































 medicine
medicineSimilar presentations:
")









Гепатолієнальний синдром у педіатрії. Сучасні підходи до діагностики, лікуваня мукополісахаридозів та іншіх орфанних захворювань
1.
ГЕПАТОЛІЄНАЛЬНИЙ СИНДРОМ УПРАКТИЦІ ПЕДІАТРА.
СУЧАСНІ ПІДХОДИ ДО ДІАГНОСТИКИ ТА ЛІКУВАНЯ
МУКОПОЛІСАХАРИДОЗІВ ТА ІНШІХ ОРФАННИХ
ЗАХВОРЮВАНЬ
К.М.Н. ДОЦЕНТ
ФОМЕНКО Н.М.
2.
ПОНЯТТЯГЕПАТОЛІЕНАЛЬНОГО
СИНДРОМУ(ГЛС)
ГЛС – це симптомокомплекс, який характеризується
одночасним збільшенням печінки та селезінки по
відношенню до вікової норми.
Чому збільшуються відразу 2 абсолютно різних за
структурою та функціями органи?
Розміщення «на одному поверсі» у черевній порожнині
Спільність іннервації (симпатичної і парасимпатичної)
Спільність венозного (система ворітної вени) та
лімфовідтіку
Належать до єдиної лімфо-макрофагоцитарної системи
3.
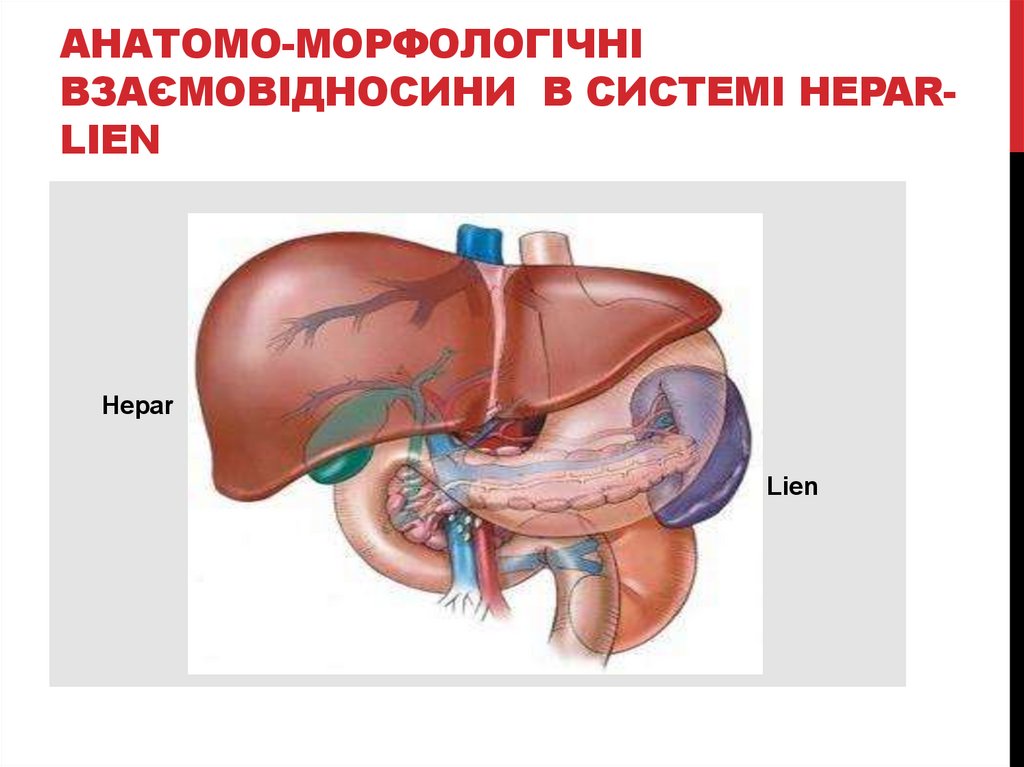
АНАТОМО-МОРФОЛОГІЧНІВЗАЄМОВІДНОСИНИ В СИСТЕМІ HEPARLIEN
Hepar
Lien
4.
ГЛС - завжди симтомокомлекс, а не конкретнанозологічна форма
ГЛС зустрічається при:
- вроджених і набутих дефектах судин портальної системи;
- Спадкових орфанних захворюваннях з порушенням обміну речовин:
Мукополісахаридози, глікогенози, галактоземія, нейроліпідози,
гепатоцеребральна дегенерація, тирозиноз та інші порушення АМК-обміну,
дефіциті альфа-1 антитрипсину, гемохроматоз тощо)
- вродженому гіпотиреозі
- тяжкому рахіті, анемії, тромбоцитопенічній пурпурі
- лімфопроліферативних захворюваннях системи крові;
- мієлопроліферативних захворюваннях системи крові;
- ревматичних захворюваннях;
- патології серцево-судинної системи(вроджені і набуті вади серця з
правошлуночковою недостатністю;
- інфекційних, паразитарних захворюваннях (гепатити, TORCH- інфекції,
СНІД, ехінококоз тощо)
N.B В цілому у 90% випадків ГЛС зумовлюється патологією печінки і у
10% патологією селезінки. Переважання спленомегалії над
гепатомегалією більше характерно для гематологічних захворювань
5.
