






























 medicine
medicineSimilar presentations:






")



Первичные иммунодефициты у детей
1.
ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России(Сеченовский Университет)
Первичные
иммунодефициты у детей
Подготовила:
студент 5 курса ИКМ
Алексеева Дарья
Игоревна.
2021
2.
Первичные иммунодефициты (ПИД)– это врожденные нарушения системы иммунитета, связанные с
генетическим дефектом одного или нескольких звеньев
иммунитета.
Распространенность ПИДС составляет 1:10 000 рождений.
3.
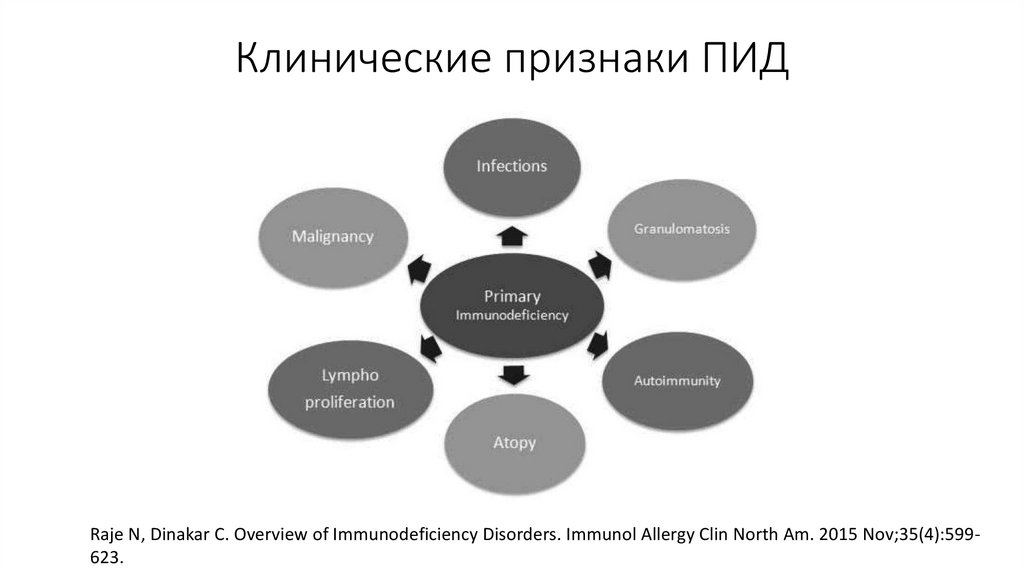
Клинические признаки ПИДRaje N, Dinakar C. Overview of Immunodeficiency Disorders. Immunol Allergy Clin North Am. 2015 Nov;35(4):599623.
4.
Особенности инфекций при ПИД• Хроническое или рецидивирующее течение,
склонность к прогрессированию
• Оппортунистический характер
• Политопность (множественные поражения
различных органов)
• Полиэтиологичность (восприимчивость ко
многим возбудителям одновременно)
• Неполная санация, неполный эффект от
лечения
5.
Связь между типом инфекции и видом ИДСНедостаточность
Ассоциированное заболевание
Т-и В-лимфоцитов (комбинированная
иммунная недостаточность)
Острые и хронические инфекции –
вирусные, бактериальные, грибковые,
протозойные
Т-лимфоцитов (преимущественно клеточная Повышенная восприимчивость к вирусным,
недостаточность)
грибковым и протозойным инфекциям
В-лимфоцитов (гуморальные ИДС)
Повторяющиеся бактериальные инфекции
(средние отиты, синуситы, пневмонии)
Фагоцитов
Пиогенные инфекции, плохое заживление
ран, системные оппортунистические
инфекции
Компонентов комплемента
Бактериальные инфекции, аутоиммунные
заболевания
6.
7.
Диагностика ПИД• Анамнез (в т.ч. семейный), физикальное обследование
• ОАК с подсчетом лейкоцитарной формулы
• Иммунограмма (определение субпопуляций лимфоцитов методом
проточной цитометрии)
• Определение в сыворотке крови уровней IgG, IgM, IgA, IgE
По показаниям:
Оценка функционального состояния фагоцитов
Оценка содержания в крови основных компонентов комплемента
Молекулярно-генетические исследования
Определение титра поствакцинальных антител
8.
Основные принципы лечения ПИД• Лечение и профилактика сопутствующих
инфекций, аутоиммунных, опухолевых
заболеваний
• Заместительная терапия
• ТГСК/ТКМ
• Генная терапия
9.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
10.
Комбинированная недостаточность клеточногои гуморального звена иммунитета (ТКИД)
Типы наследования: аутосомно-рецессивный и Х-сцепленный
Распространенность: 1:58 000 новорожденных (чаще мальчики)
Классификация:
• T-B+ ТКИД (мутация гена γ-цепи рецепторов к цитокинам – ИЛ-2,
ИЛ-4, ИЛ-7 и т.д.; мутация гена тирозинкиназы JAK3)
• Т-В- ТКИД (дефицит аденозиндезаминазы, дефицит RAG1, RAG2)
11.
Клиническая картина ТКИД• Раннее начало, прекращение прибавки массы тела,
нарастание дистрофии
• Упорная диарея в возрасте 2-6 месяцев, не
поддающаяся терапии
• Осложнения после вакцинации БЦЖ, вплоть до
диссеминированной инфекции
• Рецидивирующий кандидоз кожи и слизистых
• Интерстициальные пневмонии – грибковые и
пневмоцистные
• Кожная сыпь неясной этиологии (реакция
«трансплантат против хозяина»)
• Тяжелые формы вирусных инфекций (герпетическая,
ЦМВ, ВЭБ, аденовирусная)
• Гипоплазия лимфоидной ткани
12.
