



















































 chemistry
chemistrySimilar presentations:






")



Коллоидная химия
1.
КОЛЛОИДНАЯ ХИМИЯ2.
Историческая справкаТомас Грэм (1805-1869)
Название «Коллоидная химия» произошло от греческого
слова
- клееподобный. Термин «коллоид» ввел
шотландский химик Томас Грэм в 1861 г. Он обнаружил,
что некоторые вещества, например, желатина, агар-агар,
кремниевая кислота, при выпаривании водного раствора не
кристаллизуются, а образуют студенистые осадки. Кроме
того, скорость их диффузии через мембрану очень мала.
Некоторые из этих веществ обладают клеющим действием.
Подобные
вещества
и
были
названы
Грэмом
«коллоидами».
«Так как желатина представляет собой особый тип веществ,
предложено обозначать вещества этого типа названием
«коллоиды» и трактовать о такой форме агрегации, как о
коллоидном состоянии материи. Противоположным коллоидному
является кристаллическое состояние материи. Вещества,
принадлежащие к данной форме состояния материи, следует
обозначать названием «кристаллоиды».
(Томас Грэм)
3.
К началу XX в. было открыто много веществ с типичными коллоидными свойствами,разработаны методы получения, очистки и стабилизации разнообразных коллоидных
систем, созданы методы их изучения, в т.ч. и методы измерения размера частиц
1906-1910 гг. – профессор Горного института
Санкт-Петербурга П.П. фон Веймарн выдвинул
фундаментальный принцип универсальности
коллоидного состояния вещества:
«Коллоидное состояние не является обособленным,
обусловленным какими-либо особенностями состава
вещества. При определенных условиях каждое
вещество может быть в коллоидном состоянии»
П. П. фон Веймарн
(1879-1935)
Прямым следствием принципа универсальности стало введение понятия о
дисперсном (раздробленном) состоянии вещества и в связи с этим осознание
важнейшей роли процессов, протекающих на межфазной поверхности. С этого
времени коллоидная химия становится самостоятельной дисциплиной со своими
объектами, методами и практическими приложениями
Первая треть XX в. – период плодотворного и быстрого развития коллоидной химии
4.
Лауреаты Нобелевской премии за работыв области коллоидной химии
1925 г.
1926 г.
Р. Зигмонди
австрийский химик,
«за установление
гетерогенной природы коллоидных
растворов и за разработанные в этой
связи методы, имеющие фундаментальное значение
в современной
коллоидной химии»
Ж. Перрен
французский
физик, «за работу
по дискретной
природе материи и,
в особенности, за
открытие
седиментационнодиффузионного
равновесия»
1926 г.
Т. Сведберг
шведский ученый,
«за работы в
области
дисперсных
систем» (прежде
всего за создание
ультрацентрифуги
для определения
размеров высокодисперсных частиц
и макромолекул)
1932 г.
И. Ленгмюр
американский
физикохимик, «за
открытия и
исследования в
области химии
поверхностных
явлений»
5.
А. В. Думанский – один из основоположниковколлоидной химии в России
1904 г. – организовал первую в стране лабораторию
коллоидной химии (г. Киев)
1932 г. – по его инициативе создан Государственный
НИИ коллоидной химии (г. Воронеж)
1934 г. – состоялась Первая Всесоюзная коллоидная
конференция (г. Воронеж)
А. В. Думанский
(1880-1967)
1935 г. – основал «Коллоидный журнал»
Автор учебного пособия «Дисперсность и коллоидное
состояние вещества» (1932 и 1934 гг.), оригинального
учебника «Учение о коллоидах», который трижды
переиздавался
В 1932 г. А.В. Думанский удостоен большой
Менделеевской премии
6.
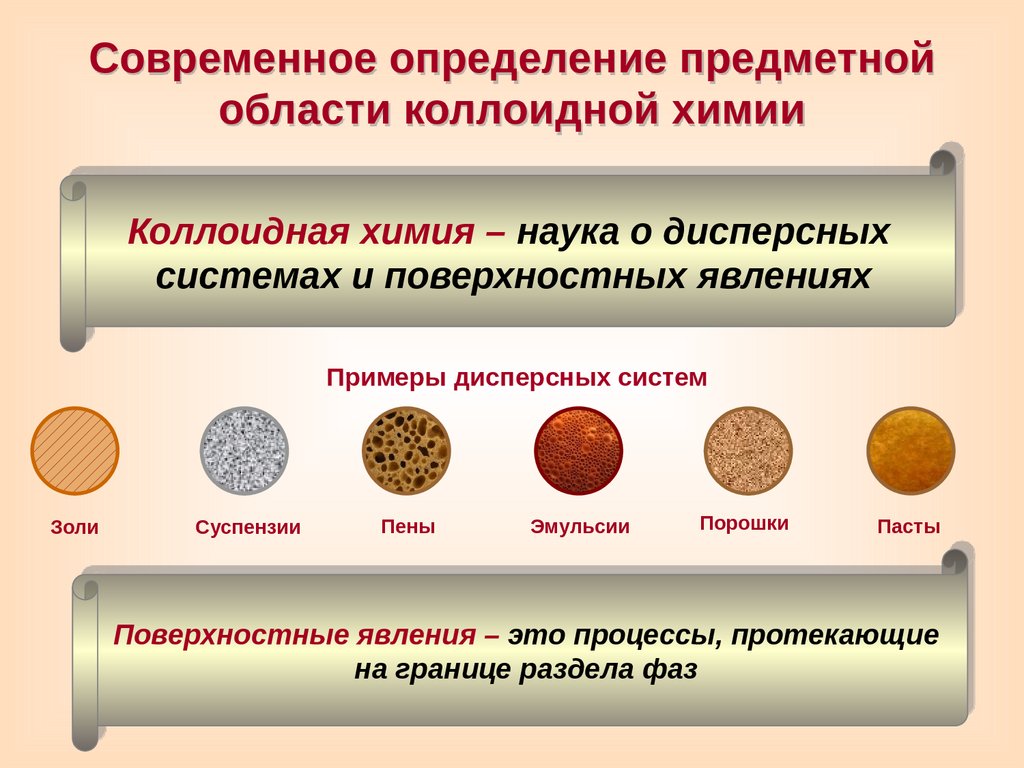
Современное определение предметнойобласти коллоидной химии
Коллоидная химия – наука о дисперсных
системах и поверхностных явлениях
Примеры дисперсных систем
Золи
Суспензии
Пены
Эмульсии
Порошки
Пасты
Поверхностные явления – это процессы, протекающие
на границе раздела фаз
7.
ХАРАКТЕРНЫЕ ПРИЗНАКИ И ОСНОВНЫЕ СВОЙСТВАДИСПЕРСНЫХ СИСТЕМ
8.
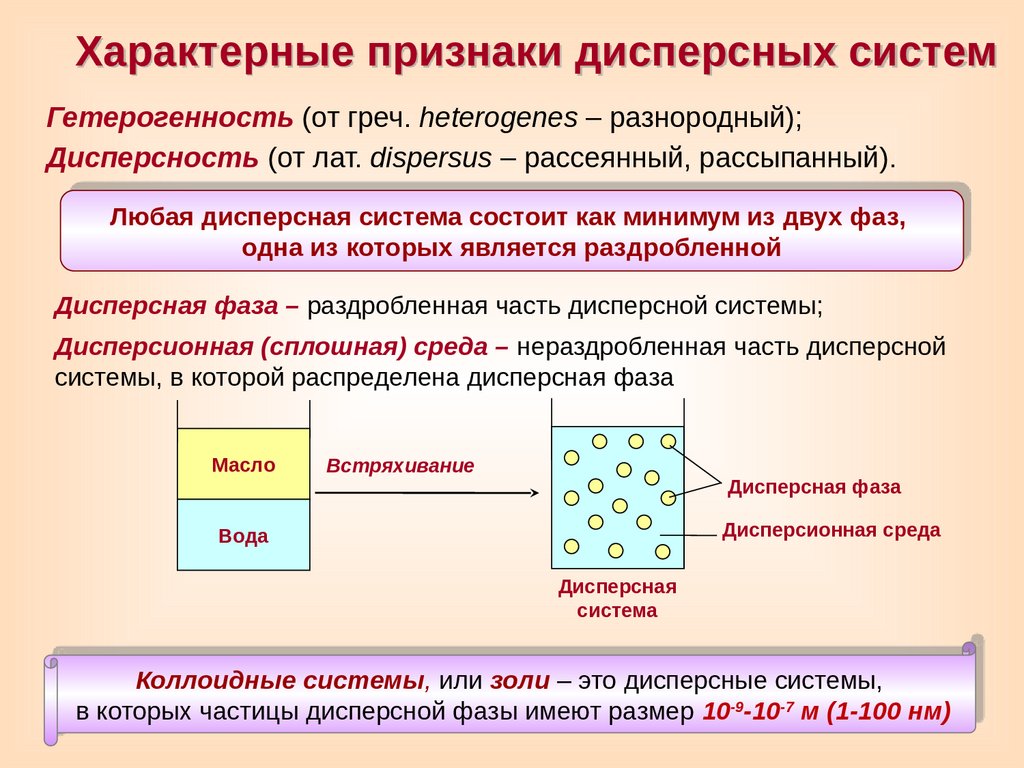
Характерные признаки дисперсных системГетерогенность (от греч. heterogenes – разнородный);
Дисперсность (от лат. dispersus – рассеянный, рассыпанный).
Любая дисперсная система состоит как минимум из двух фаз,
одна из которых является раздробленной
Дисперсная фаза – раздробленная часть дисперсной системы;
Дисперсионная (сплошная) среда – нераздробленная часть дисперсной
системы, в которой распределена дисперсная фаза
Масло
Встряхивание
Дисперсная фаза
Дисперсионная среда
Вода
Дисперсная
система
Коллоидные системы, или золи – это дисперсные системы,
в которых частицы дисперсной фазы имеют размер 10-9-10-7 м (1-100 нм)
9.
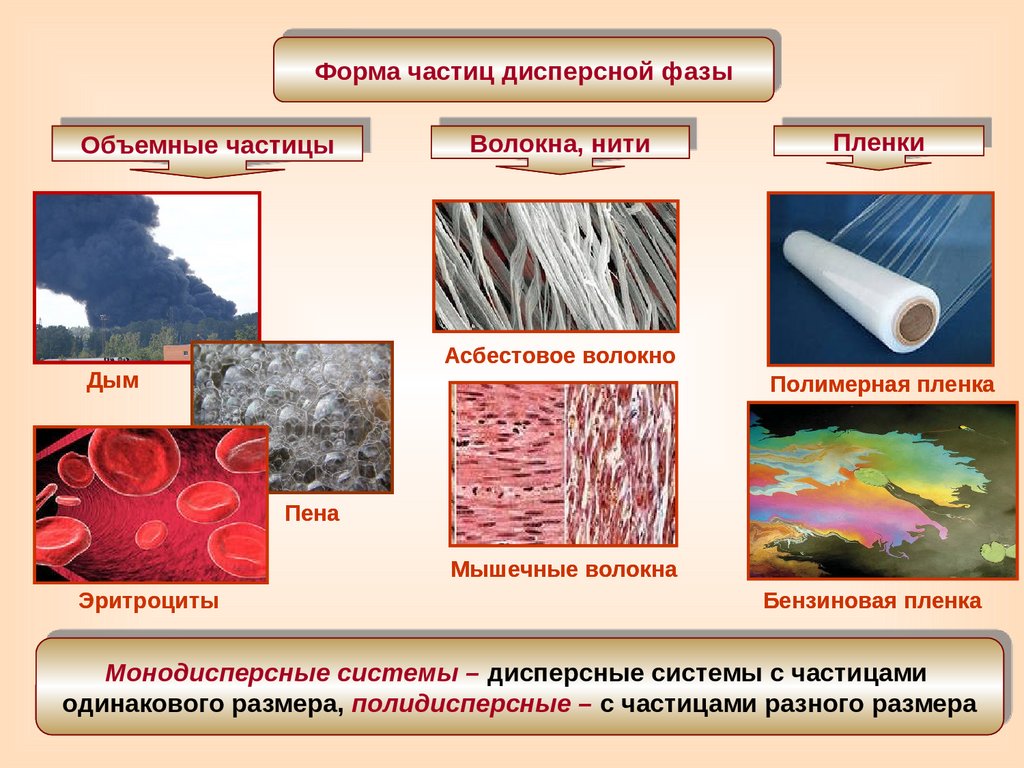
Форма частиц дисперсной фазыОбъемные частицы
Волокна, нити
Асбестовое волокно
Дым
Пленки
Полимерная пленка
Пена
Мышечные волокна
Эритроциты
Бензиновая пленка
Монодисперсные системы – дисперсные системы с частицами
одинакового размера, полидисперсные – с частицами разного размера
10.
Количественные характеристики дисперсных систем1) Дисперсность D – величина, обратная определяющему (характерному)
размеру частиц а:
D = 1/a
2) Удельная поверхность Sуд – площадь межфазной поверхности S,
отнесенная к единице массы m или объема V дисперсной фазы:
Sуд = S/m, м2/кг
Sуд = S/V, м-1
или
Для монодисперсных систем:
Sуд = k/a = kD
k – коэффициент, зависящий от формы частиц.
Для частиц кубической формы с ребром а
N S куб
а
6a 2 6
S уд
3
a
a
N Vкуб
Для частиц сферической формы с радиусом r
r
4 r 2
3 6
S уд
3
r d
4 / 3 r
3) Частичная концентрация n – число частиц дисперсной фазы в
единице объема системы, м-3
11.
Удельная поверхность и дисперсность резко возрастают! приуменьшении размера частиц дисперсной фазы.
Максимальную удельную поверхность (порядка 107 – 109 м-1) имеют
частицы дисперсной фазы в коллоидных растворах
Для частиц кубической
формы с ребром а
а, м
Sуд, м-1
1
6
10-1
60
10-5
6
105
10-7
6
107
10-9
6
109
Поэтому на поведение дисперсных систем, имеющих развитую
поверхность раздела, преобладающее влияние оказывают
процессы, протекающие на поверхности, а не внутри фазы!
12.
Основные свойства дисперсных системопределяются
НЕСКОМПЕНСИРОВАННОЙ ПОВЕРХНОСТНОЙ ЭНЕРГИЕЙ!
Раздробленность и большая удельная поверхность раздела фаз обусловливают
значительный избыток поверхностной энергии на межфазной поверхности.
Общая поверхностная энергия системы определяется
площадью поверхности S и удельной поверхностной энергией
,
называемой поверхностным натяжением:
Газ
Gs =
S
Жидкость
Межмолекулярные
взаимодействия внутри фазы и
на поверхности раздела фаз
При Т = 293 К
Поверхностное натяжение – это работа термодинамически обратимого процесса образования единицы площади новой поверхности при постоянных температуре Т, давлении р
и составе жидкости
= -
Aобр/dS
[
] = Дж/м2 = Н/м
Жидкость
,
мДж/м2
Вода
72,75
Сырое молоко
45-60
Подсолн. масло
33
Поверхностное натяжение на границе двух конденсированных фаз называет межфазовым.
Ртуть
460
Поверхностное натяжение с ростом температуры снижается.
Поверхностное натяжение характеризует различия в
интенсивности межмолекулярных взаимодействий
граничащих фаз. Чем больше эти различия, тем больше
.
13.
КЛАССИФИКАЦИЯ ДИСПЕРСНЫХ СИСТЕМ14.
Принципы классификациидисперсных систем
По степени дисперсности;
По агрегатному состоянию дисперсной фазы и
дисперсионной среды;
По характеру взаимодействия дисперсной фазы и
дисперсионной среды;
По структурно-механическим свойствам.
15.
Классификация дисперсных систем1) По степени дисперсности
Размер частиц,
м
Дисперсность,
м-1
Примеры
10-9-10-7
109-107
Гидрозоли
Среднедисперсные
10-7-10-5
107-105
Растворимый кофе,
сахарная пудра
Грубодисперсные
Более 10-5
Менее 105
Суспензии
Класс
Высокодисперсные
10-9
Среднедисперсные
системы
Высокодисперсные
системы
Истинные растворы
Sуд
10-7
Грубодисперсные
системы
(коллоидные системы)
10-5
a, м
Удельная поверхность Sуд частиц дисперсной
фазы максимальна в высокодисперсных
системах, при переходе к средне- и
грубодисперсным системам она уменьшается.
При размере частиц менее 10-9 м поверхность
раздела между частицей и средой исчезает,
образуются молекулярные или ионные растворы
(истинные растворы).
16.
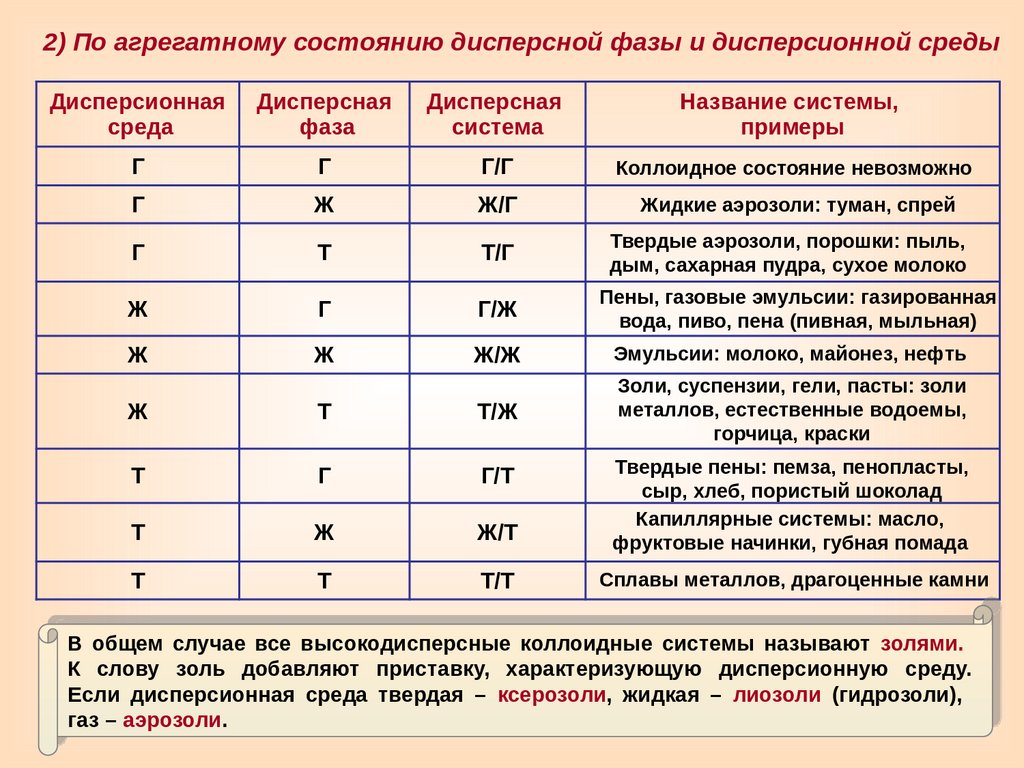
2) По агрегатному состоянию дисперсной фазы и дисперсионной средыДисперсионная
среда
Дисперсная
фаза
Дисперсная
система
Название системы,
примеры
Г
Г
Г/Г
Коллоидное состояние невозможно
Г
Ж
Ж/Г
Жидкие аэрозоли: туман, спрей
Г
Т
Т/Г
Твердые аэрозоли, порошки: пыль,
дым, сахарная пудра, сухое молоко
Ж
Г
Г/Ж
Пены, газовые эмульсии: газированная
вода, пиво, пена (пивная, мыльная)
Ж
Ж
Ж/Ж
Эмульсии: молоко, майонез, нефть
Золи, суспензии, гели, пасты: золи
металлов, естественные водоемы,
горчица, краски
Ж
Т
Т/Ж
Т
Г
Г/Т
Т
Ж
Ж/Т
Твердые пены: пемза, пенопласты,
сыр, хлеб, пористый шоколад
Капиллярные системы: масло,
фруктовые начинки, губная помада
Т
Т
Т/Т
Сплавы металлов, драгоценные камни
В общем случае все высокодисперсные коллоидные системы называют золями.