СКАРГИ ( НЕСПЕЦІФІЧНІ)болі в правому підребер'ї (тупі, ниючі, гострі,
переймоподібні);
- підвищена слабкість, втома, апатія, підвищена збудливість,
порушений сон;
- носові кровотечі;
- жовтяниця;
- шкірні висипання і геморагії;
- знижений апетит, анорексія, нудота, блювота, дисфункція
кишківника.
N.B. Скарги в першу чергу залежать від
причини ГЛС, тобто від основного
захворювання.
6.
АНАМНЕЗ- вік клінічної маніфестації захворювання;
- початок захворювання – поступовий, латентний, гострий;
- перебіг – гострий, тривалий, рецидивуючий, прогредиєнтний
- зв'язок захворювання з перенесеним вірусним гепатитом В, С, Д;
- обтяжений генеалогічний анамнез (повторні випадки в сім”ї);
- прогресуюче відставання у психомоторному розвитку (спадкові
метаболічні захворювання);
- ураження очного та слухового аналізаторів та інших органів і
систем (спадкові метаболічні захворювання);
- зв'язок захворювання з гемотрансфузіями, травмою, операцією;
-
зв'язок гепатомегалії з диспептичними симптомами, гарячкою,
суглобовим синдромом( гепатити, TORCH-інфекціїї, ревматичні
захворювання, СНІД)
-
зв'язок гепатоспленомегалії з геморагічним синдромом, анемією,
лімфоаденопатією тощо (онкогематологічні захворювання)
-
зв'язок гепатоспленомегалії зі шкірними висипаннями,
алергічними захворюваннями (паразитарні захворювання)
7.
ОБЄКТИВНЕОБСТЕЖЕННЯ ХВОРОГО
Ретельний огляд фенотипу!
Надзвичайно важливим є правильне
проведення пальпації, не ігнорувати цей дуже
простий і доступний метод!!!
УЗД органів черевної порожнини
Методи комп”ютерної та МРТ- візуалізаціїїї за
необхідності
Лапароскопічні методи з забором біопсійного
матеріалу тощо
8.
ЛАБОРАТОРНАДІАГНОСТИКА ПРИ ГЛС
При ГЛС підходи до діагностики є специфічними і
залежать від патології, яку запідозрено у кожному
конкретному випадку: при підозренні на гепатит
проводиться:
ІФА, ПЛР на ті чи інші збудники, гемограма, біохімічні маркери, а
саме :
на цитоліз - АЛТ, АСТ, гамма-глютамінтрансфераза;
на холестаз – лужна фосфатаза;
на синтетичну функцію – протеїнограма, коагулограма;
на зв’язування білірубіну – фракції білірубіну;
При підозрені на аутоімунний генез гепатиту визначаються
сироваткові антитіла: гладком'язеві (SMA), антинуклеарні (ANA),
печінково-ниркові мікросомальні (LKM-1), інші.
9.
МАРКЕРИ ГЕПАТИТІВЛабораторна діагностика гепатиту В заснована на виявленні
специфічних для нього антигенів за та відповідних антитіл у крові
тетодом ІФА, а також ДНК збуднику методом ПЛР основними з яких
є:
НВsAg
HBcAg
НВеAg
- анти-НВs (методом ІФА)
- анти-НВс класу Ig М и IgG
- анти-НВе
ДНК вірусу гепатиту В (методом ПЛР)
Основа діагностики гепатиту С – це наявність антитіл до вірусу
гепатиту С (анти-HCV), як правило, визначаються сумарні АТ.
і HCV-РНК (методом ПЛР) . Позитивні результати обох тестів
підтверджують наявність інфекції.
Антитіла (анти-ВГС) знаходять у 70% хворих при появі перших
симптомів захворювання, і у 90% хворих - протягом
трьох місяців.
10.
ГРУПА СПАДКОВИХ ПОРУШЕНЬМЕТАБОЛІЗМУ
Відомо більше 500 нозологічних форм спадкових орфанних
захворювань метаболізму (частота менше 1:2000)
Мукополісахаридози (МПС) – спадкові порушення обміну речовин, при
яких у лізосомах клітин неконтрольовано накопичуються кислі
глікозаміноглікани (ГАГ), внаслідок порушення їх розпаду (деградації).
На сьогодні відомо 7 типів, з врахуванням підтипів раціонально
виділяти 14 різновидів МПС. Всі типи МПС, окрім II типу мають
А/рецесивний тип успадкування
За фенотипом виділяють Гурлеровський фенотип та Моркіо-фенотип.
Характерний «світлий проміжок», коли біохімічні порушення є, але
клінічної маніфестації ще немає
Як правило, клінічна маніфестація наступає наприкінці 1-го року
життя, але частина дітей потрапляє у поле зору неврологів вже на 1-ому
році життя з гідроцефальним синдромом, затримкою психомоторного
розвитку.
