












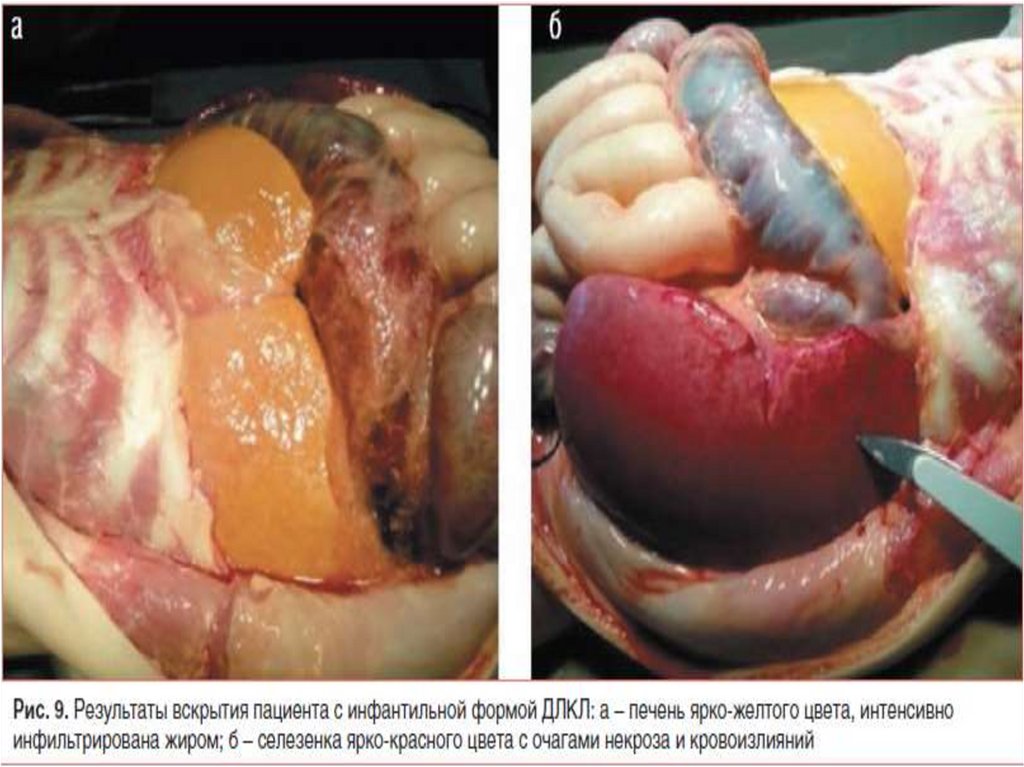

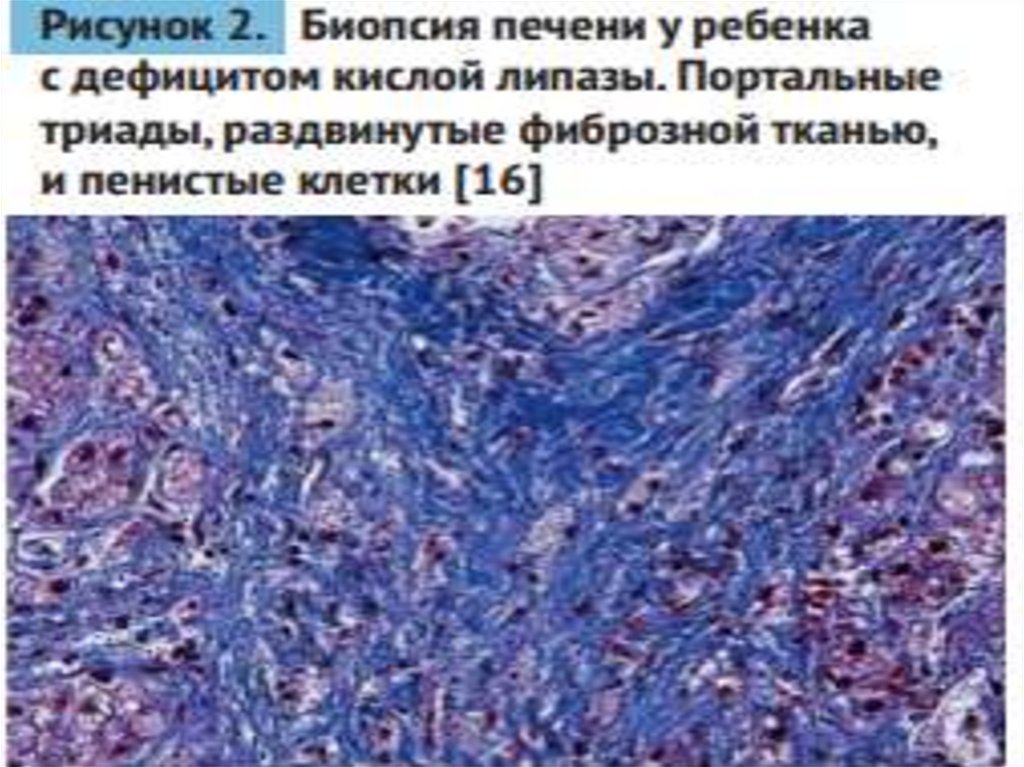














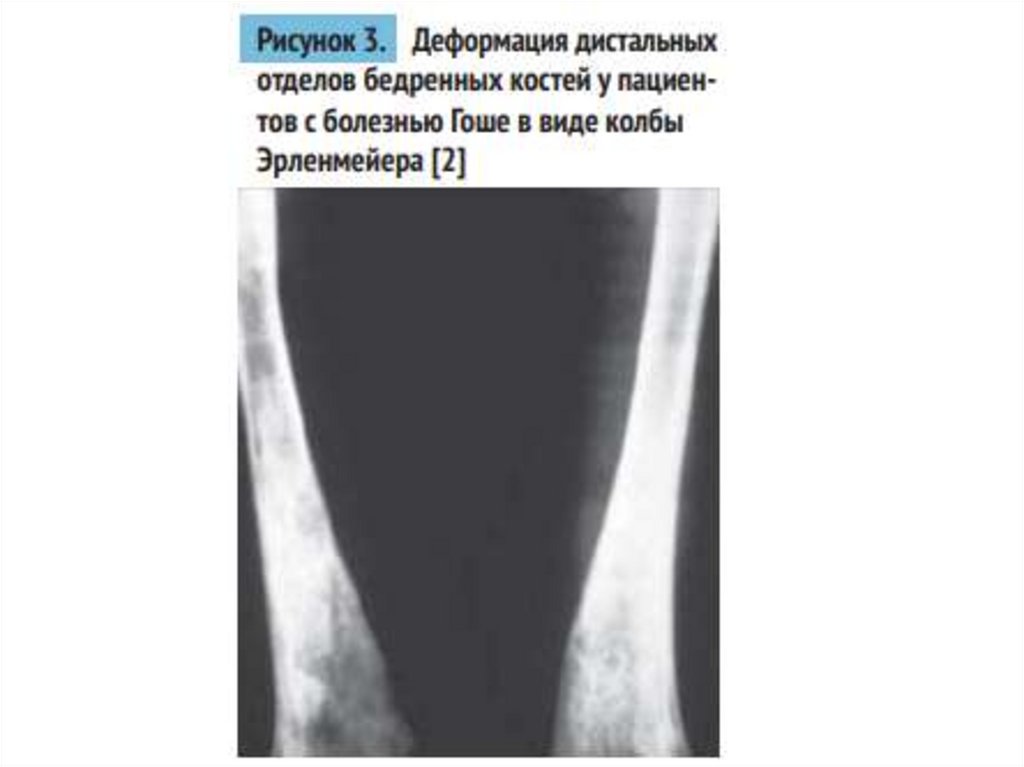











































 medicine
medicineSimilar presentations:

")








Болезни накопления липидов. Виды дислипидемий
1.
Болезни накоплениялипидов.
Виды дислипидемий.
Выполнили: Егорова А.А.,
Егоров А. Д.
Ананьев Л.С.
Гр.М-21-3-15
2.
Наиболее изученными среди болезней накопленилипидов с возможностью проведения
заместительной энзимной терапии являются:
1. болезнь Гоше
2. болезнь Ниманна – Пика
3. дефицит лизосомной кислой липазы.
• Все перечисленные заболевания имеют два
варианта дебюта:
1.на первом году жизни
2. более старшем возрасте
3.
• общность клинической картины:1.системность поражения
2.наличиегепатомегалии/гепатоспленомегалии
3.задержка роста
4. анемия, цитопения
5. развитие печеночной недостаточности в финале
болезни
• А также вероятность относительно
благоприятного и неблагоприятного (с ранним
фатальным исходом) течения.
4.
• В патогенезе лизосомных болезней накопленияв роли главных аффектированных клеток
выступают макрофаги и моноциты,
накапливающие первичный субстрат в
лизосомах и присутствующие практически во
всех органах и тканях.
5.
Дефицит лизосомнойкислой липазы
(ДЛКЛ)
6.
Дефицит лизосомной кислой липазы можноотнести к заболеваниям с мультиорганным
уровнем поражения вследствие
особенностей патогенеза: до 87% детей в
период манифестации болезни имеют
кардиоваскулярные нарушения, 86% –
изменения со стороны печени, 36% –
селезенки, 22% – желудочно-кишечного
тракта
7.
Этиология.(ДЛКЛ) связан с мутацией гена LIPA в хромосомном
локусе 10q23.31, который кодирует гидролазы,
участвующие в деградации лизосомных эфиров
холестерина и триглицеридов.
• Наследуется по аутосомно-рецессивному типу
наследования. Следовательно, с одинаковой
частотой встречается как у мужчин, так и у женщин.
Заболевание клинически манифестирует только в
случае, когда обе аутосомы, полученные по одной
от отца и матери, являются дефектными по гену LIPA