К слову золь добавляют приставку, характеризующую дисперсионную среду.
Если дисперсионная среда твердая – ксерозоли, жидкая – лиозоли (гидрозоли),
газ – аэрозоли.
17.
3) По характеру взаимодействия дисперсной фазы и дисперсионной среды4) По структурно-механическим свойствам
18.
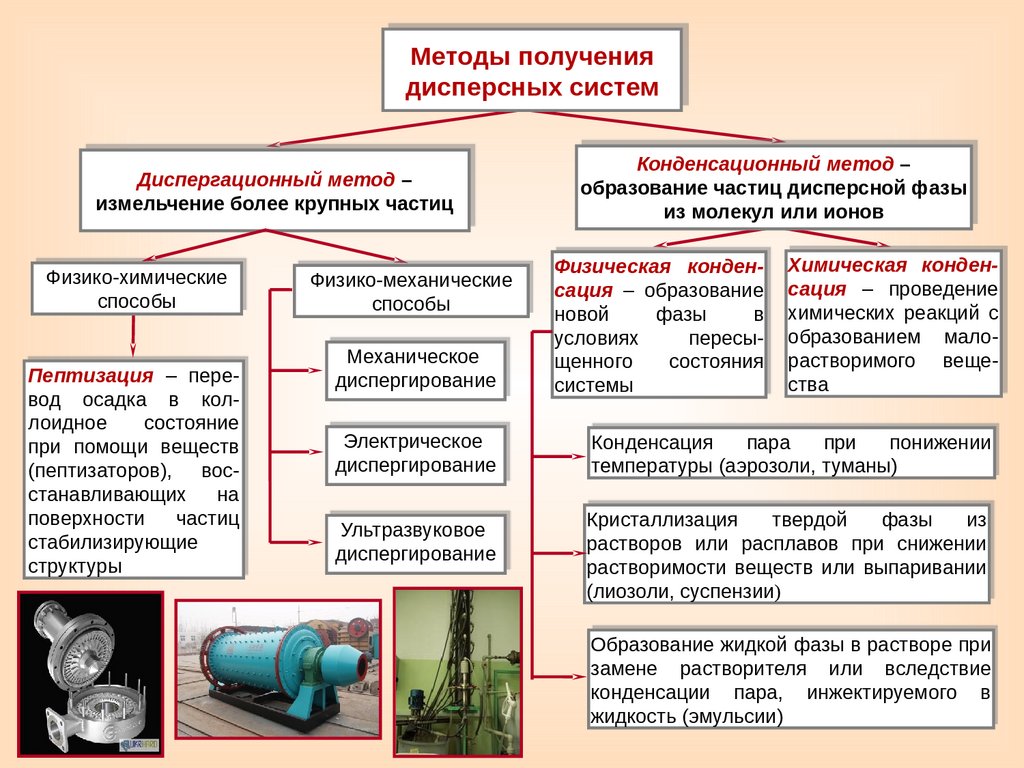
МЕТОДЫ ПОЛУЧЕНИЯ ДИСПЕРСНЫХ СИСТЕМ19.
Методы получениядисперсных систем
Диспергационный метод –
измельчение более крупных частиц
Физико-химические
способы
Пептизация – перевод осадка в коллоидное
состояние
при помощи веществ
(пептизаторов), восстанавливающих
на
поверхности
частиц
стабилизирующие
структуры
Физико-механические
способы
Механическое
диспергирование
Электрическое
диспергирование
Ультразвуковое
диспергирование
Конденсационный метод –
образование частиц дисперсной фазы
из молекул или ионов
Физическая конденсация – образование
новой
фазы
в
условиях
пересыщенного
состояния
системы
Химическая конденсация – проведение
химических реакций с
образованием малорастворимого вещества
Конденсация
пара
при
понижении
температуры (аэрозоли, туманы)
Кристаллизация
твердой
фазы
из
растворов или расплавов при снижении
растворимости веществ или выпаривании
(лиозоли, суспензии)
Образование жидкой фазы в растворе при
замене растворителя или вследствие
конденсации пара, инжектируемого в
жидкость (эмульсии)
20.
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ21.
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯЛиофобные дисперсные системы, обладающие большим избытком
поверхностной энергии, термодинамически неустойчивы; в них самопроизвольно протекают процессы, приводящие к снижению свободной энергии
Поверхностные явления – это процессы, протекающие
на границе раздела фаз и сопровождающиеся самопроизвольным снижением поверхностной энергии Gs
При p, T = const
Gs =
S
Следовательно, снижение поверхностной энергии Gs может
происходить за счет уменьшения межфазной поверхности S и
поверхностного натяжения
22.
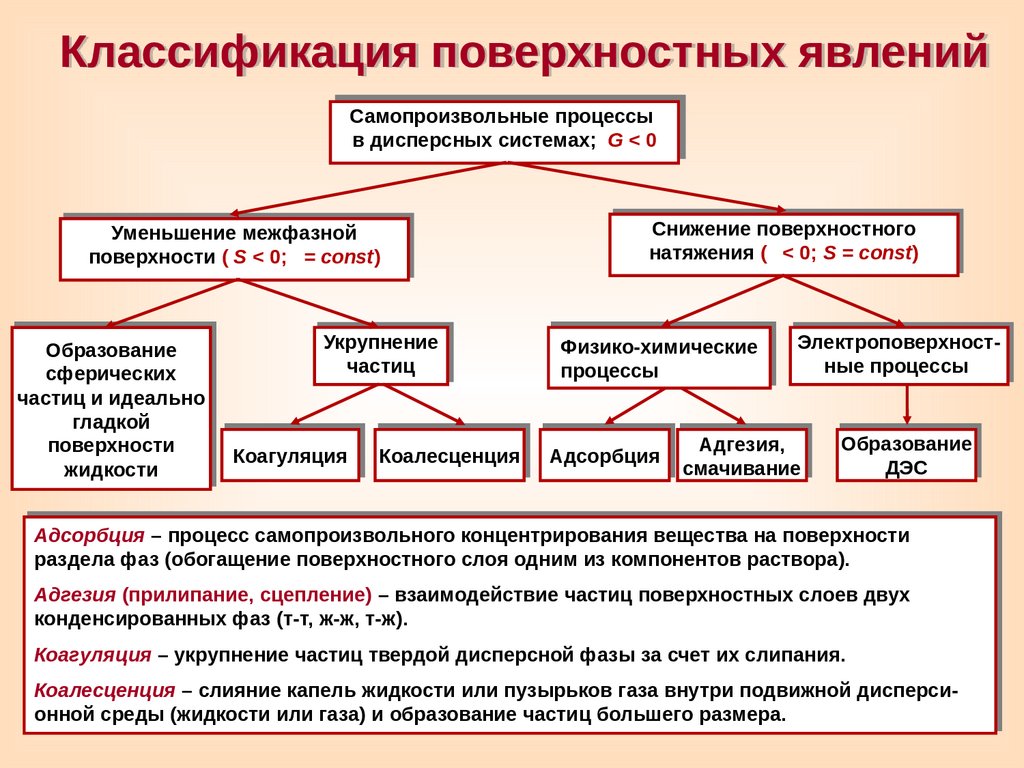
Классификация поверхностных явленийСамопроизвольные процессы
в дисперсных системах;
G<0
Уменьшение межфазной
поверхности (
S < 0; = const)
Образование
сферических
частиц и идеально
гладкой
поверхности
жидкости
Укрупнение
частиц
Коагуляция
Коалесценция
Снижение поверхностного
натяжения (
< 0; S = const)
Физико-химические
процессы
Адсорбция
Электроповерхностные процессы
Адгезия,
смачивание
Образование
ДЭС
Адсорбция – процесс самопроизвольного концентрирования вещества на поверхности
раздела фаз (обогащение поверхностного слоя одним из компонентов раствора).
Адгезия (прилипание, сцепление) – взаимодействие частиц поверхностных слоев двух
конденсированных фаз (т-т, ж-ж, т-ж).
Коагуляция – укрупнение частиц твердой дисперсной фазы за счет их слипания.
Коалесценция – cлияние капель жидкости или пузырьков газа внутри подвижной дисперсионной среды (жидкости или газа) и образование частиц большего размера.
23.
ПОВЕРХНОСТНОЕ ЯВЛЕНИЕ - АДСОРБЦИЯ24.
Основные понятия и определенияАдсорбция – процесс самопроизвольного концентрирования
вещества на межфазной поверхности
Адсорбент
фаза, на поверхности которой происходит адсорбция
(активированный уголь, силикагель, бентониты, цеолиты)
Адсорбтив – адсорбируемое вещество в
объеме фазы
Адсорбируемое
вещество имеет
2 названия
Физическая – взаимо-
действие адсорбента и
адсорбата происходит за
счет сил Ван-дер-Ваальса
и водородных связей
Адсорбция
Адсорбат – адсор-
бируемое вещество на
поверхности адсорбента
Химическая (хемосорбция) – сопровождается
установлением химических
связей между адсорбентом
и адсорбатом
Десорбция – процесс, обратный адсорбции (переход вещества из
поверхностного слоя в объем фазы)
25.
Способы выражения величины адсорбцииАбсолютная адсорбция Аi – количество i-го компонента в поверхностном
слое nsi , отнесенное к площади слоя S или к массе адсорбента m:
s
n
Ai = i , моль/м2
S
или
nsi
Ai =
, моль/кг
m
Избыточная (гиббсовская) адсорбция Гi – избыток i-го компонента в
поверхностном слое по сравнению с его количеством в таком же объеме,
но внутри фазы, отнесенный к площади слоя:
v
s
n
–
n
i
i
Ai =
, моль/м2
S
где nvi - количество i-го компонента в объеме фазы
Если концентрация адсорбируемого вещества на поверхности значительно
превышает его концентрацию в объеме, можно принять, что Г = А.
26.
Адсорбция на межфазной поверхности жидкость-газ.Поверхностно-активные вещества (ПАВ)
Поверхностно-активные вещества (ПАВ) – вещества, способные
адсорбироваться на межфазной поверхности и снижать
поверхностное (или межфазовое) натяжение
Особенность – дифильное строение молекулы
(дифильность)
Молекула ПАВ включает
2 фрагмента
полярный (гидрофильный),
обладающий значительным
дипольным моментом
(группы -ОН, -СООН, -NН2,
-SН, -NO и т.п.)
Обозначение:
Неполярная часть
Полярная часть
неполярный (гидрофобный),
обладающий слабым молекулярно-силовым полем
(углеводородный радикал)
Примеры (ПАВ относительно воды):
С5Н11ОН,
С17Н33СООNa,
С5Н11СООН
Изотерма поверхностного
натяжения для растворов ПАВ
0
0
с
Уравнение Шишковского
= Вln(1 + kc)
0 -
– поверхностное натяжение
0,
растворителя и раствора;
В, k – константы (коэффициент
В одинаков для гомологов)
27.
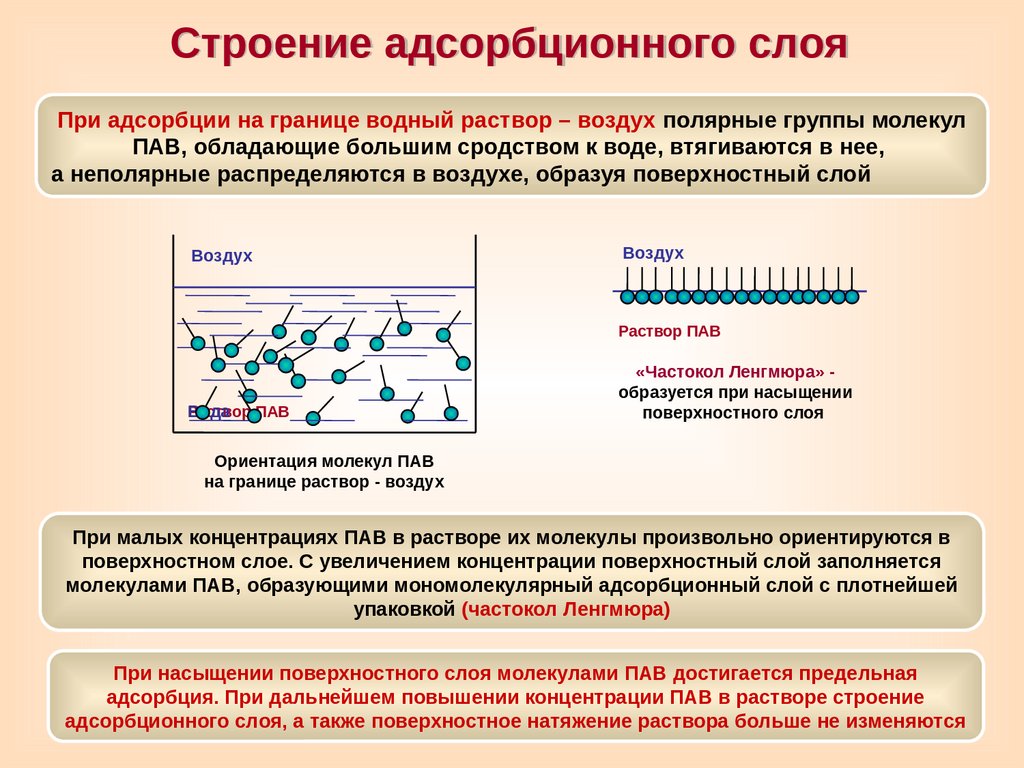
Строение адсорбционного слояПри адсорбции на границе водный раствор – воздух полярные группы молекул
ПАВ, обладающие большим сродством к воде, втягиваются в нее,
а неполярные распределяются в воздухе, образуя поверхностный слой
Воздух
Воздух
Раствор ПАВ
Раствор ПАВ
Вода
«Частокол Ленгмюра» образуется при насыщении
поверхностного слоя
Ориентация молекул ПАВ
на границе раствор - воздух
При малых концентрациях ПАВ в растворе их молекулы произвольно ориентируются в
поверхностном слое. С увеличением концентрации поверхностный слой заполняется
молекулами ПАВ, образующими мономолекулярный адсорбционный слой с плотнейшей
упаковкой (частокол Ленгмюра)
При насыщении поверхностного слоя молекулами ПАВ достигается предельная
адсорбция. При дальнейшем повышении концентрации ПАВ в растворе строение
адсорбционного слоя, а также поверхностное натяжение раствора больше не изменяются
28.
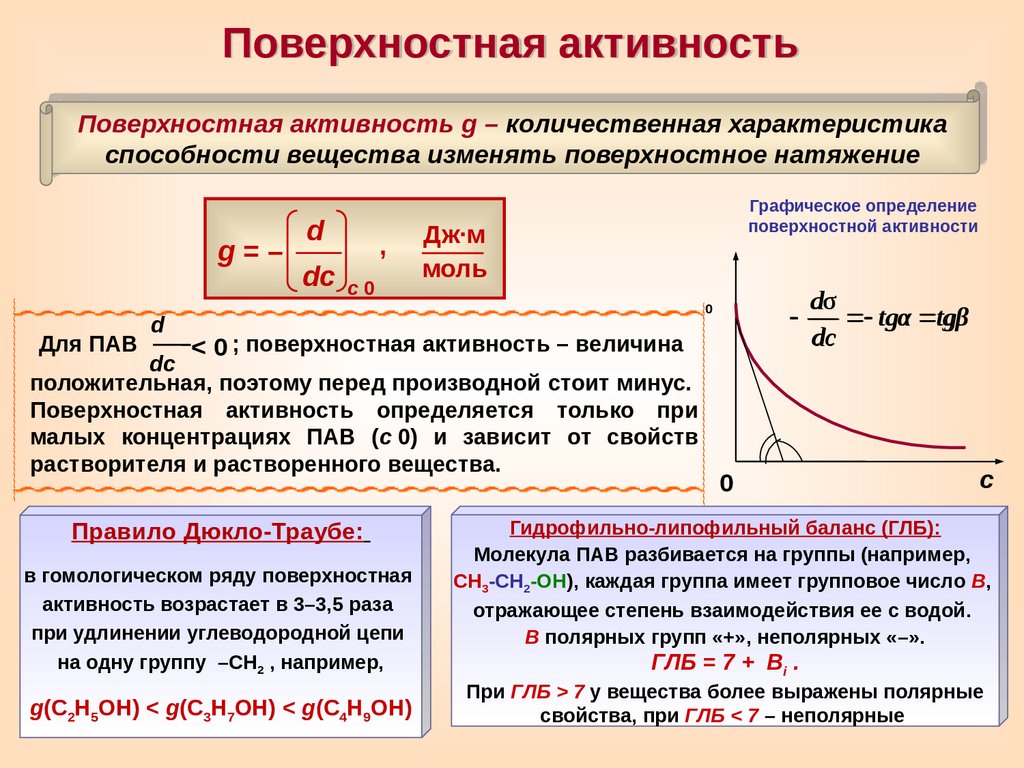
Поверхностная активностьПоверхностная активность g – количественная характеристика
способности вещества изменять поверхностное натяжение
g=–
Для ПАВ
d
d
dc c
0
,
Графическое определение
поверхностной активности
Дж∙м
моль
0
< 0 ; поверхностная активность – величина
dc
положительная, поэтому перед производной стоит минус.