11.
За 33 роки в обл. МГЦ були діагностовані різні типи МПС:МПС I-го типу (Гурлер-синдром)- 5 випадків
МПС II-го типу (с-м Хантера)
- 7випадків
МПС III-го типу (с-м Санфіліппо), - 2 випадки
МПС IV-го типу (с-м Моркіо),
- 2 випадки
Неідентифіковані типи МПС
- 7 випадків
До ензимодіагностики було практично неможливо
встановити тип МПС, тому в діагностиці допускались
певні припущення, оскільки вона грунтувалась
переважно на клініко-фенотипових ознаках
захворювання
Всім дітям оформлена інвалідність, значна кількість
хворих повмирала, і тільки в останні роки появилась
можливість патогенетично обгрунтованого лікування
даних хворих
12.
МУКОПОЛІСАХАРИДОЗИКлінічні прояви захворювання характеризуються
мультисистемними ураженнями:
Лицеві дизморфії – грубі риси обличчя: сідловидний ніс;
потовщені ціанотичні губи; макроглосія; рідкі гіпоплазовані
зуби
Рецидивуючі захворювання ВДШ, пневмонії
Деформації скелету – макроцефалія; деформації грудної
клітки та хребта; контрактури суглобів
Гепатолієнальний синдром, як правило
виражений, прогресуючий
Грижі передньої черевної стінки
Психо-інтелектуальна деградація при більшості форм МПС,
крім I-S (c-м Шайє), IV та VI типів
Значно скорочена тривалість життя
13.
ІНШІ ПРОЯВИЗАХВОРЮВАННЯ
• Потовщення та сухість шкіри, гіпертрихоз
• Ураження клапанного апарату серця (міксоматозне
потовщення стулок, стеноз або недостатність
клапанів), розвиток кардіоміопатій
• Значне відставання у рості до нанізму або субнанізму
• Синдром зап”ясного каналу (парастезії,болі та атрофії
у пальцях кистей), формування «кігтистої лапки»
• Помутніння рогівки, дистрофія сітківки
• Приглуховатість
14.
ЛАБОРАТОРНАДІАГНОСТИКА МПС
Визначення сумарної та фракційної
екскреції ГАГ з сечею
Визначення дефіцитного
ферменту(ензимодіагностика) в умовах
метаболічних центрів (Центр орфанних
захворювань ОХМАТДИТ, м. Київ)
Визначення мутантного
гену(молекулярно-генетичний аналіз) у
спеціальних лабораторіях (ДІЛА тощо)
15.
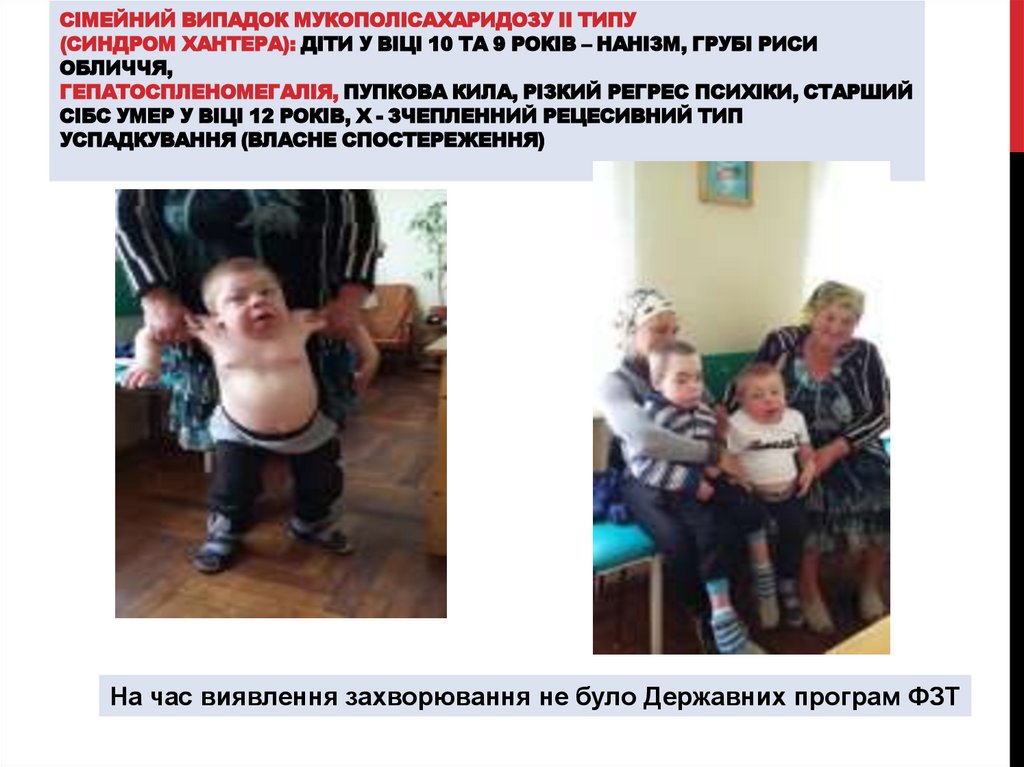
СІМЕЙНИЙ ВИПАДОК МУКОПОЛІСАХАРИДОЗУ ІІ ТИПУ(СИНДРОМ ХАНТЕРА): ДІТИ У ВІЦІ 10 ТА 9 РОКІВ – НАНІЗМ, ГРУБІ РИСИ
ОБЛИЧЧЯ,
ГЕПАТОСПЛЕНОМЕГАЛІЯ, ПУПКОВА КИЛА, РІЗКИЙ РЕГРЕС ПСИХІКИ, СТАРШИЙ
СІБС УМЕР У ВІЦІ 12 РОКІВ, Х - ЗЧЕПЛЕННИЙ РЕЦЕСИВНИЙ ТИП
УСПАДКУВАННЯ (ВЛАСНЕ СПОСТЕРЕЖЕННЯ)
На час виявлення захворювання не було Державних програм ФЗТ
16.