(повреждение обеих копий гена, находящихся на
гомологичных аутосомах 10q23.2-23.3)
8.
Клиника.• С ранней манифестацией – болезнь
Вольмана
• С поздней манифестацией – болезнь
накопления эфиров и холестерина
9.
• При ДЛКЛ фермент лизосомная кислая липаза не активна, чтоприводит к накоплению в клеточных органеллах, лизосомах
триглицеридов и эфиров холестерина.
• В норме триглицериды и эфиры холестерина разрушаются в
лизосомах, а образующиеся жирные кислоты и холестерин
усваиваются организмом до более простых молекул для
строительства новых клеток и для пополнения энергии. У
пациентов с ДЛКЛ из-за недостатка активности ЛКЛ клетки
настолько переполняются лизосомами с неразрушенными
молекулами, что они перестают выполнять свои функции.
• Больше всего таких нефункционирующих клеток находится в
печени, селезенке, стенках кровеносных сосудов,
надпочечниках, но поражаются также и другие органы и ткани.
Со временем, например, печень, значительно увеличивается в
размерах, перестает функционировать, после чего орган
зарастает соединительной тканью с переходом в цирроз. А
повышение уровня холестерина в крови сопряжено с
повышенным риском заболеваний сердца, а также рядом
других заболеваний, вызванных нарушением проходимости
артерий.
10.
• Дефект лизосомной кислой липазы проявляется вредукции гидролиза эфиров холестерина и триглицеридов
и их массивной секвестрации в лизосомах, Купферовских
клеток и гепатоцитов в клетках макрофагальномоноцитарной системы.
• Отсутствие свободного холестерина, вызванного
лизосомальной ловушкой эфиров холестерина, приводит
к редуцированию обратной ингибиции 3-гидрокси-3метилглютарил коэнзима А (HMG-CoA) редуктазы,
повышенному синтезу аполипопротеина В и рецепторов к
липидам низкой плотности на клеточных мембранах.
• Параллельно снижается уровень холестерина высокой
плотности. В итоге повышается сывороточный уровень
холестерина низкой плотности и ТГЦ, снижается уровень
холестерина высокой плотности, развивается тип IIb
гиперлипопротеинемии.
