.9 мл 0.1М раствора NaOH, то наряду со свободной уксусной кислотой в растворе
появится продукт нейтрализации уксусной кислоты – ацетат натрия. Уксусная кислота с
ее солью образует буферный раствор.
Концентрация ионов водорода в водных буферных растворах слабых кислот и их
солей без учета разбавления вычисляют по формуле (4.3)
С КИСЛ .
рН = рКкисл – lg
С СОЛИ
1. Когда прилито 50 мл NaOH
100 50
50
0.1 lg
0.1 = 4.74
100
100
pH = 4.74 – lg
2. Прилито 90 мл NaOH
pН = 4.74 lg
100 90
90
0.1 lg
0.1 5.69
100
100
3. Прилито 99 мл NaOH.
100 99
99
0.1 lg
0.1 6.69
100
100
Аналогичным путем можно рассчитать значения рН для других
промежуточных точек. Данные этих расчетов сведены в таблице № 2.
pН = 4.74 lg
25.
В точке эквивалентности вся уксусная кислота нейтрализована, и в растворебудет находиться только продукт ее нейтрализации – ацетат натрия, гидролизующийся
под влиянием воды.
рН в водных растворах гидролизующихся солей (типа СH3COONa) вычисляют по
формуле (4.3).
рН = 7 + ½ pKHAn + ½ lg С KtAn
По этой формуле и вычисляют рН в точке эквивалентности. В нашем случае
рН = 7 + ½ pKCH3COOH + ½ lgCCH3COOH
рН = 7 + ½ 4.74 + ½ lg 0.1 = 7 + 2.37 – 0.5 = 8.87
Это указывает на то, что при титровании слабой кислоты сильным
основанием точка эквивалентности не совпадает с точкой нейтрализации и
лежит в области щелочной среды. В нашем случае рН = 8.87.
После точки эквивалентности. При добавлении 100.1; 101; 110 мл, раствора едкого
натра, рН определяется концентрацией в титруемом растворе свободной щелочи NaOH.
Концентрации ионов гидроксила
[OH-] и рН растворов будут, соответственно, равны:
100.1 100
0.1 = 10-4 моль/л; [H+]= (10-14 – 10-4) = 10-10;
[OH-] =
100
pH = 10
101 100
0.1 = 10-3 моль/л;
[OH-] =
[H+]= (10-14 – 10-3) = 10-11;
100
pH = 11
110 100
0.1 = 10-2 моль/л; [H+]= (10-14 – 10-2) = 10-12;
[OH-] =
100
pH = 12
26.
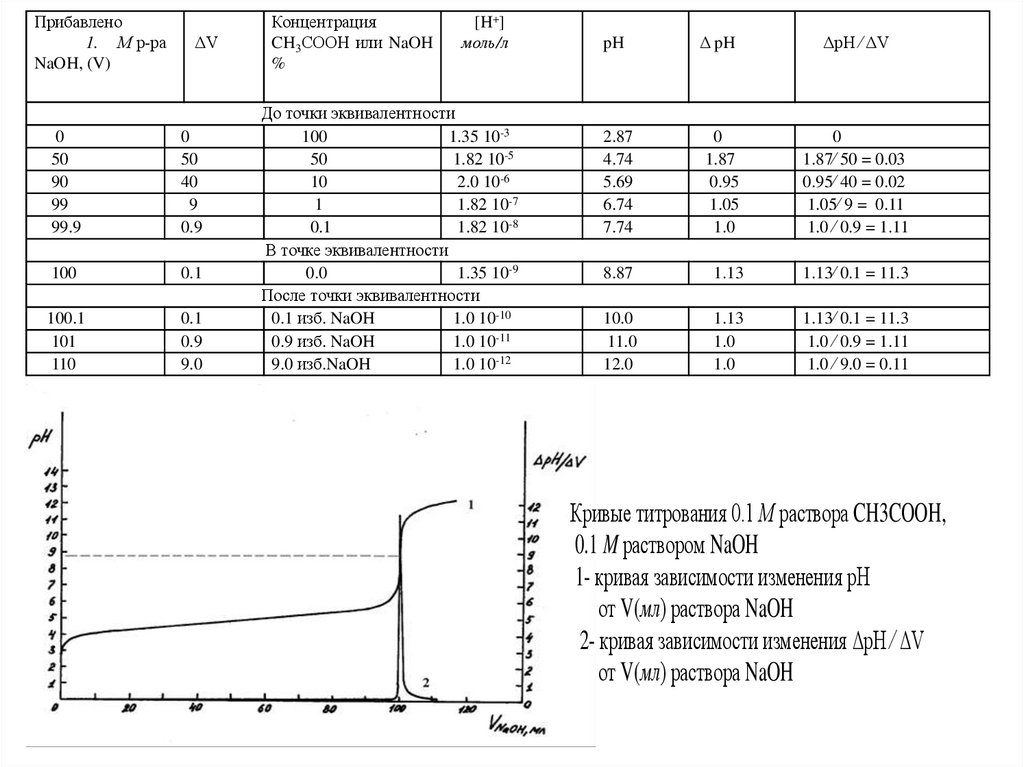
Прибавлено1. М р-ра
NaOH, (V)
ΔV
0
50
90
99
99.9
0
50
40
9
0.9
100
0.1
100.1
101
110
0.1
0.9
9.0
Концентрация
CH3СООН или NaOH
%
[H+]
моль/л
До точки эквивалентности
100
1.35 10-3
50
1.82 10-5
10
2.0 10-6
1
1.82 10-7
0.1
1.82 10-8
В точке эквивалентности
0.0
1.35 10-9
После точки эквивалентности
0.1 изб. NaOH
1.0 10-10
0.9 изб. NaOH
1.0 10-11
9.0 изб.NaOH
1.0 10-12
pH
Δ pH
ΔpH ⁄ ΔV
2.87
4.74
5.69
6.74
7.74
0
1.87
0.95
1.05
1.0
0
1.87⁄ 50 = 0.03
0.95⁄ 40 = 0.02
1.05⁄ 9 = 0.11
1.0 ⁄ 0.9 = 1.11
8.87
1.13
1.13⁄ 0.1 = 11.3
10.0
11.0
12.0
1.13
1.0
1.0
1.13⁄ 0.1 = 11.3
1.0 ⁄ 0.9 = 1.11
1.0 ⁄ 9.0 = 0.11
Кривые титрования 0.1 М раствора CH3COOH,
0.1 M раствором NaOH
1- кривая зависимости изменения рН
от V(мл) раствора NaOH
2- кривая зависимости изменения ΔpH ⁄ ΔV
от V(мл) раствора NaOH
27.
Титрование слабого основания сильной кислотойВычисление рН растворов в различные моменты титрования.
Предположим, что для титрования взято 100 мл. 0.1М раствора аммиака
(КNH4ОН= 1.76 10-5; pK = 4.76), титруемого 0.1 М раствором HCl.
До начала титрования. В данном случае концентрацию ионов
гидроксила слабого основания [OH-] нельзя приравнять к концентрации
гидроксида аммония (СNH4OH). В водном растворе слабое основание
диссоциирует согласно уравнению:
NH4OH = NH4+ + OHрН рассчитывается по формулам (3.14),
pH = 14 – ½ (рКKtOH – lg CKtOH) = 14 – ½ 4.76 + ½ lg 0.1 = 14 – 2.37 – 0.5
рН = 11.13
28.
До точки эквивалентности. Если прилить к титруемому гидроксиду аммония50; 90; 99; 99.9 мл 0.1М раствора НСl, то наряду со свободным гидроксидом
аммония в растворе появится продукт нейтрализации гидроксида аммония –
хлорид аммония. Гидроксид аммония с его солью образует буферный
раствор.
Концентрацию ионов водорода в водных буферных растворах слабого
основания и ее соли вычисляют по формуле
рН = 14 – рКосн.- lg Ссоли. + lg Сосн.
По данной формуле вычисляют рН промежуточных точек титрования,
предшествующих точке эквивалентности без учета разбавления.
Когда прилито 50 мл HCl
pH = 14 – 4.76 = 9.26
Прилито 90 мл HCl
pН = 14 – 4.76 = 8.24
Прилито 99 мл HCl.
pН = 14 – 4.76 = 7.24
Прилито 99.9 мл HCl.
pН =14 – 4.76 = 6.24
29.
В точке эквивалентности. В точке эквивалентности весь NH4ОНнейтрализован, и в растворе находится только продукт нейтрализации –
хлорид аммония, который подвергается гидролизу. рН в водных растворах
гидролизующихся солей (типа NH4Cl) вычисляют по формуле ( 5.13 )
рH = 7 – ½ рKKtOH – ½ lg С KtAn
По этой формуле и вычисляют рН в точке эквивалентности. В нашем случае:
рH = 7 – ½ 4.76 – ½ lg 0.1 = 7 – 2.37 + 0.5 = 5.12
Это указывает на то, что при титровании водного раствора
слабого основания сильной кислотой точка эквивалентности не
совпадает с точкой нейтрализации и лежит в области кислой среды ( рН
= 5.12).
После точки эквивалентности. При добавлении избытка раствора соляной
кислоты рН определяется концентрацией присутствующей в титруемом
растворе свободной кислоты (HCl). При избытке 0.1 М раствора HCl на 0.1; 1; и
10 мл концентрация ионов водорода будет, соответственно, равна:
(0.1/100) 0.1 = 10-4 моль/л; рН = 4
(1.0/100) 0.1 = 10-3 моль/л; рН = 3
(10/100) 0.1 = 10-2 моль/л; рН = 2
30.
Вычисление рН растворов в различные моменты титрования.Предположим, что для титрования взято 100 мл. 0.1М раствора аммиака
(КNH4ОН= 1.76 10-5; pK = 4.76), титруемого 0.1 М раствором HCl.
До начала титрования. В данном случае концентрацию ионов
гидроксила слабого основания [OH-] нельзя приравнять к концентрации
гидроксида аммония (СNH4OH). В водном растворе слабое основание
диссоциирует согласно уравнению:
NH4OH = NH4+ + OHрН рассчитывается по формулам (3.14),
pH = 14 – ½ (рКKtOH – lg CKtOH) = 14 – ½ 4.76 + ½ lg 0.1 = 14 – 2.37 – 0.5
рН = 11.13
До точки эквивалентности. Если прилить к титруемому гидроксиду
аммония 50; 90; 99; 99.9 мл 0.1М раствора НСl, то наряду со свободным
гидроксидом аммония в растворе появится продукт нейтрализации
гидроксида аммония – хлорид аммония. Гидроксид аммония с его солью
образует буферный раствор.
Концентрацию ионов водорода в водных буферных растворах слабого основания и
ее соли вычисляют по формуле (4.7)
рН = 14 – рКосн.- lg Ссоли. + lg Сосн.
По данной формуле вычисляют рН промежуточных точек титрования,
предшествующих точке экивалентности без учета разбавления.
Когда прилито 50 мл HCl
50
50
0.1 lg
0.1 9.26
pH = 14 – 4.76 – lg
100
100
Прилито 90 мл HCl
90
10
0.1 lg
0.1 8.24
pН = 14 – 4.76 – lg
100
100
Прилито 99 мл HCl.
99
1
0.1 lg
0.1 7.24
pН = 14 – 4.76 – lg
100
100
Прилито 99.9 мл HCl.
99.9
0.1
0.1 lg
0.1 6.24
pН =14 – 4.76 – lg
100
100
31.
В точке эквивалентности. В точке эквивалентности весь NH4ОНнейтрализован, и в растворе находится только продукт нейтрализации –
хлорид аммония, который подвергается гидролизу. рН в водных растворах
гидролизующихся солей (типа NH4Cl) вычисляют по формуле ( 5.13 )
рH = 7 – ½ рKKtOH – ½ lg С KtAn
По этой формуле и вычисляют рН в точке эквивалентности. В нашем
случае:
рH = 7 – ½ 4.76 – ½ lg 0.1 = 7 – 2.37 + 0.5 = 5.12
Это указывает на то, что при титровании водного раствора
слабого основания сильной кислотой точка эквивалентности не
совпадает с точкой нейтрализации и лежит в области кислой среды (
рН = 5.12).
После точки эквивалентности. При добавлении избытка раствора
соляной кислоты рН определяется концентрацией присутствующей в
титруемом растворе свободной кислоты (HCl). При избытке 0.1 М раствора
HCl на 0.1; 1; и 10 мл концентрация ионов водорода будет, соответственно,
равна:
(0.1/100) 0.1 = 10-4 моль/л; рН = 4
(1.0/100) 0.1 = 10-3 моль/л; рН = 3
(10/100) 0.1 = 10-2 моль/л; рН = 2
32.