Поверхностная активность определяется только при
малых концентрациях ПАВ (c
0) и зависит от свойств
растворителя и растворенного вещества.
Правило Дюкло-Траубе:
в гомологическом ряду поверхностная
активность возрастает в 3–3,5 раза
при удлинении углеводородной цепи
на одну группу –СН2 , например,
g(С2Н5ОН) < g(С3Н7ОН) < g(С4Н9ОН)
dσ
tgα tgβ
dc
0
с
Гидрофильно-липофильный баланс (ГЛБ):
Молекула ПАВ разбивается на группы (например,
СН3-СН2-ОН), каждая группа имеет групповое число В,
отражающее степень взаимодействия ее с водой.
В полярных групп «+», неполярных «–».
ГЛБ = 7 +
Вi .
При ГЛБ > 7 у вещества более выражены полярные
свойства, при ГЛБ < 7 – неполярные
29.
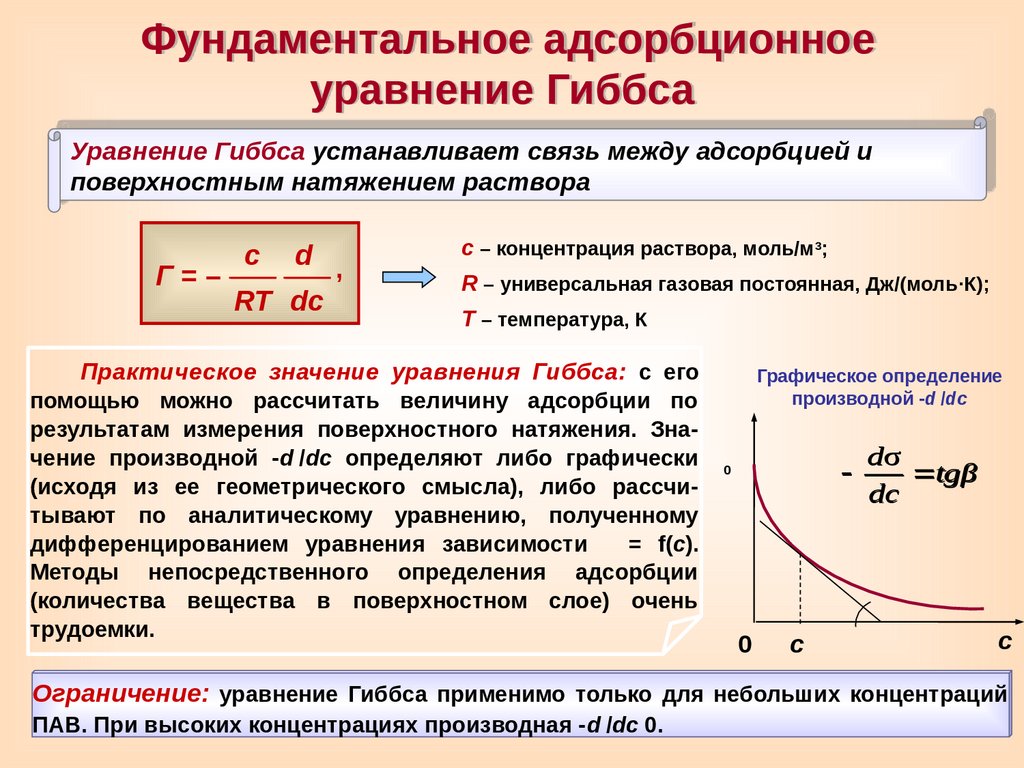
Фундаментальное адсорбционноеуравнение Гиббса
Уравнение Гиббса устанавливает связь между адсорбцией и
поверхностным натяжением раствора
Г=–
с
d ,
RT dc
с – концентрация раствора, моль/м3;
R – универсальная газовая постоянная, Дж/(моль∙К);
Т – температура, К
Практическое значение уравнения Гиббса: с его
помощью можно рассчитать величину адсорбции по
результатам измерения поверхностного натяжения. Значение производной -d
/dc определяют либо графически
0
(исходя из ее геометрического смысла), либо рассчитывают по аналитическому уравнению, полученному
дифференцированием уравнения зависимости = f(с).
Методы непосредственного определения адсорбции
(количества вещества в поверхностном слое) очень
трудоемки.
0
Графическое определение
производной -d
/dc
с
dσ
tgβ
dc
с
Ограничение: уравнение Гиббса применимо только для небольших концентраций
ПАВ. При высоких концентрациях производная -d
/dc
0.
30.
Уравнение мономолекулярной адсорбцииЛенгмюра
Общий вид
изотермы адсорбции
Г
Гmаx
с
0
Изотермы
адсорбции гомологов
Г
Уравнение Гиббса не описывает всю экспериментально
полученную изотерму адсорбции!
Уравнение Ленгмюра:
Гmаx
0
Экспериментально полученные изотермы адсорбции различных ПАВ
имели горизонтальный участок. Для гомологов все изотермы в
пределе сливаются. Это возможно, если:
1) все молекулы гомологов, независимо от длины УВ-радикала,
занимают на межфазной поверхности одинаковую площадь;
2) адсорбционный слой имеет толщину не более одной молекулы, т.е.
мономолекулярный.
с
kс
Г Г max
1 kс
Гmax – предельная адсорбция;
k – константа адсорбционно-десорбционного
равновесия
Анализ уравнения: в сильно разбавленных растворах, когда kc << 1, уравнение
принимает вид: Г=Гmaxkc (начальный (прямолинейный) участок изотермы адсорбции);
в концентрированных растворах kc >> 1, тогда Г = Гmax (горизонтальный участок изотермы)
Таким образом, уравнение Ленгмюра описывает всю изотерму
мономолекулярной адсорбции, включая ее горизонтальный участок
31.
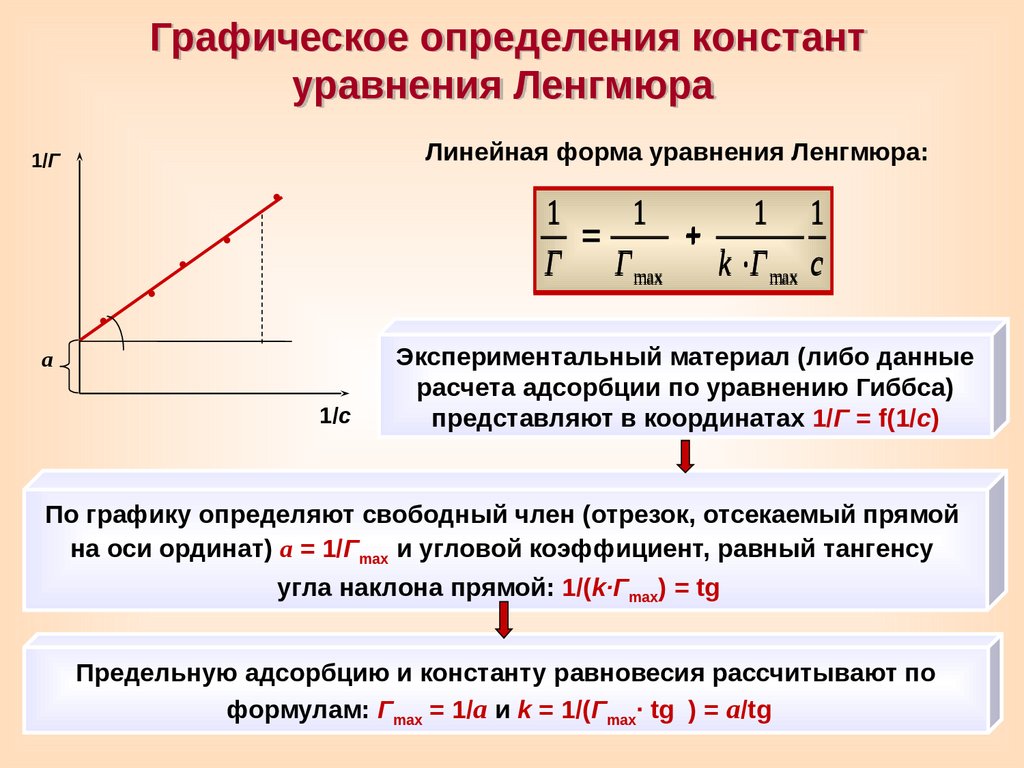
Графическое определения константуравнения Ленгмюра
Линейная форма уравнения Ленгмюра:
1/Г
1
1
1 1
Г Г max
k Г max c
а
1/с
Экспериментальный материал (либо данные
расчета адсорбции по уравнению Гиббса)
представляют в координатах 1/Г = f(1/c)
По графику определяют свободный член (отрезок, отсекаемый прямой
на оси ординат) а = 1/Гmax и угловой коэффициент, равный тангенсу
угла наклона прямой: 1/(k∙Гmax) = tg
Предельную адсорбцию и константу равновесия рассчитывают по
формулам: Гmax = 1/a и k = 1/(Гmax∙ tg
) = а/tg
32.
Определение длины молекулы ПАВ иплощади, занимаемой ею в насыщенном слое
Зная Гmax, можно рассчитать длину
молекулы ПАВ l и площадь,
занимаемую одной молекулой в
насыщенном поверхностном слое S0
l
S0
S (слоя) = 1 м2
Г max М
l
ρ
1
S0
Г max N A
М – молярная масса вещества, кг/моль;
– плотность вещества при температуре опыта, кг/м3
Na – число Авогадро, Na = 6,02∙1023 моль-1
33.
Связь между константами уравненийШишковского и Ленгмюра
Уравнение Шишковского
Уравнение Гиббса
Уравнение Ленгмюра
= Вln(1 + kc)
0 -
с d
Г=–
RT dc
kс
Г Г max
1 kс
В, k – константы
Гmax, k – константы
При малых концентрациях ПАВ расчет адсорбции по уравнениям Гиббса и Ленгмюра
дает близкие результаты, т.е. правые части этих уравнений можно приравнять:
c dσ Г max kc
RT dc
1 kc
dσ Г max RT kdc
1 kc
После интегрирования в определенных пределах от 0 до с и от
:
0 до
σ 0 σ Г max RT ln(1 kc)
Сопоставляя с уравнением Шишковского:
В Г max RT
Константа k в уравнениях Шишковского и Ленгмюра имеет один и тот же смысл
– это константа адсорбционно-десорбционного равновесия
34.
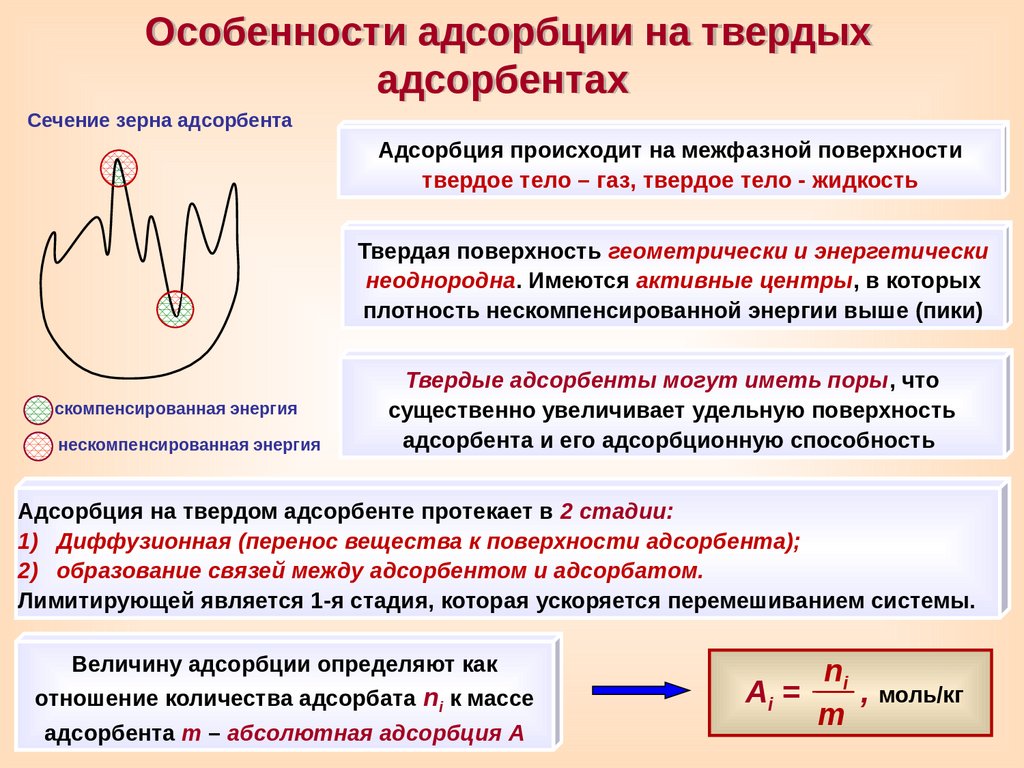
Особенности адсорбции на твердыхадсорбентах
Сечение зерна адсорбента
Адсорбция происходит на межфазной поверхности
твердое тело – газ, твердое тело - жидкость
Твердая поверхность геометрически и энергетически
неоднородна. Имеются активные центры, в которых
плотность нескомпенсированной энергии выше (пики)
скомпенсированная энергия
нескомпенсированная энергия
Твердые адсорбенты могут иметь поры, что
существенно увеличивает удельную поверхность
адсорбента и его адсорбционную способность
Адсорбция на твердом адсорбенте протекает в 2 стадии:
1) Диффузионная (перенос вещества к поверхности адсорбента);
2) образование связей между адсорбентом и адсорбатом.
Лимитирующей является 1-я стадия, которая ускоряется перемешиванием системы.
Величину адсорбции определяют как
отношение количества адсорбата ni к массе
адсорбента m – абсолютная адсорбция A
ni
Ai =
, моль/кг
m
35.
ТВЕРДЫЕ АДСОРБЕНТЫВ качестве адсорбентов наиболее часто применяют
углеродные сорбенты (древесный или костный уголь,
графитированная термическая сажа ГТС), бентонитовые
глины, силикагель, цеолиты, ионообменные смолы и др.
Ионообменная
смола
Активированный
уголь
Синтетический цеолит
Силикагель
36.
УГЛЕРОДНЫЕ СОРБЕНТЫПолучают из всевозможного сырья, которое при определенных условиях может
давать твердый углеродный остаток – ископаемых углей, торфа, древесины,
ореховой скорлупы, фруктовых косточек и животных костей. Лучшими считаются
угли, полученные из скорлупы кокосовых орехов и абрикосовых косточек
Углеродные сорбенты
используют в различной
форме: в виде порошка с
размером частиц до 0,8 мм,
гранул более крупного
размера, пленок, волокон
тканей
Углеродные сорбенты дополнительно активируют,
выдерживая при повышенной температуре в присутствии паров воды и СО2. При
этом выгорает смола, заполняющая поры углей,
удельная поверхность и
адсорбционная способность
возрастают
Порошковый
уголь
Экструдированный
уголь
Гранулированный
уголь
Ткань, пропитанная активным
углем
Формованный уголь
Удельная поверхность активированного угля, включая
поверхность всех его пор, может достигать 1000 м 2/г.
37.
справкаПервое упоминание об использовании
Применение углеродных
сорбентов
углей - в сан-скритских писаниях
Основные сферы применения:
очистка воды, пищевых масс;
очистка и разделение газов;
в хроматографии;
в медицине
Древней Индии. В них говорилось,
что «питьевую воду необходимо
предварительно пропускать через
уголь, выдерживать в медных сосудах
и подвергать действию солнечных
лучей».
1773 г. - немецкий химик Карл Шееле
сообщил об ад-сорбции газов на
древесном угле.
1785 г. - санкт-петербургский
аптекарь Ловиц Т. Е., впоследствии
ставший академиком, впервые
обратил внимание на способность угля
очищать спирт. В результате
многократных опытов он установил,
что даже простое встряхивание вина с
угольным порошком позволяет
получить намного более чистый и
качественный напиток.
1794 г. - активный уголь был
использован для осветле-ния сиропов
на сахарорафинадном заводе в
38.
СИЛИКАГЕЛЬПолучают высушиванием студня поликремниевой кислоты; по
химическому составу – это nSiO2
Товарный силикагель выпускают в виде
зерен или шаровидных гранул
размером 0,2 – 7 мм
Основные сферы применения:
осушка воздуха и промышленных
газов;
адсорбционная очистка неполярных
жидкостей;
рекуперации паров органических веществ;
в хроматографии – для разделения
спиртов, аминокислот, витаминов,
антибиотиков и др.;
крупнопористые силикагеля – в качестве носителей катализаторов
Удельная поверхность силикагеля
~ 500 м2/г
39.
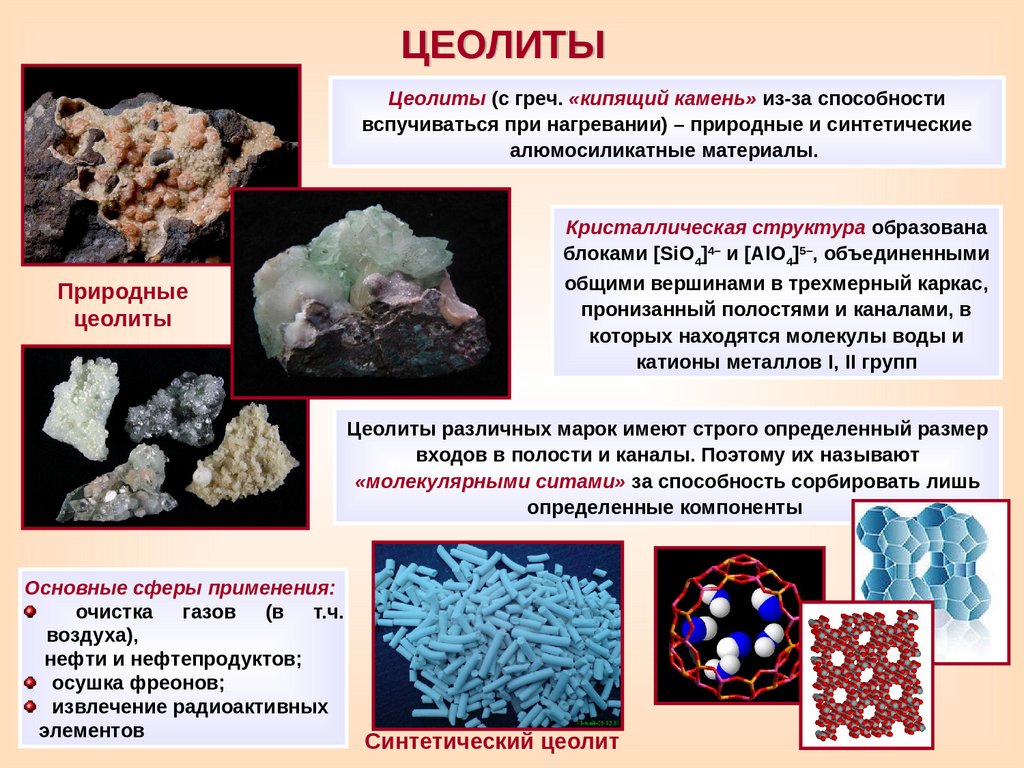
ЦЕОЛИТЫЦеолиты (с греч. «кипящий камень» из-за способности
вспучиваться при нагревании) – природные и синтетические
алюмосиликатные материалы.