Диагностика ТКИД• ОАК (лимфопения, анемия)
• БХ анализ крови (для оценки поражения органов)
• Исследование уровня иммуноглобулинов сыворотки
(гипогаммаглобулинемия)
• Иммунограмма (значительное снижение Т-лимфоцитов, число Влимфоцитов и НК-клеток зависит от генетического дефекта)
• Исследование TREC (Концентрация TREC значительно снижена)
• Молекулярно-генетическое исследование
13.
Лечение ТКИДТКИД являются неотложным состоянием в
педиатрии.
ТГСК – всем пациентам!
Консервативное – на этапе подготовки к ТГСК:
• Нахождение ребенка в стерильном боксе
• Лечение инфекций
• При отсутствии инфекционных очагов:
профилактическая антибактериальная,
противогрибковая, профилактика пневмоцистной
инфекции ко-тримоксазолом, ЦМВ-инфекции
ганцикловиром
• Лечение БЦЖита противомикобактериальными
препаратами
14.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
15.
Комбинированные иммунодефициты,ассоциированные с синдромальными
проявлениями
• Синдром Вискотта-Олдрича
• Синдром атаксии-телеангиэктазии (Луи-Бар)
• Синдром Ди-Джорджи
• Синдром Ниймеген
• Гипер-IgE-синдром
16.
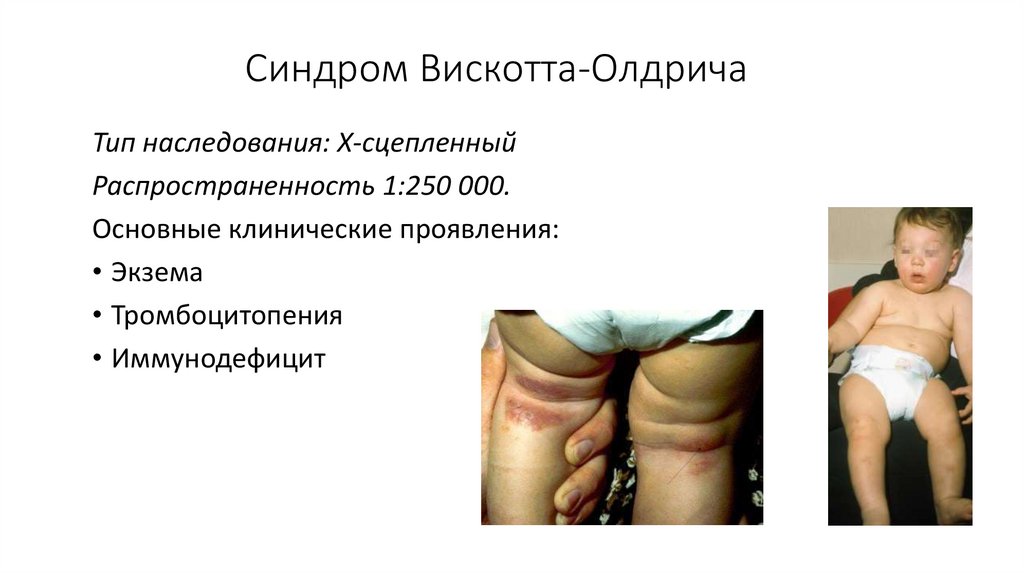
Синдром Вискотта-ОлдричаТип наследования: Х-сцепленный
Распространенность 1:250 000.
Основные клинические проявления:
• Экзема
• Тромбоцитопения
• Иммунодефицит
17.
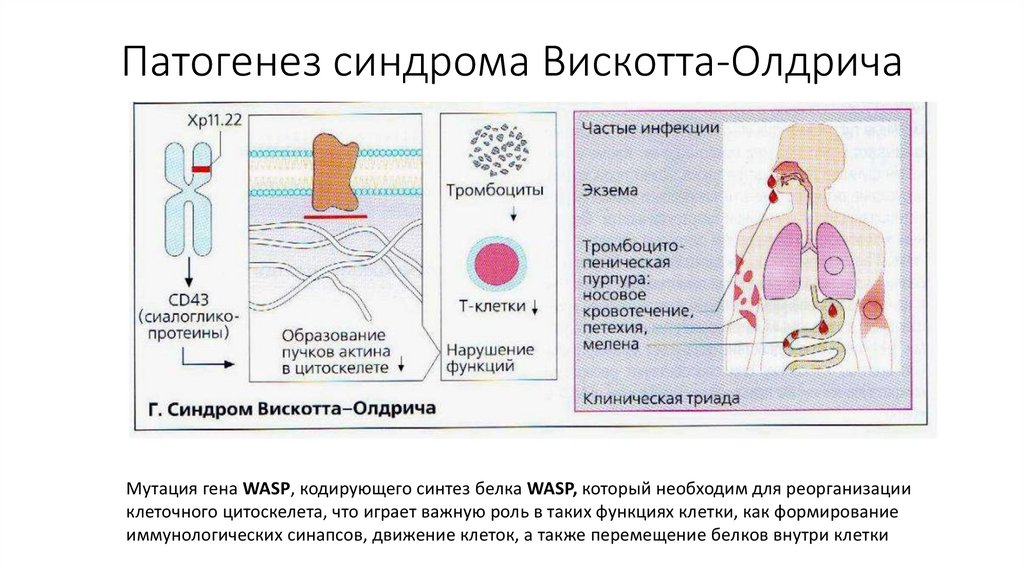
Патогенез синдрома Вискотта-ОлдричаМутация гена WASP, кодирующего синтез белка WASP, который необходим для реорганизации
клеточного цитоскелета, что играет важную роль в таких функциях клетки, как формирование
иммунологических синапсов, движение клеток, а также перемещение белков внутри клетки
18.
Лечение синдрома Вискотта-Олдрича• ТГСК
• Агонист тромбопоэтиновых рецепторов ромиплостим
• По показаниям – переливание тромбоцитов
• ВВИГ
• Лечение инфекций
oГенная терапия
19.