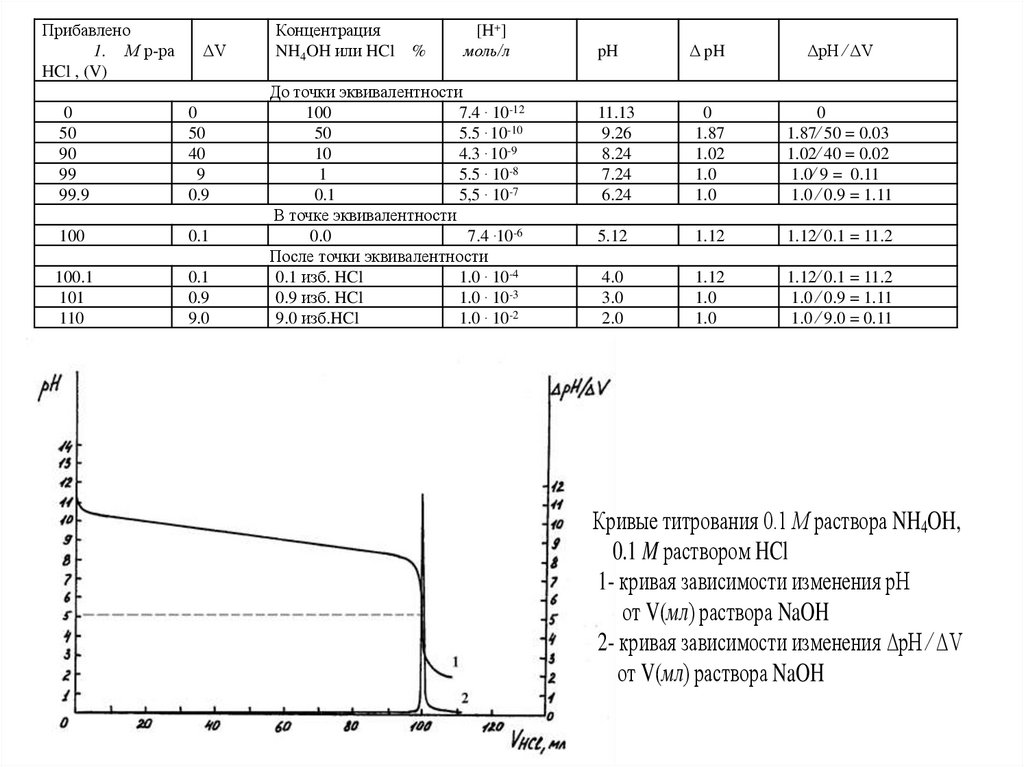
Прибавлено1. М р-ра
HCl , (V)
ΔV
0
50
90
99
99.9
0
50
40
9
0.9
100
0.1
100.1
101
110
0.1
0.9
9.0
Концентрация
NH4OH или HCl
%
[H+]
моль/л
До точки эквивалентности
100
7.4 . 10-12
50
5.5 . 10-10
10
4.3 . 10-9
1
5.5 . 10-8
0.1
5,5 . 10-7
В точке эквивалентности
0.0
7.4 .10-6
После точки эквивалентности
0.1 изб. HCl
1.0 . 10-4
0.9 изб. HCl
1.0 . 10-3
9.0 изб.HCl
1.0 . 10-2
pH
Δ pH
ΔpH ⁄ ΔV
11.13
9.26
8.24
7.24
6.24
0
1.87
1.02
1.0
1.0
0
1.87⁄ 50 = 0.03
1.02⁄ 40 = 0.02
1.0⁄ 9 = 0.11
1.0 ⁄ 0.9 = 1.11
5.12
1.12
1.12⁄ 0.1 = 11.2
4.0
3.0
2.0
1.12
1.0
1.0
1.12⁄ 0.1 = 11.2
1.0 ⁄ 0.9 = 1.11
1.0 ⁄ 9.0 = 0.11
Кривые титрования 0.1 М раствора NH4OH,
0.1 M раствором HCl
1- кривая зависимости изменения рН
от V(мл) раствора NaOH
2- кривая зависимости изменения ΔpH ⁄ ΔV
от V(мл) раствора NaOH
33.
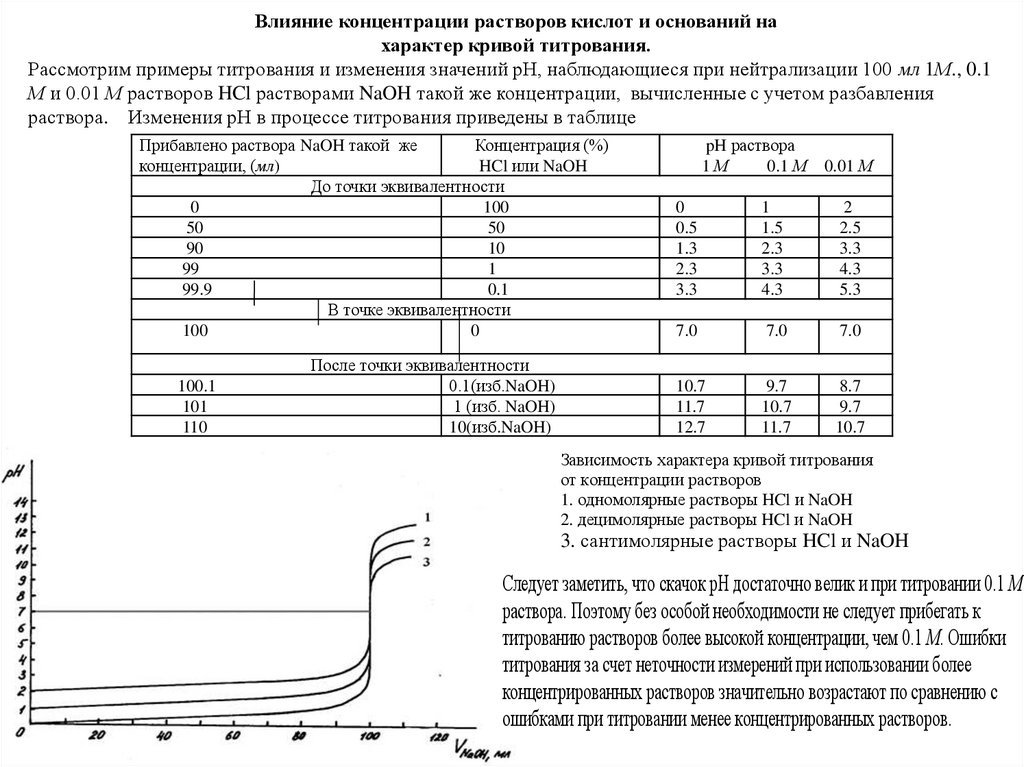
Влияние концентрации растворов кислот и оснований нахарактер кривой титрования.
Рассмотрим примеры титрования и изменения значений рН, наблюдающиеся при нейтрализации 100 мл 1М., 0.1
М и 0.01 М растворов HCl растворами NaOH такой же концентрации, вычисленные с учетом разбавления
раствора. Изменения рН в процессе титрования приведены в таблице
Прибавлено раствора NaOH такой же
Концентрация (%)
концентрации, (мл)
HCl или NaOH
До точки эквивалентности
0
100
50
50
90
10
99
1
99.9
0.1
В точке эквивалентности
100
0
100.1
101
110
После точки эквивалентности
0.1(изб.NaOH)
1 (изб. NaOH)
10(изб.NaOH)
pH раствора
1М
0.1 М
0.01 М
0
0.5
1.3
2.3
3.3
1
1.5
2.3
3.3
4.3
2
2.5
3.3
4.3
5.3
7.0
7.0
7.0
10.7
11.7
12.7
9.7
10.7
11.7
8.7
9.7
10.7
Зависимость характера кривой титрования
от концентрации растворов
1. одномолярные растворы HCl и NaOH
2. децимолярные растворы HCl и NaOH
3. сантимолярные растворы HCl и NaOH
Следует заметить, что скачок рН достаточно велик и при титровании 0.1 М
раствора. Поэтому без особой необходимости не следует прибегать к
титрованию растворов более высокой концентрации, чем 0.1 М. Ошибки
титрования за счет неточности измерений при использовании более
концентрированных растворов значительно возрастают по сравнению с
ошибками при титровании менее концентрированных растворов.
34.
35.
Титрование многоосновных кислотДиссоциация многоосновных кислот совершается по ступеням с
образованием кислых и средних ионов. Каждой ступени нейтрализации
соответствует своя точка эквивалентности. Например, при нейтрализации
трехосновной фосфорной кислоты сильным основанием наблюдаются три точки
эквивалентности, соответствующие добавлению к одному молю кислоты одного,
двух и трех молей NaOH. Происходит как бы нейтрализация смеси трех кислот:
H3PO4 (HAn1), H2PO4- (HAnII) и HPO42- (HAnIII). Сначала нейтрализуется более
сильная кислота H3PO4, отличающаяся большей константой электролитической
диссоциации (К1=1.1.10-2). Во вторую очередь нейтрализуется H2PO4- (К2 = 2.0 .
10-7). И наконец нейтрализуется HPO42-, с наименьшей константой диссоциации
(К3 = 3.6 . 10-12).
36.
Вычисление рН раствора в различные моменты титрованиямногоосновных кислот сильными основаниями.
Многоосновные кислоты можно рассматривать и как смеси кислот. При
нейтрализации смеси кислот гидроксидом натрия в начале титрования
практически нейтрализуется только более сильная кислота (HAnI).
[ HAn I ]
[ H ][ AnI ]
+
[ H ]I K I
HAnI H + AnI ; K I
откуда,
,
[ An I ]
[ HAn I ]
Когда концентрация ионов водорода [H+]1 сильной кислоты (HAnI)
станет равной концентрации ионов водорода [H+]II слабой кислоты
(HAnII), диссоциирующей по уравнению
[ H ][ An II
]
[ HAn II ]
+
K
[ H ] II K II
HAnII H + AnII ;
где,
,
II
[ HAn II ]
[ An II
]
начнется нейтрализация второй кислоты (HAnII). В этот момент в
растворе
[H+]1 = [H+]2,
[ HAn I ]
[ HAn II ]
KI
K II
из чего следует:
[ An I ]
[ An II
]
Отношение концентраций ионов водорода будет определяться
отношением констант диссоциации соответствующих кислот
[ H ]1
[ HAn I ]
[ HAn II ]
KI
(
:
)
=
(6.1)
[ AnI ]
[ An II
]
[H ]2
K II
Чем больше отношение КI : KII , тем точнее можно определить
количество каждой из кислот в смеси. Опыт показывает, что
дифференцированное (раздельное) титрование в водном растворе смеси
кислот или многоосновной кислоты может быть выполнено в случае,
если отношение констант различается более, чем в 10 000 раз. Так для
фосфорной кислоты:
KI
K II
1.1 10 2
2.0 10 7
4
5.5 10
3.3 10 5
;
13
7
K III
K II
3.6 10
2.0 10
Это значит, что фосфорную кислоту можно дифференцированно
титровать. А такие кислоты, как щавелевая (KI/KII = 1000) или винная
(KI/KII = 10) титруются как одноосновные.
37.
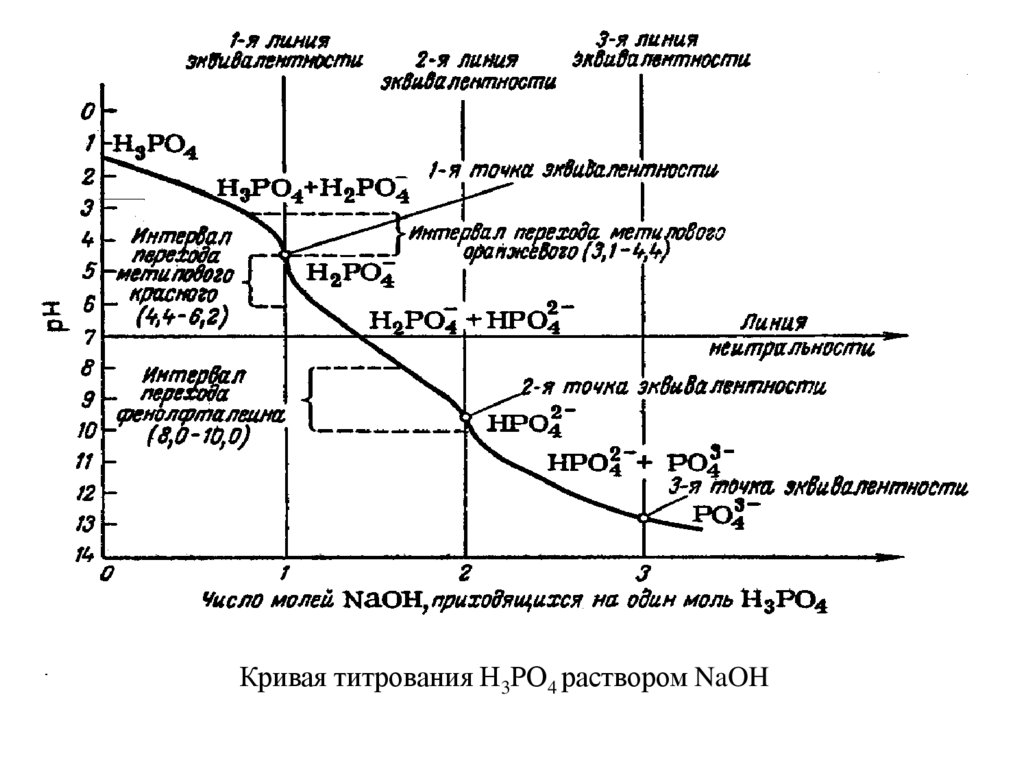
pН в точке эквивалентности при титровании многоосновных кислот или смеси кислотопределяют по формуле
pН= ½(рК1 + рК2)
Например, для фосфорной кислоты: рК1 = 1,96; рК2 = 6,70; рК3 =12,44
В первой точке эквивалентности
рН = ½(1,96 + 6,70) = 4,33.
Следовательно, первая точка эквивалентности лежит вблизи области окраски
индикатора метилового оранжевого (рТ = 3,1- 4,4).
Во второй точке эквивалентности
рН = ½(6,70 + 12,44) = 9,57.
Следовательно, вторая точка эквивалентности лежит вблизи области изменения окраски
фенолфталина (рТ = 8,0 – 10,0).
В третьей точке эквивалентности для трехосновных кислот, когда остается не
нейтрализованной только одна слабая кислота НРО4-2, рН вычисляют так же, как при
титровании слабых кислот
pН = 7 + ½ рК(кисл.) + ½ lgC(соли) .
В нашем случае
рН=7+ ½ 12,44 + ½ lg0,1 = 12,72
Так как рН в третьей точке эквивалентности больше 10, ее нельзя оттитровать
обычным способом. Обычно HPO42- ионы осаждают в виде малорастворимых солей,
например, концентрированным раствором хлорида кальция;
2HPO42- + 3Ca2+ → Ca3(PO4)2 + 2H+
Освобождающиеся при этом протоны титруют щелочью в присутствии
фенолфталина или тимолового синего.
38.
Кривая титрования Н3РО4 раствором NaOH39.
Титрование солей, образованных катионами сильных основанийи анионами слабых многоосновных кислот.
Водные растворы гидролизующихся солей, образованных катионами сильных
оснований и анионами слабых многоосновных кислот, имеют щелочную реакцию.