Природные
цеолиты
Кристаллическая структура образована
блоками [SiO4]4– и [AlO4]5–, объединенными
общими вершинами в трехмерный каркас,
пронизанный полостями и каналами, в
которых находятся молекулы воды и
катионы металлов I, II групп
Цеолиты различных марок имеют строго определенный размер
входов в полости и каналы. Поэтому их называют
«молекулярными ситами» за способность сорбировать лишь
определенные компоненты
Основные сферы применения:
очистка газов (в т.ч.
воздуха),
нефти и нефтепродуктов;
осушка фреонов;
извлечение радиоактивных
элементов
Синтетический цеолит
40.
БЕНТОНИТОВЫЕ ГЛИНЫБентониты (от названия месторождения Бентон, США)
– тонкодисперсные глины, состоящие на 60-70 % из
минералов группы монтмориллонита (Al2O3∙4SiO2∙H2O). В
зависимости от состава цвет глины может быть желтым,
коричневым, серым, зеленым или голубым.
Глины
различных
месторождений
Монтмориллонит
Основные сферы применения:
в пищевой промышленности – для осветления и
стабилизации виноматериалов и вин, и частично фруктовых
соков, очистки растительных масел;
в биотехнологии – для очистки белков, ферментов;
в косметологии
41.
Пористость адсорбентовПористые
Твердые
адсорбенты
Непористые
Пористость адсорбента определяется
отношением суммарного объема пор
Vп к общему объему адсорбента Vадс
П = Vп/Vадс.
Классификация пористых адсорбентов
Тип
Диаметр пор,
Примеры
адсорбентов
нм
Макропористые
более 50
Асбест, мука, древесина
Мезопористые
2-50
Микропористые
менее 2
Бентониты, силикагель
Активированный уголь,
цеолиты, пористые стекла
Пористость адсорбента имеет большое значение для адсорбции: чем она выше, тем больше
удельная поверхность и выше адсорбционная способность. Однако это справедливо только в
том случае, если молекулы адсорбата невелики и легко могут проникать в поры
42.
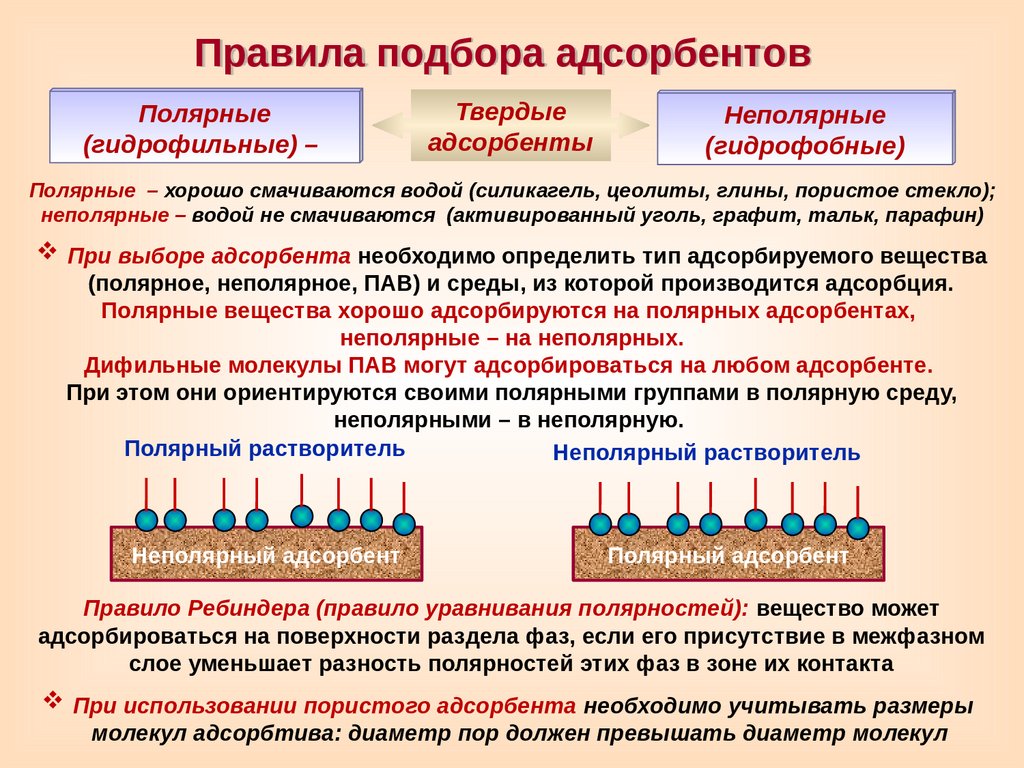
Правила подбора адсорбентовПолярные
(гидрофильные) –
Твердые
адсорбенты
Неполярные
(гидрофобные)
Полярные – хорошо смачиваются водой (силикагель, цеолиты, глины, пористое стекло);
неполярные – водой не смачиваются (активированный уголь, графит, тальк, парафин)
При выборе адсорбента необходимо определить тип адсорбируемого вещества
(полярное, неполярное, ПАВ) и среды, из которой производится адсорбция.
Полярные вещества хорошо адсорбируются на полярных адсорбентах,
неполярные – на неполярных.
Дифильные молекулы ПАВ могут адсорбироваться на любом адсорбенте.
При этом они ориентируются своими полярными группами в полярную среду,
неполярными – в неполярную.
Полярный растворитель
Неполярный растворитель
Неполярный адсорбент
Полярный адсорбент
Правило Ребиндера (правило уравнивания полярностей): вещество может
адсорбироваться на поверхности раздела фаз, если его присутствие в межфазном
слое уменьшает разность полярностей этих фаз в зоне их контакта
При использовании пористого адсорбента необходимо учитывать размеры
молекул адсорбтива: диаметр пор должен превышать диаметр молекул
43.
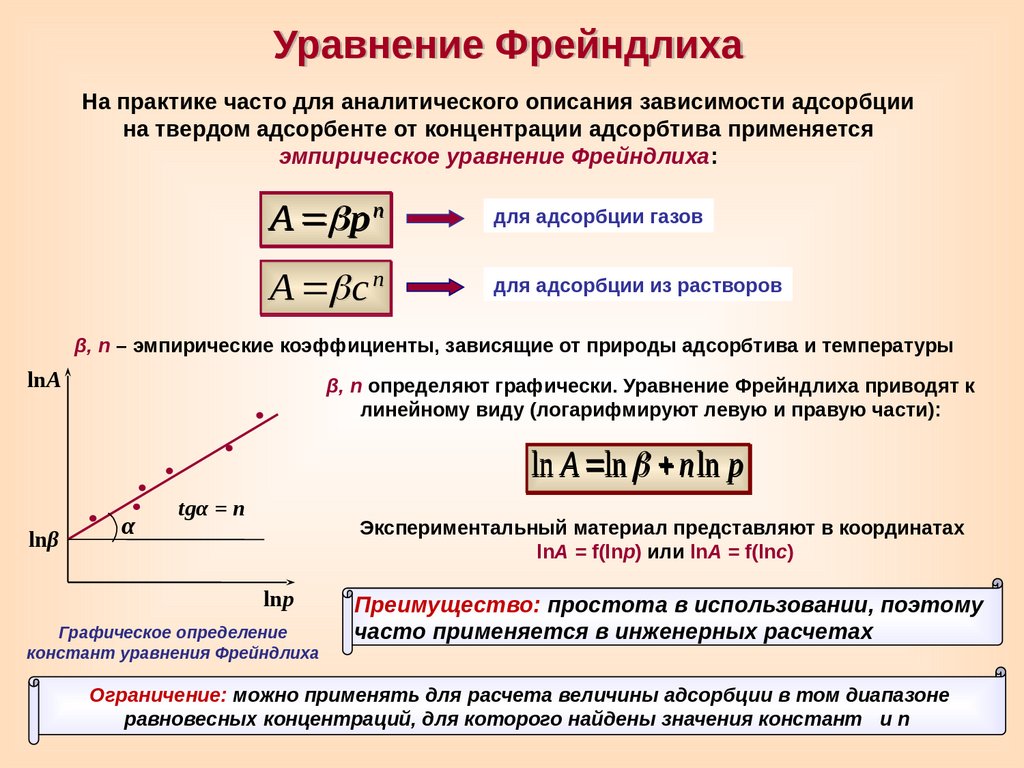
Уравнение ФрейндлихаНа практике часто для аналитического описания зависимости адсорбции
на твердом адсорбенте от концентрации адсорбтива применяется
эмпирическое уравнение Фрейндлиха:
А p n
для адсорбции газов
А c n
для адсорбции из растворов
β, n – эмпирические коэффициенты, зависящие от природы адсорбтива и температуры
lnА
β, n определяют графически. Уравнение Фрейндлиха приводят к
линейному виду (логарифмируют левую и правую части):
ln A ln n ln р
lnβ
α
tgα = n
Экспериментальный материал представляют в координатах
lnA = f(lnр) или lnA = f(lnс)
lnp
Графическое определение
констант уравнения Фрейндлиха
Преимущество: простота в использовании, поэтому
часто применяется в инженерных расчетах
Ограничение: можно применять для расчета величины адсорбции в том диапазоне
равновесных концентраций, для которого найдены значения констант и n
44.
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМ.ЭЛЕКТРОКИНЕТИЧЕСКИЕ ЯВЛЕНИЯ
45.
Влияние электрического потенциала поверхности наповерхностное натяжение
В дисперсных системах, обладающих избытком свободной поверхностной энергии Gs
Gs =
S,
самопроизвольно протекают процессы, приводящие к снижению Gs (∆Gs < 0).
Экспериментально установлено, что поверхностное натяжение зависит от электрического
потенциала поверхности
: максимальное значение
наблюдается при потенциале
нулевого заряда
по
н.з., когда поверхность не имеет заряда; увеличение
абсолютному значению вызывает снижение
Историческая справка:
Первые исследования
влияния электрического поля на поверхностное натяжение
жидкости выполнены еще во второй половине XIX в.
французским физиком Габриэлем Липпманом. В своих
опытах он измерял поверхностное натяжение на границе
ртуть - разбавленная серная кислота при различных
значениях электрического потенциала ртутной поверхности
Термодинамическое соотношение между
и выражает 1-е уравнение Липпмана:
d
d
qss
qs – поверхностная плотность заряда
н.з.
Вывод: появление электрического заряда
на поверхности, сопровождающееся
уменьшением поверхностного натяжения,
– процесс самопроизвольный
46.
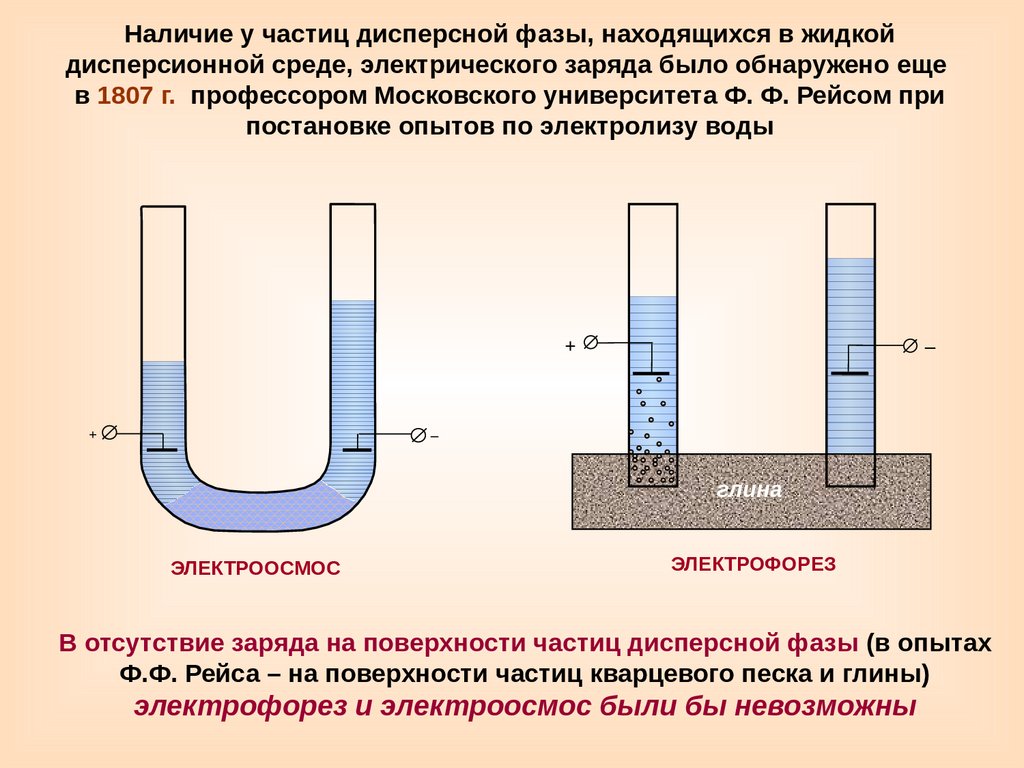
Наличие у частиц дисперсной фазы, находящихся в жидкойдисперсионной среде, электрического заряда было обнаружено еще
в 1807 г. профессором Московского университета Ф. Ф. Рейсом при
постановке опытов по электролизу воды
+
+
–
–
глина
ЭЛЕКТРООСМОС
ЭЛЕКТРОФОРЕЗ
В отсутствие заряда на поверхности частиц дисперсной фазы (в опытах
Ф.Ф. Рейса – на поверхности частиц кварцевого песка и глины)
электрофорез и электроосмос были бы невозможны
47.
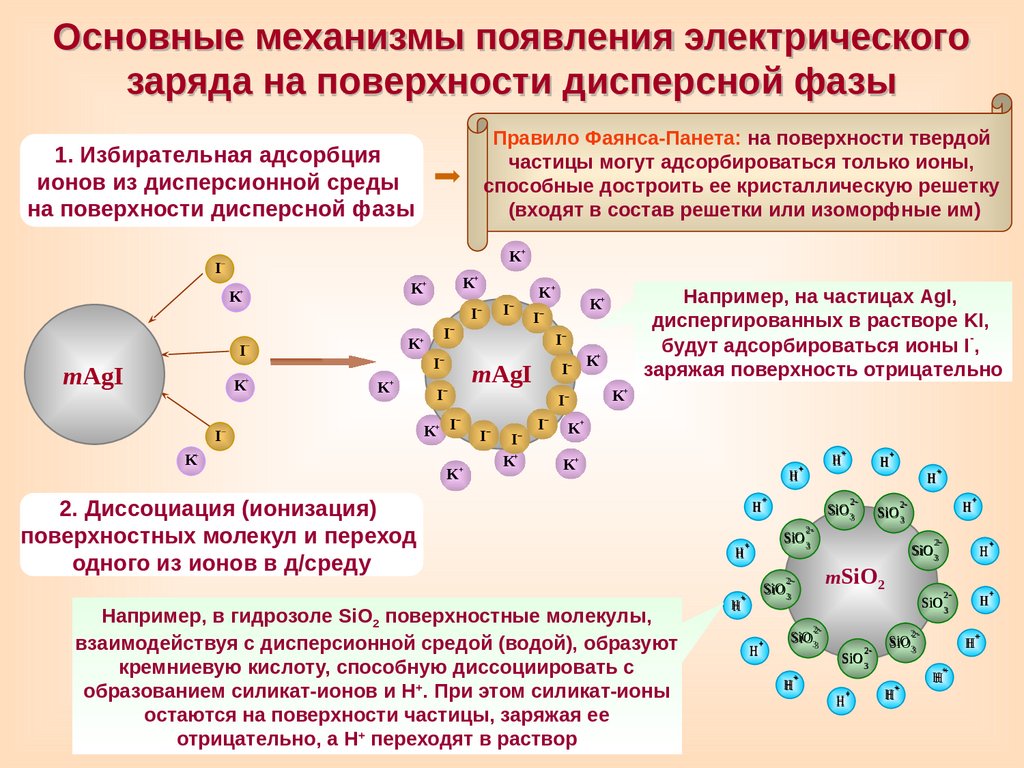
Основные механизмы появления электрическогозаряда на поверхности дисперсной фазы
Правило Фаянса-Панета: на поверхности твердой
частицы могут адсорбироваться только ионы,
способные достроить ее кристаллическую решетку
(входят в состав решетки или изоморфные им)
1. Избирательная адсорбция
ионов из дисперсионной среды
на поверхности дисперсной фазы
I
K+
–
K
I
mAgI
K+
K+
+
K+
–
K+
K+
I–
K+
I
I–
I
K+
–
Например, на частицах AgI,
диспергированных в растворе KI,
будут адсорбироваться ионы I-,
заряжая поверхность отрицательно
K+
I–
I–
I–
mAgI
I–
K+ I
–
–
K
I–
+
I
K+
–
K+
I–
K+
I–
I–
+
K
K+
2. Диссоциация (ионизация)
поверхностных молекул и переход
одного из ионов в д/среду
Например, в гидрозоле SiO2 поверхностные молекулы,
взаимодействуя с дисперсионной средой (водой), образуют
кремниевую кислоту, способную диссоциировать с
образованием силикат-ионов и Н+. При этом силикат-ионы
остаются на поверхности частицы, заряжая ее
отрицательно, а Н+ переходят в раствор
Н
Н
Н
Н
22
НН
Н
22
Н
2
Н
SiO 3
22
Н
SiO 33
mSiO2
22
SiO 33
SiO 3
SiO 3
Н
2
SiO 33
2
Н
SiO
SiO33
Н
22
2
SiO 3
Н
SiO 33
Н
НН
НН
48.