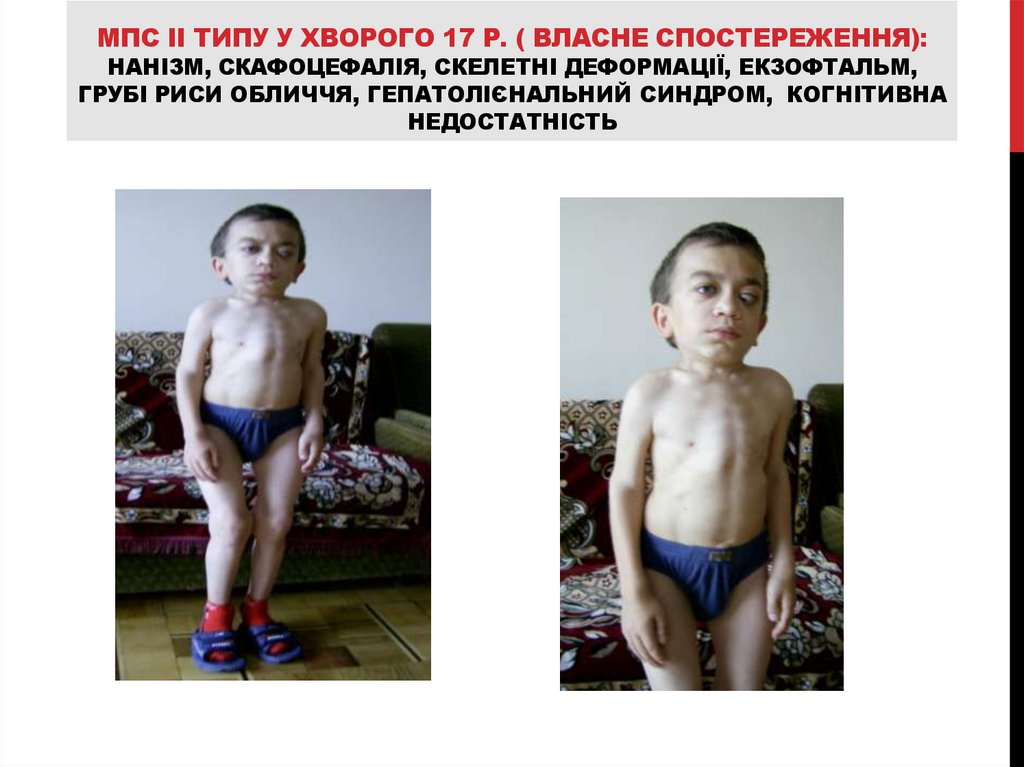
МПС II ТИПУ У ХВОРОГО 17 Р. ( ВЛАСНЕ СПОСТЕРЕЖЕННЯ):НАНІЗМ, СКАФОЦЕФАЛІЯ, СКЕЛЕТНІ ДЕФОРМАЦІЇ, ЕКЗОФТАЛЬМ,
ГРУБІ РИСИ ОБЛИЧЧЯ, ГЕПАТОЛІЄНАЛЬНИЙ СИНДРОМ, КОГНІТИВНА
НЕДОСТАТНІСТЬ
17.
ПРОФІЛЬНЕ ФОТОХВОРОГО
Зрозуміло, чому раніше
це захворювання
називалось
гаргоїлізмом
18.
ПАРАКЛІНІЧНА ДІАГНОСТИКАПри обстеженні у Львівському інституті
спадкової патології НАМНУ виявлено
високий рівень сумарних ГАГ- 718 од ЦПХ
(норма до 176)
Від ензимодіагностики мати відмовилась, у
даний час хворому 25 років
Найбільш вірогідний в даному випадку
II – тип МПС
Хворий отримує симптоматичну терапію
19.
МПС II ТИПУ, ДІАГНОСТОВАНИЙ ДО1,5 РОКУ, 2016Р.
КОМЕНТАР:
Захворювання
діагностовано нами,
генетиками, скерований
пульмонологом у
зв”язку з частими
захворюваннями ВДШ.