Титрование карбоната натрия. Карбонат натрия в водном растворе диссоциирует на
катион сильного основания Na+ и анион слабой кислоты CO32-, который гидролизуется
по двум ступеням, согласно уравнениям:
CO32- + НОН НCO3- + ОН- (I ступень)
НCO3- + НОН Н2CO3 + ОН- (II ступень)
То есть в растворе находятся соли анионов слабой (CO32-) и очень слабой (НCO3-)
кислот. Константы гидролиза по I и II ступеням, согласно закону действия масс, равны:
КI =
КII =
Умножив и разделив оба уравнения на величину [H+] получим выражения
для I ступени гидролиза:
КI =
или
для II ступени гидролиза:
КII =
или
Следовательно, щелочность раствора обуславливается преимущественно I
ступенью гидролиза так как KI больше KII в 10000 раз, и характеризоваться
электролитической диссоциации более слабой кислоты. НCO3- = Н+ +CO3+ Константа
которой равна = 4.7.10-11.
40.
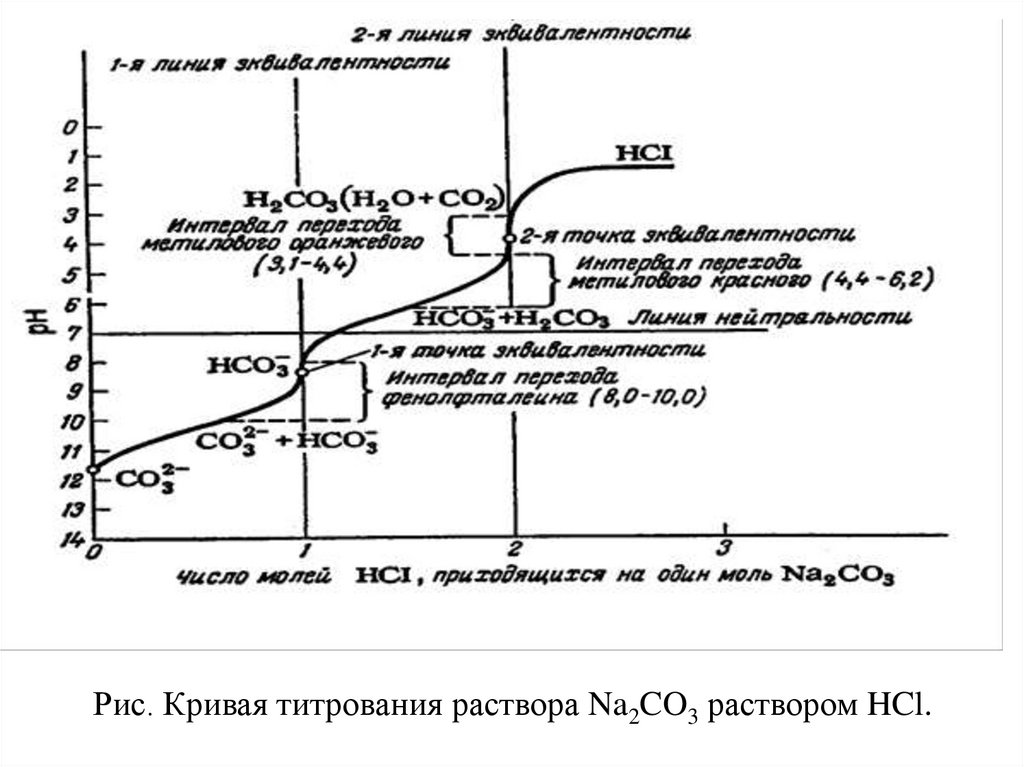
До начала титрования в водных растворах средних солей слабыхкислот рН
вычисляют по формуле. pН = 7 + ½ рК (НCO3-) + ½ lgC(Na2CO3)
В нашем случае для 0,1 М раствора Na2CO3
рН = 7 + 5.15 – 0.5 = 11.6
В первой точке эквивалентности
pН = ½(КI + KII)
рН = ½(6.4+10.3) = 8.35
Этот момент может быть зафиксирован с помощью фенолфталина
(рТ изменения окраски 8.0 – 10.0) .
Во второй точке эквивалентности значение рН вычисляют так же, как для слабых
одноосновных кислот:
pН = ½ рК (Н2CO3) – ½ lgC(Н2CO3)
рН= ½ 6.4 – ½ lg 0,1 = 3.2 + 0.5 = 3.7
Этот момент фиксируется метилоранжем ( pT = 3.1 – 4.4).
После достижения второй точки эквивалентности раствор будет кислым. Значение рН
соответствует концентрации избыточной соляной кислоты.
41.
Рис. Кривая титрования раствора Na2CO3 раствором HCl.42.
Анализ смеси 0.1 М р-ра Na(OH) и 0.1 М р-ра Na2CO3 в присутствии двухиндикаторов фенолфталина и метилоранжа
Рассмотрим гидролиз соли Na2CO3, так ка соль образована сильным
основанием и слабой кислотой, гидролиз идет полностью и в две ступени
CO32- + НОН НCO3- + ОН- (I ступень) К1= 0.21.10-3
НCO3- + НОН Н2CO3 + ОН- (II ступень) К2=0.22.10-7
Первую ступень можно представить как образование слабой кислота НCO3CO32-+Н+=НCO3- Это уравнение ассоциации, а диссоциации НCO3- =CO32-+Н+.
Константа диссоциации которой:
К НCO3- = [CO32-] [Н+]/[ НCO3-] и ровна 4.8.10-11. (рК=10.3)
По второй ступени образование кислоты Н2CO3. Константа диссоциации
которой Н2CO3=НCO3- + Н+. ровна:
К Н2CO3 = [НCO3-] [Н+]/[ Н2CO3] и ровна 4.5.10-7. (рК=6,4)
Разность констант чуть больше 10-4, следовательно, будет два скачка.
Первый скачек титрования будет при значении рН равным:
рН=1/2(рК1+рК2); рН = ½(10.3+6,4); рН=8.35
Этот может быть зафиксировано с помощью фенолфталина (рТ= 8.0-10.0)
43.
Но в растворе есть щелочь Na(OH), Точке эквивалентности рН=7.Кривая титрования Na(OH) и Na2CO3 показывает, что обесцвечивание
фенолфталина произойдет в тот момент, когда гидроксид натрия будет
полностью оттитрован соляной каслотой, а карбонат натрия на ½ и перейдет
в гидрокарбонат, то есть:
ОН-+ Н+ = Н2О
(1)
2+
CO3 + Н НCO3
(2)
На все это расходуется объем кислоты V1мл. При дальнейшем добавлении
титранат (соляной кислоты) реакция протекает по уравнению:
НCO3- + Н+ Н2CO3 (3)
рН в точке эквивалентности рассчитывается как для слабых кислот
Во второй точке эквивалентности значение рН в точке эквивалентности
рассчитывают так как для слабых одноосновных кислот:
pН = ½ рК (Н2CO3) + ½ lgC(Н2CO3)
рН= ½ 6.4 – ½ lg 0,1 = 3.2 + 0.5 = 3.7
Окончание этой реакции устанавливается по изменению цвета метилоранжа
(pT = 3.1 – 4.4), а общий объем титранат (соляной кислоты) равный V2.
Разность V2 – V1 равна объему соляной кислоты, затраченному на реакцию
(3). Так как на реакции (2) и (3) расходуется одно и тоже количество соляной
кислоты, то 2(V2 – V1) равно всему объему соляной кислоты
израсходованному на реакцию Na2CO3. Следовательно, на реакцию с Na(OH)
приходится объем соляной кислоты равный: V1 - 2(V2 – V1) = 2V1 – V2.
По этим данным рассчитывается масса Na2CO3
m(Na2CO3)=C(HCl)* 2(V2 – V1)*M(1/2 Na2CO3)/1000
m(NaOH)=C(HCl)*( 2V1 – V2)*M(NaOH)/1000
Аналогично проводят определение смесей и других композиций, например
серной и фосфорной кислот.
44.
В пяти колбах без этикеток с названиями, находятся различные кислоты и ихсмеси:1( HCl, H3PO4) 2. (CH3COOH, H3PO4), (HCl), (H2CO3), (NaHCO3). Как
методом титриметрии определить в какой колбе, какая смесь?
Проба, которая может содержать Na(OH), Na2CO3 и NaНCO3 и
индифферентные примеси массой 0.2582 г растворили в воде. При
добавлении фенолфталина раствор остался бесцветным . При титровании с
метиловым оранжевым расходуется 29.30 мл раствора серной кислоты
С=0.1020 моль.экв./л. Какое вещество содержится в пробе? Вычислите его
массовую долю.
Навеску образца, содержащего Na(OH), Na2CO3 и индифферентные
примеси, массой 0.6075 г. растворили в мерной колбе вместимостью 100 мл.
Аликвотную долю полученного раствора объемом 10.00 мл оттитровали
раствором HCl с молярной концентрацией 0.1022 моль/л. В присутствии
фенолфталина было затрачено12.09 мл титранат, а в присутствии
метилового оранжевого – 13.49 мл. Вычислить массовую долю (%) Na(OH)
и Na2CO3 в навеске.
45.
ИНДИКАТОРЫИндикаторами
называются
такие
вещества,
которые дают возможность с известной точностью
установить конечную точку титрования. При правильном
выборе индикатора точка эквивалентности совпадает с
конечной точкой титрования.
46.
Классификация индикаторов. В зависимости от типа используемой прититровании реакции индикаторы подразделяют на следующие группы:
Кислотно-основные индикаторы, реагирующие на изменение рН
раствора. Эти индикаторы широко применяются в методах нейтрализации.
К ним относятся фенолфталин, метилоранж, лакмус и др.
Окислительно-восстановительные
реагирующие
на
изменение
(ред-окс)
окислительно
-
индикаторы,
восстановительного
потенциала системы. К ним относятся дифениламин, азокрасители и т.д.
Комплексонометрические индикаторы, реагирующие на изменение
концентрации катиона комплексообразователя. К ним относятся эриохром
черный, мурексид и др.
Адсорбционные индикаторы, реагирующие на изменение
концентрации ионов, осаждаемых в виде малорастворимых соединений
(например, галогенидов серебра). К ним относятся флуоресцеин и эозин
47.
Индикаторы, применяемые в методе нейтрализации, представляют собойорганические вещества, слабые электролиты, обладающие кислыми или
основными свойствами, которые диссоциирует в растворе согласно
уравнению:
HInd H+ + Indкислота (донор протона)
основание
(акцептор протонов)
где HInd – молекулярная форма; Ind- – ионная форма индикатора .
Окраска кислотных форм этих индикаторов отличается от окрасок
основных форм. Эти изменения окраски зависят от степени изменения
концентрации ионов водорода ( рН раствора).
Например,
нейтральный
и
щелочной
растворы
метилового
оранжевого, являющегося относительно сильной кислотой (рК = 3.7),
имеют желтую окраску, так как в растворе преобладает ионная форма
HInd H+ + Indкрасная форма
желтая форма
Например: ОН + HInd Ind- + H2O
В кислой среде они имеют красную окраску, так как равновесие
смещено влево и в растворе преобладает молекулярная форма.
H+ + Ind- = HInd,
В нейтральном растворе фенолфталин – слабая кислота (рК=9.2)
равновесие сдвинуто влево, и молекулярная форма преобладает над
ионной (так же и в кислой), поэтому раствор бесцветен
HInd H+ + Indбесцветная форма
красная форма
В щелочной среде равновесие будет смещено вправо и цвет раствора
поменяется на красный. Например: ОН- + HInd Ind- + H2O
48.
Выбор индикатораСамым важным условием, соблюдаемым при титровании, является правильный
выбор индикатора.
Интервал перехода окраски выбранного индикатора должен по возможности
совпадать со скачком рН (скачком титрования) т.е. показатель титрования
индикатора - рТ с рН в точке эквивалентности. Поэтому при выборе идикатора
сначала теоретически рассчитывают изменение рН титруемого раствора, в котором
наблюдается скачок рН, а затем выбирают такой индикатор, у которого интервал
перехода окраски (рТ) совпадал бы с вычисленным значением скачка рН. Например,
если рН = 6 в точке эквивалентности, то подходящими индикаторами могут быть:
метиловый красный рТ = (4.4 – 6.2) и лакмус рТ = (5.0 – 8.0).
49.
Для получения точных и воспроизводимых результатов анализа необходимособлюдать определенные условия при титровании:
1. Следует установить титр стандартного раствора и применять установленный
для титрования раствор в присутствии одного и того же индикатора.
2. Для титрования следует брать всегда одно и то же количество индикатора и
повторять титрование определяемого вещества несколько раз до тех пор, пока не
будут получены три близко сходящихся результата.
3. Необходимо брать, как правило, не более 1-2 капель индикатора, не забывая о
том, что индикаторы, применяемые в методе нейтрализации, сами являются
кислотами или основаниями. На нейтрализацию их также расходуется некоторое
количество раствора титранта.
4. Всегда следует титровать до появления одного и того же оттенка окраски
раствора, используя для титрования по возможности одинаковые объемы титруемого
раствора.
5. Необходимо выбирать такой индикатор, который изменяет свой цвет вблизи
точки эквивалентности.
50.
ОШИБКИ ТИТРОВАНИЯЕсли изменение цвета индикатора происходит не точно в
точке эквивалентности, а раньше или позже, то и точка
эквивалентности будет фиксироваться раньше или позже
требуемого момента, допуская некоторую ошибку. Такая
ошибка называется индикаторной ошибкой. Следовательно,
если индикатор выбран неправильно, то ошибка может
превышать допустимые погрешности. Индикаторные ошибки –
это
следствие
недотитрования
или
перетитрования
исследуемого раствора. В методе нейтрализации различают
несколько типов индикаторных ошибок.
51.
. Водородная ошибка титрования H+(ошибка) связана с наличием втитруемом растворе, по окончании титрования, избытка ионов
водорода, остающихся в растворе в результате недотитрования
сильной кислоты сильной щелочью или перетирования сильного
основания сильной кислотой.
Например, для титрования сильной кислоты сильным основанием
выбран индикатор метиловый оранжевый
(рТ = 4). Титрование
+
-4
заканчивается при рН = рТ = 4, т.е. при [H ] = 10 моль/л (в кислой среде).