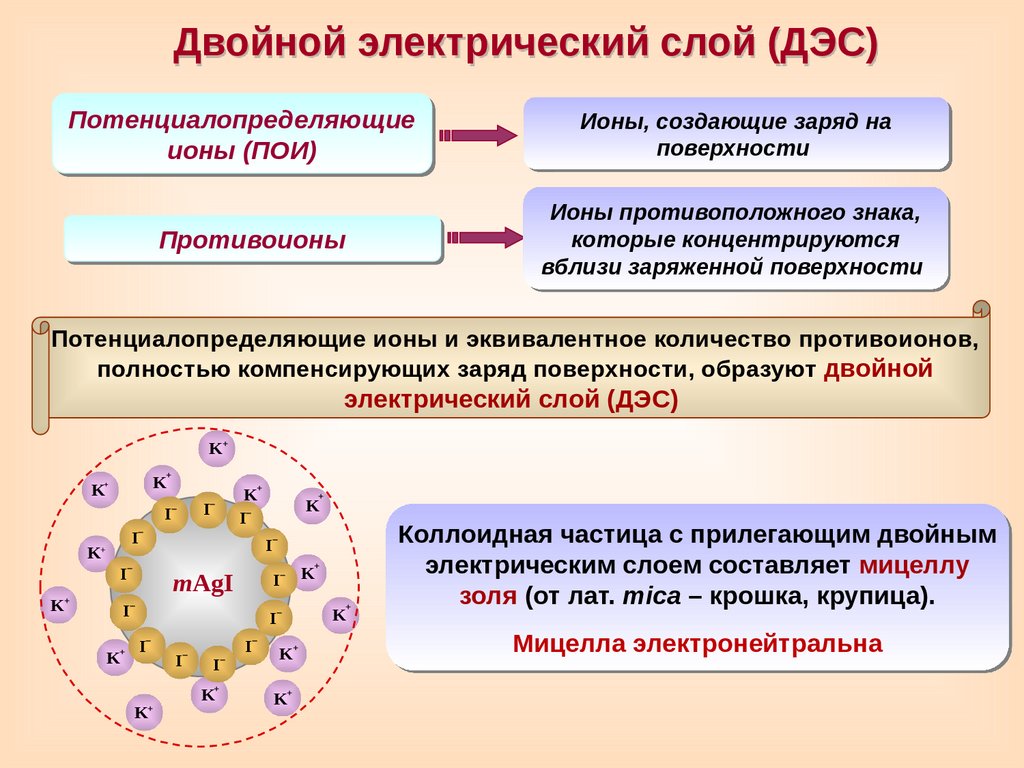
Двойной электрический слой (ДЭС)Потенциалопределяющие
ионы (ПОИ)
Ионы, создающие заряд на
поверхности
Противоионы
Ионы противоположного знака,
которые концентрируются
вблизи заряженной поверхности
Потенциалопределяющие ионы и эквивалентное количество противоионов,
полностью компенсирующих заряд поверхности, образуют двойной
электрический слой (ДЭС)
K+
+
K
K+
+
I
–
I
K
I–
–
–
K+
K+
I
I–
I–
+
I– K
mAgI
I–
K+
+
K
I–
I–
K
+
–
I
–
I
K+
I–
K+
K+
K
+
Коллоидная частица с прилегающим двойным
электрическим слоем составляет мицеллу
золя (от лат. mica – крошка, крупица).
Мицелла электронейтральна
49.
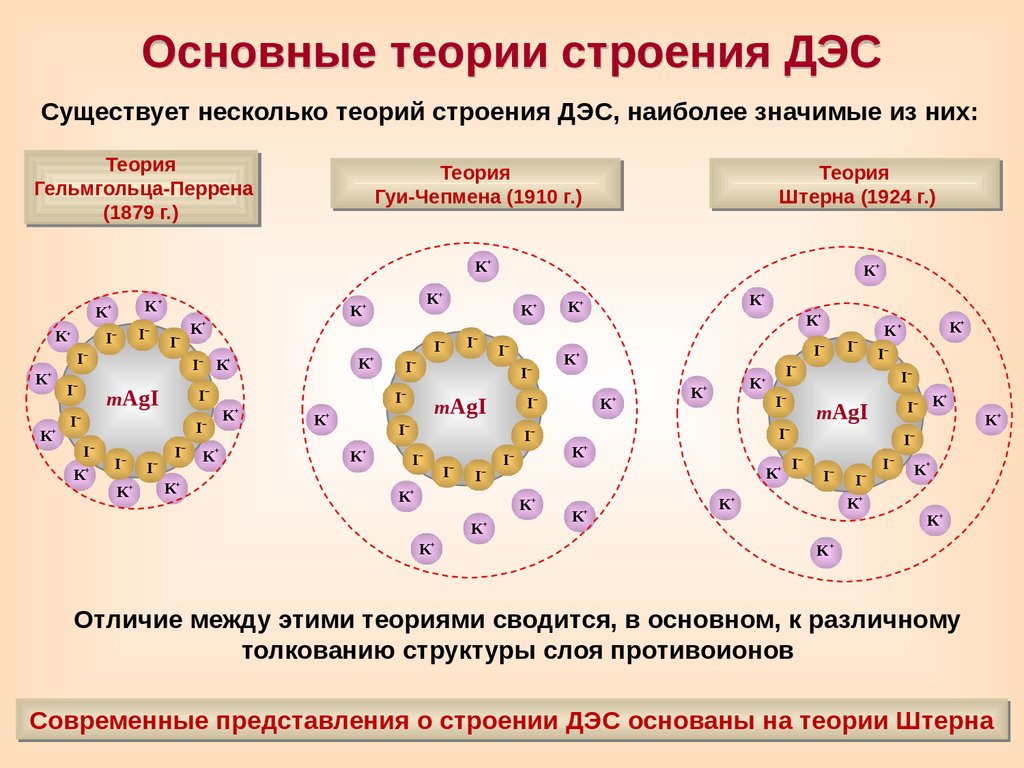
Основные теории строения ДЭССуществует несколько теорий строения ДЭС, наиболее значимые из них:
Теория
Гельмгольца-Перрена
(1879 г.)
Теория
Гуи-Чепмена (1910 г.)
Теория
Штерна (1924 г.)
K+
K
+
K+
K+
I
I–
–
I–
K+
K
+
I–
I
–
I–
K+
I
–
I
mAgI
K
–
K+
I–
I–
K+
I–
I–
K
+
K+
+
–
I
K+
K+
K+
+
K
I
I
K
+
I
K
K+
–
I
–
–
I–
I
–
mAgI
I–
+
K+
–
I
I–
I–
K+
I
K+
K+
K+
K+
–
I
K+
K+
K+
I–
I
I–
I–
+
I– K
mAgI
–
K+
K+
–
–
I–
I
–
K+
K+
K+
K+
I–
I
–
K+
K+
I–
I–
I–
I
–
K+
K+
I–
K+
+
K
K+
K+
Отличие между этими теориями сводится, в основном, к различному
толкованию структуры слоя противоионов
Современные представления о строении ДЭС основаны на теории Штерна
50.
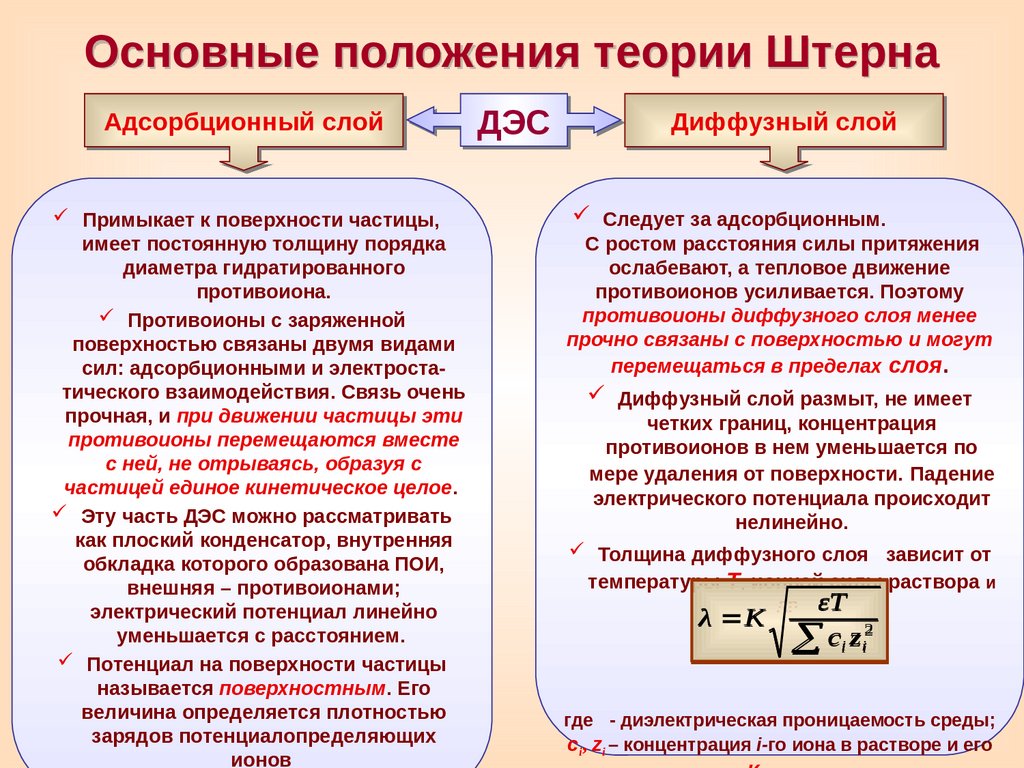
Основные положения теории ШтернаАдсорбционный слой
Примыкает к поверхности частицы,
имеет постоянную толщину порядка
диаметра гидратированного
противоиона.
Противоионы с заряженной
поверхностью связаны двумя видами
сил: адсорбционными и электростатического взаимодействия. Связь очень
прочная, и при движении частицы эти
противоионы перемещаются вместе
с ней, не отрываясь, образуя с
частицей единое кинетическое целое.
Эту часть ДЭС можно рассматривать
как плоский конденсатор, внутренняя
обкладка которого образована ПОИ,
внешняя – противоионами;
электрический потенциал линейно
уменьшается с расстоянием.
Потенциал на поверхности частицы
называется поверхностным. Его
величина определяется плотностью
зарядов потенциалопределяющих
ионов
ДЭС
Диффузный слой
Следует за адсорбционным.
С ростом расстояния силы притяжения
ослабевают, а тепловое движение
противоионов усиливается. Поэтому
противоионы диффузного слоя менее
прочно связаны с поверхностью и могут
перемещаться в пределах слоя.
Диффузный слой размыт, не имеет
четких границ, концентрация
противоионов в нем уменьшается по
мере удаления от поверхности. Падение
электрического потенциала происходит
нелинейно.
Толщина диффузного слоя
зависит от
температуры Т, ионной силы раствора и
λ K
εT
ci zi2
др.
где
- диэлектрическая проницаемость среды;
сi, zi – концентрация i-го иона в растворе и его
51.
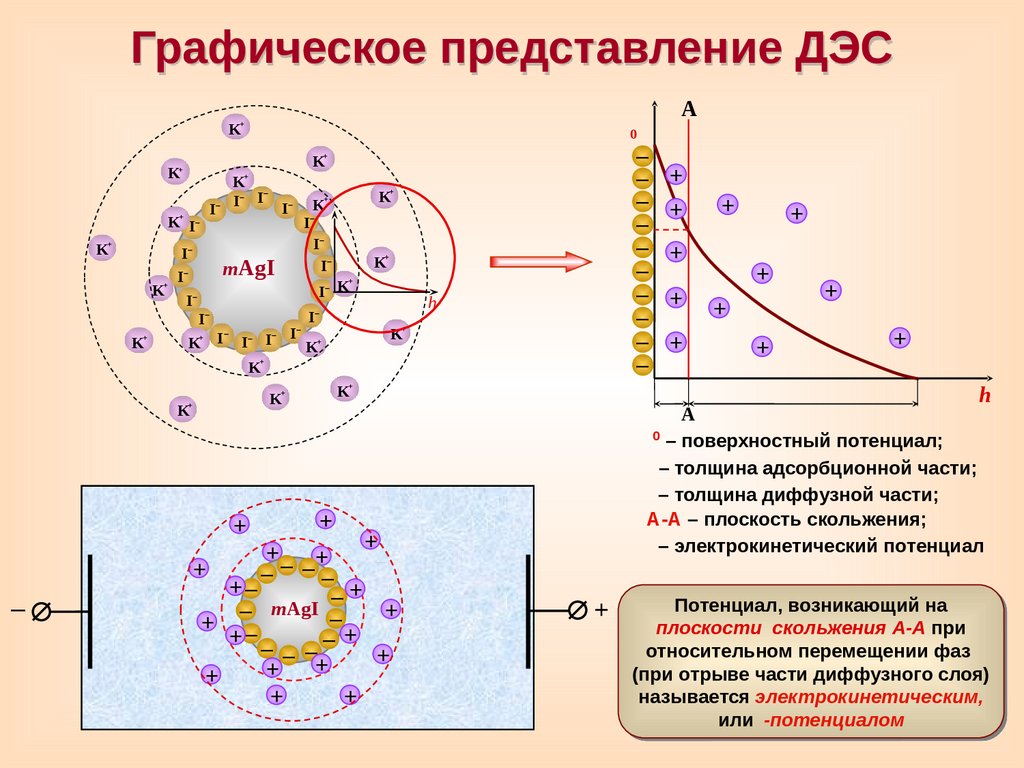
Графическое представление ДЭС+
K
0
–
–
–
–
–
–
–
–
–
–
+
K+
I–
K+ I–
K+
I–
I–
+
K
I–
+
K
K+
–
I– I I–
mAgI
I
+
K
–
I
–
I
–
I
–
K+
K
+
+
+
K
I
I–
I–
+
I– K
K
–
I
K+
h
–
K+
K+
K+
K+
+
–
I–
K
+
+
+
+
+
+
А
+
+
+
+
+
+
h
0
– поверхностный потенциал;
– толщина адсорбционной части;
– толщина диффузной части;
+
+
+
+
+
–– ––
+–
–+
– mAgI – +
+ –
+
+
– – ––
+
+
+
+
+
+
А
А-А – плоскость скольжения;
– электрокинетический потенциал
+
Потенциал, возникающий на
плоскости скольжения А-А при
относительном перемещении фаз
(при отрыве части диффузного слоя)
называется электрокинетическим,
или
-потенциалом
52.
Строение мицеллы гидрофобного золяМицелла гидрофобного золя (от лат. mica – крошка, крупица) –
частица дисперсной фазы, окруженная ДЭС
AgI
AgI
I–
AgI
AgI
AgI
K
+
-
NO3
AgNO3 + KI = AgI + KNO3
избыток
Условие получения устойчивого золя –
избыток одного из реагентов (стабилизатор золя)
AgI
mAgI
AgI
AgI
AgI
AgI
AgI
AgI
AgI
Агрегат
Ядро
Частица
Мицелла
Основа мицеллы – нерастворимое в воде
вещество – имеет кристаллическое
строение и размер порядка 1 – 100 нм
АГРЕГАТ
Агрегат + Потенциалопределяющие ионы
ЯДРО
Ядро + противоионы адсорбционного слоя,
прочно удерживаемые поверхностью ДФ
ЧАСТИЦА
Мицеллярная формула золя АgI, стабилизированного KI
Ядро
(mAgI nI (n-x)K+ )–x xK+
–
Агрегат
Частица
Мицелла
53.
ВЛИЯНИЕ ЭЛЕКТРОЛИТОВ НА ПОВЕРХНОСТНЫЙ ИЭЛЕКТРОКИНЕТИЧЕСКИЙ ПОТЕНЦИАЛЫ
54.
0Влияние электролитов на
-и
-потенциалы
Индифферентные
(безразличные)
Неиндифферентные
(не безразличные)
ЭЛЕКТРОЛИТЫ
Не имеют ионов, способных
достраивать кристаллическую
решетку агрегата. Поэтому такие
электролиты не могут существенно
изменить поверхностный потенциал
0
(потенциал ядра)
0
Вызывают изменение
. Один из
ионов неиндифферентного
электролита (неиндифферентный
ион) либо входит в состав
кристаллической решетки агрегата,
либо способен образовывать
малорастворимое соединение с
потенциалопределяющим ионом
Например, для положительного золя AgI
x
3
{m AgI n Ag (n x) NO } x NO
3
электролиты KNO3, NaNO2 являются индифферентными,
а KI, NaCl, AgNO3 – неиндифферентными (из-за ионов I–, Cl–и Ag+)
Изменение
-потенциала вызывают все электролиты
55.
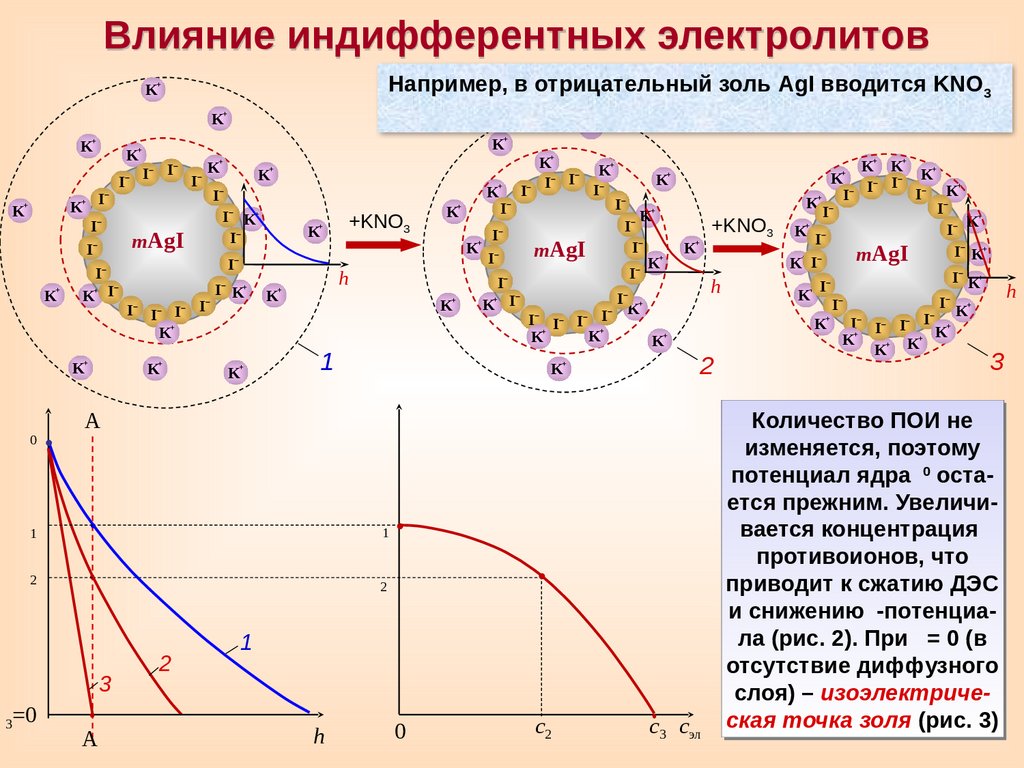
Влияние индифферентных электролитовНапример, в отрицательный золь AgI вводится KNO3
K+
K+
K+
K
K
K+
I–
I–
I–
K+
K+ I
K+
0
I
I–
+
+
–
K
I– I
+
–
I
–
K
I–
I– K+
I–
mAgI
I
I
–
K+
K+
I
–
I
–
I– K+
K+
+KNO3
K+
I–
–
–
K+
h
K+
1
1
2
2
А
2
1
h
0
с2
с3 сэл
K+ K+
K+
–
K
–
I
+
I
–
I– –K
K+ – I
I
I
K+
+
I–
K –
I
I– K+
m
AgI
+ –
K I
I– K+
–
+ I
K
I– K+
I–
–
+
–
–
I I– I I +
K
K
+
K+
K+ K
+
+KNO3
h
2
А
3
K+
K+
–
–
I
K+
I
I– –
K+ I–
I
I–
+
K+
– K
I
I–
I–
K+ –
K+
mAgI
+
I
K
I–
I–
–
I– +
K+ I
K+
K
–
–
I I – I– I
+
+
K
K
K+
K+
1
3=0
K+
+
h
3
Количество ПОИ не
изменяется, поэтому
0
потенциал ядра
остается прежним. Увеличивается концентрация
противоионов, что
приводит к сжатию ДЭС
и снижению
-потенциала (рис. 2). При = 0 (в
отсутствие диффузного
слоя) – изоэлектрическая точка золя (рис. 3)
56.