Синдром атаксии-телеангиэктазии (Луи-Бар)Тип наследования: аутосомно-рециссивный.
Распространенность: 1 случай на 40 000 – 88 000 живых новорожденных
Основные клинические проявления:
• Неврологические нарушения (мозжечковая атаксия, снижение сухожильных
рефлексов, расходящееся косоглазие, утрата двигательной функции)
• Конъюнктивальные или лицевые телеангиэктазии
• Синопульмональные инфекции (бронхиты, пневмонии с развитием
бронхоэктазов, легочного фиброза)
• Гипоплазия лимфоидной ткани
• Повышение АФП, лимфопения, снижение количества CD3+, CD4+, CD8+,
снижение IgA и в ряде случаев IgG
20.
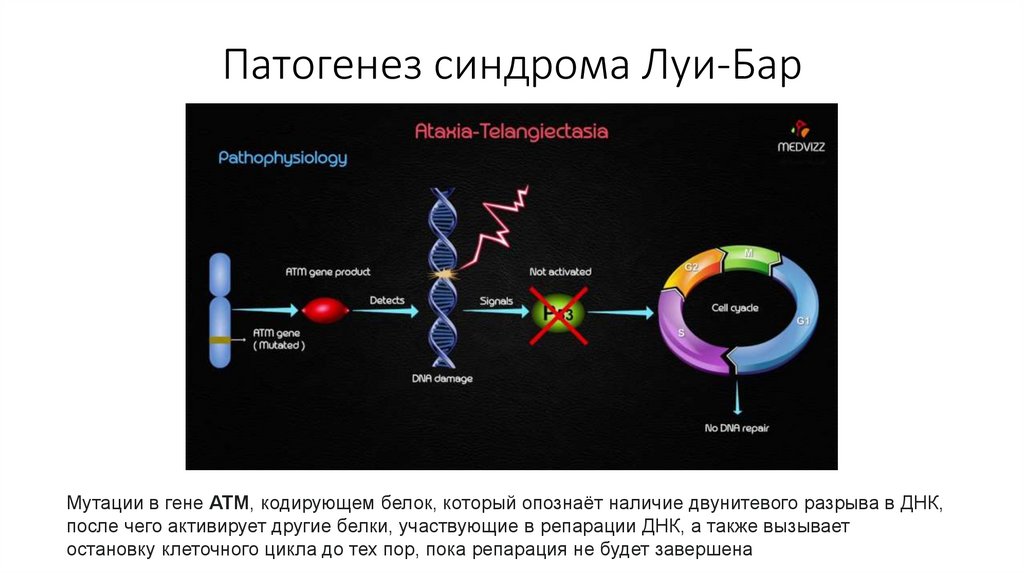
Патогенез синдрома Луи-БарМутации в гене АТМ, кодирующем белок, который опознаёт наличие двунитевого разрыва в ДНК,
после чего активирует другие белки, участвующие в репарации ДНК, а также вызывает
остановку клеточного цикла до тех пор, пока репарация не будет завершена
21.
Клиническая картина синдрома Луи-Бар22.
Лечение синдрома Луи-Бар• Лечение инфекционных заболеваний
• ВВИГ
• Физиотерапия для поддержания мышечного тонуса
• Ограничение рентгеновского облучения
• Химиотерапия при возникновении лимфопролиферативных
заболеваний.
23.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
24.
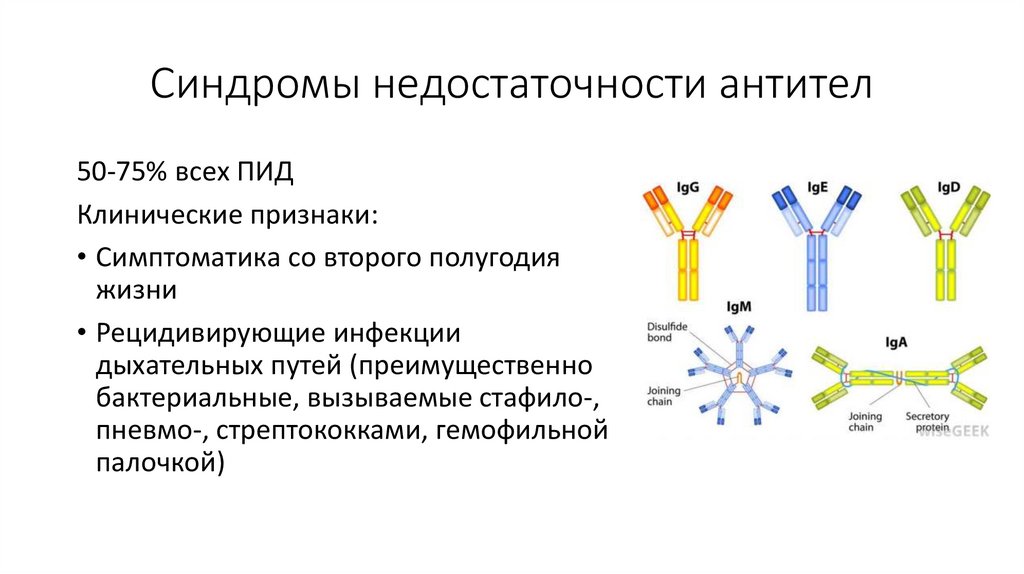
Синдромы недостаточности антител50-75% всех ПИД
Клинические признаки:
• Симптоматика со второго полугодия
жизни
• Рецидивирующие инфекции
дыхательных путей (преимущественно
бактериальные, вызываемые стафило-,
пневмо-, стрептококками, гемофильной
палочкой)
25.