• Нарастание уровня холестерина низкой плотности
способствует быстрому развитию атеросклероза, ишемии,
инсульта, необходимости коронарного шунтирования.
11.
Болезнь Вольмана.• Манифестирует в неонатальном периоде
• Отличается фульминантным течением.
• При этом или полностью отсутствует активность
лизосомной кислой липазы, или ее активность
составляет менее 1%.
• Результатом является массивное накопление
эфиров холестерина и триглицеридов
(цероидного материала) в печени, селезенке,
надпочечниках, костном мозге, лимфатических
узлах и макрофагах во всех органах и тканях, в
ворсинках кишечника
12.
• В возрасте 2–4 мес. жизни у детей отмечаютсярвота, диарея и выраженная
гепатоспленомегалия.
• Около 50% пациентов имеют кальцификацию
надпочечников.
• Трудности вскармливания и мальабсорбция
ведут к мальнутриции, задержке роста, кахексии,
что в сочетании с тяжелым заболеванием печени
приводит к раннему летальному исходу в
возрасте до года
13.
• Основным клиническим проявлением ДЛКЛявляется прогрессирующее поражение печени с
развитием гепатомегалии, повышением уровня
трансаминаз и/или микровезикулярного или
смешанного стеатоза вследствие накопления
эфиров ХС и ТГ в гепатоцитах и клетках Купфера.
Осложнениями являются:
• — развитие микровезикулярного или смешанного
стеатоза;
• — прогрессирование в фиброз и цирроз;
• — портальная гипертензия;
• — печеночная недостаточность.
• Для 87% пациентов с ДЛКЛ характерны ДЛП,
обусловливающая раннее развитие и быстрое
прогрессирование атеросклероза, ИБС, инфаркта
миокарда, аневризмы или инсульта
14.
15.
16.
17.
Болезнь накопления эфировхолестерина (БНЭХ)
• часто остается нераспознанной
• может проявить себя в любом возрасте (от
младенческого периода до взрослого)
• характеризуется снижением активности
лизосомной кислой липазы до 1–12% от нормы.
• От уровня активности фермента зависят сроки
манифестации и выраженность клинических
проявлений.
18.
Прогрессирующее лизосомное накопление эфировхолестерина и триглицеридов приводит к
характерным изменениям в печени, повышению
уровня трансаминаз, сывороточного холестерина
низкой плотности и триглицеридов, при этом
отмечается нормальный или низкий уровень
холестерина высокой плотности (тип IIb
гиперлипопротеинемии). Точная
распространенность болезни накопления эфиров
холестерина до настоящего времени не
установлена.
19.
Клиника.В период младенчества БНЭХ имеет клиническую
картину, сходную с болезнью Вольмана:
1.задержку роста
2. Рвоту
3. увеличение в объеме живота
4.кальцификацию надпочечников
Продолжительность жизни при данной патологии
намного больше: до взрослого периода
20.
• К ранним признакам повреждения печениотносятся повышение сывороточных
аминотрансфераз и гепатомегалия.
• При биопсии ткань печени ярко-желтооранжевого цвета, гистологически выявляются
большие, заполненные липидами гепатоциты и
Купферовские клетки с характерным
микровезикулярным стеатозом.
• Патогномоничным признаком считается наличие
кристаллов эфиров холестерина или их осколков
в клетках печени.
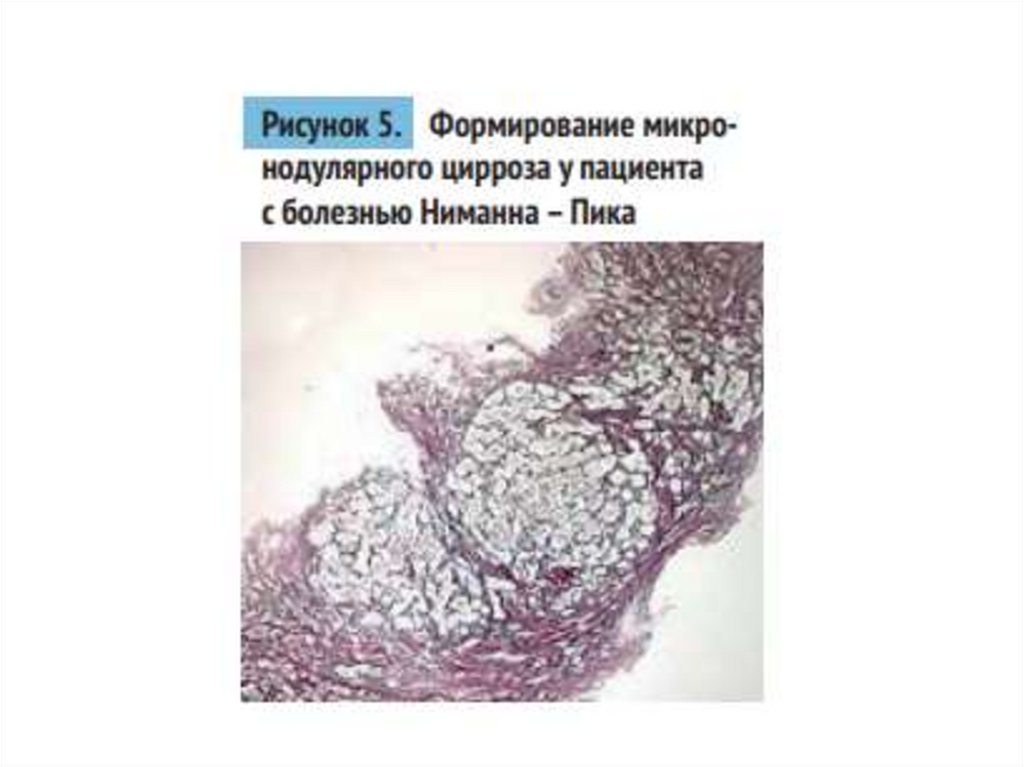
21.
• Увеличение липидных депозитов ведет кфиброзу, микронодулярному циррозу и
печеночной недостаточности, которая является
главной причиной фатального исхода. В 8%
случаев причиной летального исхода может быть
кровотечение из варикозно расширенных вен
пищевода
22.