Фенотип:
• крупна голова;
• диспропорційність;
• збільшення живота;
• гепатолієнальний с-м
• незначне
відставання у
психомоторному
розвитку.
20.
Діагноз верифіковано за допомогоюензимодіагностики в Центрі орфанних захворювань «
ОХМАТДИТ» м. Києва
Хворий протягом 1,5 року на ферментзамісній терапії
(ФЗТ) рекомбінантним препаратом дефіцитного
ферменту ідуронат-сульфатази – Елапраза
Препарат вводиться в/в повільно через лініамат, тривалість
інфузії 4-5 годин, кратність введення щотижнево, лікування
необхідно проводити пожиттєво
Наявна позитивна динаміка: відзначається
збільшення активності дитини, зменшились
розміри печінки і селезінки, покращились когнітивні
функції.
21.
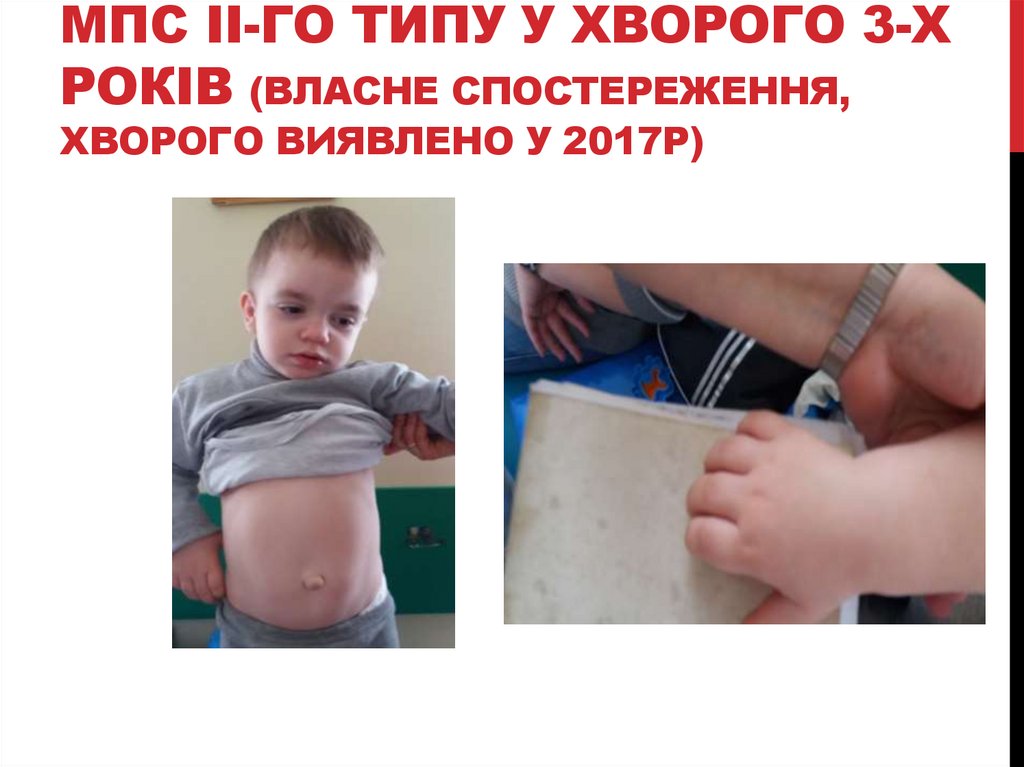
МПС II-ГО ТИПУ У ХВОРОГО 3-ХРОКІВ (ВЛАСНЕ СПОСТЕРЕЖЕННЯ,
ХВОРОГО ВИЯВЛЕНО У 2017Р)
22.
КОМЕНТАР ДО ДАНОГОВИПАДКУ
Хлопчик 3-х років скерований неврологом до генетика у зв’язку з
відставанням у психомоторному розвитку та особливостями
фенотипу.
Уже на клінічному рівні нами практично зі 100% вірогідністю був
встановлений діагноз МПС, гурлеровський фенотип (крупна
голова, грубоваті риси обличчя, контрактури, гепатолієнальний см, пупкова кила, операція по видаленню пахових кил в анамнезі.)
Рівень ТТГ,Т3,Т4 – N;
УЗД щитовидної залози – N
УЗД ОЧП: гепатоспленомегалія
Дитина скерована на ензимодіагностику у Центр орфанних
захворювань М. Києва
23.
ЗА ДАНИМИЕНЗИМОДІАГНОСТИКИ:
Активність ферменту ідуронатсульфатази – 0 при нормі 1882онмоль/мл/24год
Т.ч підтверджено діагноз МПС II- типу
Цікавим виявився генеалогічний анамнез: у матері пробанда був
хворий брат, якому нами був встановлений діагноз МПС, на той час
ензимодіагностика не проводилась. Хворий помер у віці 10років,
тобто наявна класична ситуація щодо Х-зчепленого рецесивного
успадкування. Саме таким чином успадковується II тип МПС
Дитина - пробанд включена у Державну програму замісної
ферментотерапії (ФЗТ): отримує елапразу протягом 2018 року.