Синдромы недостаточности антител• Болезнь Брутона (Х-сцепленная агаммаглобулинемия)
• Общая вариабельная иммунная недостаточность (ОВИН)
• Селективный дефицит иммуноглобулина А
• Транзиторная младенческая гипогаммаглобулинемия
(преходящая гипогаммаглобулинемия детей, поздний
иммунологический старт)
26.
Болезнь Брутона(Х-сцепленная агаммаглобулинемия, наследственная
агаммаглобулинемия)
Тип наследования: Х-сцепленное
Распространенность: 1-5:1000 0000
Основные клинические проявления:
• Повторные синопульмональные инфекции
• Персистирующая диарея
• Гнойные инфекции кожи и мягких тканей
(фурункулезы, абсцессы, флегмоны)
• Гипоплазия небных миндалин, лимфоузлов
• Стойкое снижение концентраций всех
иммуноглобулинов – IgG, IgA, IgM, IgD, IgE
• Резкое снижение фракции гамма-глобулинов (<1-2%)
• Глубокий дефицит В-лимфоцитов (CD19+<1-2%)
27.
Патогенез болезни БрутонаХаитов P.M., Ярилин А.А., Пинегин Б.В. И53 Иммунология : атлас. — М .: ГЭОТАРМедиа, 2011. — 624 с . : ил.
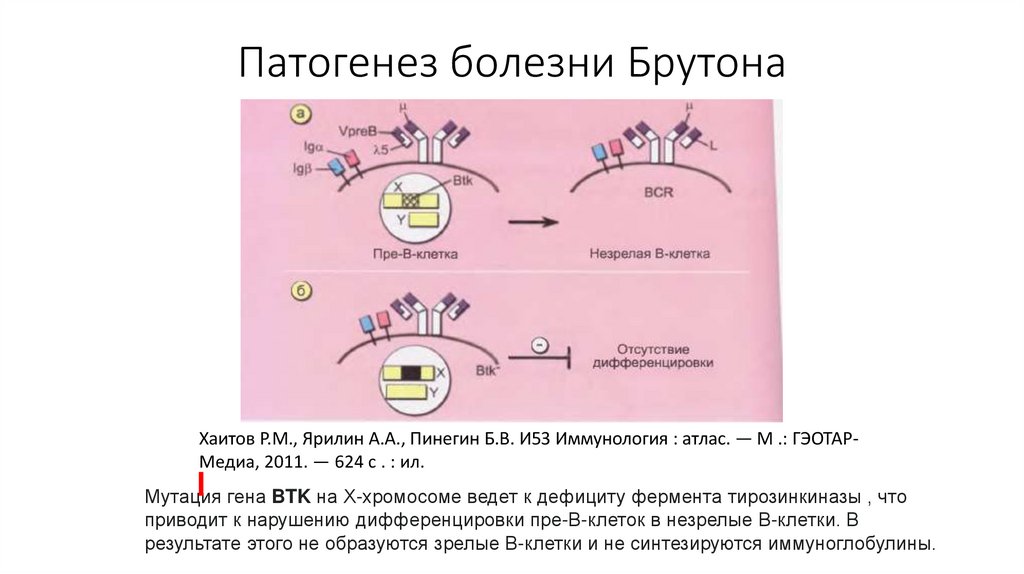
Мутация гена BTK на Х-хромосоме ведет к дефициту фермента тирозинкиназы , что
приводит к нарушению дифференцировки пре-В-клеток в незрелые В-клетки. В
результате этого не образуются зрелые В-клетки и не синтезируются иммуноглобулины.
28.
Лечение болезни Брутона• Пожизненная заместительная терапия препаратами донорских
иммуноглобулинов
• Лечение инфекционных заболеваний - антибактериальная
терапия
• При хронических бронхолегочными инфекциями - ежедневный
вибрационный массаж и постуральный дренаж
29.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
30.
Заболевания иммунной дисрегуляции:Аутоиммунная полиэндокринопатия —
кандидоз-эктодермальная дистрофия (АПЭКЭД)
Тип наследования: аутосомно-рецессивный
Распространенность: 1:100 000
Основные клинические проявления:
• Кожно-слизистый кандидоз, онихомикоз
• Повторные бактериальные инфекции респираторного тракта, кожи, ЛОРорганов
• Эктодермальная дисплазия (тотальный кариес, алопеция)
• Эндокринопатии (гипопаратиреоз, надпочечниковая недостаточность,
гипогонадизм)
• Витилиго, алопеция
• Аутоиммунный гепатит
31.
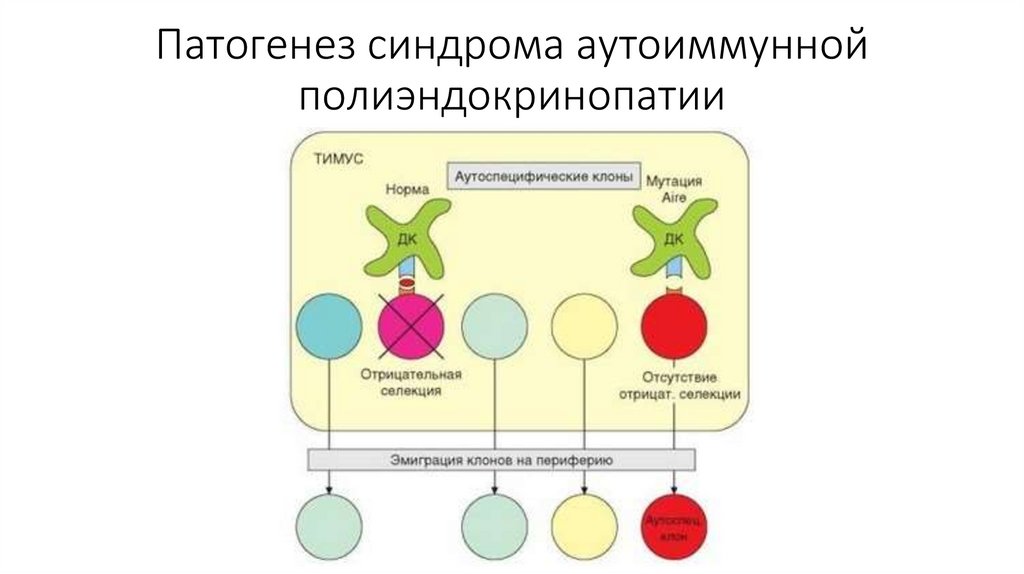
Патогенез синдрома аутоиммуннойполиэндокринопатии
32.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
33.