Диагностика.• Диагноз ДЛКЛ подтверждается на основании
обнаружения значительного дефицита
активности фермента. Активность ЛКЛ
оценивается путем определения активности
фермента в сухих пятнах крови на фильтрах.
Такой способ определения практически
однозначно указывает на ДЛКЛ, так как у
больных активность ЛКЛ снижена в десятки и
сотни раз, а иногда, при тяжелых формах, вовсе
отсутствует. Неизвестно больше никаких других
состояний, когда активность ЛКЛ настолько
низкая.
23.
Диагностика.• Анализ крови на ЛКЛ может выступать в качестве
инструмента в программах скрининга и крупных
популяционных исследованиях ДЛКЛ, а также
может быть адаптирован для скрининга
новорожденных.
• ДНК-диагностика для обнаружения мутации гена
LIPA является дополнительным методом
исследования, так как стандартными методами
исследований мутация в гене LIPA не всегда
выявляются. При этом некоторые пациенты
имеют тяжелые клинические симптомы и
существенную недостаточность ЛКЛ.
24.
Лечение.• На сегодняшний день жизнеспасающей признана
единственная патогенетическая
ферментзаместительная терапия себелипазой альфа
— рекомбинантной лизосомной кислой липазой. В
такой форме ЛКЛ проникает непосредственно в
лизосомы клеток, где замещает
нефункционирующий фермент пациента. В ряде
клинических исследований высоко уровня
доказательности продемонстрировано, что на фоне
лечения себелипазой альфа происходит улучшение
по многим параметрам заболевания, в том числе,
нормализация биохимических показателей,
уменьшение объема жира в печени, нормализация
липидограммы и улучшения выживаемости у
младенцев с тяжелыми формами заболевания.
25.
Болезнь Гоше.26.
Определение.наследственное заболевание, является
самой распространённой из лизосомных
болезней накопления. Развивается в
результате
недостаточности фермента глюкоцереброз
идазы, которая приводит к
накоплению глюкоцереброзида во многих
тканях,
включая селезёнку, печень, почки, лёгкие,
мозг и костный мозг.
27.
Этиология.• Заболевание связано с рецессивной мутацией в
гене GBA, расположенном в 1-й хромосоме, и
поражает как мужчин, так и женщин.
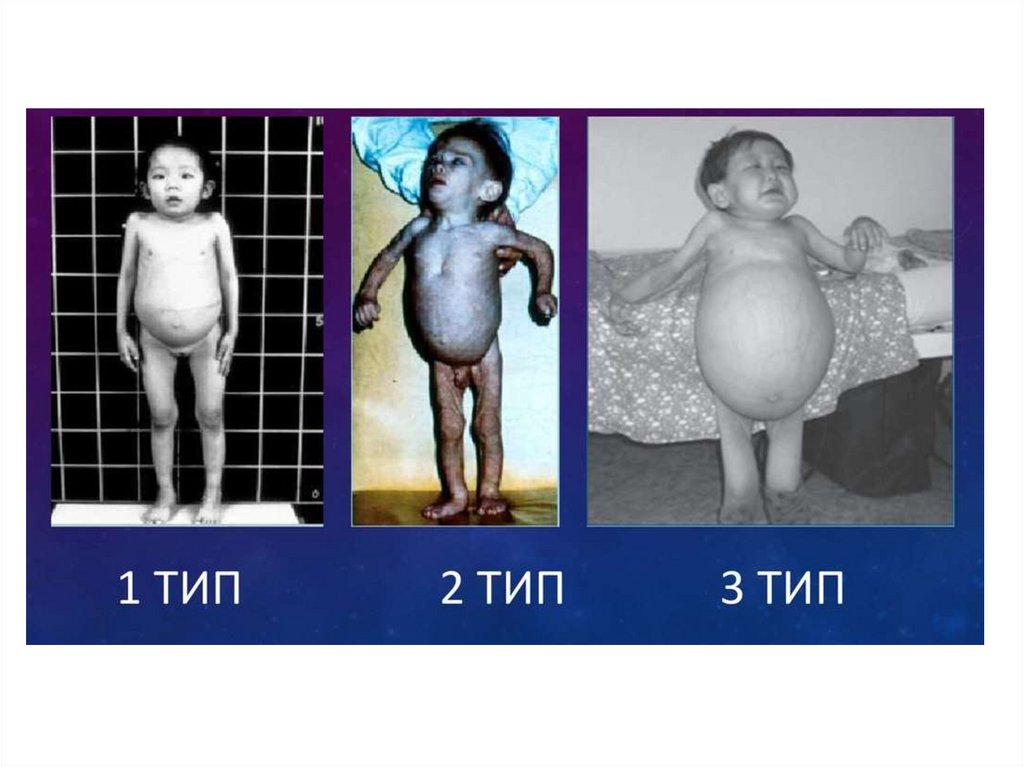
• Болезнь Гоше подразделяется на три основных
типа.
• Нарушения функции β-глюкозидазы обусловлено
более чем 300 мутациями в гене GBA1,
приводящими к продукции кислой βглюкозидазы с изменением функциональных,
кинетических, транспортных свойств и
нарастанию аккумуляции глюкозилцерамида.
28.
Тип 1.Болезнь Гоше I (ненейронопатического) типа встречается с
частотой 1/50000. Наиболее часто встречается
среди ашкеназских евреев. Проявление симптомов
начинается в детстве или во взрослом возрасте и
включают увеличенную печень и сильно увеличенную
селезёнку (что может приводить к её разрыву и
дополнительным повреждениям). Возможны слабость
костей и выраженные костные заболевания. Изменённые
селезёнка и костный мозг
вызывают анемию, тромбоцитопению и лейкопению. Хотя
мозг при этом типе не повреждается, могут быть
нарушения в лёгких и почках. Больные страдают от
частых гематом, вызванных тромбоцитопенией, и от
постоянной усталости (из-за пониженного числа
эритроцитов). Больные могут доживать до взрослого
возраста и при умеренной форме симптомы могут
отсутствовать.
29.
Тип 2.Тип II представляет собой нейронопатическую
инфантильную форму. Средний возраст
заболевания 3—5 мес. Неврологические
осложнения (тяжелые судорожные приступы,
гипертонус, апноэ, выраженная задержка
умственного развития) проявляются к 6 мес.