На тлі ФЗТ незначне покращення психоємоційних функцій,
зменшення розмірів печінки, покращився функціональний стан
суглобів китиць.
24.
МПС - I ТИП У ДИТИНИВІКОМ 4,5 РОКІВ, ФЗТ
АЛЬДУРАЗИМОМ З 1 РОКУ
Фенотип без
виражених ознак
захворювання.
Відносно задовільний
психоінтелектуальний
розвиток, регрес
гепатоспленомегалії.
25.
В ДАНИЙ ЧАС МИ ВИЯВИЛИ ЩЕ2 ВИПАДRB МПС II ТИПУ
Діагноз підтверджено за допомогою ензимодіагностики та
молекулярно-генетичного аналізу ( МПС-II типу)
Діагноз було встановлено відразу після огляду дитини
генетиком
Але батьки з дитиною скеровані тільки у віці 3р.9міс., що
є великим недоліком, дитина буде включена у Державну
програму ФЗТ.
Т.ч. ФЗТ у нас в області отримують 3 дітей з МПС II типу,
ще 1 дитина запланована на наступний рік та 2 хворих з
МПС I типу
Річна потреба у ФЗТ для хворих у грошовому виразі
складає близько 42 млн. гривень.
26.
ГЕПАТОЛІЕНАЛЬНИЙ СИНДРОМ ПРИ ВРОДЖЕНОМУГІПОТИРЕОЗІ У ДИТИНИ 1 МІС 1 ТИЖДЕНЬ
Захворювання виявлено за
допомогою неонатального скринінгу.
Але батьки з”явились тільки
після повторного активного
виклику:
Наявні грубі риси обличчя;
Макроглосія
Погана прибавка у вазі;
Збільшення і гіпотонія живота;
Пупкова кила;
Затяжна жовтяниця;
!!! Гепатоспленомегалія;
Закрепи
Дитина отримує замісну терапію
L-тироксином, відмічається значне
покращення стану
27.
НЕЙРОЛІПІДОЗИ(НЛ)велика, гетерогенна група СПМ, в основі яких лежить
порушення обміну мембранних ліпідів нейронів (хвороба ТеяСакса, хвороба Німана- Піка, хвороба Гоше
В клініці характерно:
регрес психомоторного розвитку з 4-6 міс, судоми, м’язова
дистонія,
!!! гепатолієнальний синдром,
ураження очного аналізатора (симптом вишневої кісточки на
очному дні),
рання летальність
Лабораторно: в метабол. центрах визначається рівень
дефіцитних ферментів, на окремі НЛ - розроблена
молекулекулярно - генетична діагностика.
28.
ДЕКІЛЬКА СЛІВ ПРО Х-БУ ГОШЕ(ДЕФІЦИТ
ГЛЮКОЦЕРЕБРОЗИДАЗИ).
Виділяють ІІІ типи:
Тип І - хронічна форма без неврологічної
симптоматики
Тип ІІ - гостро злоякісна інфантильна форма з
неврологічними симптомами
Тип ІІІ - підгостра ювенільна форма з
неврологічними симптомами.
29.
Найбільш важким з ранньою маніфестацією є ІІтип, дуже характерна спленомегалія, деколи
колосальних розмірів, печінка збільшується
менше, болі у кістках, кровотечі
Виражена тромбоцитопенія, панцитопенія у
наслідок витиснення всіх паростків крові у
кістковому мозку.
В пунктаті кісткового мозку знаходять клітини
Гоше (великі клітини в цитоплазму яких
«вплетені» шари фібрилл).
Ензимодіагностика: виявляють низький рівень
ферменту глюкоцереброзидази
30.
ЛІКУВАННЯХворі отримують замісну терапію
рекомбінантним препаратом дефіцитного
ферменту - церезим).
В нашій області таку терапію отримує 1
дитина та 1 доросла хвора з 1-м типом
захворювання.
У дитини захворювання діагностовано у віці 4,5
роки, ФЗТ протягом 4-х років отримано дуже
позитивний ефект.
31.
МУКОВІСЦИДОЗНа сьогодні в області 30 дітей і 9 дорослих хворих
Тяжкі рецидивуючі обструктивні бронхіти, пневмонії з ранньою
маніфестацією і розвитком бронхоектазів. Формування легеневого серця.
Необхідність постійного прийому муколітиків, кінезітерапії, А/Б,
Синдром мальабсорбції, необхідність постійної замісної ферментотерапії
(креон) у великих дозах: 6-10 тис од ліпази/кг маси тіла
!!!Ураження печінки за рахунок густої та в’язкої жовчі (холестатичний
гепатит, жировий гепатоз, фіброз, цироз), необхідність прийому
препаратів урсодезоксихолевої к-ти,
Лабораторно:
високий рівень хлоридів поту (більше 60мекв/дл,)
низький рівень панкреатичної еластази калу(менше 200),
молекулярна діагностика на найбільш поширені мутації гену ТРБМ (
відомо біля 2000 мутацій, в Україні проводиться визначення біля 25
мажорних мутацій, на які припадає близько 85% випадків МВ)
Підвищений рівень ЛФ та гамма-глютамінтрансферази;
32.