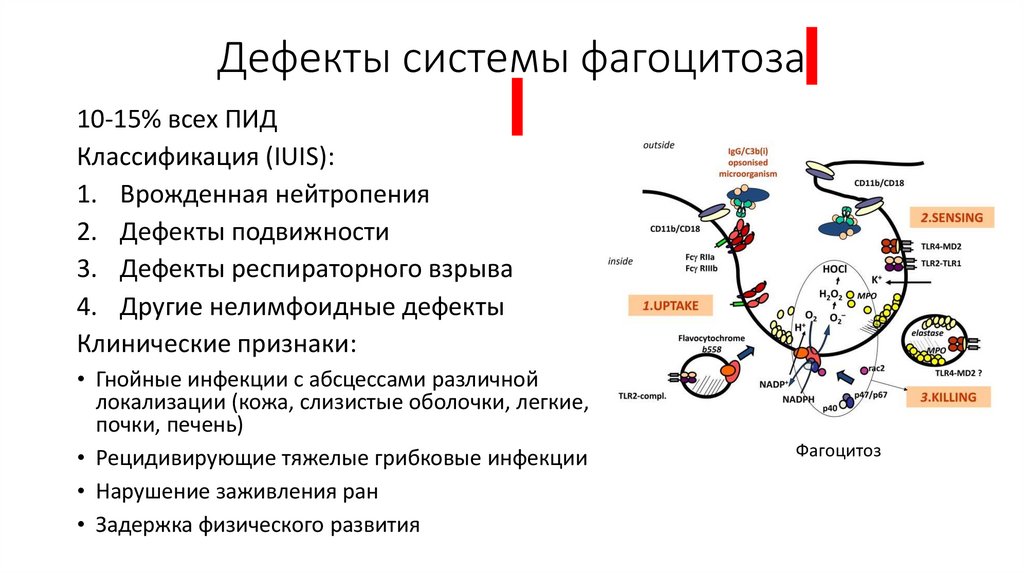
Дефекты системы фагоцитоза10-15% всех ПИД
Классификация (IUIS):
1. Врожденная нейтропения
2. Дефекты подвижности
3. Дефекты респираторного взрыва
4. Другие нелимфоидные дефекты
Клинические признаки:
• Гнойные инфекции с абсцессами различной
локализации (кожа, слизистые оболочки, легкие,
почки, печень)
• Рецидивирующие тяжелые грибковые инфекции
• Нарушение заживления ран
• Задержка физического развития
Фагоцитоз
34.
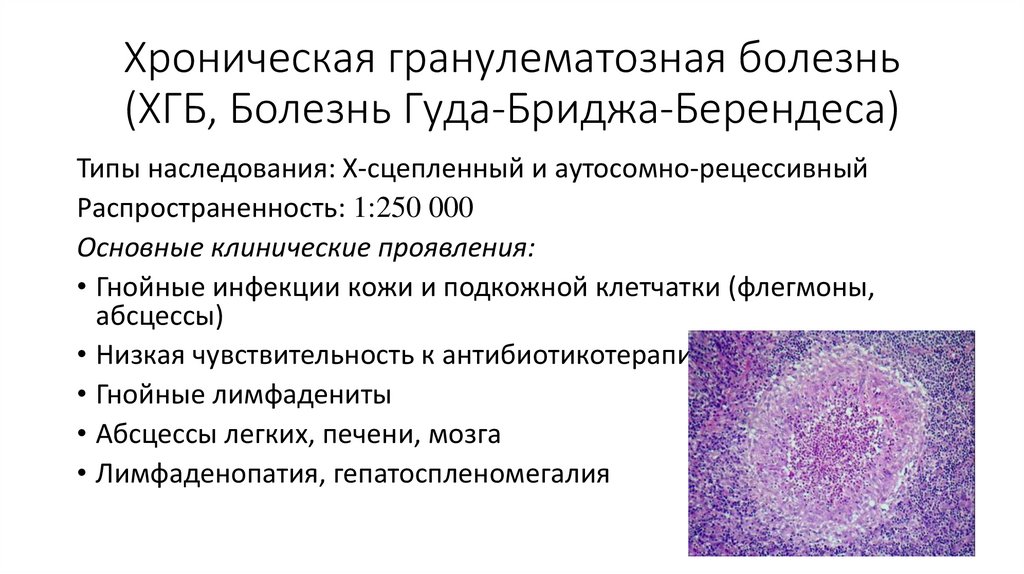
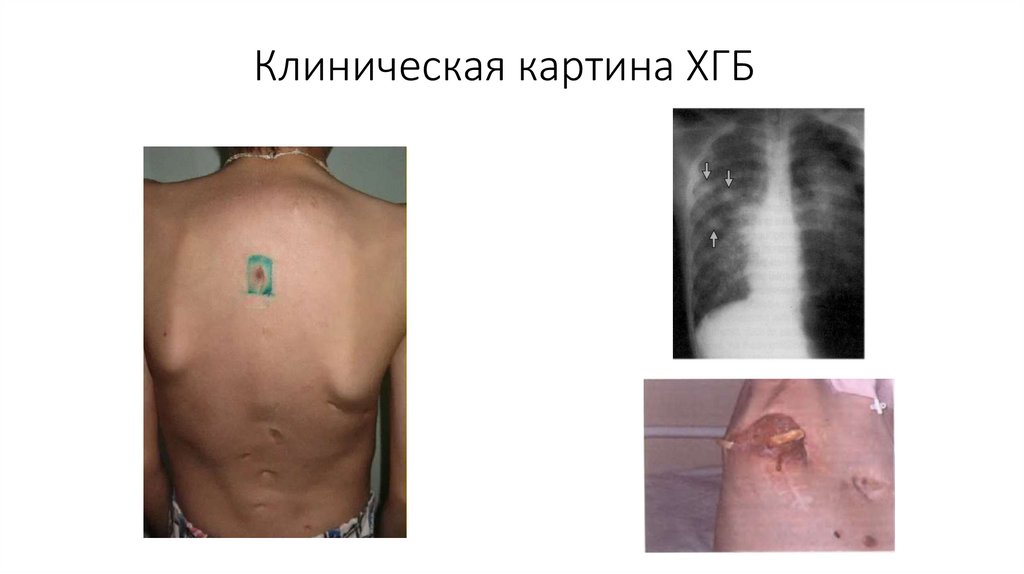
Хроническая гранулематозная болезнь(ХГБ, Болезнь Гуда-Бриджа-Берендеса)
Типы наследования: Х-сцепленный и аутосомно-рецессивный
Распространенность: 1:250 000
Основные клинические проявления:
• Гнойные инфекции кожи и подкожной клетчатки (флегмоны,
абсцессы)
• Низкая чувствительность к антибиотикотерапии
• Гнойные лимфадениты
• Абсцессы легких, печени, мозга
• Лимфаденопатия, гепатоспленомегалия
35.