Симптомы включают гепатоспленомегалию,
широкое прогрессирующее повреждение мозга,
нарушенную моторику глаз, спастичность, судороги,
ригидность конечностей. Больные дети плохо сосут
и глотают; обычно умирают в возрасте от одного до
двух лет. Частота встречаемости 1/100000,
этнической предрасположенности не имеет.
30.
Тип 3.• Тип 3 может начинаться как в детстве, так и у взрослых с частотой
встречаемости 1/100000. У большинства характеризуется
медленным прогрессированием и умеренностью неврологических
симптомов. Первым неврологическим признаком является, как
правило, окуломоторная апраксия, расстройство глазодвигательных
функций. По мере прогрессирования заболевания присоединяются
атаксия, мышечная спастичность и слабоумие. Наряду с
гепатоспленомегалией в патологический процесс вовлекаются и
другие органы и системы. Спленомегалия безболезненная и обычно
выявляется случайно. Больные доживают до подросткового и
взрослого возраста.
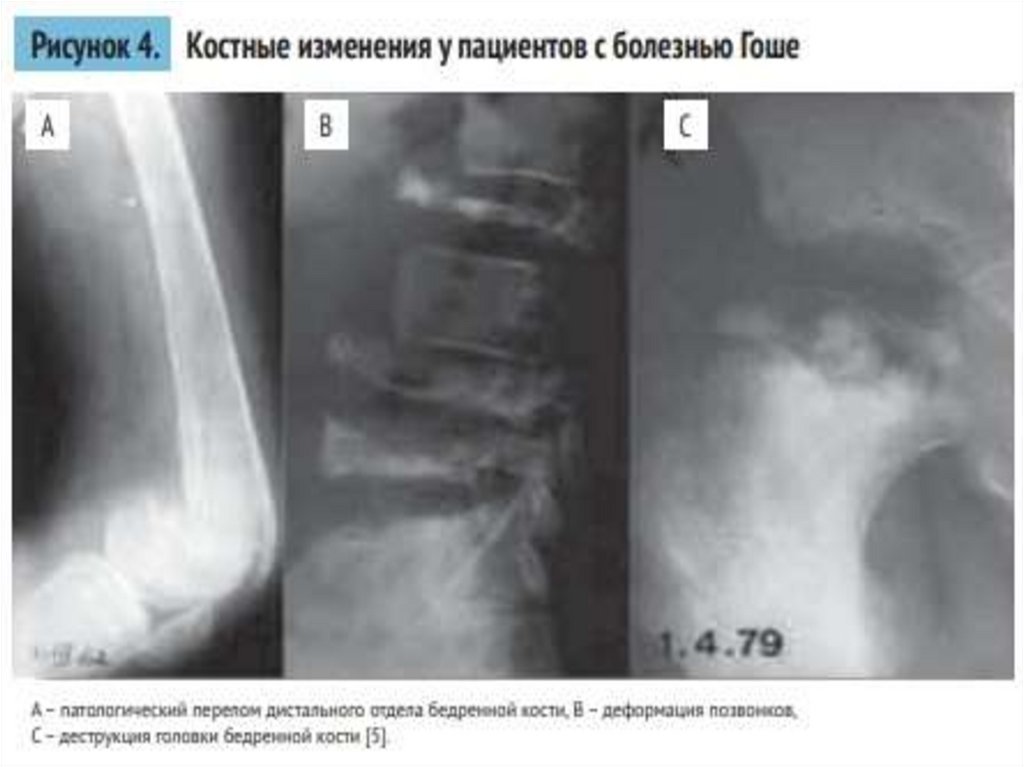
• Одна из главных причин инвалидизации при 1 и 3 типе болезни
Гоше —- поражение костной ткани. Нарушение нормальных
физиологических процессов происходит из-за накопления липидов
в остеокластах и замещении инфильтратами клеток Гоше
нормальных элементов костного мозга. Несмотря на увеличение
печени и её дисфункцию, случаи тяжёлой печёночной
недостаточности встречаются редко. Чаще встречается
относительная портальная гипертензия как следствие фиброза
31.
32.
33.
34.
35.
36.
Патогенез.При болезни Гоше к главным мишеням поражения
относятся селезенка, печень, легкие, а также
костная ткань и костный мозг. Принципиально
аффектированными клетками считаются
производные макрофагально-моноцитарного
звена, клетки миелоидного и лимфоидного ряда. В
основе патогенеза болезни Гоше лежит первичный
дефицит лизосомного фермента кислой βглюкозидазы, который ведет к накоплению
неразрушенного субстрата глюкозилцерамида в
органах, содержащих большое количество клеток,
относящихся к макрофагам
37.
Дефицит кислой β-глюкозидазы не являетсяабсолютным, у каждого пациента с болезнью
Гоше, независимо от тяжести течения, имеется
свой уровень дефицита и остаточная активность
фермента во всех ядерных клетках. Если же
фермент отсутствует вообще, отмечается
неблагоприятный исход во время беременности
или в периоде новорожденности
38.
Первичным источником для глюкозилцерамида ворганах являются стареющие форменные
элементы крови (лейкоциты, эритроциты и
тромбоциты), мембраны которых содержат
гликосфинголипиды [1]. Аккумулированный
глюкозилцерамид стимулирует классическую и
альтернативную активацию макрофагов, точный
механизм которой и последующей за ней
продукции цитокинов непонятен до настоящего
времени. Про- и противовоспалительные
эффекты цитокинов способствуют необратимым
повреждениям тканей и прогрессирующему
течению болезни.
39.
Диагностика.• Диагностика болезни Гоше включает комплексную оценку клинической
картины, лабораторные тесты и другие исследования (например, УЗИ печени и
селезёнки).
• Для диагностики болезни Гоше применяются следующие лабораторные
исследования:
1. Определение активности бета-глюкоцереброзидазы (ферментная
диагностика)
• При болезни Гоше — снижена.
2. Определение активности хитотриозидазы
• При болезни Гоше — повышена.