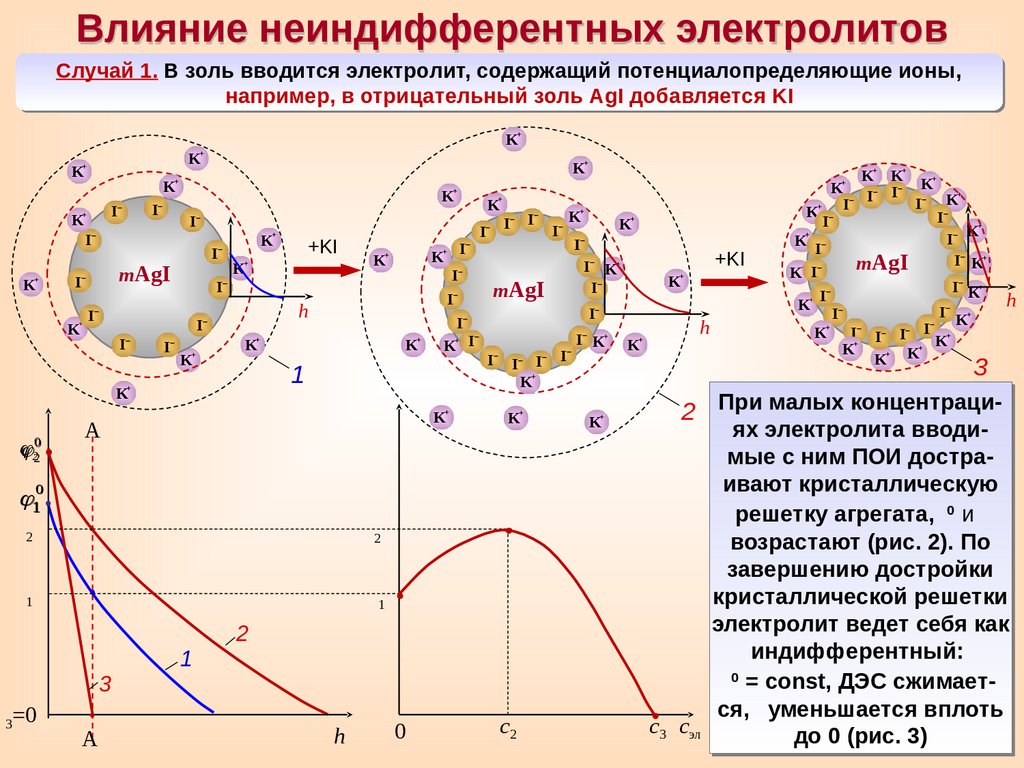
Влияние неиндифферентных электролитовСлучай 1. В золь вводится электролит, содержащий потенциалопределяющие ионы,
например, в отрицательный золь AgI добавляется KI
K+
+
K
K+
K
I
+
K
+
K
I
K+
+
I–
–
I
I–
–
I
mAgI
–
I–
K+
I–
K+
K
2200
K+
K+
1
K
+
–
I– I I – K
K+
I
–
I–
+ I
K
I– K+
I–
I–
mAgI
I–
I–
I–
–
I– K+ K+
K+ K+ I
–
I– I – I – I
K+
h
+
+KI
+
–
K+
–
K+
–
I–
I
I–
K+
А
K+
K+
0
10
2
2
1
1
3=0
3
А
1
2
h
0
с2
K+
+KI
+
K
h
K+ K+
K+
–
K+
–
I
+
I
–
I– –K
I
+
K –
I
I
K+
+
I–
K –
I
I– K+
mAgI
+ –
K I
I– K+
–
+ I
K
I– K+
I–
–
K+ I– I– I– I +
K
+
K+
+
K
K
h
3
2 При малых концентраци-
ях электролита вводимые с ним ПОИ достраивают кристаллическую
0
решетку агрегата,
и
возрастают (рис. 2). По
завершению достройки
кристаллической решетки
электролит ведет себя как
индифферентный:
0
= const, ДЭС сжимается, уменьшается вплоть
с3 сэл
до 0 (рис. 3)
57.
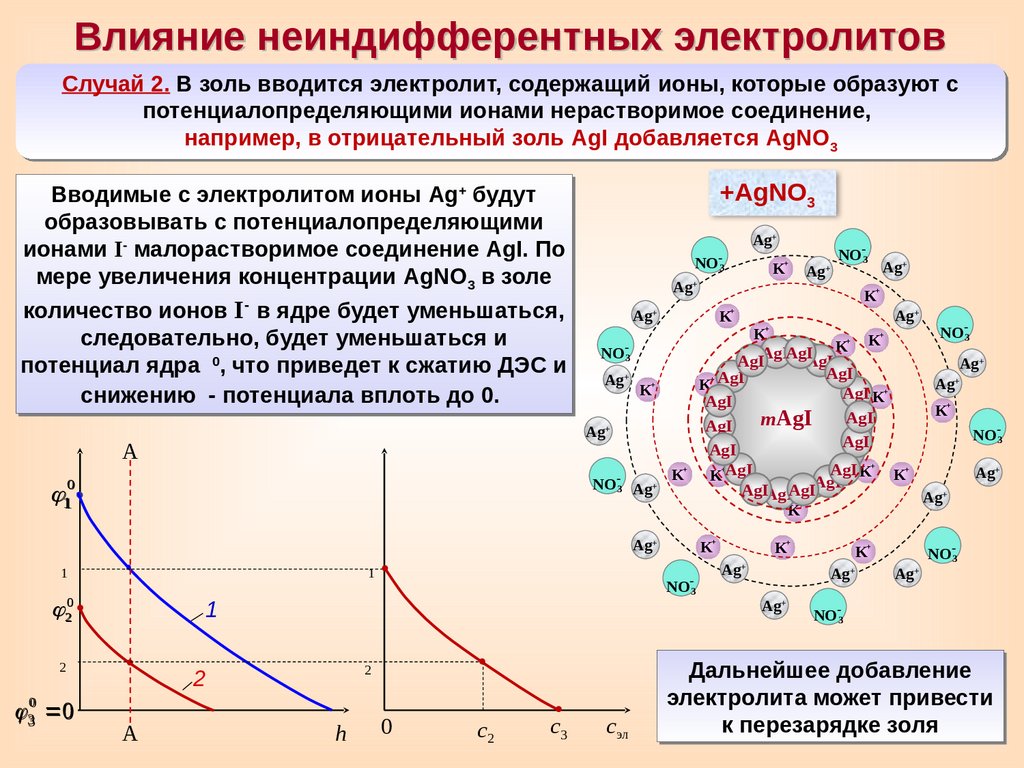
Влияние неиндифферентных электролитовСлучай 2. В золь вводится электролит, содержащий ионы, которые образуют с
потенциалопределяющими ионами нерастворимое соединение,
например, в отрицательный золь AgI добавляется AgNO3
Вводимые с электролитом ионы Ag+ будут
образовывать с потенциалопределяющими
ионами I- малорастворимое соединение AgI. По
мере увеличения концентрации AgNO3 в золе
количество ионов I- в ядре будет уменьшаться,
следовательно, будет уменьшаться и
0
потенциал ядра
, что приведет к сжатию ДЭС и
снижению
- потенциала вплоть до 0.
А
Ag
Ag+
-
NO 3
Ag+
1
2
2
2
0
30 0
А
h
0
с2
с3
сэл
K
Ag
+
NO-3
K+
K+
Ag+
Ag+
NO-3
+
K+
K
I I I–
I– AgIAgIAgI
AgI
Ag+
–
AgI
I–
+ AgI
I
+
Ag
K
AgI
I– K+
AgI
I–
–
K+
I
AgI
m
AgI
AgI
I–
NO-3
–
AgI
I
–
AgI
I
–
+
+AgI
I– K+ K+
AgI
Ag+
K
K I
–
AgI
–
–
I
–
I
AgIAgI
I
I AgI
Ag+
K+
+
K
K+
NO-3
1
+
+
Ag+
1
20
K+
NO-3 Ag+
0
Ag+
NO-3
Ag+
1
+AgNO3
–
–
K+
Ag
+
Ag
+
Ag+
NO-3
K+
Ag
+
NO-3
Дальнейшее добавление
электролита может привести
к перезарядке золя
58.
ЭЛЕКТРОКИНЕТИЧЕСКИЕ ЯВЛЕНИЯЭлектрокинетические явления – это явления, которые возникают при
воздействии электрического поля на дисперсную систему (электрофорез,
электроосмос) или в результате перемещения частиц дисперсной фазы или
дисперсионной среды (потенциалы течения и оседания).
Все электрокинетические явления обусловлены наличием ДЭС на
межфазной поверхности, интенсивность их определяется величиной
-потенциала
59.
ЭлектрофорезЭлектрофорез – направленное движение частиц
дисперсной фазы относительно дисперсионной
среды под действием внешнего электрического поля
При наложении внешнего электрического поля происходит разрыв
мицеллы по плоскости скольжения: коллоидная частица перемещается к одному электроду, а противоионы диффузного слоя, энергетически менее прочно связанные с поверхностью, – к другому
Электрофоретическая скорость
0
E
+
–
Е – напряженность (градиент) внешнего электрического
поля; - динамическая вязкость;
,
0 – диэлектрическая
проницаемость среды и электрическая проницаемость;
- фактор формы (для сферы = 0,66)
Электрофоретический эффект:
под действием электрического поля противоионы
передвигаются в направлении, противоположном
движению частицы. За счет гидратации они увлекают за собой и окружающую их жидкость, создавая противоток движению частицы
+
+
+
+
+
–– ––
+–
–+
mAgI
–
– +
+ –
+
+
– – ––
+
+
+
+
+
+
Релаксационный эффект:
вследствие движения фаз в противоположных
направлениях симметрия диффузной части ДЭС
нарушается. Возникает добавочное электрическое
поле, направленное против внешнего и стремящееся двигать частицу в обратном направлении
+
60.
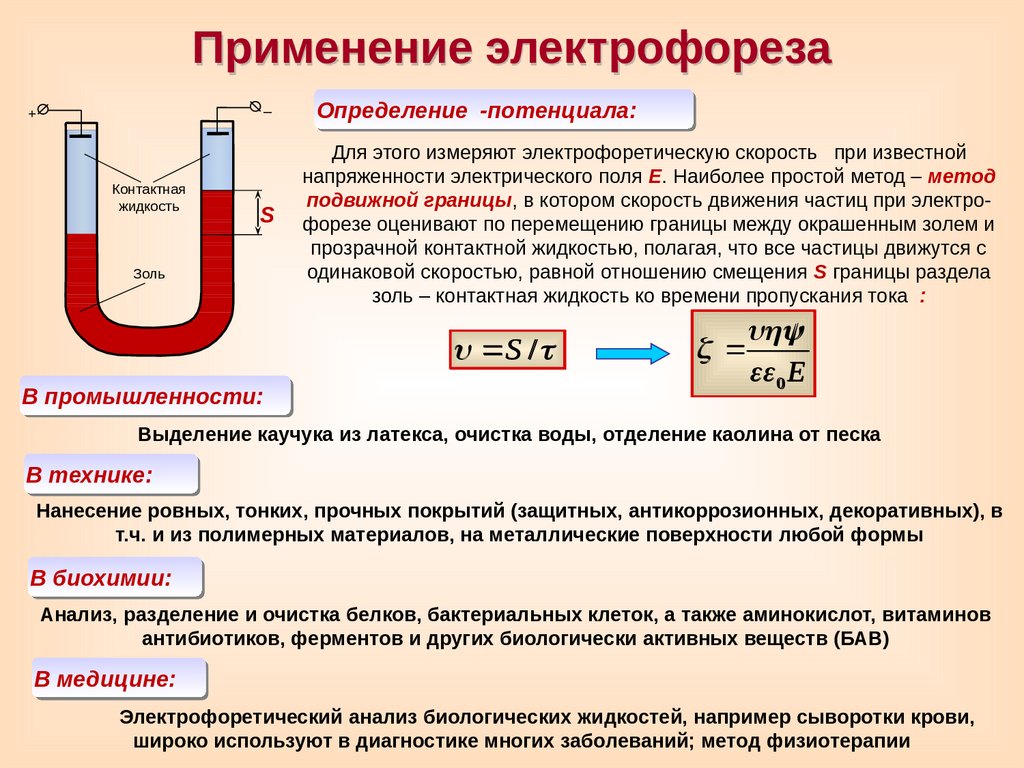
Применение электрофореза–
+
Контактная
жидкость
S
Золь
Определение
-потенциала:
Для этого измеряют электрофоретическую скорость при известной
напряженности электрического поля Е. Наиболее простой метод – метод
подвижной границы, в котором скорость движения частиц при электрофорезе оценивают по перемещению границы между окрашенным золем и
прозрачной контактной жидкостью, полагая, что все частицы движутся с
одинаковой скоростью, равной отношению смещения S границы раздела
золь – контактная жидкость ко времени пропускания тока
:
S /
В промышленности:
0 E
Выделение каучука из латекса, очистка воды, отделение каолина от песка
В технике:
Нанесение ровных, тонких, прочных покрытий (защитных, антикоррозионных, декоративных), в
т.ч. и из полимерных материалов, на металлические поверхности любой формы
В биохимии:
Анализ, разделение и очистка белков, бактериальных клеток, а также аминокислот, витаминов
антибиотиков, ферментов и других биологически активных веществ (БАВ)
В медицине:
Электрофоретический анализ биологических жидкостей, например сыворотки крови,
широко используют в диагностике многих заболеваний; метод физиотерапии
61.
ЭлектроосмосЭлектроосмос – движение дисперсионной среды через
неподвижную капиллярно-пористую перегородку под
действием внешнего электрического поля
–
+
Капиллярно-пористое
тело
–+–
+
+
–+–
–+ –
–+ –
+
+
+
– +–
+
– +–
– + –
+
+
– + –
– + –
+
– + –
Движение противоионов
в капилляре
Электроосмотический перенос жидкости происходит в
капиллярно-пористых системах и обусловлен наличием ДЭС
на внутренней поверхности капилляров пористого тела.
Рассмотрим сечение отдельного капилляра, например,
кварцевого, поверхность которого заряжена отрицательно.
Пленка жидкости вместе с противоионами адсорбционного
слоя «прилипает» к стенке капилляра и остается
– неподвижной, а противоионы диффузной части ДЭС, слабо
связанные с поверхностью, под действием внешнего
электрического поля перемещаются к противоположно
заряженному электроду. Противоионы гидратированы,
поэтому они увлекают за собой и окружающую их жидкость.
Применение электроосмоса:
1. Определение
-потенциала на поверхности капиллярно-пористых тел.
2. Удаление избыточной влаги из почв при прокладке транспортных магистралей и
гидротехническом строительстве, обезвоживания древесины и пористых материалов,
осушки зданий, понижения уровня грунтовых вод
62.
рПотенциал течения
Потенциал течения (эффект Квинке) –
возникновение разности потенциалов при протекании
жидкости, находящейся под давлением, через неподвижную
капиллярно-пористую перегородку
Капиллярнопористое тело
р
– +–
+
– +–
–+ –
–+ –
+
+
+
–+ –
+
–+–
– + –
+
+
– + –
– + –
+
– + –
Движение противоионов
в капилляре
При течении жидкости по капиллярам под действием разности
давлений за нею увлекаются противоионы диффузного слоя. На
концах капилляра возникает разность потенциалов, которая, в
свою очередь, вызывает встречный объемный поток ионов
противоположного знака. По разные стороны от капиллярнопористой
перегородки
будут
накапливаться
ионы
противоположного знака. При установившемся равновесии
потоки ионов станут равными, а разность потенциалов примет
постоянное значение, равное потенциалу течения.
Потенциалы течения возникают, например, при движении подземных вод через грунт и
горные породы и могут служить источником информации для геологов и геофизиков,
при циркуляции крови по капиллярам кровеносной системы, являясь одним из
источников биопотенциалов в организме, перекачке технологических растворов и
жидкого топлива по трубопроводам. При транспортировке жидкого топлива
возникающие потенциалы течения могут стать причиной пожаров и взрывов. Например,
при перекачке нефти потенциал течения может достигать сотен вольт.
63.