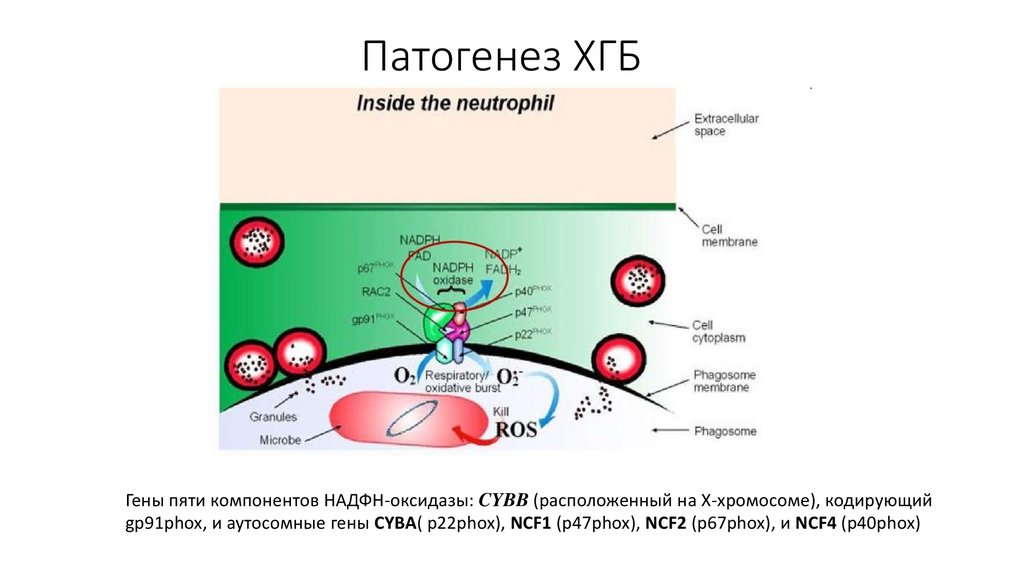
Патогенез ХГБГены пяти компонентов НАДФН-оксидазы: CYBB (расположенный на Х-хромосоме), кодирующий
gp91phox, и аутосомные гены CYBA( p22phox), NCF1 (p47phox), NCF2 (p67phox), и NCF4 (p40phox)
36.
Клиническая картина ХГБ37.
Лечение ХГБ• Антибактериальная и противогрибковая терапия пожизненно
• Ко-тримоксазол
• Интерферон-гамма, под действием которого увеличивается
продукция активных радикалов кислорода.
• ТКМ по показаниям
38.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
39.
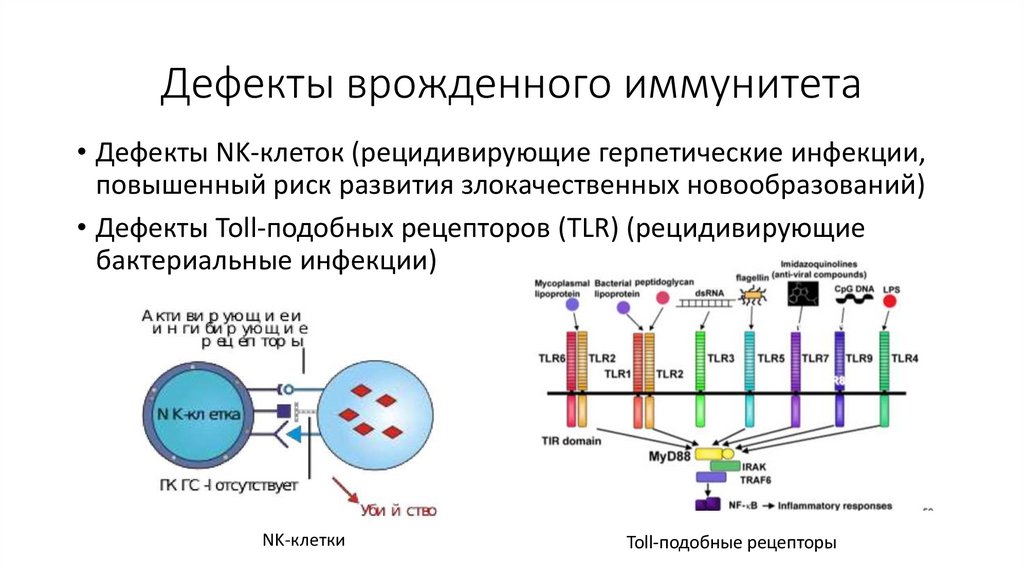
Дефекты врожденного иммунитета• Дефекты NK-клеток (рецидивирующие герпетические инфекции,
повышенный риск развития злокачественных новообразований)
• Дефекты Toll-подобных рецепторов (TLR) (рецидивирующие
бактериальные инфекции)
NK-клетки
Toll-подобные рецепторы
40.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
41.