Следовательно, часть кислоты будет недотитрована, что приведет к
водородной ошибке. Вычислим ее величину.
Пусть концентрация титруемой кислоты С = 0.1 моль./л, начальный
объем кислоты V1= 25.0 мл; объем раствора в конце титрования V2= 2V1=
50.0 мл.
Количество
вещества
соляной
кислоты,
соответственно,
и
.
количество ионов водорода равно С V1 которые можно принять за 100%.
Так как рТ- это обратный логарифм концентрации ионов водорода, при
котором
данный
индикатор
меняет
свою
окраску,
количество
+
неоттитрованных ионов водорода [H ] в точке эквивалентности будет
равно 10-рТ.V2. (т.е [H+] = 10-4 моль/л )Это и будет составлять водородную
ошибку титрования.
CV1 – 100%
10 pT V 2
-рТ.
+
+
100 %
10 V2 – % ([Н ]- ошибка); Н (ошибка)=
CV1
2 10 pT
100 %
C
В нашем случае Н+(ошибка) = (2 . 10-4 :10-1 ) . 100 = 0.2%
Так как V2 = 2V1 Н+(ошибка) =
52.
Гидроксильная ошибка титрования (ОН-(ошибка)) связана с наличием втитруемом растворе по окончании титрования избытка гидроксидионов, остающихся в растворе в результате недотитрования
сильного основания сильной кислотой или перетитрованиия сильной
кислоты сильным основанием.
Рассмотрим пример. Предположим, что для титрования сильной кислоты
сильным основанием выбран индикатор с рТ = 9 (например,
фенолфталин). В этом случае титрование закончится при рН = рТ = 9, т.е.
при [H+] = 10-9 (в щелочной среде). Следовательно, при титровании будет
прилит некоторый избыток щелочи, что приведет к ОН - - ошибке.
Вычислим ее величину.
Так как [H+] . [OH-] = Kw = 10-14 , то в конце титрования количество
ионов гидроксила будет составлять [OH-] = 10-14/10-9 = 10-5 или
[OH-] =10-(14-рТ). Количество гидроксид-ионов в растворе в конце
титрования будет равно: [OH-] =10-(14 - рТ) . V2.
Эта величина и составляет гидроксильную ошибку титрования. Если
общее количество гидроксид-ионов, равное Сосн.. V1, принять за 100% , то
гидроксильную ошибку титрования в процентах можно рассчитать по
уравнению:
(14 pT )
10
V2
100 %
OH (ошибка) =
CV1
В конце титрования объем увеличился в два раза V2 = 2V1
2 10 (14 pT )
100 %
OH (ошибка) =
C
В нашем случае при V1 = 25 мл, V2 = 50 мл, С = 0.1 моль-экв/л, рТ = 9
ОН-(ошибка) = (2 . 10-(14 – 9) /10-1 ) . 100% = 2 . 10-2 = 0.02%.
53.
Кислотнаяошибка
титрования.
(HАn-ошибка)
обусловлена
присутствием в титруемом растворе по окончании титрования
нейтральных молекул недотитрованной слабой кислоты.
Предположим, что для титрования была взята слабая кислота с
константой диссоциации равной КHAn. Титрование ведут в присутствии
индикатора с определенным
значением рТ. Так как кислота слабая,
константу диссоциации можно записать в виде:
[ H ][ An ]
[ HAn ]
[H ]
KHAn =
или
[ HAn]
K HAn
[ An ]
Для слабой кислоты [HАn] можно приравнять СHАn, а
уравнение примет вид:
[An-] = СKtAn,
С HAn
[ HAn ]
[H ]
C KtAn
K HAn
[ An ]
отношение
А
[ HAn ]
и есть ни что иное, как отношение концентрации
[ An ]
неоттитрованной части слабой кислоты к ее оттитрованной части, это
отношение и можно считать кислотной ошибки титрования. Процент
неоттитрованной части кислоты будет равен:
C HAn
100 %
C KtAn
или
[H ]
100 %
HАn (ошибка) =
K HAn
Титрование заканчивается при значении
[H+]=10-pT, а КHAn = 10-рК ,
и, следовательно,
10 pT
[H ]
100 %
100 % =
HАn (ошибка) =
K HAn
10 pK
НАn (ошибка) = 10рК-рТ .100 %.
54.
Если задаться целью и ограничить HАn (ошибку) титрования 0.1%, то[H ]
100 % = 0.1%. Разделим обе части уравнения на 100% и получим
K HAn
[H ]
10 3 . Это означает, что доля неоттитрованной части кислоты
K HAn
составляет 0.001 от количества оттитрованной кислоты. Для правильного
подбора индикатора титрования запишем выражение
[H ]
10 3 в виде 10 рК - рТ = 10-3 откуда рТ = рК+3.
K HAn
Следовательно, для титрования слабой кислоты,
имеющей константу диссоциации равной Ккис, будут пригодны
только те индикаторы, у которых рТ - индикатора на 3 единицы
превышают значение рКкис. Например, для титрования уксусной
кислоты могут быть использованы индикаторы, у которых рТ = 4.76 + 3 =
7.76 (фенолфталин).
55.
Щелочнаяошибка
титрования
(KtOH-ошибка)
возникает
в
результате присутствия в титруемом растворе по окончании
титрования
нейтральных
недотитрованных
молекул
слабого
основания.
Предположим, что для титрования было взято слабое основание с
константой диссоциации равной КKtOH. Титрование ведут в присутствии
индикатора с определенным значением рТ. Так как основание слабое,
константу диссоциации можно записать в виде:
[ KtOH ] [OH ]
[ Kt ][OH ]
K KtOH
или
K KtOH
[ Kt ]
[ KtOH ]
Для слабого основания [KtOH] можно приравнять СKtOH, а [Kt+] = СKtAn,
[ KtOH ] C KtOH
[OH ]
т.е. можно записать:
C KtAn
K KtOH
[ Kt ]
Это отношение и есть ни что иное, как отношение концентрации
неоттитрованной части слабого основания к ее оттитрованной части, это
отношение и можно считать щелочной ошибкой титрования, а процент
неоттитрованного основания равен
C KtOH
[OH ]
KtOH ( ошибка)
100%
100 %
или
K KtOH
C KtAn
+
Титрование заканчивается при значении [H ]=10-pT, или [OH-] = 10-(14-pT),
а КKtOH = 10-рК и, следовательно,
[OH ]
10 (14 pT )
KtOH ( ошибка)
100%
100%
K KtOH
10 pK
Или
KtOH-ошибка = 10 pK+ рT-14 . 100%
56.
Если задаться целью и ограничить KtOH-ошибку титрования 0.1%, то[ KtOH ]
100% 0.1%.
[ Kt ]
Разделим обе части уравнения на 100% получим
[OH ]
[ KtOH ]
3
10
0
.
001
или
.
K KtOH
[ Kt ]
Это означает, что доля неоттитрованной части кислоты составляет
0.001 от количества оттитрованной кислоты. Для правильного подбора
индикатора титрования запишем выражение [HО-]/КKtOH = 10-3 в виде:
10 pK+ рT-14 = 10-3
откуда
рТ = 11 – рК.
Следовательно, для титрования слабого основания, имеющего
константу диссоциации Косн, будут пригодны только те индикаторы,
рТ которых меньше или равно значению 11- рКосн.
Например, у аммиака рК = 4.75, поэтому для титрования аммиака будут
пригодны индикаторы, у которых рТ меньше или равно 11 – 4.75 = 6.25.
Этому условию не удовлетворяют ни фенолфталин, у которого рТ = 9.0,
ни бромтимоловый синий, имеющий рТ = 7.2. Удовлетворяет метиловый
оранжевый, у которого рТ = 4.0.
57.
%Н+(ошибка)%=
2 10 pT
100
C
(14 pT )
2
10
100 %
OH-(ошибка) =
C
НАn (ошибка) = 10рК-рТ .100 %
рТ = рК+3
KtOH-ошибка = 10 pK+ рT-14 . 100%
рТ = 11 – рК
58.
ТИТРОВАНИЕ ПО МЕТОДУ ОСАЖДЕНИЯНаиболее важными требованиями, которые
предъявляются к реакциям осаждения в
титриметрическом анализе, являются:
1. достаточно малая растворимость осадка
2. быстрое его образование при добавлении
титранта,
3. минимальное содержание примесей
4. наличие индикатора, позволяющего фиксировать
конец реакции.
Наибольшее значение приобрели те методы
осаждения, которые связаны с образованием
малорастворимых соединений серебра, бария, ртути,
свинца, цинка и других элементов.
59.
Аргентометрияметод титриметрического анализа, основанный на
применении стандартного раствора нитрата серебра.
При титровании раствора, содержащего ионы хлора,
стандартным раствором нитрата серебра образуется
осадок AgCl. Зная произведение растворимости ПРAgCl,
концентрации титруемого хлорид иона [Cl-] и
стандартного растворов AgNO3, можно легко
вычислить изменения [Ag+] и [Cl-] в любой момент
титрования.
60.
Предположим, что 100 мл 0.1 M раствора NaCl титруют 0.1 M раствором AgNO3. Доначала титрования 0.1 M раствор NaCl полностью диссоциирует на ионы:
[Cl-] = CNaCl; pCl = -lg 10-1 = 1
Если прилить к первоначальному раствору NaCl 50; 90; 99; 99.9 мл 0.1 M раствора
AgNO3, концентрация Cl- будет уменьшаться, а рСl – увеличиваться.
1. При прибавлении 50 мл. 0.1 M раствора AgNO3 в растворе останется не
осажденным 50 % NaCl, т.е. концентрация ионов хлора (без учета разбавления)
уменьшится в два раза:
рСl = 1.3
2. При добавлении 90 мл 0.1 M раствора АgNO3 останется не осажденным 10%
ионов хлора, т.е. концентрация ионов хлора уменьшится в 10 раз
рСl = 2.0
При прибавлении 99 мл. 0.1 M раствора AgNO3
[Cl ]
1
0.1 1 10 3 моль / л ;
100
рСl = 3.0
При прибавлении 99.9 мл. 0.1 M раствора AgNO3
[Cl ]
0.1
0.1 1 10 4 моль / л ;
100
рСl = 4.0
61.
При прибавлении 100 мл. 0.1 M раствора нитрата серебра наступитточка эквивалентности, и концентрация ионов хлора и серебра будет
определяться, исходя из растворимости трудно растворимой соли
хлорида серебра:
ПРAgCl = [Ag+] [Cl-] = 1.7.10-10;
[Cl ] 1.7 10 10 1.304 10 5
-lg [Cl-] = pCl = 4.885
62.
В процессе дальнейшего прибавления AgNO3 в растворе появляется избыток ионовсеребра, вследствие чего концентрация хлорид ионов уменьшается, так как: ПРAgCl =
[Ag+] [Cl-] = 1.7.10-10 - есть величина постоянная, и с увеличением концентрации
одного из ионов трудно растворимой соли концентрация другого иона уменьшается, а
произведение концентраций остается постоянным
При добавлении 0.1 мл избытка 0.1 M раствора АgNO3
0.1
[ Ag ]
0.1 1 10 4 моль / л ; pAg = 4
100
10
ПР
1
.
7
10
6
1
.
7
10
[Cl-] =
[ Ag ] 1 10 4
pCl- = -lg[Cl-] = -lg 1,7 . 10-6 = 5.77 или pCl- = (9.77 – 4) = 5.77
Аналогично, при добавлении 1 мл избытка 0.1 М раствора АgNO3
1
[ Ag ]
0.1 1 10 3 моль / л ; pAg = 3 или pCl- = (9.77 – 3) = 6.77
100
При добавлении 10 мл избытка 0.1 М раствора АgNO3
10
[ Ag ]
0.1 1 10 2 моль / л ; pAg = 2 или pCl = (9.77 – 2) = 7.77
100
По полученным данным составим таблицу и построим график
зависимости рСl- от V(мл) раствора AgNO3
63.
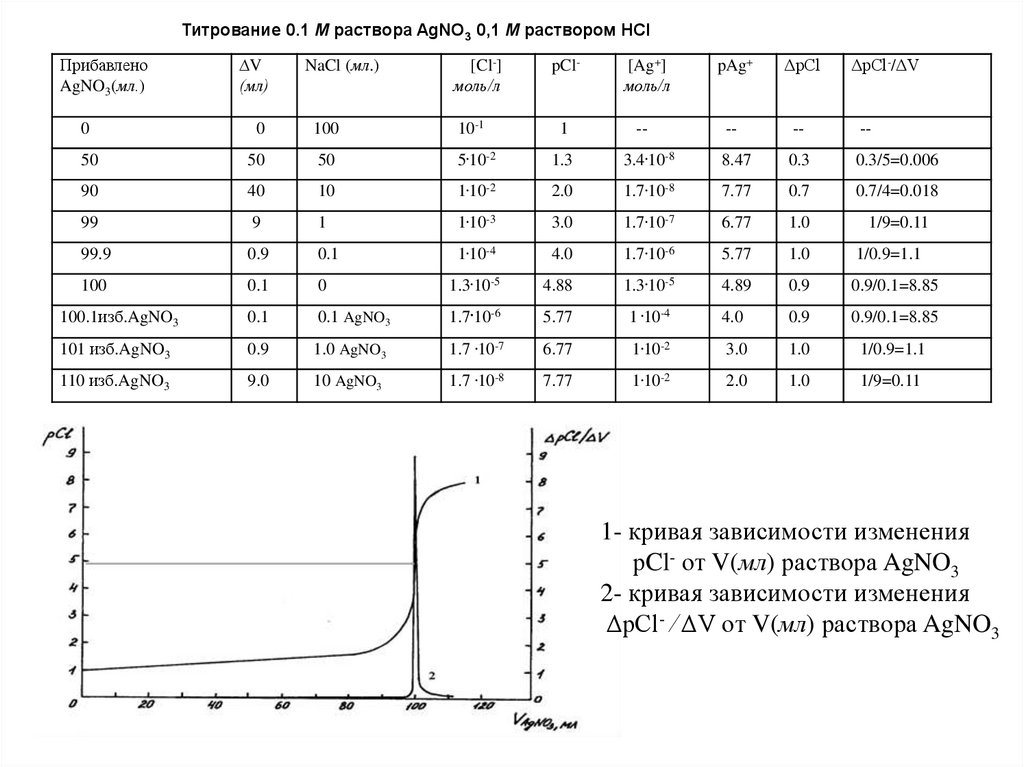
Титрование 0.1 М раствора AgNO3 0,1 М раствором HClПрибавлено
AgNO3(мл.)