3. Секвенирование экзонов и приэкзонных участков интронов гена GBA (ДНКдиагностика)
• Идентифицировано значительное количество мутаций гена GBA, приводящих
к развитию болезни Гоше.
• 4.Диагностика болезни Гоше в России
В России ферментную и генетическую диагностику болезни Гоше выполняют в
Лаборатории наследственных болезней обмена веществ МГНЦ РАМН и в
Лаборатории молекулярной генетики и медицинской геномики Научного
медицинского исследовательского центра здоровья детей минздрава России.
40.
Лечение.• Современное лечение БГ заключается в назначении
пожизненной ферментной заместительной терапии
(ФЗТ) рекомбинантной глюкоцереброзидазой,
которая купирует основные клинические
проявления заболевания, улучшая качество жизни
больных с БГ и не оказывая выраженных побочных
эффектов. ФЗТ показана для длительной
заместительной ферментотерапии у больных с
подтвержденным диагнозом БГ тип 1 без
поражения нервной системы (1B) или с
хроническим поражением нервной системы (БГ тип
3), у которых имеются клинически значимые
неневрологические проявления заболевания
41.
• До последнего времени единственнымлекарственным препаратом для лечения БГ был
Церезим (имиглюцераза)синтезируемый клеточной
линией, полученной из яичников китайских
хомяков.
• Под действием имиглюцеразы происходит гидролиз
гликолипида глюкоцереброзида до глюкозы и
церамида по обычному пути метаболизма
мембранных липидов.
• Препарат выпускается во флаконах в дозировке 400
ЕД. У детей ФЗТ начинают в стационаре. Препарат
вводится в/в капельно в течение 1,5-2 часов, частота
введения - 1 раз в 14 дней. В связи с
гетерогенностью БГ доза препарата для каждого
пациента должна подбираться индивидуально.
42.
У детей с БГ начальная доза имиглюцеразысоставляет:
— при типе 1 БГ, протекающей без поражения
трубчатых костей скелета - 30 ЕД/кг на 1
введение (60 ЕД/кг/мес);
— при типе 1 БГ, протекающей с поражением
трубчатых костей скелета (костные кризы,
патологические переломы, очаги литической
деструкции, асептический некроз головок
бедренных костей) - 60 ЕД/кг на 1 введение (120
ЕД/кг/мес);
— при типе 3 БГ - 120 ЕД/кг на 1 введение (240
ЕД/кг/мес)..
43.
С января 2013 г. в России с целью проведения ФЗТпациентам с БГ зарегистрирован новый препарат
для внутривенного введения ВПРИВ (велаглюцераза
альфа) (Код АТХ: A16AB11). Велаглюцераза альфа единственный препарат для ФЗТ, производящийся с
использованием человеческой линии клеток.
Аминокислотная последовательность у препарата
идентична эндогенной глюкоцереброзидазе и
имеет более длинные цепи гликанов, содержащие
большее количество маннозы, что облегчает его
интернализацию макрофагами. Велаглюцераза
альфа хорошо переносится, не оказывает
выраженных побочных эффектов. Ни у одного
пациента, получавшего велаглюцеразу альфа не
были выявлены антитела к препарату.
Рекомендуемая доза составляет 60 ЕД/кг 1 раз в 2
недели.
44.
Симтоматическая терапия.При развитии проявлений остеопороза для
замедления и прекращения потери костной
массы, повышения ее прочности,
предотвращения переломов костей, в
комплексной терапии применяют:
1.альфакальцидол
2.соли кальция
3.В качестве симптоматической терапии скелетных
осложнений БГ типа 1 назначаются анальгетики
(во время костных кризов), антибактериальная
терапия.
45.
Болезнь Ниманна –Пика
46.
Определение.Это гетерогенная группа аутосомно-рецессивных
нарушений, проявляющихся накоплением
липидов, общими клиническими признаками
которых являются гепатоспленомегалия и
отложение сфингомиелина в
ретикулоэндотелиальных и паренхиматозных
тканях, с вовлечением или без вовлечения ЦНС.
47.
Патогенез.В основе болезни лежит дефицит кислой
сфингомиелиназы, ведущий к патологическому
накоплению сфингомиелина в печени, селезенке,
легких. Причиной дефицита кислой
сфингомиелиназы являются мутации гена SMPD1.
Патологическое накопление сфингомиелина в
клетках моноцитарно-макрофагального ряда
ретикулоэндотелиальной системы проявляется
гепато спленомегалией, интерстициальной
болезнью легких, инфильтрацией костного мозга.
Накопление сфингомиелина происходит еще
внутриутробно, обнаруживается на 12-й нед.
гестации.
48.
Биопсия печени у пациентов с болезньюНиманна – Пика выявляет перипортальный
и перицеллюларный фиброз, окружающий
аффектированные Купферовские клетки,
отложения сфингомиелина и
инфильтрацию макрофагами. При средней
степени изменений биоптата
сфингомиелин аккумулируется только в
Купферовских клетках, при тяжелых – в
Купферовских клетках и гепатоцитах
49.
50.
51.
52.
• В биоптате кожи накопление сфингомиелинаотмечается в дермальных фибробластах,
макрофагах, клетках сосудов (эндотелиальных,
гладкомышечных и клетках Шванна)
• Классификация болезни Ниманна – Пика:
1. Тип А характеризуется тяжелым ранним
поражением ЦНС и массивными висцеральными
и церебральными отложениями сфингомиелина.
2. Тип В имеет хроническое течение с поражением
висцеральных органов, но без явного
вовлечения ЦНС
3. Тип С отличается подострым вовлечением ЦНС с
медленным или средним прогрессированием,
умеренными висцеральными отложениями.
53.
Тип А.Тип А (инфантильная форма) имеет признаки
быстропрогрессирующего
нейродегенеративного заболевания и
проявляется выраженной
гепатоспленомегалией, прогрессирующими
психомоторными нарушениями и
фатальным исходом в первые годы жизни.
Активность сфингомиелиназы при типе А
составляет от 1 до 10% по сравнению с
нормой
54.
Тип В.Тип В характеризуется преимущественно
висцеральными нарушениями с развитием
гепатоспленомегалии, поражением легких.