Аутовоспалительные заболеванияГенетически детерминированная активация врожденного
иммунитета, в первую очередь NOD-рецепторов.
Основные клинические проявления:
• эпизоды гектической лихорадки, сопровождающиеся
высыпаниями, миалгиями, артралгиями
• возможно тяжелое поражение опорно-двигательного аппарата,
ЦНС, ЖКТ
42.
Классификация ПИДInternational Union of Immunological Societies (IUIS)
1. Комбинированная недостаточность клеточного и гуморального звена
иммунитета (тяжелый комбинированный иммунодефицит –ТКИД)
2. Комбинированные иммунодефициты,
ассоциированные с синдромальными проявлениями
3. Синдромы недостаточности антител (гуморальные иммунодефициты)
4. Заболевания иммунной дисрегуляции
5. Дефекты системы фагоцитоза
6. Дефекты врожденного иммунитета
7. Аутовоспалительные заболевания
8. Дефекты системы комплемента
9. Недостаточность костного мозга
43.
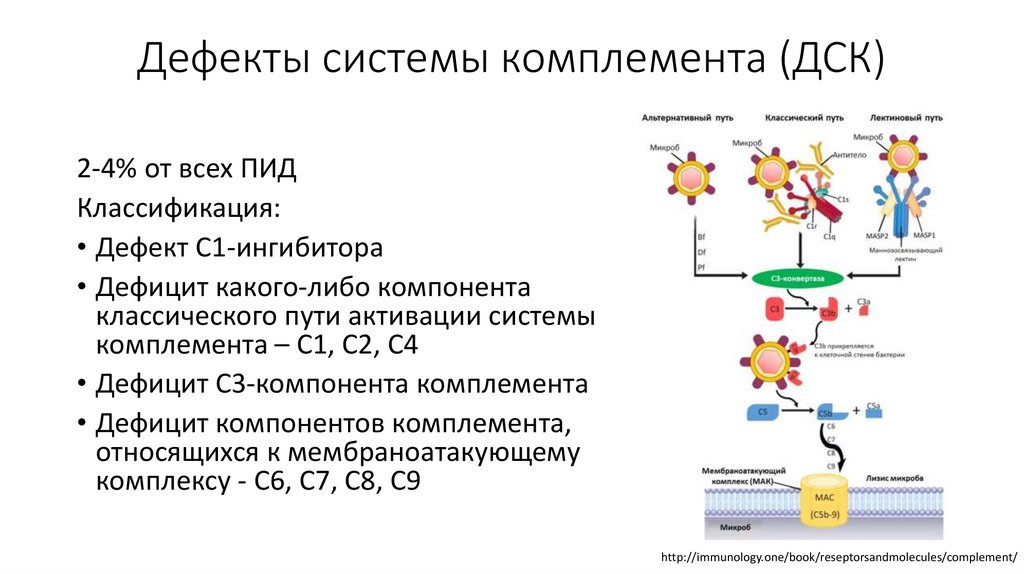
Дефекты системы комплемента (ДСК)2-4% от всех ПИД
Классификация:
• Дефект С1-ингибитора
• Дефицит какого-либо компонента
классического пути активации системы
комплемента – С1, С2, С4
• Дефицит С3-компонента комплемента
• Дефицит компонентов комплемента,
относящихся к мембраноатакующему
комплексу - С6, С7, С8, С9
http://immunology.one/book/reseptorsandmolecules/complement/
44.
Наследственный ангионевротический отек(НАО)
Тип наследования: аутосомно-доминантный
Распространенность: 1:50 000
Основные клинические проявления:
• Семейный анамнез отеков различной локализации
• Отеки различной локализации, без зуда, не купируются
антигистаминными средствами и ГКС
• Снижение количества/функциональной активности С1ингибитора
45.
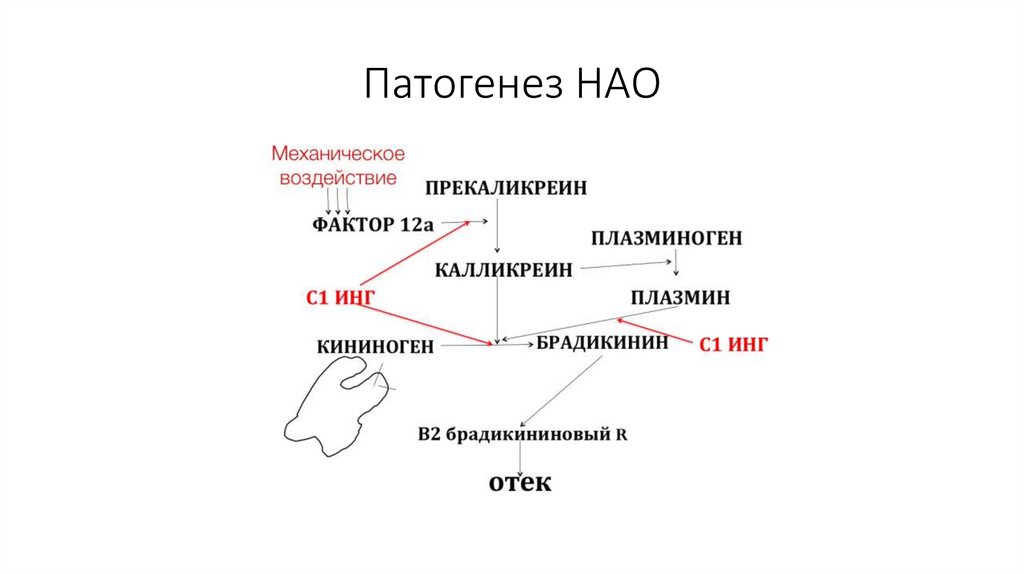
Патогенез НАО46.
