











 medicine
medicine biology
biologySimilar presentations:
")









Фенілкетонурія. Класична форма захворювання
1.
ФенілкетонуріяВиконала:
Студентка 2 медичного факультету
15 групи
Остапова Ірина
2.
• Фенілкетонурія -спадкове захворювання,пов'язане з порушенням метаболізму
амінокислот, головним чином фенілаланіну.
Супроводжується накопиченням
фенілаланіну і його токсичних продуктів, що
призводить до важкого ураження ЦНС, що
виявляється, зокрема, у вигляді порушення
розумового розвитку.
• Виділено в самостійну нозологіч• ну форму А. Фелінгом (1934).
3.
• Класична форма захворювання(фенілкетонурія I) пов’язана з дефіцитом
ферменту фенілаланін-4-гідроксилази, що
перетворює фенілаланін (ФА) на тирозин.
Ген ФКУ знаходиться в довгому плечі 12-ї
хромосоми (12q22-24).
• У гені описано більше 250 мутацій. Найча• стішою є мутація R408W (заміна аргініну на
триптофан у 408-му кодоні).
4.
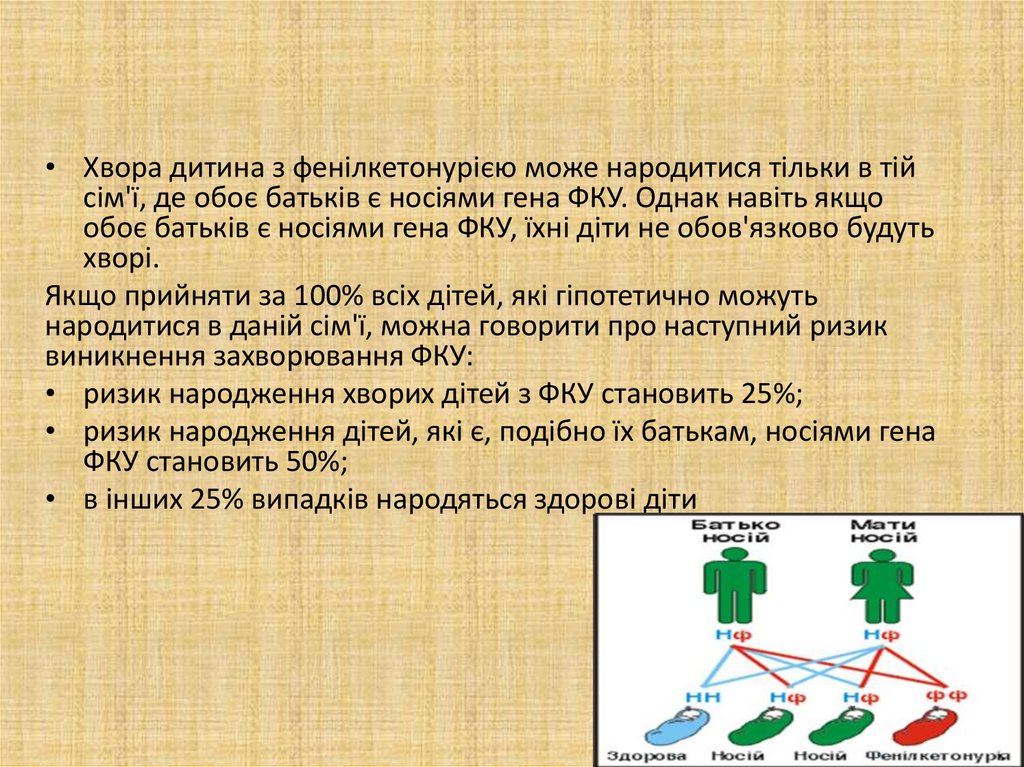
• Хвора дитина з фенілкетонурією може народитися тільки в тійсім'ї, де обоє батьків є носіями гена ФКУ. Однак навіть якщо
обоє батьків є носіями гена ФКУ, їхні діти не обов'язково будуть
хворі.
Якщо прийняти за 100% всіх дітей, які гіпотетично можуть
народитися в даній сім'ї, можна говорити про наступний ризик
виникнення захворювання ФКУ:
• ризик народження хворих дітей з ФКУ становить 25%;
• ризик народження дітей, які є, подібно їх батькам, носіями гена
ФКУ становить 50%;
• в інших 25% випадків народяться здорові діти
5.
Патогенез• 1. Фенілаланін у надлишку накопичується в крові й
екскретується з сечею. У нормі концентрація ФА в крові
= 1–2 мг/дл. У хворих рівень ФА досягає 15 мг/дл і
вище.
• 2. Утворюються інші метаболіти ФА
(фенілпіровиноградна, фенілоцтова, фенілмолочна
кислоти), які є токсичними для ЦНС. Більша частина
фенілоцтової кислоти з’єднується в печінці з глутаміном
і екскретується з сечею у формі фенілацетилглутаміну.
Метаболіти фенілаланіну виділяються з сечею, потом.
Виділення аномального метаболіту — фенілоцтової
кислоти -зумовлює специфічний «мишачий» запах сечі,
тіла і особливо голови дитини.
6.
Патогенез• 3. Зменшення вмісту тирозину спричинює
порушення синтезу меланіну (слабка пігментація
очей, волосся), адреналіну, тироксину.