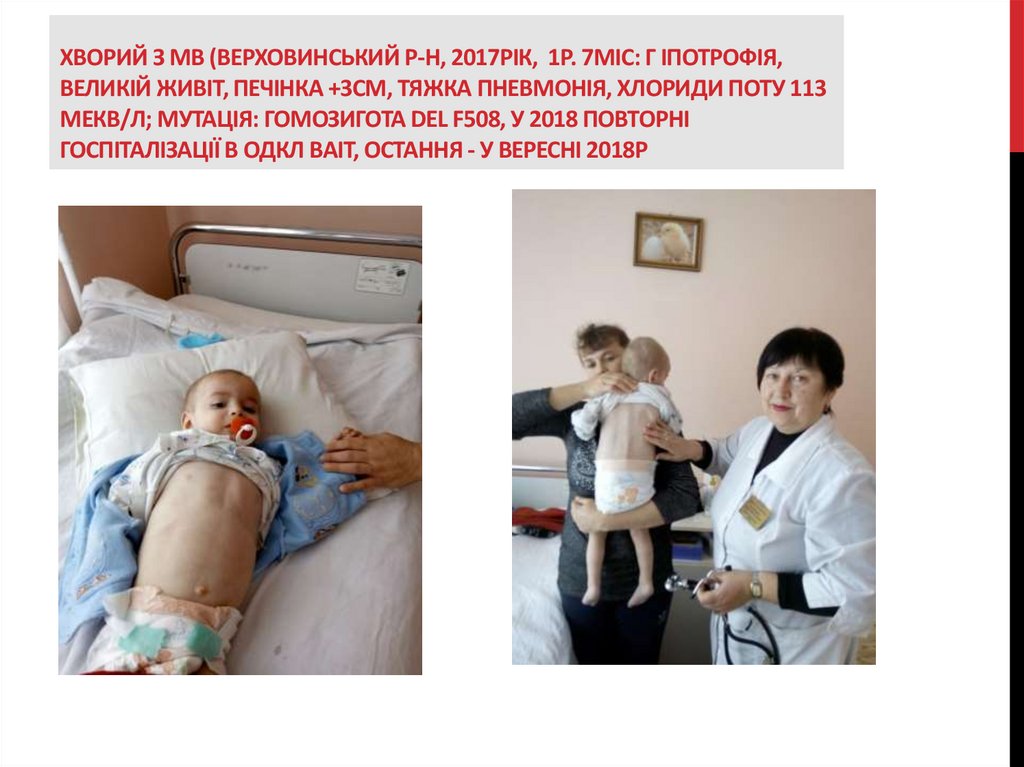
ХВОРИЙ З МВ (ВЕРХОВИНСЬКИЙ Р-Н, 2017РІК, 1Р. 7МІС: Г ІПОТРОФІЯ,ВЕЛИКІЙ ЖИВІТ, ПЕЧІНКА +3СМ, ТЯЖКА ПНЕВМОНІЯ, ХЛОРИДИ ПОТУ 113
МЕКВ/Л; МУТАЦІЯ: ГОМОЗИГОТА DEL F508, У 2018 ПОВТОРНІ
ГОСПІТАЛІЗАЦІЇ В ОДКЛ ВАІТ, ОСТАННЯ - У ВЕРЕСНІ 2018Р
33.
ВИПАДОК МВ У ДИТИНИ У ВІЦІ 1Р 5МІС.(2018Р),
маніфестація у вигляді набряковоанемічного синдрому (лікувалась у
нефрологічному відділенні);
гепатомегалія, великій живіт,
нестійкий стілець;
субнанізм ;
заг білок 38-44 г/л
хлориди поту 114мекв/л
фекальна еластаза 61 (N>200)
гомозигота Del F508 ;
Колонізація Ps. Aeruginosa
!!! Ранній розвиток фіброзу печінки,
не дивлячись на протокольну
терапію.
Дитина проходила стацлікування як
в ОДКЛ м. Івано-Франківська, так і
Західно-Украінському
спеціалізованому дитячому центрі м.
Львова, де вдалось покращити та
стабілізувати стан дитини
34.
ВИПАДОК ПІЗНЬОЇ ДІАГНОСТИКИМВ 2017Р
Нажаль, ми не можемо говорити тільки про
позитивні моменти
У нас у 2017 році був випадок пізнього виявлення
МВ у дитини 10 років, хоча була наявна характерна
клінічна картина. В ОДКЛ встановлено діагноз: МВ з
панкреатичною недостатністю, хронічний
обструктивний бронхіт з формуванням
бронхоектазів, холестатичний гепатит з
розвитком фіброзу(F2). БЕН 2ст( ДВ21%)
Генотип: delF508/2184insA
35.
