




















 medicine
medicine informatics
informaticsSimilar presentations:



")

")




Компьютерное моделирование в медицине. Лекция 10. Определение филогенетического расстояния
1.
Федеральное государственное бюджетное образовательное учреждение высшего образования"Красноярский государственный медицинский университет имени профессора В.Ф. Войно-Ясенецкого"
Министерства здравоохранения Российской Федерации
Кафедра медицинской кибернетики и информатики
Дисциплина:
КОМПЬЮТЕРНОЕ МОДЕЛИРОВАНИЕ В МЕДИЦИНЕ
Лекция 10
Шадрин Константин Викторович
2.
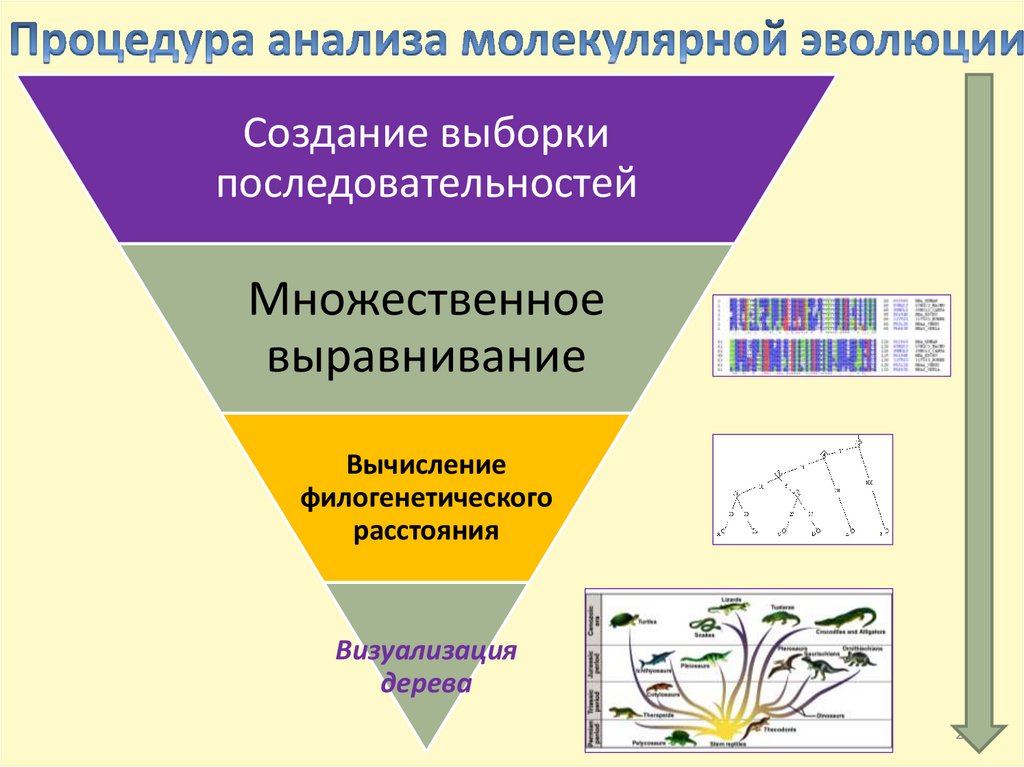
Создание выборкипоследовательностей
Множественное
выравнивание
Вычисление
филогенетического
расстояния
Визуализация
дерева
2
3.
1. Глобальное выравнивание(последовательности одинаковой длины)
2. Локальное выравнивание
3
4.
Поскольку в настоящее времярасшифровывать структуру ДНК
достаточно просто,
возникает вопрос:
можем ли мы реконструировать
эволюционные отношения
между несколькими современными видами
путем сравнения последовательностей ДНК
или вариантов определенного гена?
4
5.
1. Вероятность замены нуклеотида.2. События и условная вероятность.
3. Матричные модели замены нуклеотидов (ДжуксаКантора и Кимуры).
4. Филогенетическое расстояние.
5. Вычисление
филогенетического
расстояния
методами Джукса-Кантора и Кимуры.
5
6.
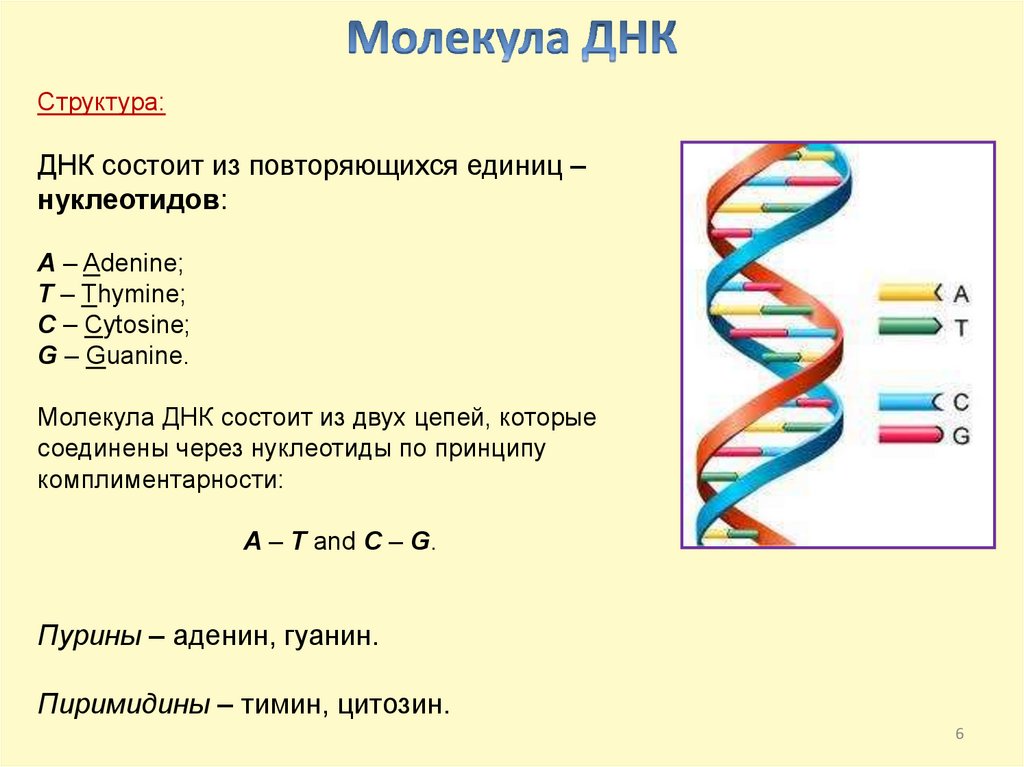
Структура:ДНК состоит из повторяющихся единиц –