Пациенты могут доживать до подросткового и
взрослого периода. Промежуточный фенотип,
названный тип А/В, может включать
неврологические нарушения, умеренную
задержку интеллектуального развития,
изменения на глазном дне по типу «вишневой
косточки» и висцеральные нарушения,
характерные для типа
55.
Тип С.Тип С является совершенно другим заболеванием, в основе
которого лежат мутации в генах NPC1 и NPC2 и нарушение
транспорта холестерина. Ген NPC1 (Gene ID 4864) имеет
очень широкий спектр мутаций, отсюда нет возможности
выявить четкую связь между уровнем мутации,
клинической картиной, сроками манифестации болезни и
тяжестью течения. Болезнь может манифестировать с
неонатального периода до 39 лет . NPC1-ген кодирует
трансмембранный протеин, участвующий в
интрацеллюлярном транспорте холестерина. Мутации
гена приводят к интрацеллюлярному накоплению
неэстерифицированного холестерина в поздних
эндосомальных/лизосомальных структурах (рис. 8, 9),
накоплению глико сфинголипидов в тканях, в первую
очередь нейрональной. Итогом таких нарушений является
гепатоспленомегалия и неврологическая деградация,
ведущие к раннему неблаго приятному исходу.
56.
Диагностика третьего типа представляет большиетрудности в клиническом плане. Это связано с
большим разбросом времени манифестации
болезни: от раннего периода младенчества до
взрослого; разнообразием клинической
картины. N. Bergamin et al. предлагают строить
классификацию болез ни Ниманна – Пика, тип C,
на сроках манифестации неврологических
симптомов:
1.тяжелая инфантильная форма – в первые 2 года
жизни
2. поздняя инфантильная форма – 3–5 лет жизни,
3.ювенильная форма – от 5 до 16 лет, взрослая
форма – старше 16 лет
57.
В типичных случаях в список основныхневрологических нарушений входят :
1.атаксия,
2. дизартрия,
3.дисфагия
4. прогрессирующая деменция
5. вертикальный паралич взора.
К другим достаточно общим симптомам относятся
катаплексия (аффективная адинамия), судороги,
дистония.
У пациентов с поздней манифестацией болезни
нередко возникают психические нарушения
58.
В перинатальном периоде возможно развитиеводянки плода, пролонгированная неонатальная
желтуха, обусловленная холестазом и
появляющаяся в первые дни и недели жизни,
прогрессирующая гепатоспленомегалия [13]. В
подавляющем большинстве случаев желтуха
спонтанно уходит к 2–4 мес. жизни, остается
гепатоспленомегалия в течение весьма
вариабельного периода до появления
неврологической симптоматики. Примерно у
10% пациентов развивается печеночная
недостаточность в первые 6 мес. жизни.
59.
В раннем инфантильном периоде (2 мес. – 2 года)основным системным признаком болезни является
изолированная гепатоспленомегалия. Первым
признаком неврологических нарушений является
задержка моторного развития, которая появляется с
8–9 мес. жизни и становится явной в возрасте 1–2
лет. У пациентов отмечается прогрессирующая
утрата моторных навыков, прогрессирующее
снижение умственного развития. Многие из этих
детей никогда не начинают ходить. Нередким
признаком является интенсивный тремор,
супрануклеарный паралич взора (который, как
правило, не распознается), редко отмечаются
судороги. Магнитно-резонансная томография
выявляет признаки лейкодистрофии и
церебральной атрофии. Продолжитель ность жизни
редко превышает 5 лет
60.
• В позднем инфантильном периоде (2 года – 6 лет) у многихпациентов первыми признаками болезни являются
гепатоспленомегалия или изолированная спленомегалия.
Признаками неврологических нарушений являются задержка
развития речи, нарушение походки, дети часто падают,
неуклюжи, что связано с развитием атаксии. Паралич взора
есть, но не распознается на ранней стадии. Возможно
снижение слуха. Достаточно частым симптомом и первым
признаком манифестации болезни может быть катаплексия.
Ухудшается моторика, становится явной задержка умственного
развития.
• У большойчасти пациентов развиваются судороги, которые
могут быть парциальными, генерализованными или
смешанными. В большинстве случаев дети отвечают на
стандартное лечение противосудорожными средствами, но
могут быть и случаи рефрактерности. Параллельно с
прогрессированием атаксии развиваются дисфагия, дизартрия
и деменция. На поздних стадиях у пациентов развиваются
спастичность, прогрессирующее нарушение речи. Большин
ству требуется гастростомия. При этой форме заболевания
фатальный исход наступает на 7—12-й год жизни.
61.
• В возрасте 6–15 лет развивается классическая форма болезниНиманна – Пика, тип C, с неврологическим дебютом, которая является
самой частой.
• Умеренная спленомегалия или гепатоспленомегалия выявляется
намного раньше неврологических симптомов, практически может
отмечаться начиная с неонатального периода.
• Школьные проблемы выступают на первый план и характеризуются
нарушением письма и внимания. Часто первым признаком
заболевания может быть вертикальный паралич взора, который
появляется в разные сроки. Ребенок становится неуклюжим,
демонстрирует нарастающую неспособность к обучению.
• Другой типичный общий симптом – катаплексия, которая
индуцируется смехом, может быть с нарколепсией или без нее.
• Появляется явная атаксия с частыми падениями, трудностями при беге,
развиваются дизартрия и дисфагия. Около половины детей реализуют
судороги различной степени выраженности и тяжести.
• На более поздних стадиях болезни пациенты перестают говорить,
появляются пирамидальные признаки и спастичность, проблемы с
глотанием, которые требуют гастростомии. Пациенты живут до 30 лет,
иногда больше
62.
Диагностика.1.Нейровизуализация
Магнитнорезонансная томография (МРТ) у пациентов
с НП-С с поздним началом неврологических
расстройств часто выявляет церебральную и/или
мозжечковую атрофию, а также гиперинтенсивность
белого вещества головного мозга в режимах Т2 и
FLAIR у пациентов с началом заболевания в раннем
детском возрасте. К другим областям головного
мозга, которые преимущественно поражаются по
данным нейровизуализации, относятся гиппокамп,
таламус и полосатое тело. Кроме того, часто
наблюдается истончение мозолистого тела и
некоторое уменьшение области среднего мозга на
срединном сагиттальном срезе.