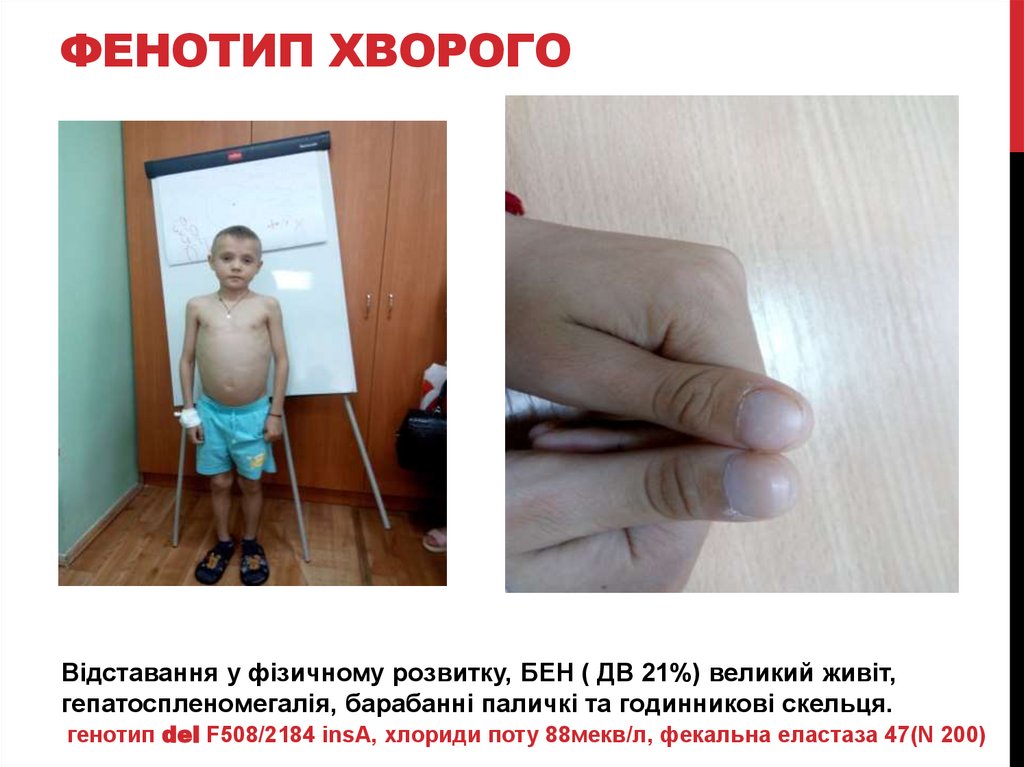
ФЕНОТИП ХВОРОГОВідставання у фізичному розвитку, БЕН ( ДВ 21%) великий живіт,
гепатоспленомегалія, барабанні паличкі та годинникові скельця.
генотип del F508/2184 insA, хлориди поту 88мекв/л, фекальна еластаза 47(N 200)
36.
ОСТАННІ 2 ВИПАДКИ ДІАГНОСТОВАНОГО МВНА ПРИКАРПАТТІ ЗА ДОПОМОГОЮ СКРИНІНГУ
I. Випадок. Захворювання виявлено за допомогою неонатального
скринінгу: Рівень IRT -113(N до 60мекв/л) Фекальна еластаза 12(N200)
I
Діагноз верифіковано за допомогою молекулярнр-генетичного аналізу:
компаунд мутацій del F508/с.1086Т>А
Генеалогічний анамнез: з приводу МВ нами спостерігається двоюрідний
сібс пробанда з аналогічними мутаціями гену ТРБМ (тяжкий перебіг
захворювання)
Дитина консультована в Західно-Украінському спеціалізованому дитячому
центрі.
Отримує протокольну терапію: стан відносно задовільний, прибавка у вазі
за 2 міс - 2,3 кг
У віці 3,5 міс лікувався в стаціонарі з приводу пневмонії (хлориди поту
120мекв/л)
II. Випадок. Двічи високий рівень IRT 88 і 109 мекв/л, хлориди поту
78мекв/л, фекальна еластаза 40 (N200)
Клінічно:затяжна жовтяниця, печінка+2,5см прибавка у вазі за 1місяц
700г. Отже, без скринінгу поставити діагноз досить складно.
Взято кров на молекулярно- генетичний аналіз на мутації гену ТРБМ( в
роботі)
37.
ЗАКЛЮЧЕННЯГепатолієнальний синдром не є самостійним
захворюванням, а лише симптомокомплексом
багатьох захворювань
Надзвичайно важливою є обізнаність широкого кола практикуючих
лікарів щодо основних груп та нозологічних форм патології, що
супроводжуються гепатолієнальним синдромом.
Абсолютно слушним є акцент на групі орфанних метаболічних
захворювань, які сьогодні діагностуються і лікуються за сучасними
протоколами.
Своєчасна діагностика даної групи патології та призначення
патогенетично обгрунованої, в першу чергу ФЗТ, покращує якість і
тривалість життя хворих,а також психологічний клімат у сім”ях.
Безсумнівним є позитивний досвід використання ФЗТ при МПС I таII
типів, хворобі Гоше вже й на нашому рівні.