∆V
(мл)
NaCl (мл.)
[Cl-]
моль/л
pCl-
[Ag+]
моль/л
pAg+
ΔpCl
ΔpCl-/ΔV
0
0
100
10-1
1
--
--
--
--
50
50
50
5.10-2
1.3
3.4.10-8
8.47
0.3
0.3/5=0.006
90
40
10
1.10-2
2.0
1.7.10-8
7.77
0.7
0.7/4=0.018
99
9
1
1.10-3
3.0
1.7.10-7
6.77
1.0
1/9=0.11
99.9
0.9
0.1
1.10-4
4.0
1.7.10-6
5.77
1.0
1/0.9=1.1
100
0.1
0
1.3.10-5
4.88
1.3.10-5
4.89
0.9
0.9/0.1=8.85
100.1изб.AgNO3
0.1
0.1 AgNO3
1.7.10-6
5.77
1 .10-4
4.0
0.9
0.9/0.1=8.85
101 изб.AgNO3
0.9
1.0 AgNO3
1.7 .10-7
6.77
1.10-2
3.0
1.0
1/0.9=1.1
110 изб.AgNO3
9.0
10 AgNO3
1.7 .10-8
7.77
1.10-2
2.0
1.0
1/9=0.11
1- кривая зависимости изменения
рCl- от V(мл) раствора AgNO3
2- кривая зависимости изменения
ΔpCl- ⁄ ΔV от V(мл) раствора AgNO3
64.
Анализируякривую
осаждения,
можно
титрования
рСl-
титрования
в
методе
в
начале
изменяется
очень
заметить,
раствора
что
медленно, а вблизи точки эквивалентности – очень
быстро, и скачок тем больше, чем меньше
значение
произведения растворимости трудно
растворимой
соли,
образующейся
титровании в методе осаждения.
при
65.
Определение конечной точки титрованияПо способу установления конечной точки титрования (точки
эквивалентности) различают методы:
а. Метод Гей – Люссака (или метод равного помутнения).
При титровании хлорида по этому методу вблизи точки
эквивалентности отбирают небольшие порции прозрачного
раствора и добавляют к одной порции AgNO3, а к другой NaCl. Если
достигнута точка эквивалентности, помутнение в обеих порциях
будет одинаковым. В недотитрованных растворах помутнение будет
происходить только при добавлении AgNO3, а в перетитрованных
при добавлении NaCl.
66.
Метод Мора (индикатор-хромат калия). Идея метода основана наобразовании кирпично-красного осадка хромата серебра Ag2CrO4 в
конечной точке титрования хлорид-иона нитратом серебра. В
определенных условиях Ag2CrO4 начнет выпадать лишь после того, как
определяемые Cl- - ионы будут практически полностью осаждены из
раствора в виде AgCl. И связано это с различной растворимостью
хлорида и хромата серебра.
Предположим, что 0.1 М раствор NaCl, содержащий также индикатор
K2CrO4 c концентрацией [CrO42-] равным 10-2 М, титруют раствором
AgNO3. При этом каждый из осадков (AgCl и Ag2CrO4) начинает
выпадать только после того, как будет превышено произведение
растворимости каждого из них. Так как величина ПРAgCl ≈ 10-10, для
достижения ее требуется, чтобы концентрация Ag+ в растворе составляла:
ПРAgCl
10
10
9
[ Ag ]
10
моль / л
1
[Cl ] 10
67.
Предположим, что 0.1 М раствор NaCl, содержащий также индикаторK2CrO4 c концентрацией [CrO42-] равным 10-2 М, титруют раствором
AgNO3. При этом каждый из осадков (AgCl и Ag2CrO4) начинает
выпадать только после того, как будет превышено произведение
растворимости каждого из них. Так как величина ПРAgCl ≈ 10-10, для
достижения ее требуется, чтобы концентрация Ag+ в растворе составляла:
ПР AgCl
10 10
[ Ag ]
10 9 моль / л
1
[Cl ]
10
Рассчитаем теперь, при какой концентрации ионов серебра начинается
осаждение Ag2CrO4, произведение растворимости которого равно:
ПРAg2CrO4 = (2[Ag+])2 [CrO42-] = 0.11. 10-11 Отсюда:
11
ПР Ag2CrO4
0
.
11
10
3
2
[ Ag ] 2
0
.
17
10
моль / л
2
2
4[CrO4 ]
4 10
Таким образом, произведение растворимости AgCl достигается
раньше, т.е. при меньшей концентрации [Ag+] = (10-9 моль/л). Поэтому и
осаждается, в первую очередь, AgCl. Поскольку произведение [Ag+][Cl-]
остается все время постоянным, то по мере прибавления [Ag+] и
осаждения Cl- в виде AgCl концентрация [Cl-] в растворе постепенно
понижается, а [Ag+] повышается. При этом в конце концов окажется
достигнутой концентрация ионов серебра [Ag+] равная 0.17.10-3 моль/л,
которая необходима для начала осаждения Ag2CrO4.
68.
С этого момента наряду с AgCl начнет осаждаться и Ag2CrO4, и осадокприобретает красновато-бурую окраску, что может служить индикатором
точки эквивалентности титрования. Из уравнения [Ag+][Cl-] = ПРAgCl
нетрудно вычислить, какова будет концентрация [Cl-] -ионов в растворе в
19
ПР
1
10
AgCl
7
[
Cl
]
5
.
88
10
моль / л
этот момент:
3
[ Ag ] 0.17 10
Таким образом, в указанных условиях выпадение осадка Ag2CrO4
действительно начинается только после практически полного осаждения
Cl- -ионов в виде AgCl. Для визуального обнаружения осадка хромата
серебра достаточно перетитровать анализируемый раствор на одну каплю
AgNO3.
Титрование с хроматом в качестве индикатора, проводится в нейтральной или
слабощелочной среде, когда рН раствора больше 6.5, но менее 10.5. В более кислой
области происходит протонирование хромата (CrO42- + Н+ = НCrO4-) и
чувствительность индикатора падает, а в щелочных растворах (рН более чем 10.5)
гидроксид серебра может выпадать ранее хромата.
Метод Мора обычно применяют для определения хлоридов и бромидов. Иодиды и
тиоцианаты не определяются, так как вследствие адсорбции установление точки
эквивалентности становится затруднительным, и погрешность анализа возрастает.
69.
Метод Фольгарда [индикатор–тиоцианатные комплексы железа]Реакцию взаимодействия серебра с тиоцианатом используют для
определения галогенидов методом обратного титрования и выполняют его в
присутствии индикатора- иона железа (III). По этому методу к
анализируемому раствору галогенида (хлорида или бромида), добавляют
избыток титрованного раствора нитрата серебра. Не вошедшее в реакцию
количество [Ag+] оттитровывают, приливая по каплям из бюретки
стандартный раствор тиоцианата калия или аммония в присутствии Fe3+. При
этом вначале образуется малорастворимый осадок AgSCN:
Ag+ + SCN- = ↓AgSCN
Образование AgSCN продолжается до эквивалентного соотношения
количеств ионов серебра и тиоцианата. Лишняя капля раствора тиоцианатиона, прибавленная после достижения точки эквивалентности, вызывает
появление кроваво-красного окрашивания вследствие образования Fe(SCN)3:
Fe3+ + 3SCN- = Fe(SCN)3
По методу Фольгарда могут быть оттитрованы и другие анионы,
образующие малорастворимые соединения с ионом серебра (С2О42-, РО43-)
Существенным достоинством метода Фольгарда является возможность
определения галогенидов в кислой среде.
70.
Метод Фаянса (адсорбционные индикаторы).Адсорбционными индикаторами называют соединения, которые
при адсорбции на осадке изменяют свой цвет. Установлено, что в
первую очередь на коллоидной частице адсорбируются ионы,
одноименные с осадком. Например, при титровании хлорида калия
нитратом серебра до точки эквивалентности в растворе находится
избыток одноименного с осадком AgCl - хлорид иона, который и будет
адсорбироваться на его поверхности, придавая частице
отрицательный заряд:
K+ + Cl- + Ag+ +NO3- = ↓AgCl + K+ + NO3- + Cl-(изб.)
[(AgCl) . nCl-]nПосле точки эквивалентности в растворе будет избыток ионов
серебра, которые и будут адсорбироваться на поверхности коллоидной
частицы хлорида серебра, придавая ему положительный заряд:
K+ + Cl- + Ag+ +NO3- = ↓AgCl + K+ + NO3- + Ag+(изб.)
[(AgCl) . nAg+]n+
71.
Образование заряженных коллоидных частиц, и изменение знака в процессеосадительного тирования позволяет использовать для определения точки
эквивалентности так называемые адсорбционные индикаторы.
Адсорбционные индикаторы представляют собой органические соединения,
являющиеся слабыми кислотами, диссоциирующими согласно уравнению:
HInd H++ IndАнионы этих индикаторов, адсорбируясь на поверхности положительно
заряженных коллоидных частиц, выпадающих в процессе титрования осадков,
вызывают изменение цвета поверхности этих осадков.
[(AgCl) . nAg+]n+ + nInd- = {[(AgCl) . nAg+]n+ . nInd-}
Отличным адсорбционным индикатором для титрования хлорида является
флуоресцеин, имеющий в растворе желто-зеленую окраску. В точке эквивалентности,
адсорбируясь на поверхности положительно заряженной коллоидной частицы
хлорида серебра, он придает осадку красный цвет. Титрование с флуоресцеином
происходит при рН от 7 до 10, поскольку индикатор является слабой кислотой.
72.
ОКИСЛИТЕЛЬНО – ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИРассматриваемые методы основаны на использовании определенных типов
окислительно – восстановительных реакций. Эти методы наряду с методами кислотноосновного, осадительного, комплексонометрического титрования очень широко
применяются в аналитической химии. Этому способствует их большая точность,
хорошая воспроизводимость и простота.
Окислительно – восстановительные, или ред-окс, процессы включают перенос
электронов от одного реагирующего вещества к другому. Титриметрические методы,
основанные на реакциях переноса электронов, наиболее многочисленны и
разнообразны по сравнению с методами, основанными на реакциях любого другого
типа.
В процессе окисления происходит отдача, а в процессе восстановления –
присоединение электронов. В любой окислительно – восстановительной реакции
молярное отношение между окисляющимся и восстановляющимся веществами таково,
что число электронов, отданных одним веществом, равно числу электронов, принятых
другим веществом. Это следует учитывать при составлении уравнений окислительновосстановительных реакций.
Для того чтобы указать, которое из веществ присоединяет электроны, а какое
отдает, удобно разделить окислительно-восстановительную реакцию на две части, то
есть на две полуреакции.
Так, суммарная реакция 5Fe2+ + MnO4- + 8H+ = 5Fe3+ + Mn2+ + 4H2O получается при
объединении полуреакций окисления Fe2+ c полуреакцией восстановления MnO4-:
73.
Так, суммарная реакция 5Fe2+ + MnO4- + 8H+ = 5Fe3+ + Mn2+ + 4H2O получается приобъединении полуреакций окисления Fe2+ c полуреакцией восстановления MnO4-:
Fe2+ – e- = Fe3+
5
+
2+
MnO4 + 8H + 5e = Mn + 4H2O 1
5Fe2+ + MnO4- + 8H+ = 5Fe3+ + Mn2+ + 4H2O
В процессе титрования по методу окисления – восстановления происходит
изменение окислительно–восстановительных потенциалов взаимодействующих друг с
другом систем.
Количественная зависимость окислительно–восстановительного потенциала
системы (E) от концентрации (активности) реагирующих веществ и температуры
описывается уравнением Нернста:
RT [окисл.]a
E E
ln
nF [восст.]в
0
(11.1)
где Ео – стандартный окислительно-восстановительный потенциал;
R – универсальная газовая постоянная, равная 8.312 Дж/(моль . К);
Т – абсолютная температура (К);
F – постоянная Фарадея, равная 96500 Кл;
n – число электронов, принимающих участие в электронном
процессе;
[окисл] – концентрация окисленной формы;
[восст] – концентрация восстановленной форм;
(а-, в-) – показатели степени, равные стехиометрическим коэффициентам, в
уравнениях полуреакций.
74.
Если заменить константы их числовыми значениями и перейти от натуральных логарифмов кдесятичным (коэффициент перевода 2,303), то при Т=298К (+25оС) уравнение 11.1 примет вид:
0.059 [окисл.]a
E E
lg
n
[восст.]в
0
Если в уравнение полуреакции входят [Н+] или [ОН-] ионы, то их также следует
включать в уравнение Нернста. Например, для полуреакции
MnO4- + 8H+ + 5e = Mn2+ + 4H2O
потенциал редокс – пары окисления перманганата вычисляется по уравнению
0.059 [ MnO4 ][ H ]8
E E
lg
5
[ Mn 2 ]
0
Рассматривая данное уравнение, можно прийти к следующим выводам:
Изменение отношения концентраций (активностей) окислителя и восстановителя
приводит к изменению значения потенциала системы (Е). В случае, когда
концентрации (активности) всех веществ, участвующих в реакции, равны единице (в
данном случае [MnO4-]=[H+] =[Mn2+] = 1 моль/л), за логарифмическое выражение
также равно единице, а значение логарифма равно нулю (lg 1 = 0). Потенциал
системы в этом случае равен стандартному нормальному окислительно–
восстановительному потенциалу (Е=Ео).
С повышением температуры (Т) потенциал системы (Е) увеличивается, так как
значение RT/nF увеличивается по мере возрастания температуры.
В тех случаях, когда в реакции принимают участие ионы водорода или гидроксила,
потенциал (Е) увеличивается по мере возрастания концентрации (активности) ионов
водорода или гидроксила.