нуклеотидов:
A – Adenine;
T – Thymine;
C – Cytosine;
G – Guanine.
Молекула ДНК состоит из двух цепей, которые
соединены через нуклеотиды по принципу
комплиментарности:
A – T and C – G.
Пурины – аденин, гуанин.
Пиримидины – тимин, цитозин.
6
7.
• Простая замена одного основания наопределенном месте в последовательности.
другое
в
• Если AATCGC (последовательность предка) становится
у потомка AATGGC, это значит, что
у потомка в
четвертой позиции произошла замена С → G.
Транзиция – замена основания, при которой происходит
замена одного пурина на другой пурин или пиримидина на
пиримидин. Такие замены наблюдаются гораздо чаще, чем
трансверсия.
Трансверсия – замена основания, при которой
происходит замена пурина на пиримидин или наоборот.
7
8.
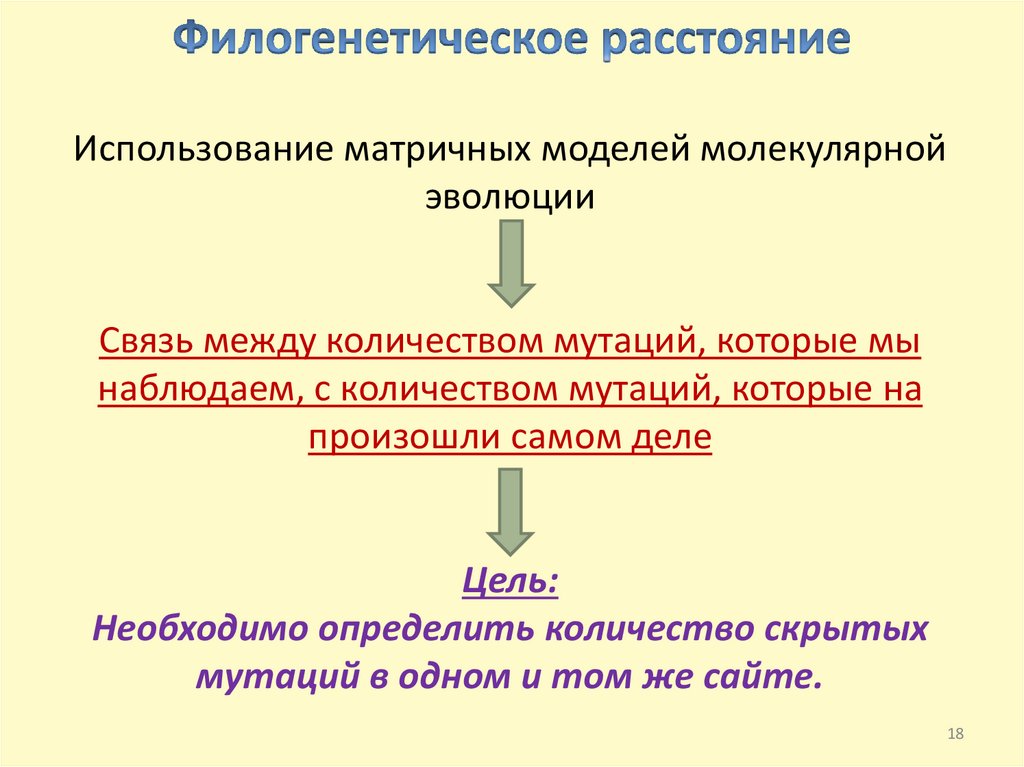
Цель: определение количества мутаций, которые произошли входе эволюционного образования ДНК-последовательностей.
Пусть, например мы знаем, что потомок S2 произошел от
промежуточного вида S1, который в свою очередь произошел от
вида S0.
S0: ACCTGCGCTА…
S1: ACGTGCACTА…
S2: ACGTGCGCTА…
• Настоящая мутация была скрыта, так как произошла
обратная мутация, в результате чего окончательное
основание стало таким же, как и было первоначально.
• Сравнивая S0 с S1, а затем S1 с S2 можно обнаружить три
3
мутации среди первых 10 сайтов, т.е. , что уже не так мало.
10
КАК НАЙТИ ЧИСЛО СКРЫТЫХ МУТАЦИЙ???
8