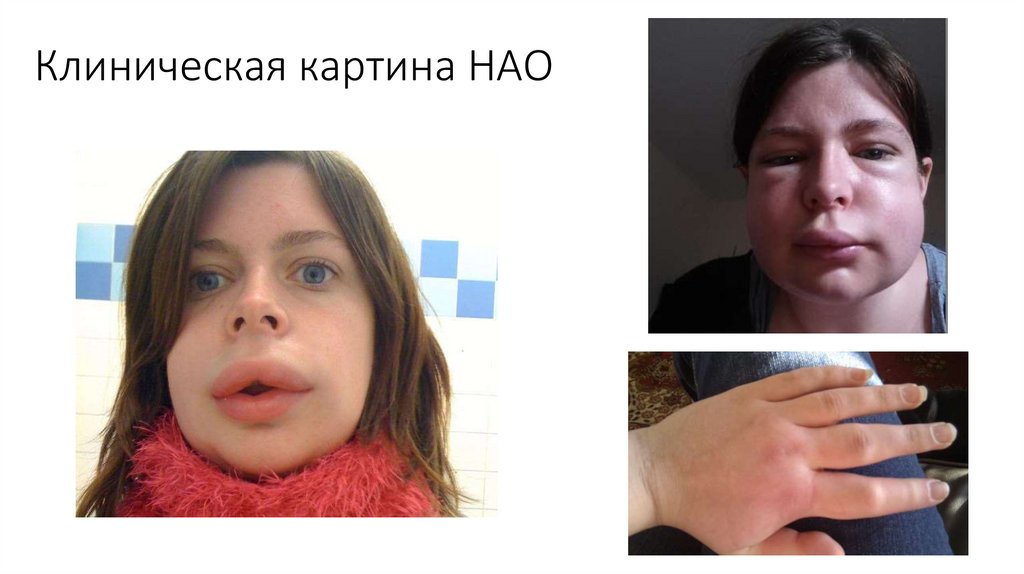
Клиническая картина НАО47.
Источники информацииМЕТОДИЧЕСКАЯ РАЗРАБОТКА к клиническому практическому занятию для студентов 6 курса специальности 31.05.02. Педиатрия по учебной дисциплине по
выбору «Клиническая иммунология, аллергология» Тема 3. Первичные и вторичные иммунодефициты ФГБОУ ВО «Ставропольский государственный
медицинский университет» МЗ РФ, 2020г.
https://www.uptodate.com/contents/inborn-errors-of-immunity-primary-immunodeficienciesclassification?search=primary%20immunodeficiency&topicRef=3914&source=see_link#H86894852
https://www.uptodate.com/contents/primary-disorders-of-phagocyte-number-and-or-function-anoverview?search=primary%20immunodeficiency&topicRef=117758&source=see_link#H3483866396
Tangye, Stuart G et al. “Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert
Committee.” Journal of clinical immunology vol. 40,1 (2020): 24-64.
Тяжелая комбинированная иммунная недостаточность у детей. Клинические рекомендации. 2016
Клинические рекомендации Первичные иммунодефициты преимущественно с недостаточностью антител 2018
Первичные иммунодефициты с дефектом системы фагоцитоза. Клинические протоколы МЗ РК – 2016
Федеральные клинические рекомендации по диагностике и лечению детей с синдромом Вискотта-Олдрича. 2015
https://www.uptodate.com/contents/chronic-mucocutaneouscandidiasis?sectionName=Autoimmune%20regulator%20deficiency&search=primary%20immunodeficiency&topicRef=117758&anchor=H571991&source=see_link#H57
1991
Roos, Dirk. “Chronic granulomatous disease.” British medical bulletin vol. 118,1 (2016): 50-63.
Сураев В.К. КЛИНИЧЕСКИЙ СЛУЧАЙ СИНДРОМА ЛУИ-БАР В СОЧЕТАНИИ С ЭПИЛЕПСИЕЙ У ПОДРОСТКА
А.С. Прилуцкий и соавт. КЛИНИЧЕСКИЕ И ЛАБОРАТОРНЫЕ ОСОБЕННОСТИ СЛУЧАЯ АТАКСИИ – ТЕЛЕАНГИЭКТАЗИИ 2013
Клинические рекомендации Наследственный ангионевротический отёк (НАО). 2019
48.
Источники информации• Хаитов Р.М. Иммунология: учебник / Р.М. Хаитов. - 2-е изд., перераб. и доп. - 2013. - 528 с.: ил.
• Хаитов P.M., Ярилин А.А., Пинегин Б.В. И53 Иммунология : атлас. — М .: ГЭОТАР-Медиа, 2011. — 624 с . : ил.
• https://rae.ru/ru/publishing/mono07_58.html
• Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review.
Orphanet J Rare Dis. 2016 Nov 25;11(1):159.
• Raje N, Dinakar C. Overview of Immunodeficiency Disorders. Immunol Allergy Clin North Am. 2015 Nov;35(4):599-623.
• Raje, Nikita, and Chitra Dinakar. “Overview of Immunodeficiency Disorders.” Immunology and allergy clinics of North America vol.
35,4 (2015): 599-623.
• Прилуцкий А.С и соавт. Аутоиммунные полиэндокринные синдромы: классификация, клиника, диагностика, лечение. 2014
• Robert A Schwartz Pediatric Wiskott-Aldrich Syndrome Workup. 2021