75.
Константа равновесия реакций окисления - восстановленияВозможность изменения направления реакций окисления – восстановления
является следствием обратимости этих реакций. Обратимые реакции, как известно,
приводят к установлению химического равновесия. Константу равновесия нетрудно
рассчитать, зная стандартные потенциалы обеих окислительно –восстановительных
пар. Сделаем такой расчет на примере реакции:
Sn2+ + 2Fe3+ = Sn4+ + 2Fe2+
(11.7)
Исходя из закона действия масс, константа равновесия равна произведению
концентраций продуктов реакции на произведение концентраций исходных веществ:
[ Sn 4 ][ Fe 2 ] 2
K
[ Sn 2 ][ Fe 3 ] 2
(11.8)
Теперь напишем выражения двух потенциалов полуреакций восстановления для
данной окислительно – восстановительной реакции.
Sn4+ + 2e = Sn2+; (Eo = 0.15 в);
0.059 [ Sn 4 ]
E1 0.15
lg
2
[ Sn 2 ]
(11.9)
Fe3+ + 1e = Fe2+; (Eo = 0.77 в);
[ Fe 3 ]
E 2 0.77 0.059 lg
[ Fe 2 ]
(11.10)
76.
. Константа равновесия реакций окисления - восстановленияВозможность изменения направления реакций окисления – восстановления
является следствием обратимости этих реакций. Обратимые реакции, как известно,
приводят к установлению химического равновесия. Константу равновесия нетрудно
рассчитать, зная стандартные потенциалы обеих окислительно –восстановительных
пар. Сделаем такой расчет на примере реакции:
Sn2+ + 2Fe3+ = Sn4+ + 2Fe2+
(11.7)
Исходя из закона действия масс, константа равновесия равна произведению
концентраций продуктов реакции на произведение концентраций исходных веществ:
[ Sn 4 ][ Fe 2 ] 2
K
[ Sn 2 ][ Fe 3 ] 2
(11.8)
Теперь напишем выражения двух потенциалов полуреакций восстановления для
данной окислительно – восстановительной реакции.
Sn4+ + 2e = Sn2+; (Eo = 0.15 в);
0.059 [ Sn 4 ]
E1 0.15
lg
2
[ Sn 2 ]
(11.9)
Fe3+ + 1e = Fe2+; (Eo = 0.77 в);
[ Fe 3 ]
E 2 0.77 0.059 lg
[ Fe 2 ]
(11.10)
77.
Из этих уравнений видно, что по мере увеличения концентраций продуктов реакции(Sn4+- и Fe2+- ионов) и уменьшения концентрации исходных веществ (Fe3+- и Sn2+ионов) в результате течения реакции потенциал первой пары, который был
первоначально меньше, должен постепенно увеличиваться, а потенциал второй пары
– уменьшаться. В конце концов эти потенциалы сравняются, и установится
равновесие.
Е1 = Е2
(11.11)
Подставляя в это равенство их значения (11.8 и 11.9), получим:
0.059 [ Sn 4 ]
[ Fe3 ]
0.15
lg 2 = 0.77 0.059 lg
2
[ Sn ]
[ Fe 2 ]
(11.12)
Второй член этого равенства можно преобразовать, разделив и умножив коэффициент
0.058 на два:
0.059 [ Fe 3 ]2
[ Fe3 ]
0.059
[ Fe3 ]
0.77
lg
0.77 0.059 lg
0.77
2 lg
2 2 (1.13)
2 =
2 =
2
[ Fe ]
2
[ Fe ]
[ Fe ]
78.
Из этих уравнений видно, что по мере увеличения концентраций продуктов реакции(Sn4+- и Fe2+- ионов) и уменьшения концентрации исходных веществ (Fe3+- и Sn2+ионов) в результате течения реакции потенциал первой пары, который был
первоначально меньше, должен постепенно увеличиваться, а потенциал второй пары –
уменьшаться. В конце концов эти потенциалы сравняются, и установится равновесие.
Е1 = Е2
(11.11)
Подставляя в это равенство их значения (11.8 и 11.9), получим:
3
0.059 [ Sn 4 ]
[
Fe
] Второй член равенства можно разделить
0.15
lg
0
.
77
0
.
059
lg
=
2
[ Sn 2 ]
[ Fe 2 ] и умножить коэффициент 0.058 на 2:
[ Fe3 ] =
0.77 0.059 lg
[ Fe 2 ]
0.059
[ Fe3 ]
0.77
2 lg
2
[ Fe 2 ]
=
0.059 [ Fe 3 ] 2
0.77
lg
2
[ Fe 2 ] 2
Подставив в уравнение (11.12) значение уравнения (11.13) получим:
3 2
0.059 [ Sn 4 ]
0
.
059
[
Fe
]
0.15
lg
=
0
.
77
lg
преобразуем уравнение
2 2
2
[ Sn 2 ]
2
[ Fe ]
0.059 [ Fe 3 ]2
0.059 [ Sn 4 ]
lg
–
= 0.77 – 0.15 Коэффициент (0.059/2) можно
lg
2
2
[ Fe 2 ]2
2
[ Sn ]
вынести за скобки:
0.059
2
[ Sn 4 ]
[ Fe 3 ]2
( lg [Sn 2 ] – lg [ Fe 2 ]2 ) = 0.77 – 0.15
79.
Так как выражение, стоящее за знаком логарифма, есть константа равновесия даннойреакции (11.8), то уравнение (11.16) можно записать в виде:
2 (0.77 0.15)
lg K =
≈ 21,
0.059
(11.17)
откуда константа окислительно-восстановительной реакции будет равна
К ≈1021
Найденный результат показывает, что в состоянии равновесия произведение
концентраций продуктов реакции (Sn4+- и Fe2+- ионов) в 1021 раз превышает
произведение концентраций исходных веществ (Fe3+- и Sn2+- ионов).
Другими словами, большое числовое значение константы равновесия
свидетельствует о том, что соответствующая реакция протекает практически до
конца.
Используя приведенные вычисления, можно получить значение константы
равновесия для любого обратимого окислительно – восстановительного процесса
0
0
n( Eок
Е
.
вос. )
lg K
0.059
80.
МЕТОДЫ ОКИСЛИТЕЛЬНО – ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯВсе окислительно-восстановительные методы классифицируются в
зависимости от характера основного титранта, используемого для данного
конкретного случая титрования. Например, метод основанный на окислении
перманганатом, называется перманганатометрией, на окислении иодом –
иодометрией,
Перманганатометрия.
Перманганометрия – один из наиболее часто применяемых методов
окислительно – восстановительного титрования. В качестве титранта используют
раствор перманганата калия, окислительные свойства которого можно
регулировать в зависимости от кислотности среды.
К достоинствам перманганатометрического метода относят:
Возможность титрования раствором KMnO4 в любой среде
Стехиометричность и высокая скорость большинства реакций с участием KMnO4;
Возможность титрования без индикатора;
Доступность перманганата калия.
Наряду с достоинствами метод перманганометрии имеет ряд недостатков:
1.Титрант KMnО4 готовят как вторичный стандарт, поскольку исходный реагент перманганат калия - трудно получить в химически чистом состоянии;
2.Стандартные растворы перманганата не устойчивы, со временем меняют свой
титр, поэтому их необходимо периодически проверять;
3.Реакции с участием KMnO4 возможны в строго определенных условиях (рН,
температура и т.д.);
81.
Задачи, решаемые с использованием перманганометрии.а. Определение восстановителей. Если окислительно –
восстановительная реакция между определяемым восстановителем и перманганат
ионом MnO4– протекает быстро, то титрование проводят прямым способом. Так
определяют оксалаты, нитриты, перекись водорода, железо (II), ферроцианиды и др.
Например:
5H2O2 + 2MnO4– + 6H+ = 5O2 + 2Mn2+ + 8H2O
υ (1/5MnO4–) = υ (1/2H2O2)
5[Fe(CN)6]4– + MnO4– + 8H+ = 5[Fe(CN)6]3– + Mn2+ + 4H2O
υ (1/5MnO4–) = υ (1[Fe(CN)6]4– )
б.Определение окислителей. Добавляют избыток стандартного раствора
восстановителя и затем титруют его остаток раствором KMnO4 (Способ обратного
титрования). Например, хроматы, персульфаты, хлориты, хлораты и другие окислители
можно определить перманганометрическим методом, подействовав сначала избытком
стандартного раствора Fe2+, а затем оттитровав непрореагировавшее количество Fe2+
раствором перманганата калия:
Cr2O72– + 6Fe2+(избыток) +14H+ = 2Cr3+ + 6Fe3+ + 7H2O + Fe2+(остаток)
5Fe2+(остаток) + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O
υ (1/6Cr2O72– ) = υ (Fe2+(избыток)) – υ (1/5MnO4–)
82.
в. Определение веществ, не обладающих окислительно-восстановительнымисвойствами. Определение проводят косвенным способом, титрованием по заместителю.
Для этого определяемый компонент переводят в форму соединения, обладающего
восстановительными или окислительными свойствами, а затем проводят титрование.
Например, ионы кальция, цинка, кадмия, никеля, кобальта осаждают в виде
малорастворимых оксалатов
Мe2+ + С2О42– = ↓МС2О4
Осадок отделяют от раствора, промывают и растворяют в H2SO4:
МeС2О4 + H2SO4 = H2С2О4 + MeSO4
Затем выделившуюся щавелевую кислоту H2С2О4 (заместитель) титруют раствором
KMnO4:
5С2O42– + 2MnO4– + 16H+ = 2Mn2+ + 10CO2 + 8H2O
υ (Мe2+) = υ (1/2С2О42–) = υ (1/5MnO4–)
г. Определение органических соединений. Отличительной особенностью реакций
органических соединений с MnO4– является их малая скорость. Определение возможно,
если использовать косвенный способ. Анализируемое соединение предварительно
обрабатывают избытком крепкого щелочного раствора перманганата и дают
возможность реакции протекать необходимый период времени. Остаток перманганата
титруют раствором оксалата натрия:
C3H5(OH)3+14MnO4–(избыток)+20OH–=3CO32–+14MnO42–+14H2O+MnO4–(ост)
2MnO4–(остаток) + 5C2O42– + 16H+ = 2Mn2+ + 10CO2 + 8H2O
υ (C3H5(OH)3) = υ (1/5MnO4–(избыток)) – υ (1/2C2O42–)
Этим методом можно определить также муравьиную, винную, лимонную,
салициловую и другие кислоты, глицерин, фенол, формальдегид и другие органические
соединения.
83.
На титрование раствора FeSO4 израсходовано 15,00 мл раствора KMnO4,С(1/5MnO4–) = 0,09 M. Определить массу железа в растворе.
Решение.
5Fe2+ + MnO4– +8 H+ ⇄ Fe 3+ + Mn2+ +4H2O, oтсюда: n(Fe2+) = n(1/5MnO4–).
m(Fe2+) = n(1/5KMnO4) ∙ M(FeSO4) = C(1/5KMnO4) ∙ V(KMnO4) ∙ M(FeSO4) =
15,00 ∙10–30,09 ∙55,85 = 0,07540 г.
84.
Иодометрия.В основе метода иодометрии лежат реакции, в которых молекулы
свободного иода при взаимодействии с восстановителями, отнимая у них
электроны, превращаются в отрицательно заряженные иодид-ионы. Или
реакции, в которых иодид-ионы при взаимодействии с окислителями,
отдавая им электроны, превращаются в молекулы свободного иода:
I2 + 2e- = 2I– или 2I- - 2e- = I2
Свободный иод даже в самых незначительных концентрациях с
раствором крахмала дает синюю окраску, тогда как иодид-ион с крахмалом
не дает окраски. На этом основано применение в иодометрии крахмального
раствора в качестве индикатора.
Стандартный потенциал пары I2/2I– сравнительно невелик, он равен
Ео = + 0.54 в. Отсюда возникает возможность двоякого использования
окислительно-восстановительных свойств пары I2/2I– в титриметрическом
анализе:
1) определения восстановителей окислением их раствором иода (I2)
2) определения окислителей восстановлением их раствором иодида (I–).
85.
Задачи, решаемые с использованием метод иодометрии.а. Определение восстановителей. Если на раствор тиосульфата натрия подействовать
свободным иодом, то происходит реакция:
2S2O32- + I2= S4O62- + 2IИз ионного уравнения реакции видно, что два тиосульфат – иона (2S2O32-)
превращаются в один тетратионат – ион (S4O62-).
2S2O32- – 2е = S4O62Следует запомнить, что фактор эквивалентности тиосульфата равен единице, так как
2 электрона равноценны двум тиосульфат-ионам согласно данной полуреакции
окисления.
При титровании раствора Na2S2O3 раствором иода присущая свободному иоду
темно – бурая окраска моментально исчезает из–за реакции восстановления
I2 + 2e = 2IКогда же весь Na2S2O3 будет окислен, одна лишь капля раствора свободного иода
окрасит титруемую жидкость в бледно-желтый цвет. Однако окраска иода,
получающаяся в конце титрования, слаба, и это затрудняет фиксирование точки
эквивалентности. Поэтому в качестве индикатора на иод применяют крахмал,
который образует с иодом комплексные соединения темно-синего цвета. При
титровании в присутствии крахмала конец реакции определяют по появлению синей
окраски.
86.
Можно также титровать раствор иода тиосульфатом до обесцвечиваниясинего раствора в точке эквивалентности от одной капли тиосульфата. В
этом случае раствор крахмала прибавляют в самом конце титрования,
когда свободного иода останется очень мало, и изменение окраски
титруемого раствора будет от синего до бледно – желтого.
Зная молярную концентрацию эквивалента раствора иода и
затраченные на титрование объемы растворов иода и тиосульфата, можно
найти нормальность и титр тиосульфата. Наоборот, по известной
нормальности раствора тиосульфата Na2S2O3 можно подсчитать
нормальность и титр иода.
υ(1/2I2) = υ (S2O32–)
Аналогично определяют ряд других восстановителей, например:
SO32- + I2 + H2O = SO42- + 2I- + 2H+
H2S + I2 = ↓S + 2I- + 2H+
Sn2+ + I2 = Sn4+ + 2I-
87.