• 4. Високі концентрації фенілаланіну порушують
метаболізм інших сполук:
• — пригнічується перетворення триптофану на
серотонін, що сприяє наростанню розумової
відсталості;
• — пригнічується перетворення глутатіонової
кислоти на (ГАМК); порушення синтезу ГАМК
призводять до судом;
• — порушується синтез нікотинової кислоти, що
призводить до дерматитів і стійких попрілостей
7.
Види• Фенілкетонурія I типу - класична форма хвороби (98%
випадків). В основі виникнення - дефіцит ферменту
фенілаланін-4-гідроксилази, що забезпечує
перетворення амінокислоти фенілаланіну в тирозин.
• Фенілкетонурія II типу – атипова. Описана I.Smith, 1974р.
Зустрічається у (1-2%) і характеризується дефіцитом
ферменту дігідроптеридинредуктази. Захворювання
успадковується аутосомно-рецесивно. Генний дефект
локалізується в короткому плечі 4 хромосоми, ділянці 4р
15.3.
Фенілкетонурія III типу - дієторезистентна.Описав S.
Kaufman в 1978р.Захворювання успадковується аутосомнорецесивно
і
пов'язано
з
недостатністю
6пірувоілтетрагідроптерін синтетази,
8.
Клініка• У періоді новонародженості дитина виглядає здоровою, перші симптоми
фенілкетонурії частіше з'являються у віці від 2-х до 6-ти місяців. Це виражена
млявість, відсутність інтересу до навколишнього, можливо - підвищена
дратівливість, неспокій, блювота.
• У другому півріччі життя у дитини виражено відставання в психічному
розвитку: менш ніж 10% -це слабка ступінь олігофренії, а приблизно у 60%
дітей, які страждають ФКУ формується ідіотія.
• Зростання у дитини може бути нормальним, а може бути і зниженим.
Відзначається пізніше прорізування зубів, деяке зменшення розмірів черепа,
діти пізно починають сидіти і ходити. У таких дітей своєрідні поза і хода: вони
стоять широко розставивши ноги, зігнуті в колінних і тазостегнових суглобах,
опустивши голову і плечі, ходять дрібними кроками, похитуючись. Сидять
вони підібгавши ноги (положення кравця), це пов'язано з підвищеним
м'язовим тонусом.
• Ці діти зазвичай світловолосі, блакитноокі, шкіра майже повністю
позбавлена пігменту, характерний «мишачий» запах, у частини хворих
бувають епілептичні припадки, але з віком вони проходять. Виражені
пітливість, дермографізм і синюшність кінцівок (акроціаноз), внаслідок цього
підвищена чутливість до сонячних променів і травм. Значно частіше у таких
дітей зустрічається дерматит, важка екзема, артеріальна гіпотонія, схильність
до закрепів.
9.
Діагностика• Для скринінгу у всіх новонароджених на 4–5-й
день життя перед виписуванням з пологового
будинку проводять взяття крові на спеціальні
бланки хроматографічного паперу. Плями
крові висушуються і потім пересилаються в
спеціальну лабораторію.
• У разі отримання результату аналізу з рівнем
фенілаланіну в крові вище за допустимий (2
мг/дл) необхідний терміновий виклик дитини
в генетичну лабораторію для повторного
обстеження і проведення медико-генетичної
консультації сім’ї.
10.
• З другого місяця життя дитини стає позитивноюпроба з трихлорним залізом (реакція Фелінга): до 3
мл сечі додають 1 краплю 1 М розчину HCl,
струшують і додають кілька крапель 10%-го розчину
FeCl3. Якщо в сечі міститься фенілпіровиноградна
кислота, з’являється стійке забарвлення зеленого
кольору. Фенілпіровиноградна кислота в сечі
визначається тільки після того, як рівень ФА в крові
перевищує 10–15 мг/дл, тому тест стає
інформативним тільки з другого місяця життя.
• Для верифікації діагнозу можна використовувати
кількісні високотехнологічні методи —кількісну
рідинну хроматографію, амінокислотний
аміноаналізатор, молекулярно-генетичні методи.
11.
Лікування• Лікування проводиться у вигляді суворої дієти від
виявлення захворювання як мінімум до статевого
дозрівання .
• Дієта виключає м'ясні, рибні, молочні продукти та інші
продукти, що містять тваринний і, частково, рослинний
білок. Дефіцит білка заповнюється амінокислотними
сумішами без фенілаланіну.
• роводиться під контролем рівня фенілаланіну.
• Хворі потребують додаткового введення вітамінів,
мінеральних речовин, мікроелементів. У разі потреби
проводиться симптоматичне лікування ноотропними
засобами, антиконвульсантами,препаратами з
нейромедіаторною дією та ін. Необхідні курси масажу і
лікувальної фізкультури
12.
• Деякі (м'які) форми захворювання піддаються лікуваннюкофактором (тетрагідробіоптерином) ураженого ферменту
(фенілаланінгідроксилази).
• Розробляються нові підходи до лікування фенілкетонурії використання замісної терапії фенілаланінлиазою - рослинним
ферментом, який перетворює фенілаланін в нешкідливі
метаболіти, і генотерапія на основі введення в організм
вірусного вектора, що містить ген фенілаланінгідроксилази. Ці
методи поки не вийшли зі стін лабораторій.
• Атипові форми не піддаються дієтотерапії і лікуються тільки
введенням препаратів тетрагідробіоптерину або його
синтетичних аналогів (сапроптерин).