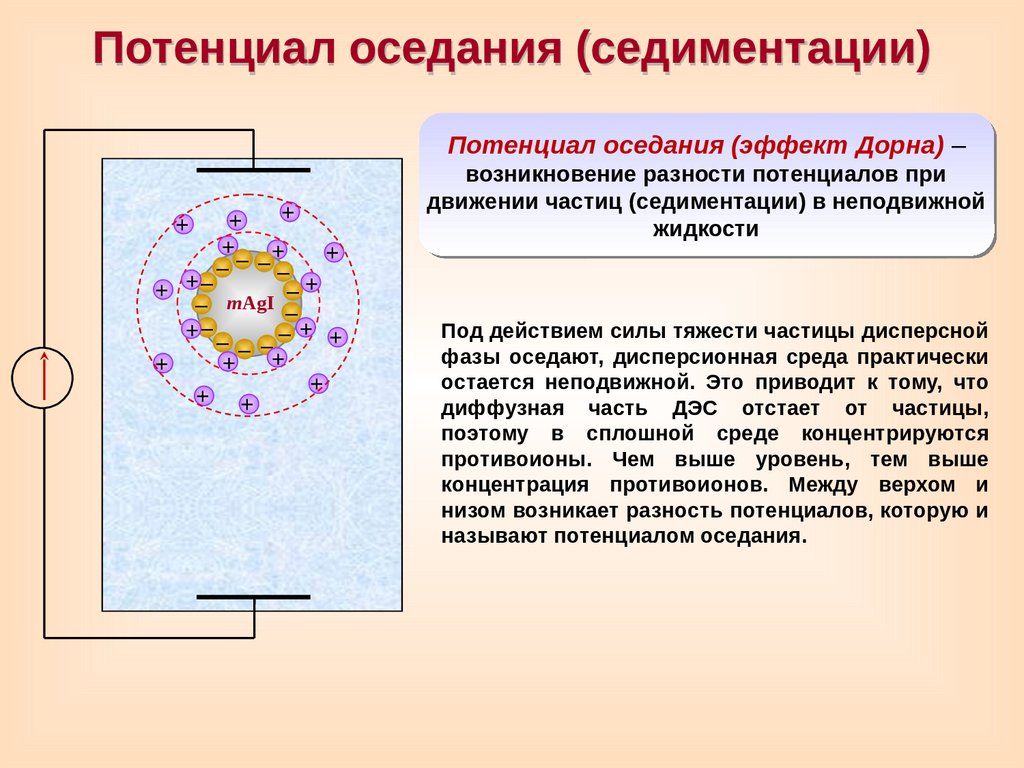
Потенциал оседания (седиментации)Потенциал оседания (эффект Дорна) –
+
+
+
возникновение разности потенциалов при
движении частиц (седиментации) в неподвижной
жидкости
+
+
+
–– ––
+–
–+
– mAgI –
+
+–
– – ––
+
+
+
+
+
+
+
+
Под действием силы тяжести частицы дисперсной
фазы оседают, дисперсионная среда практически
остается неподвижной. Это приводит к тому, что
диффузная часть ДЭС отстает от частицы,
поэтому в сплошной среде концентрируются
противоионы. Чем выше уровень, тем выше
концентрация противоионов. Между верхом и
низом возникает разность потенциалов, которую и
называют потенциалом оседания.
64.
УСТОЙЧИВОСТЬ И НАРУШЕНИЕ УСТОЙЧИВОСТИЛИОФОБНЫХ ЗОЛЕЙ
65.
Проблема устойчивости дисперсных систем – одна изцентральных проблем коллоидной химии
Лиофобные дисперсные системы термодинамически неустойчивы.
Для них характерно протекание процессов, приводящих к их
разрушению, вплоть до разделения на макрофазы.
Масло
Вода
Эмульсия
Например, эмульсия без эмульгатора разрушается в
течение нескольких секунд с момента ее образования.
Поэтому можно говорить только об относительной
устойчивости, под которой понимается способность
дисперсной системы сохранять неизменными во
времени численную концентрацию частиц дисперсной
фазы и их распределение по объему системы
На практике, часто приходится решать две противоположные задачи сохранить дисперсную систему или разрушить ее.
Например, устойчивыми должны быть дисперсные пищевые
системы; очистка воды природных водоемов от примесей связана с
разрушением дисперсной системы.
Различают 2 вида устойчивости – седиментационную и агрегативную
66.
Седиментационная устойчивость дисперсных системСедиментационная устойчивость – это способность дисперсной
системы противодействовать оседанию частиц
Седиментация (оседание) происходит под действием силы тяжести Fт:
Fт = mg =
Vg,
где m,
, V – масса, плотность и объем частицы; g – ускорение свободного
падения
Противодействие силе тяжести зависит от размеров частиц. Для относительно крупных частиц (средне- и грубодисперсные системы) – это архимедова сила FА и сила трения Fтр, для частиц коллоидных размеров –
броуновское движение, следствием которого является их диффузия,
стремящаяся выровнять концентрацию частиц по всему объему.
Сила трения пропорциональна вязкости дисперсионной среды
:
Fтр
,
а интенсивность броуновского движения – температуре Т:
Fдиф Т
67.
Для повышения седиментационной устойчивости необходимо:уменьшить силу тяжести за счет уменьшения размера
частиц с помощью специальных устройств – коллоидных
мельниц, гомогенизаторов и др.
увеличить вязкость среды введением специальных добавок (сиропов, ВМС)
обеспечить хранение дисперсной системы
при температуре, не ниже установленной
нормы
Для нарушения седиментационной устойчивости необходимо:
увеличить силу тяжести, например, применением
центробежного поля (сепарирование,
центрифугирование). Так, при сепарировании
молока эмульсия разрушается и выделяется
молочный жир, капельки которого являются
дисперсной фазой
68.
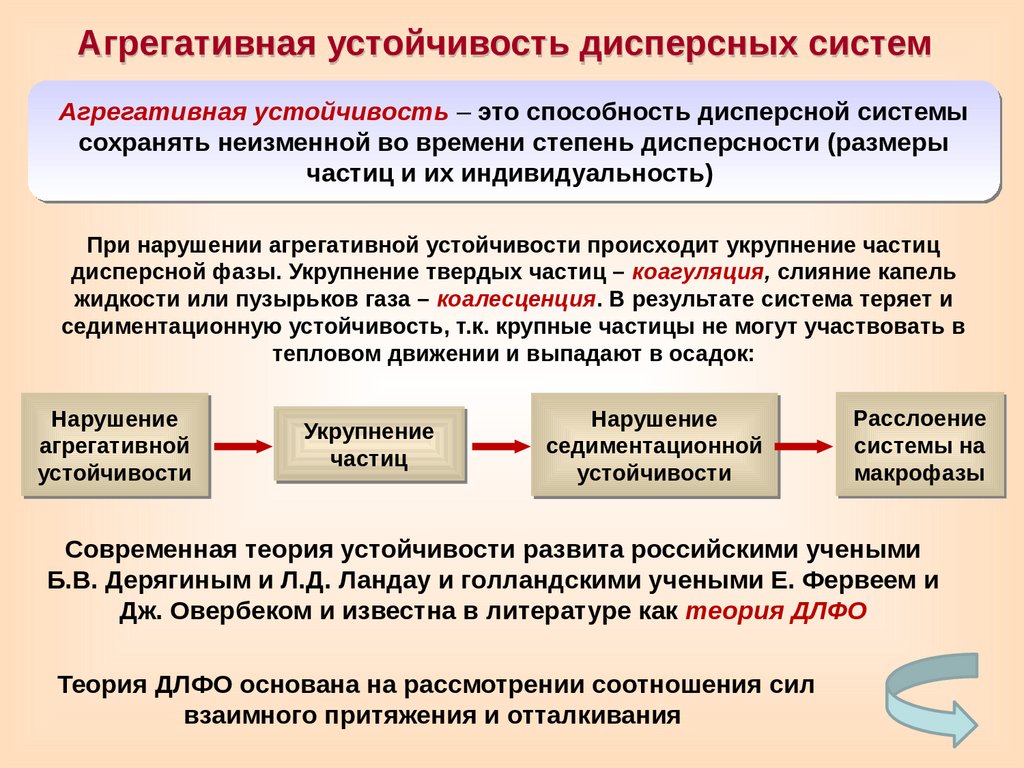
Агрегативная устойчивость дисперсных системАгрегативная устойчивость – это способность дисперсной системы
сохранять неизменной во времени степень дисперсности (размеры
частиц и их индивидуальность)
При нарушении агрегативной устойчивости происходит укрупнение частиц
дисперсной фазы. Укрупнение твердых частиц – коагуляция, слияние капель
жидкости или пузырьков газа – коалесценция. В результате система теряет и
седиментационную устойчивость, т.к. крупные частицы не могут участвовать в
тепловом движении и выпадают в осадок:
Нарушение
агрегативной
устойчивости
Укрупнение
частиц
Нарушение
седиментационной
устойчивости
Расслоение
системы на
макрофазы
Современная теория устойчивости развита российскими учеными
Б.В. Дерягиным и Л.Д. Ландау и голландскими учеными Е. Фервеем и
Дж. Овербеком и известна в литературе как теория ДЛФО
Теория ДЛФО основана на рассмотрении соотношения сил
взаимного притяжения и отталкивания
69.
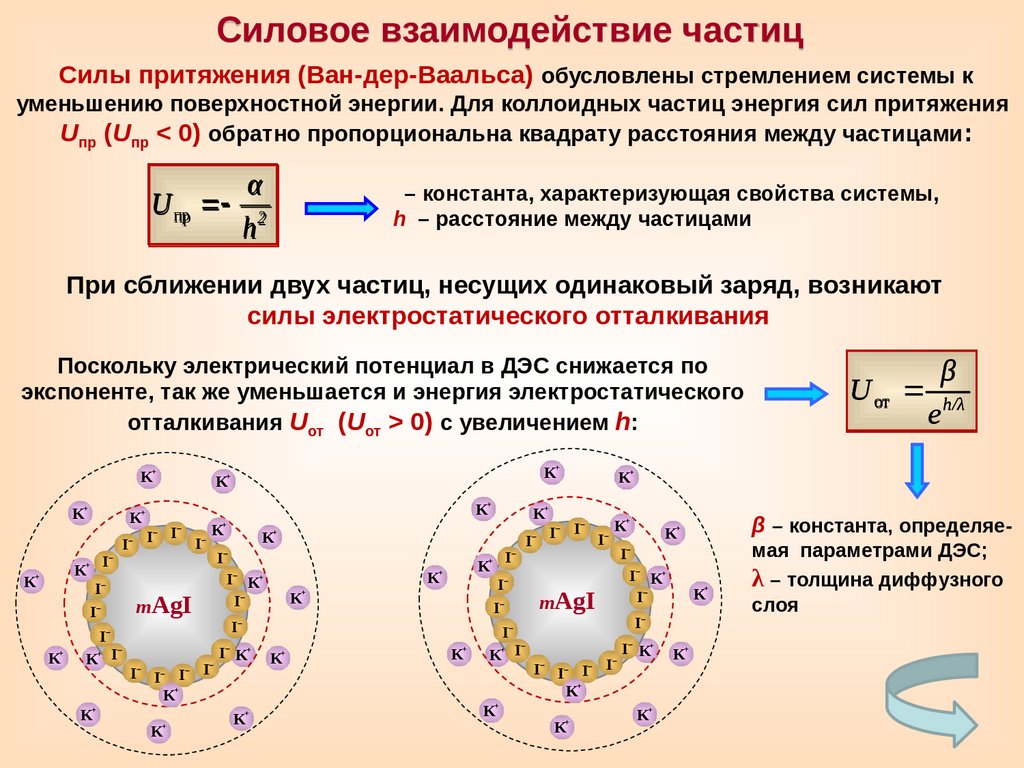
Силовое взаимодействие частицСилы притяжения (Ван-дер-Ваальса) обусловлены стремлением системы к
уменьшению поверхностной энергии. Для коллоидных частиц энергия сил притяжения
Uпр (Uпр < 0) обратно пропорциональна квадрату расстояния между частицами :
U пр
α
h2
– константа, характеризующая свойства системы,
h – расстояние между частицами
При сближении двух частиц, несущих одинаковый заряд, возникают
силы электростатического отталкивания
Поскольку электрический потенциал в ДЭС снижается по
экспоненте, так же уменьшается и энергия электростатического
отталкивания Uот (Uот > 0) с увеличением h:
K+
K+
K+
K+
K+
K+
+
–
I– I I– K
K+
I
–
I
I–
K+
+
I– K+
K
I–
K+
I–
–
m
I
I–
I–
–
I– K+ K+
K+ K+ I
–
I – I– I – I
K+
K+
K+
+
K
K+
K+
AgI
I–
I–
I–
I–
I
K+
K+
K+
–
K+
+
–
I– I I– K
K+
I–
I– K+
–
K+
I
m
AgI
I–
–
K+ I
–
I
–
I– I
K+
K+
β
U от h/λ
е
–
I
–
I– K+
K+
K+
β – константа, определяемая параметрами ДЭС;
λ – толщина диффузного
слоя
70.
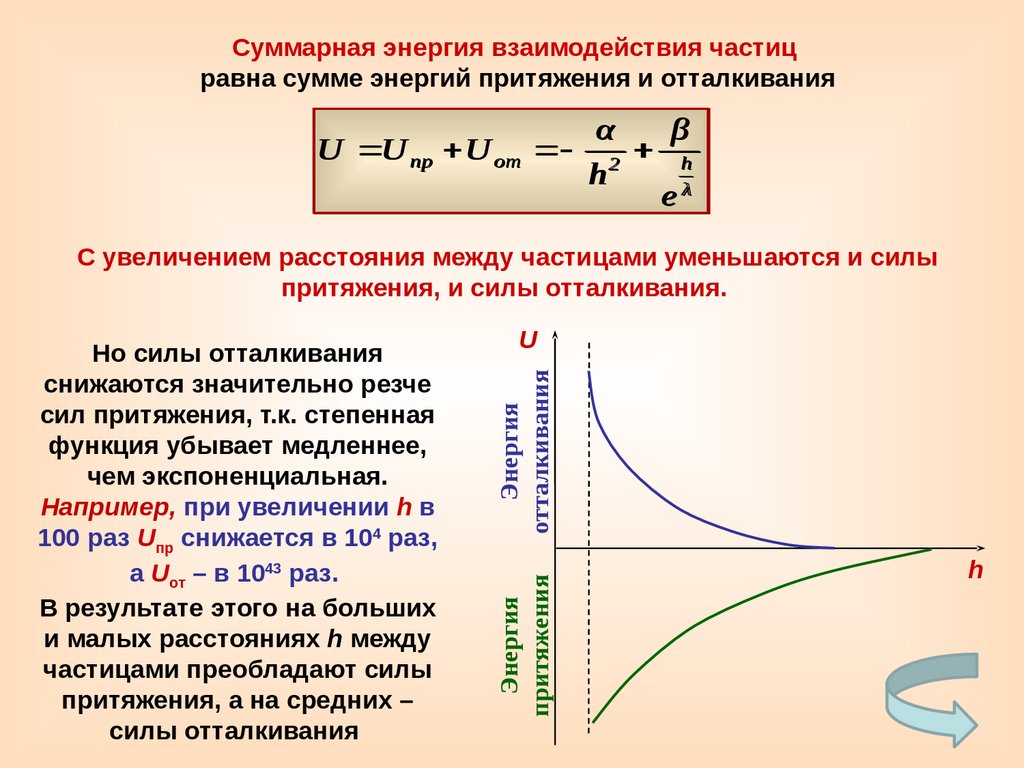
Суммарная энергия взаимодействия частицравна сумме энергий притяжения и отталкивания
α
β
U U пр U от 2 h
h
eλ
С увеличением расстояния между частицами уменьшаются и силы
притяжения, и силы отталкивания.
Энергия
отталкивания
U
Энергия
притяжения
Но силы отталкивания
снижаются значительно резче
сил притяжения, т.к. степенная
функция убывает медленнее,
чем экспоненциальная.
Например, при увеличении h в
100 раз Uпр снижается в 104 раз,
а Uот – в 1043 раз.
В результате этого на больших
и малых расстояниях h между
частицами преобладают силы
притяжения, а на средних –
силы отталкивания
h
71.
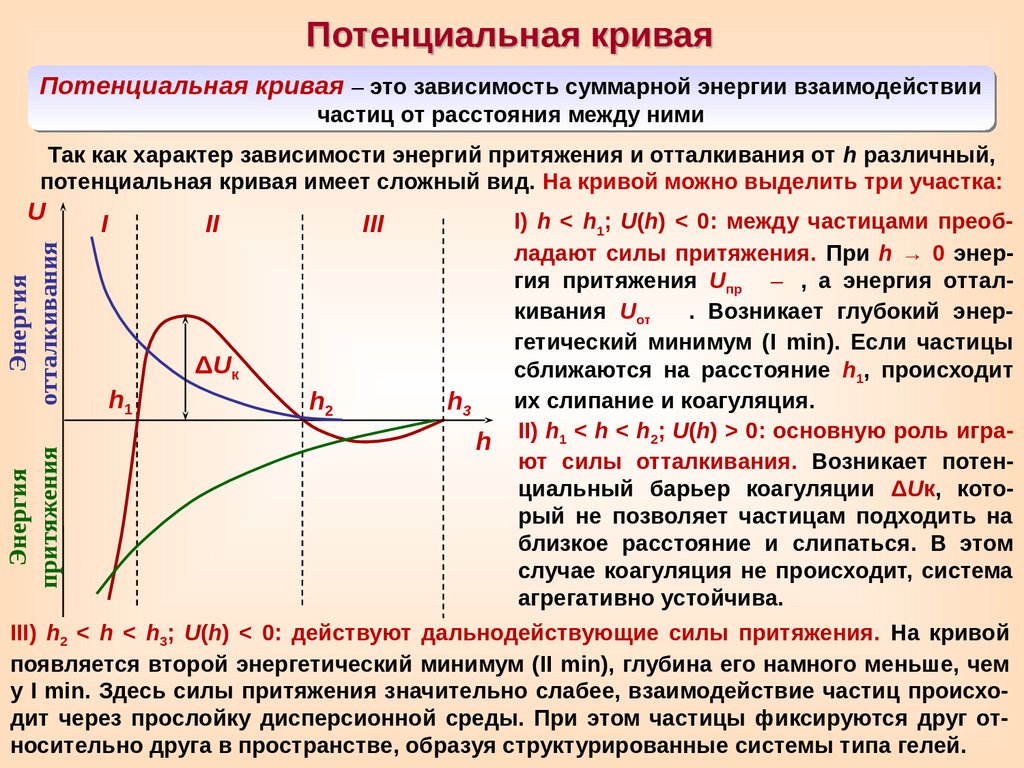
Потенциальная криваяПотенциальная кривая – это зависимость суммарной энергии взаимодействии
частиц от расстояния между ними
Так как характер зависимости энергий притяжения и отталкивания от h различный,
потенциальная кривая имеет сложный вид. На кривой можно выделить три участка:
Энергия
притяжения
Энергия
отталкивания
U
I
II
h1
III
ΔUк
h2
I) h < h1; U(h) < 0: между частицами преобладают силы притяжения. При h → 0 энергия притяжения Uпр –
, а энергия отталкивания Uот
. Возникает глубокий энергетический минимум (I min). Если частицы
сближаются на расстояние h1, происходит
их слипание и коагуляция.
h3
h II) h1 < h < h2; U(h) > 0: основную роль играют силы отталкивания. Возникает потенциальный барьер коагуляции ΔUк, который не позволяет частицам подходить на
близкое расстояние и слипаться. В этом
случае коагуляция не происходит, система
агрегативно устойчива.
III) h2 < h < h3; U(h) < 0: действуют дальнодействующие силы притяжения. На кривой
появляется второй энергетический минимум (II min), глубина его намного меньше, чем
у I min. Здесь силы притяжения значительно слабее, взаимодействие частиц происходит через прослойку дисперсионной среды. При этом частицы фиксируются друг относительно друга в пространстве, образуя структурированные системы типа гелей.
72.