б) Определение окислителей. Определение окислителей основано нареакции восстановления их ионами I– (например, иодидом калия). Однако
трудность фиксации точки эквивалентности усложняет подобное
титрование. Действительно, при титровании окислителя, например,
дихромата калия раствором KI:
Cr2O72- + 6I- + 14H+ = 3I2 + 2 Cr3+ + 7H2O
Конец реакции характеризовался бы прекращением образования свободного
иода. Но этот момент заметить нельзя, так как первые же молекулы иода
(при наличии крахмала) окрашивают раствор в темно- синий цвет.
Поэтому применяют косвенный метод – метод замещения. К смеси
растворов иодида калия и кислоты (взятых в избытке) прибавляют точно
отмеренный объем раствора определяемого окислителя (например,
K2Cr2O7). Выделившийся иод оттитровывают тиосульфатом. Количество
вещества эквивалентов тиосульфата равно количеству вещества
эквивалентов иода, а последнее – количеству вещества эквивалентов
окислителя (K2Cr2O7). Таким образом, хотя при данном определении
тиосульфат и дихромат ионы непосредственно друг с другом не реагируют,
тем не менее их количества эквивалентны, и оба они эквивалентны
количеству иода:
υ(1/6Сr2O72-) = υ(1/2I2) = υ (S2O32–)
88.
Иодометрически можно определять многие окислители, способные окислятьI– до I2. Таковы, например, Cl2, Br2, KMnO4, KClO3, белильная известь CaOCl2,
нитриты, перекись водорода, соли железа(III), соли меди(II) и др. Приведем
уравнения этих реакций:
Br2 + 2I– = 2Br– + I2
2MnO4– + 10I– + 16H+ = 5I2 + 2Mn2+ + 8H2O
ClO3– + 6I– + 6H+ = 3I2 + Cl– + 3H2O
2NO2 + 2I– + 4H+ = I2 + 2NO↑ + 2H2O
H2O2 + 2I– + 2H+ = I2 + 2H2O
2Fe3+ + 2I– + 6H+ = 3I2 + 3H2O + Fe2+
в) Определение кислот. Иодометрическим методом определяют также
кислоты:
IO3– + 5I– + 6H+ = 3I2 + 3H2O
Как видно из уравнения, при реакции расходуются Н+ - ионы определяемой
кислоты и выделяется эквивалентное количество свободного иода.
Выделившийся иод оттитровывают тиосульфатом и по затраченному объему и
нормальности его раствора вычисляют нормальность и титр соответствующей
кислоты.
υ(Н+) = υ(1/2I2) = υ(S2O32–)
89.
г) Определение органических веществ проводят, как правило, способом обратноготитрования; добавляют избыток стандартного раствора иода к пробе, после того как
реакция между иодом и органическим соединением пройдет до конца (окисление
проводят в щелочной среде), непрореагировавший иод титруют стандартным
раствором тиосульфата, создав слабокислую среду. Так определяют, например,
формальдегид и спирты:
RCOH + I2(избыток) + 3OH– = RCOO– + 2I– + 2H2O + I2(остаток)
I2(остаток) + 2S2O32- = S4O62- + 2Iυ(RCOH) = υ(1/2I2 избыток) – υ(S2O32–)
Иодометрический метод титриметрического анализа имеет широкое применение.
Важным преимуществом его является большая точность, связанная с высокой
чувствительностью применяемого индикатора – раствора крахмала. Наименьшая
концентрация свободного иода, которую можно обнаружить с помощью иод –
крахмальной реакцией, составляет при комнатной температуре от 2.10-5 до 1.10-6 моль
эквивалентов при условии, что в растворе присутствует немного (хотя бы 0.001 мольэквивалентов) I- – ионов. При отсутствии их реакция менее чувствительна.
Следует помнить, что кристаллический иод трудно растворим в воде, но хорошо
растворяется в концентрированных растворах KI.
90.
ДихроматометрияСущность метода: окисление восстановителей.
Основное уравнение метода:
Сr2O7 2– + 14H+ 6e ⇄ 2Cr 3+ +7H2O, E0Cr O2–ΤCr3+ = 1,33 B, f
2 7
(Сr2O7 2–) = 1/6.
Индикатор – дифениламин, в точке
эквивалентности окрашивается в сине-фиолетовый цвет.
Применяется дихроматометрия для решения тех же
задач, что и перманганатометрия.
91.
РЕШЕНИЕ ТИПОВЫХ ЗАДАЧ ПО ТЕМЕ «ОКИСЛИТЕЛЬНОВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ»Задача 1. Определить молярную массу эквивалента окислителя и восстановителя в
реакции 5НАsО2 + 2МnО4- + 6Н+ + 2Н2О → 5Н3АsО4 + 2Мn2+
Решение.
Запишем уравнения полуреакций, соответствующих превращениям окислителя
МnО4- + 8Н+ + 5е- → Мn2+ + 4Н2О и восстановителя НаsО2 + 2Н2О → Н3АsО4 + 2Н+ + 2еКак можно видеть из уравнения, перманганат-ион восстанавливается до Мn2+, принимая
пять электронов. Следовательно, одному электрону соответствует частица 1/5 МnО4-, т.е.
fэ(МnО4-) = 1/5. Молярная масса эквивалента окислителя равна
М(1/5МnО4-) =М(MnO4-)/5 = 118,936/5 = 23,787 г/моль.
Аналогично из уравнения следует, что одному электрону соответствует частица ½ НАsО2,
т.е. fэ(НАsО2) = ½. Молярная масса эквивалента восстановителя равна
М(1/2 НAsО2) =М(HАsO2)/2 =107,928/2 = 53,964 г/моль.
Ответ: 23,787 г/моль; 53,964 г/моль
Задача 2. Рассчитать окислительно-восстановительный потенциал в процессе титрования
Fе2+ раствором КMnО4, если взято 50 мл 0,1 н. раствора соли Fе2+ и добавлено 49 мл 0,1 н.
раствора КМnО4, [H+]= 1 моль/л.
Решение.
Находим стандартные потенциалы:
= 1,51 В;
= 0,77 В.
Запишем уравнение реакции 5Fe2+ + 8Н+ + МnО4- → 5Fe3+ + Мn2+ + 4Н2О
Рассматриваемый момент титрования предшествует точке эквивалентности, поэтому
электродный потенциал рассчитаем, используя уравнение Нернста для полуреакции Fe2+ e → Fe3+. Найдем концентрацию Fe2+, условно считая, что объем раствора при титровании
не изменяется:
[Fe2+] = (50∙0,1 – 49∙0,1)/1000 = 1∙10-4 моль/л.
Концентрация Fe3+, образовавшегося, будет равна [Fe2+]= 49×0,1/1000= 4,9×10-3 моль/л.
Рассчитываем значение потенциала в данной точке титрования
Е = 0,77 +0,059/1∙lg(4,9∙10-3/1∙10-4)=0,87 В.
Ответ: Е = 0,87 В.
92.
93.
Задачи на 23.11.К 25 мл раствора KMnO4 с титром по О2, равным 0,0008112 г/мл, прибавили KI и
кислоту, полученный раствор оттитровали тиосульфатом, V = 21,14 мл. Определите титр
тиосульфата по иоду.
Из 0,8842 г оксалата аммония (NH4)2C2O4∙ H2O) приготовили 250 мл раствора.
Рассчитайте молярную концентрацию эквивалентоа оксалата аммония, его титр, титр по
перманганату калия
5,0000 г пероксида водорода разбавили до 500 мл в мерной колбе. На титрование 25,00
мл этого раствора расходуется 37,43 мл раствора перманганата калия. Определить степень
чистоты препарата, если молярная концентрация эквивалента раствора перманганата равна
0,1124 M.
0,0921 г хромового ангидрида растворили, обработали раствором иодида калия в кислой
среде. Выделившийся иод оттитровали 23,75 мл раствора тиосульфата натрия, титр
которого по иоду равен 0,01354 г/мл. Определите массовую долю оксида хрома в образце.
Сколько граммов хлороводорода содержится в 200 мл соляной кислоты, если на
титрование иода, выделившегося из 20 мл ее при иодометрическом определении
израсходовано 18,25 мл тиосульфата натрия с концентрацией 0,01965 M.
. К смеси, состоящей из 9,73 мл 0,1096 М раствора иода и 10,70 мл 0,1015 М раствора
тиосульфата натрия, прибавили каплю раствора крахмала. Каков цвет раствора?
94.
РЕШЕНИЕ ТИПОВЫХ ЗАДАЧ ПО ТЕМЕ «ОКИСЛИТЕЛЬНОВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ»Задача 1. Определить молярную массу эквивалента окислителя и восстановителя в
реакции
5НАsО2 + 2МnО4- + 6Н+ + 2Н2О → 5Н3АsО4 + 2Мn2+
Решение.
Запишем уравнения полуреакций, соответствующих превращениям окислителя
МnО4- + 8Н+ + 5е- → Мn2+ + 4Н2О и восстановителя НаsО2 + 2Н2О → Н3АsО4 + 2Н+ + 2еКак можно видеть из уравнения, перманганат-ион восстанавливается до Мn2+, принимая
пять электронов. Следовательно, одному электрону соответствует частица 1/5 МnО4-, т.е.
fэ(МnО4-) = 1/5. Молярная масса эквивалента окислителя равна
М(1/5МnО4-) =М(MnO4-)/5 = 118,936/5 = 23,787 г/моль.
Аналогично из уравнения следует, что одному электрону соответствует частица ½ НАsО2,
т.е. fэ(НАsО2) = ½. Молярная масса эквивалента восстановителя равна
М(1/2 НAsО2) =М(HАsO2)/2 =107,928/2 = 53,964 г/моль.
Ответ: 23,787 г/моль; 53,964 г/моль
Задача 2. Рассчитать окислительно-восстановительный потенциал в процессе титрования
Fе2+ раствором КMnО4, если взято 50 мл 0,1 н. раствора соли Fе2+ и добавлено 49 мл 0,1 н.
раствора КМnО4, [H+]= 1 моль/л.
Решение.
Находим стандартные потенциалы:
= 1,51 В;
= 0,77 В.
Запишем уравнение реакции
5Fe2+ + 8Н+ + МnО4- → 5Fe3+ + Мn2+ + 4Н2О
Рассматриваемый момент титрования предшествует точке эквивалентности, поэтому
электродный потенциал рассчитаем, используя уравнение Нернста для полуреакции Fe2+ e → Fe3+. Найдем концентрацию Fe2+, условно считая, что объем раствора при титровании
не изменяется:
[Fe2+] = (50∙0,1 – 49∙0,1)/1000 = 1∙10-4 моль/л.
Концентрация Fe3+, образовавшегося, будет равна [Fe2+]= 49×0,1/1000= 4,9×10-3 моль/л.
Рассчитываем значение потенциала в данной точке титрования
Е = 0,77 +0,059/1∙lg(4,9∙10-3/1∙10-4)=0,87 В.
Ответ: Е = 0,87 В.
Задача 3. Найти константу равновесия реакции
Н3АsО4 + 2I- + 2H+ → HAsO2 + I2 + 2H2O
Решение.
Найдем значения стандартных потенциалов полуреакций:
H3AsO4 + 2H+ + 2e- → HAsO2 + 2H2O
E0 = 0,56В (рН=0)
I2 + 2e- = 2IЕ0 = 0,54В.
Рассчитаем константу равновесия окислительно-восстановительной реакции:
lgК = 2∙(0,56-0,54)/0,059 = 0,66
К=100,66=4,57.
Рассматриваемая реакция характеризуется сравнительно небольшой константой
равновесия, поэтому не может быть использована в аналитической практике при рН=0.
Ответ: К=4,57
Задача 4. Какая масса (г) пероксида водорода содержится в пробе, если при титровании
израсходовано 14,50 мл перманганата калия с Т(КMnО4/Fe) = 0,08376 г/мл ?
Решение.
При прямом титровании число молей эквивалента определяемого вещества (пероксида
водорода) равно числу молей эквивалента титранта (перманганата калия). При
перманганатометрическом титровании пероксида водорода протекает реакция
95.
КОМЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕОбразовавшиеся координационные соединения могут быть положительно
заряженными, нейтральными или отрицательно заряженными. Так, например, медь(II)
с координационным числом равным четырем может образовывать положительно
заряженный катионный аммиачный комплекс Cu(NH3)42+, нейтральный глициновый
комплекс Cu(NH2CH2COO)2 и отрицательно заряженный анионный комплекс с
хлорид-ионами CuCl42-.
Равновесие в реакциях комплексообразования характеризуется константой
устойчивости К (уст.), или константой нестойкость К (нест.).
К ( нест.)
1
К ( уст.)
Поскольку образование и распад комплексов в растворе происходят ступенчато,
каждой ступени отвечает определенная величина константы устойчивости
(образования) характеризующиеся частными константами.
96.
Частные константы устойчивости, например, аммиаката меди(II), можно записать:2+
Cu
2+
+
NH3 = [Cu(NH3) ];
2+
2+
[Cu ( NH 3 ) 2 ]
K1
[Cu 2 ][ NH 3 ]
[Cu ( NH 3 ) 22 ]
[Cu(NH3) ] + NH3 = [Cu(NH3)2 ];
K2
[Cu(NH3)22+] + NH3 = [Cu(NH3)32+];
[Cu ( NH 3 ) 32 ]
K3
[Cu ( NH 3 ) 22 ][ NH 3 ]
[Cu ( NH 3 ) 2 ][ NH 3 ]
K1, K2, K3 – частные константы образования отдельных ступеней реакций
комплексообразования. Легко показать, что общая константа устойчивости К уст.
комплекса равна произведению частных констант К1, К2, К3, отвечающих отдельным
стадиям комплексообразования. Это общая константа устойчивости, приводимая в
справочниках в виде логарифмов, которая обозначается К 1,2 = К1.К2; К1,2,3 = К1.К2.К3.
Общие константы образования аммиаката меди (К 1; К1,2; К1,2,3) можно выразить,
исходя из уравнений реакций:
2+
Cu
2+
Cu
2+
Cu
+
NH3 = [Cu(NH3) ];
[Cu ( NH 3 ) 2 ]
K1
[Cu 2 ][ NH 3 ]
2+
[Cu ( NH 3 ) 22 ]
K 1, 2
[Cu 2 ][ NH 3 ] 2
2+
[Cu ( NH 3 ) 32 ]
К 1, 2,3
[Cu 2 ][ NH 3 ]3
2+
+ 2NH3 = [Cu(NH3)2 ];
+ 3NH3 = [Cu(NH3)3 ];
Из сказанного выше следует, что логарифмы констант отдельных ступеней
комплексообразования (т.е. частные константы устойчивости), можно найти по
разностям:
lgK2 = lgK1,2 – lgK1; lgK3 = lgK1,2,3 – lgK1,2
97.
Важной характеристикой комплексного соединения является координационноечисло (к.ч.), показывающее число атомов (ионов) или атомных группировок
(лиганд), непосредственно связанных с центральным атомом. Наиболее часто
встречаются координационные числа шесть и четыре. Лиганды характеризуются
дентатностью (от лат. dentatus – зубчатый), т.е. способностью занимать
определенное число координационных мест около центрального атома. Моно –
или однодентатные лиганды занимают одно координационное место, например:
(ОН-, F-, NH3 и др.), би – или двудентатные – два (этилендиамин, С2О42- и др.).
Полидентатные лиганды при реакции с ионами металлов обычно образуют
координационные соединения, содержащие цикл – замкнутую группировку
атомов. Например, в результате реакции
Cu2+ + NH2CH2COO- = CuNH2CH2COO+
образующееся соединение содержит один пятичленный цикл. Нередко в
молекуле координационного соединения насчитывается два, три и большее
число циклов. Координационные соединения с одним или несколькими циклами
называются хелатными (от англ. сhelate – клешня) или просто хелатами.
Типичными хелатами являются соединения с органическими лигандами
полиаминополикарбоновых кислот, которые называются комплексонами.
98.
Широкоизвестным
представителем
этилендиаминтетрауксусная кислота ЭДТА,
HOOCCH2
комплексонов
является
CH2COOH
N – CH2 – CH2 – N
HOOCCH2
CH2COOH
Двунатриевая соль ЭДТА известна под названием комплексон III или трилон Б.
Молекула ЭДТА, содержит две амино- и четыре карбоксильные группы и,
следовательно, способна образовывать с ионами металла комплексообразователя
максимум шесть связей. Поэтому ЭДТА – гексадентантный лиганд
Наиболее ценным свойствам ЭДТА является его способность реагировать с ионами
металла в отношении 1:1(когда один моль ЭДТА взаимодействует с одним молем
ионом металла-комплексообразователя) независимо от заряда иона металла.
99.
Например, при взаимодействии с магнием (II), железом (III) или торием (IV)ЭДТА образует металлокомплексные соединения хелатного типа, где стрелкой
обозначены координационные связи, сплошной линией - валентные.
Электронные
пары, осуществляющие координационную и валентную связи, различаются по своему
происхождению. Так, электронная пара, отвечающая за координационную связь,
является парой р – электронов атомов азота. Электронные же пары валентных связей
образуются за счет спаривания неспаренных электронов атомов кислорода
карбоксильных групп и электронов, находящихся на соответствующих орбиталях
атома металла-комплексообразователя.
100.
ЭДТА -четырехосновная кислота - H4Y. В ее водном растворе имеются следующиепротолитические реакции:
H4Y ↔ H+ + H3YpK1 =2.00
+
2H3Y ↔ H + H2Y
pK2 =2.76
2+
3H2Y ↔ H + H1Y
pK3 =6.16
3+
4HY ↔ H + Y
pK4 =10.26
Из приведенных значений рКА следует, что ионные равновесия в растворе ЭДТА и
области существования различных ионных форм действительно сильно зависят от рН
раствора. Кроме того, практически при любых значениях рН, в растворе имеется
смесь различных форм ЭДТА, однако с преобладанием одной из них. Например, при
рН = 4 – 5 в растворе преобладает двухзарядный анион H2Y2-. При рН = 7 – 9 –
трехзарядный HY3-, а четырехзарядный анион Y4- в ощутимых количествах имеется
только в сильнощелочном растворе, при рН >10.
Рис. 9. Состав раствора ЭДТА при различных значениях рН
101.
Металлоиндикаторы.Для титрования с помощью ЭДТА и других
хелатообразующих реагентов используют металлоиндикаторы. Как правило, эти
индикаторы представляют собой органические красители, образующие с катионами
металла растворимые в воде окрашенные комплексные соединения, которые менее
устойчивы, чем соединения катионов данного металла с ЭДТА. Поэтому в процессе
титрования с ЭДТА в точке эквивалентности происходит разрушение комплекса
металл-индикатора с образованием более прочного комплекса катиона металла с
ЭДТА, и выделяется индикатор в свободном виде. Так как окраска комплексного
соединения металл-индикатора отличается от окраски свободного индикатора, то
происходит изменение окраски титруемого раствора в точке эквивалентности.
Схематично это можно представить следующим образом:
Kt2+(бесцветный) + H2Ind(окрашен) = KtInd(окрашен в другой цвет) + 2H+
KtInd(окрашен в другой цвет) + H2Y2- = KtY2-(бесцветный) + H2Ind(окрашен)
Чаще всего используют металлоиндикаторы: эриохром черный (С20H13O7N3S-) и
мурексид (C8H6N6O6-). Комплекс металла с эриохромом черным в основном красного
цвета, сам индикатор синего. Изменение окраски индикатора в конечной точке
титрования можно представить следующим образом:
Ме2+ + НInd2-(синий) = МеInd-(красный) + 2H+
МеInd-(красный) + H2Y2- = MeY2-(бесцветный) + HInd-(синий) + H+
Для мурексида:
Me2+ + Ind-(синий) = MeInd+(красный)
MeInd+(красный) + H2Y2- = MeY2-(бесцветный) + Ind-(синий) +2Н+
Таким образом, металл индикатор реагирует на изменение концентрации катиона
аналогично тому, как кислотно-основной индикатор реагирует на изменение рН
титруемого раствора.
102.
Прямое титрование –это титрование при котором ионы металла
комплексообразователя непосредственно взаимодействуют с комплексоном.
Прямое титрование применяют для определения ионов металлов, быстро
реагирующих с ЭДТА, при условии, что существует подходящий индикатор для
определении конечной точки титрования. Сущность метода:
υ(Ме2+) = υ(Н2У2-)
Ме2+ + Н2Ind = МеInd + 2H+
MeInd + H2Y2- = MeY2- + H2Ind
(Изменение окраски металл-индикатора MeInd ↔ H2Ind)
Соединения металла с индикатором должны быть достаточно устойчивыми,
чтобы в растворе не было свободных ионов металла. Однако это соединение должно
быть менее устойчивым по сравнению с комплексным соединением металла с
ЭДТА.
КMeInd ‹ КMeY2Методом прямого комплексонометрического титрования определяют: Cu2+, Cd2+,
Pb2+, Ni2+, Co2+, Fe3+, Zn2+, ThIV, Al3+, Ba2+, Sr2+, Ca2+, Mg2+ и др.
103.
Обратное титрование. Обратное титрование менее удобно, однако к немуприходится прибегать, когда для прямого титрования нельзя подобрать
соответствующий индикатор, или когда катионы металлов очень медленно
взаимодействуют с титрантом. Во всех этих случаях к титруемому раствору
прибавляют избыток ЭДТА, раствор оставляют на некоторое время для достижения
полноты реакции. Затем остаток ЭДТА титруют раствором ионов другого металла,
например, Mg2+, для которого реакция с комплексоном соответствует всем
требованиям комплексонометрического титрования. Сущность метода:
υ(Me2+) = υ(H2Y2-) – υ(Mg2+)
Ме2+ + Н2Y2-(изб.) = МеY2- + Н2Y2-(остаток) + 2H+
Mg2+ + Н2Y2-(остаток) + H2Ind = МgY2- + H2Ind + 2H+
В точке эквивалентности: Mg2+ + H2Ind = МgInd + 2H+
(Изменение окраски металл-индикатора H2Ind ↔ MgInd)
Комплексное соединение определяемого металла с ЭДТА должно быть более
устойчивым, нежели соединение магния с ЭДТА, которое, в свою очередь, должно
быть более устойчивым, чем соединение магния с металл-индикатором
КMeY2- › КMgY2- › КMgInd
104.
Вытеснительное титрование (титрование заместителя). При вытеснительномтитровании в анализируемый раствор вводят избыток ЭДТА в виде комплекса с
магнием или цинком. Если катионы определяемого металла образуют с ЭДТА более
устойчивый комплекс, чем магний или цинк, протекает реакция вытеснения:
МgY2- + Ме2+ = МеY2- + Mg2+
Выделившийся магний затем титруют стандартным раствором ЭДТА. Этот метод
удобен при отсутствии подходящего индикатора для титрования определяемого
катиона. Сущность метода:
υ(Me2+) = υ(H2Y2-) = υ(Mg2+)
МgY2-(избыток) + Ме2+ = МеY2- + Mg2+ + МgY2-(остаток)
Mg2+ + H2Ind = MgInd + 2H+
MgInd + Н2Y2- = MgY2- + H2Ind
Изменение окраски металл-индикатора в точке эквивалентности происходит при
MgInd ↔ H2Ind.
При титровании по заместителю, так же должно выполняться условие:
КMeY2- › КMgY2- › КMgInd
Кислотно-основное титрование. В методе кислотно-основного титрования
избыток ЭДТА добавляют к нейтральному раствору иона металла:
Mе2+ + Н2Y2- = Mе2+ + 2H+
Высвободившиеся ионы водорода титруют стандартным раствором шелочи.
Сущность метода:
υ(Me2+) = υ(H+) = υ(OH-).
105.
106.
7. Исходя из величин произведений растворимости хлорида и бромида серебра,подсчитать разницу между количествами серебра, находящимися в 200 мл насыщенного
раствора той и другой соли. ПР(AgCl)=1.78.10-10. ПР(AgBr)=5.10-13
8. Сколько граммов BaSO4 (ПР =10-10) остается в 200 мл раствора при осаждении ВаСl2
эквивалентным количеством H2SO4? Можно ли считать осаждение в этих .условиях
практически полным?
9. Какoва будет потеря от растворимости BaSO4, если в условиях задачи 10 прибавлением
избытка H2SO4 концентрацию ионов SO42- повысить до 0,001 г-ион/л?
10. Для осаждения серебра из 100 мл раствора, содержащего 0,3398 г AgNO3,
израсходовано 17 мл 0,1 М раствора НС1. Вычислить количество серебра (в молях),
оставшееся не осажденным, и ошибку (в процентах). Ответ: около 3 .10-4М; около 15%.
11. Какова была бы ошибка (в процентах), если бы в условиях задачи 22 употребили при
осаждении вместо 17 мл 21 мл 0,1 М раствора НС1?
Ответ: около 0,08%.
107.
Для приготовления титровавнного растворасерной кислоты взяли 2.9 мл
концентрированной (98%) плотностью 1.84 г/мл и разбавили ее водой до объема 1 л.
Определить титр, молярную и нормальную концентрации, а также титр H2SO4 по
Na2CO3
9. Для установки титра раствора H2SO4 25 мл раствора Na2CO3 с титром
0.0053 г/мл оттитровали 24.5 мл раствора H2SO4 . Вычислить титр раствора H2SO4.
Ответ: Титр серной кислоты равен 0.005003 г/мл.
10.Чему равен титр HCl, если при прибавлении к 20 мл этого раствора
избытка раствора AgNO3 получено 0.2868 г AgCl?
Ответ: 0.003646 г/мл.
11. Как проводят титриметрические определения по способу
пипетирования? По способу отдельных навесок?
12. Сколько граммов H2SO4 содержится в 5 л раствора, если на титрование 25 мл
этого раствора израсходовано 22.5 мл 0.095 молярного
раствора эквивалентов КОН.
Ответ: 20.97 г.
13. Сколько процентов H2C2O4.2H2O содержит данный препарат щавелевой кислоты,
если на титрование навески 0.15 г его, растворенной в произвольном объеме воды,
изрсходовано 25.6 мл раствора КОН с молярной концентрацией эквивалентов равной
0.09
Ответ: 96.97%.
108.
Какова константа ионизации уксусной кислоты, если 0,0035 н. раствор ее имеет рН3,61? От в ет: К = 1,74 • 10-5.
22. Чему равен рН 0,01 н. раствора муравьиной кислоты, если известно, что Кнсоон
= 1,8 • 10-4? О т в е т: р Н 2,87.
23. Чему равен рН смеси, содержащей 0,01 моль СНзСООН и 0,1 моль CH3COONa.
От в е т: р Н 5,76.
26. В каких случаях точка эквивалентности лежит при рН 7При рН > 7? При рН < 7?
27. Рассчитать и построить график титрования 0.1 н. раствора муравьиной кислоты
НСООН 0.1 н. раствором КОН. Подобрать индикатор. Можно ли применить
бромтимоловый синий (рТ=3.0 – 4.6), нейтральный красный (рТ = 6.8 – 8.0)?
28. При каком рН достигается точка эквивалентности титрования 0.1 н.
раствора NH4OH 0.1 н. раствором НСООН ?
Ответ: рН 6.51
30. Чему равна индикаторная ошибка титроания: а) 0.1н. раствора HCl 0.1н.
раствором NaOH с метиловым красным (рТ = 5.5); б) 0.1 н. раствора NaOH 0.1 н.
раствором HCl с индикатором нитрамином (рТ = 12).
Ответ: а) - 0.0064% ; б) +20%
31.Чему равна индикаторная ошибка титроания: а) 0.1н.раствора НСООН 0.1н.
раствором NaOH с тимолфталином (рТ = 10); б) 0.1 н. раствора NН4OH 0.1 н.
раствором HCl с метиловым красным (рТ = 5.5).
Ответ: а) +0.2% ; б) – 0.018%.