Выводы:Если кинетическая энергия частиц мала, то при достаточно
глубоком II min они фиксируются друг относительно друга
на некотором расстоянии, образуя структурированные системы.
Если глубина II min невелика, а потенциальный барьер коагуляции высокий, частицы не могут сблизиться на критические расстояния, так как действуют мощные силы отталкивания.
Система агрегативно устойчива.
Если частицы обладают высокой кинетической энергией, то при
небольших II min и потенциальном барьере частицы слипаются,
происходит коагуляция.
Таким образом, чтобы началась коагуляция, частицы должны
преодолеть потенциальный барьер, высота которого определяется
величиной
-потенциала
73.
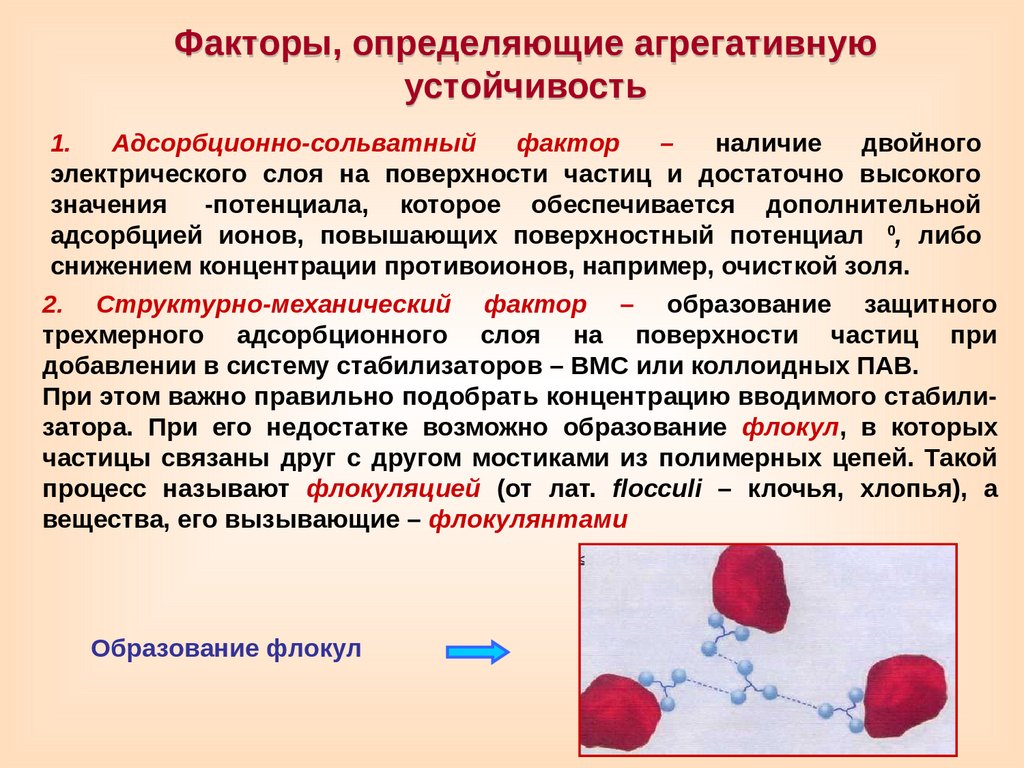
Факторы, определяющие агрегативнуюустойчивость
1.
Адсорбционно-сольватный
фактор
–
наличие
двойного
электрического слоя на поверхности частиц и достаточно высокого
значения
-потенциала, которое обеспечивается дополнительной
0
адсорбцией ионов, повышающих поверхностный потенциал
, либо
снижением концентрации противоионов, например, очисткой золя.
2. Структурно-механический фактор – образование защитного
трехмерного адсорбционного слоя на поверхности частиц при
добавлении в систему стабилизаторов – ВМС или коллоидных ПАВ.
При этом важно правильно подобрать концентрацию вводимого стабилизатора. При его недостатке возможно образование флокул, в которых
частицы связаны друг с другом мостиками из полимерных цепей. Такой
процесс называют флокуляцией (от лат. flocculi – клочья, хлопья), а
вещества, его вызывающие – флокулянтами
Образование флокул
74.
Коагуляция лиофобных золейКоагуляция – процесс слипания частиц дисперсной фазы с образованием
крупных агрегатов (от лат. coagulation – свертывание, сгущение)
Коагуляция сопровождается уменьшением межфазной поверхности, а,
следовательно, и свободной поверхностной энергии Gs. Система
переходит в более устойчивое состояние.
Для начала коагуляции частицы должны преодолеть потенциальный
барьер ΔUк и сблизиться на критические расстояния – попасть в
I энергетический минимум потенциальной кривой, что возможно
при снижении электрокинетического потенциала.
Коагуляция может происходить под действием различных факторов в
зависимости от способа стабилизации системы, например,
воздействием ультразвуком, электрическим полем, высокими и низкими
температурами. Наиболее часто коагуляцию вызывают добавлением
электролитов – это электролитная коагуляция.
75.
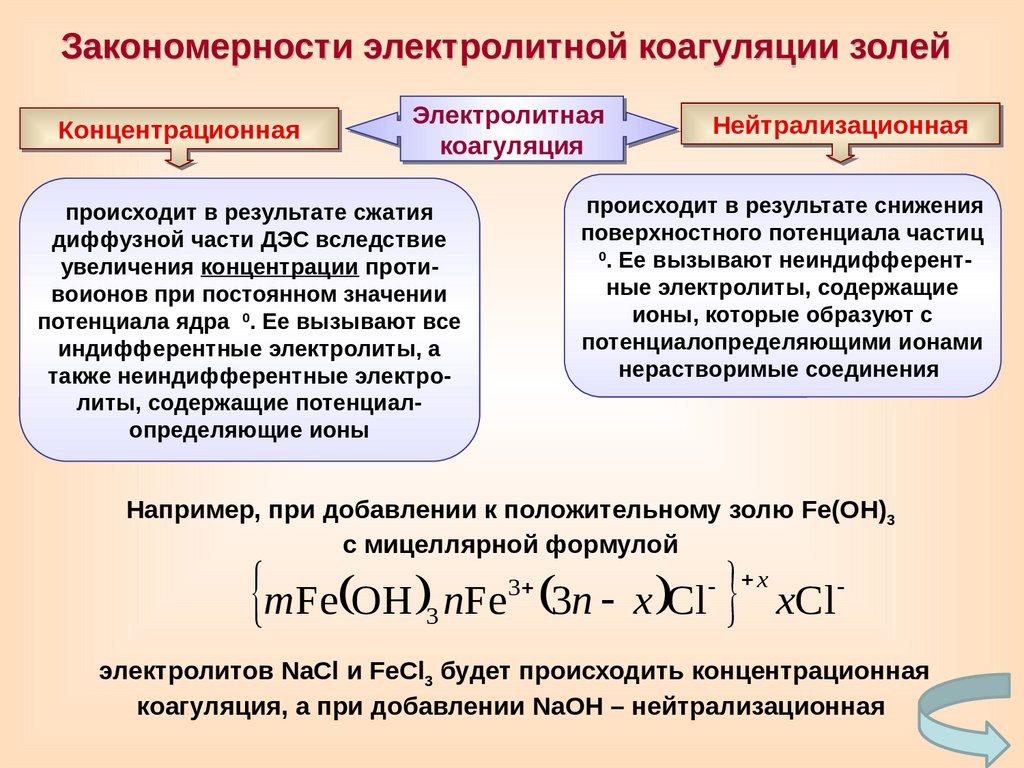
Закономерности электролитной коагуляции золейКонцентрационная
Электролитная
коагуляция
Нейтрализационная
происходит в результате снижения
поверхностного потенциала частиц
0
. Ее вызывают неиндифферентные электролиты, содержащие
ионы, которые образуют с
потенциалопределяющими ионами
нерастворимые соединения
происходит в результате сжатия
диффузной части ДЭС вследствие
увеличения концентрации противоионов при постоянном значении
0
потенциала ядра
. Ее вызывают все
индифферентные электролиты, а
также неиндифферентные электролиты, содержащие потенциалопределяющие ионы
Например, при добавлении к положительному золю Fe(OH)3
с мицеллярной формулой
mFe OH nFe 3n x Cl xCl
3
3
x
электролитов NaCl и FeCl3 будет происходить концентрационная
коагуляция, а при добавлении NaOH – нейтрализационная
76.
Стадии коагуляции гидрозоля Fe(OH)377.
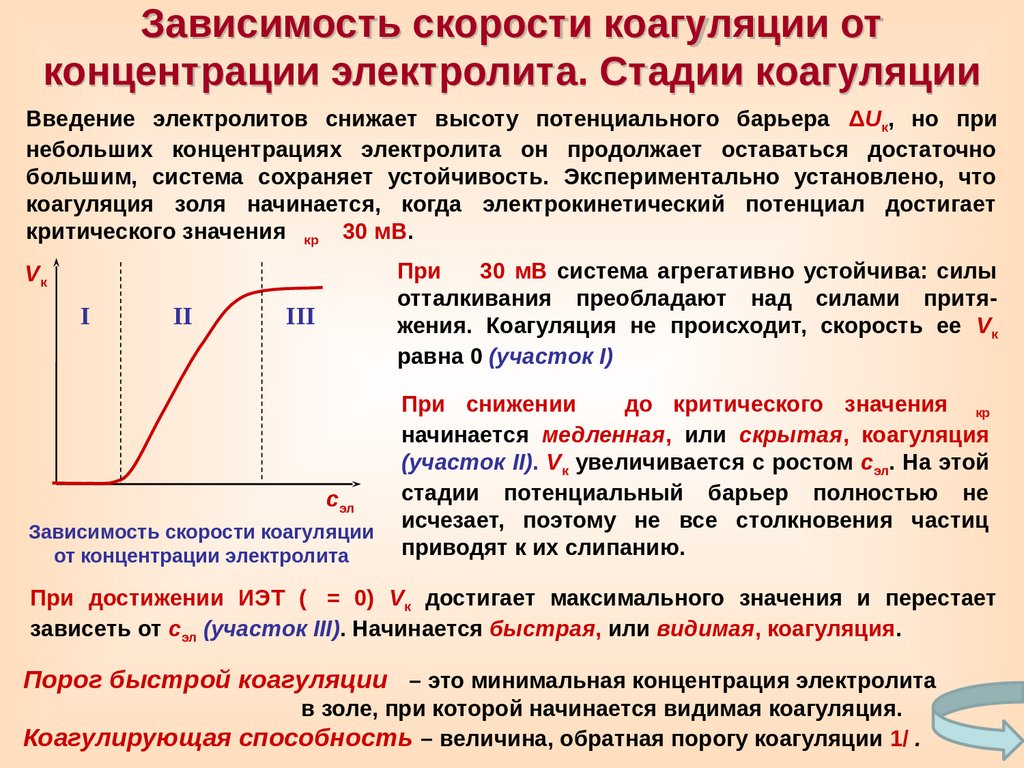
Зависимость скорости коагуляции отконцентрации электролита. Стадии коагуляции
Введение электролитов снижает высоту потенциального барьера ΔUк, но при
небольших концентрациях электролита он продолжает оставаться достаточно
большим, система сохраняет устойчивость. Экспериментально установлено, что
коагуляция золя начинается, когда электрокинетический потенциал достигает
критического значения
30 мВ.
кр
При 30 мВ система агрегативно устойчива: силы
отталкивания преобладают над силами притяжения. Коагуляция не происходит, скорость ее Vк
равна 0 (участок I)
Vк
I
II
III
сэл
Зависимость скорости коагуляции
от концентрации электролита
При снижении до критического значения
кр
начинается медленная, или скрытая, коагуляция
(участок II). Vк увеличивается с ростом сэл. На этой
стадии потенциальный барьер полностью не
исчезает, поэтому не все столкновения частиц
приводят к их слипанию.
При достижении ИЭТ ( = 0) Vк достигает максимального значения и перестает
зависеть от сэл (участок III). Начинается быстрая, или видимая, коагуляция.
Порог быстрой коагуляции – это минимальная концентрация электролита
в золе, при которой начинается видимая коагуляция.
Коагулирующая способность – величина, обратная порогу коагуляции 1/
.
78.
Правила электролитной коагуляции золейКоагулирующим действием обладает не весь электролит, а только тот ион,
заряд которого противоположен по знаку заряду коллоидной частицы. Этот ион
называют ионом-коагулянтом. Таким образом, коагуляцию положительных
золей вызывают анионы, отрицательных – катионы.
Коагулирующее действие иона-коагулянта тем сильнее, чем больше его заряд
(правило значности Шульце-Гарди). При электролитной коагуляции по
концентрационному механизму порог коагуляции обратно пропорционален
заряду z противоионов в шестой степени, т.е.
1
6
z
α – постоянная для данной системы величина
Таким образом, коагулирующие способности одно-, двух и трехзарядных ионов
связаны соотношением:
1 1 1
: : 16 : 26 : 36
1 2 3
При одинаковом заряде коагулирующая способность ионов увеличивается с
ростом их кристаллического радиуса (или массы иона ).
Например,
Li+ < Na+ < K+ < Rb+ < Cs+
Mg2+ < Ca2+ < Sr2+ < Ba2+
рост коагулирующей способности
79.
Определение порога коагуляцииПорог коагуляции золя по конкретному электролиту устанавливается
экспериментально добавлением к фиксированному объему золя Vз
небольших объемов раствора электролита известной концентрации
cэл. до появления первых признаков коагуляции (помутнение,
возможно изменение окраски). В этом случае порог коагуляции
рассчитывают с учетом разбавления золя раствором электролита:
cэлVэл.
Vз Vэл.
Vэл. – объем электролита, израсходованный на
коагуляцию золя
Если известен порог коагуляции золя по одному электролиту
1 с зарядом
иона-коагулянта z1, пользуясь правилом значности, можно рассчитать
порог коагуляции этого же золя по другому электролиту
2 с зарядом ионакоагулянта z2:
6
1
z1
6
2
z2
z16
2 1 6
z2
80.
ОПТИЧЕСКИЕ СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМ81.
ОПТИЧЕСКИЕ СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМПри падении луча света на дисперсную систему могут наблюдаться прохождение
света через систему, преломление, отражение, рассеивание и поглощение света
частицами дисперсной фазы. Преобладающий характер наблюдаемых явлений зависит от
размеров частиц дисперсной фазы и их соотношения с длиной волны падающего света
Видимый спектр
400
500
600
Длина волны (
), нм
700
Ограничимся рассмотрением видимой
области спектра, длина световой волны
которого колеблется от 400 нм (фиолетовый
свет) до 700 – 750 нм (красный свет).
Прохождение света характерно для прозрачных систем (истинных растворов, чистых
жидкостей). Размер частиц в них (молекул, ионов) намного меньше длины волны падающего света, поэтому частицы не создают препятствий для прохождения лучей видимого света.
Преломление и отражение света характерно для средне- и грубодисперсных систем
(суспензий, эмульсий), содержащих частицы дисперсной фазы, размер которых намного
превышает длину волны падающего света. Попадая на поверхность частиц, световые лучи
отражаются под различными углами и выходят из системы в разных направлениях.
Прямому прохождению лучей через дисперсную систему препятствуют также многократные
отражения и преломления при переходах частица – дисперсионная среда и наоборот. Это
обусловливает мутность таких систем, видимую невооруженным глазом.
Рассеяние света во всех направлениях наблюдается для систем, в которых размер частиц
дисперсной фазы меньше, но соизмерим с длиной волны падающего света. Именно такое
соотношение выполняется для коллоидных систем с размером частиц от 1 до 100 нм.
Поглощение света (абсорбция) характерно для окрашенных коллоидных растворов
82.
Рассеяние света коллоидными частицами. Эффект ТиндаляЯвление рассеяния света коллоидными системами впервые изучено М.
Фарадеем в 1857 г. на примере золей золота. Более подробно это
явление было описано Дж. Тиндалем в 1869 г. Тиндаль обнаружил, что
если направить на золь пучок света, то при наблюдении освещаемого
сосуда сбоку внутри золя можно увидеть светящийся голубым светом
конус (эффект Тиндаля). Это свечение было названо опалесценцией.
Джон Тиндаль
(1820-1893)
Схема эффекта
Тиндаля
Подобное явление можно наблюдать при прохождении луча света
через темное запыленное помещение, при свете автомобильных
фар и фонарей в туманную погоду.
В истинных растворах и чистых жидкостях
эффект Тиндаля не наблюдается. Поэтому им
часто пользуются для того, чтобы отличить
коллоидный раствор от истинного (например,
золь берлинской лазури от раствора медного
купороса), по внешнему виду это не всегда
возможно сделать.
83.
Уравнение Релеяnд/ф nд/с
Теорию светорассеяния создал английский ученый Релей. Он
вывел уравнение, связывающее интенсивность рассеянного света
Iр с интенсивностью падающего света I0:
n n
I p I 0 24
n 2n
3
Джон Уильям Стрэт
(лорд Рэлей)
(1842-1919)
2
д/ф
2
д/ф
2
д/с
2
д/с
2
V 2
4
где nд/ф, nд/с – показатели преломления дисперсной фазы и дисперсионной
среды; – численная концентрация частиц; V – объем частицы; λ – длина
волны падающего света
Уравнение Релея применимо для коллоидных систем, в которых частицы дисперсной
фазы имеют сферическую форму, не поглощают свет и не проводят электрический
ток, а расстояние между частицами больше длины волны падающего света.
Анализ уравнения:
Интенсивность рассеянного света тем больше, чем сильнее различаются показатели преломления дисперсной фазы и дисперсионной среды. Если nд/ф nд/с, то
светорассеяние отсутствует и в неоднородной среде
Интенсивность рассеянного света обратно пропорциональна λ4. Следовательно,
если источник падающего света содержит волны различной длины (белый свет),
то наиболее сильно будут рассеиваться самые короткие волны (синей и фиолетовой частей спектра). Поэтому бесцветный золь в рассеянном свете имеет голубоватую окраску, а в проходящем – красноватую.
84.
Голубой цвет неба обусловлен рассеянием светамельчайшими частицами пыли и капельками воды в
атмосфере.
Оранжевый или красный цвет неба при
восходе или заходе солнца объясняется тем,
что при расположении солнца вблизи
горизонта наблюдается, главным образом,
свет, прошедший через атмосферу.
Зависимость интенсивности рассеянного света от λ
имеет практическое значение. Красный цвет выбран
сигналом опасности, так как он виден даже в туманную
погоду на больших расстояниях вследствие малого
рассеяния. Лампы синего цвета применяются для
светомаскировки: при прохождении через толстый слой
атмосферы синие лучи почти полностью рассеиваются.
85.
СПАСИБО ЗА ВНИМАНИЕ!ЛЕКЦИЯ ОКОНЧЕНА!