63.
2. Ультразвуковое исследование (УЗИ) внутренних органов УЗИвнутренних
органов должно проводиться всем пациентам с подозрением на НП-С для
выявлениясплено/гепатоспленомегалии.
3. Хитотриозидаза
Активность фермента хитотриозидазы в плазме крови
повышается при некоторых лизосомных болезнях накопления
и, в том числе, у пациентов с болезнью НП-С. Так, при болезни
Гоше – она превышает норму в 10-20 раз, при НП-С повышение
активности не столь значительно, но высокая активность
хитотриозидазы у пациента с изолированной
гепатоспленомегалией и каким-либо неврологическим
симптомом, описанным в Разделе 2, является важным
критерием вероятности НП-С. Нормальная активность
фермента не исключает наличие заболевания, кроме того, у
10% населения в Европейскких странах присутвуют мутации в
гене самой хитотриозидазы, что, не вызывая клинических
последствий, является причиной ее крайне низкой активности.
Это следует учитывать при проведении биохимического
тестирования.
64.
4.Повышение уровня продуктов окисленияхолестерина (холестан-3,5,6-триола и 7кетостерола) у животных и людей с мутациями в
генах NPC1 и NPC2, является чувствительным и
специфическим маркером для скрининга НПС.
Уровень данных метаболитов не повышается у
пациентов с другими нейродегенеративными
заболеваниями. Определить эти соединения можно
с помощью метода газовой хроматографии массспектрометрии (ГХ-МС) или высокоэффективной
жидкостной хроматографии тандемной массспектрометрии (ВЭЖХ-МС/МС). Последний метод
является предпочтительным, поскольку позволяет
определять эти соединения в небольшом
количестве биологического материала и не требует
длительной пробоподготовки, как при ГХ-МС
65.
5.Неспецефические методыВ течение первых месяцев может отмечаться
повышение биохимических маркеров холестаза:
билирубина за счет прямой фракции, холестерина,
триглицеридов, ГГТ и ЩФ, синдром цитолиза
(повышение АЛТ и АСТ). В редких случаях при
развитии печеночной недостаточности характерно
снижение в крови показателей, отражающих
синтетическую функцию печени: альбумина,
фибриногена, ПТИ или ПТВ, ХЭ, холестерина и др.
В более старшем возрасте лабораторные изменения
могут отсутствовать или наблюдается умеренное
повышение трансаминаз (АЛТ, АСТ), триглицеридов,
снижение ЛПНП и ЛПВП.
В любом возрасте может наблюдаться развитие
тромбоцитопении, что обусловлено
гиперспленизмом.
66.
6.Гистологическое исследованиеСветовая микроскопия может определить
характерные пенистые клетки в различных тканях,
но данные изменения не являются специфическими
для болезни Ниманна-Пика, тип С.
Биопсия костного мозга. При возможности
проведения данного анализа, аспирация костного
мозга может выявить пенистые клетки. Следует
помнить, что инфильтрация костного мозга
пенистыми клетками коррелирует со степенью
тяжести заболевания и может быть минимальной у
пациентов на ранних этапах заболевания.
Множественные плашки костного мозга требуются
для исключения ошибки отбора пробы; в первую
очередь следует проводить менее инвазивные
процедуры.
67.
7. Цитохимический анализДоказательство нарушенного внутриклеточного
транспорта холестерина путем окраски филипином
в культуре фибробластов из биопсии кожи пациента
остается ключевым диагностическим тестом для НПС в ряде стран. При окрашивании культуры клеток
фибробластов в 80-85% случаев НП-С наблюдаются
интенсивные флюоресцирующие области,
сконцентрированные вокруг ядра клетки, которые
соответствуют накоплению неэстерифицированного
холестерина. В большинстве других случаев с
«вариантным биохимическим фенотипом»
наблюдается менее выраженное, более
вариабельное накопление холестерина. Это
исследование проводится только в небольшом
числе специализированных лабораторий. В России
данный метод не используется, для подтверждения
диагноза применяется генетическое тестирование.
68.
8. Генетическое тестированиеБолезнь НПС обусловлена мутациями генов
NPC1 и NPC2. 95% случаев связаны c
мутациями гена NPC1 (локус 18q11–q12),
около 4% случаев — с мутациями гена NPC2
(локус 14q24), примерно в 1% случаев
молекулярно-генетический дефект
идентифицировать не удается .
69.
Специфическое лечение.Миглустат - является небольшой молекулой иминосахара,
действующая как конкурентный ингибитор фермента,
глюкозирцерамидсинтазы, который катализирует первый
фиксированный этап синтеза гликосфинголипидов (ГСЛ) .
Миглустат может проникать через гематоэнцефалический
барьер, и было показано, что он уменьшает накопление
ГСЛ в головном мозге, замедляет развитие
неврологической симптоматики и удлиняет
выживаемость при доклинических исследованиях.
Он может также непрямым образом регулировать
внутриклеточный гомеостаз кальция, связанный с
накоплением сфингозина – предполагаемым пусковым
фактором в патогенезе НПС1 – путем влияния на
концентрацию глюкозилцерамида.
70.
Доза у пациентов в возрасте 4-12 лет назначается,исходя из площади поверхности тела:
>1,25 --------------------200 мг три раза в день
>0,88–1,25 -------------200 мг два раза в день
>0,73–0,88 --------------100 мг три раза в день
>0,47–0,73 ---------------100 мг два раза в день
≤0,47 ----------------------100 мг один раз в день
71.
Симтоматическая терапия.Катаплексия Трициклические …антидепрессанты или стимуляторы ЦНС
Дистония и тремор …………………..Холинолитические средства
Двигательные нарушения …………Физиотерапия
Эпилептические приступы …………Антиконвульсанты
Нарушения сна……. Мелатонин или седативные препараты перед сном
Нарушения питания ……………..Зондовое питание, гастростомия
Респираторные осложнения ……..Физиотерапия с бронходилятаторами;
антибиотикотерапия
Слюнотечение ……………Атропин перорально, инъекции ботулотоксина в
околоушную или подчелюстную железу, гиосциновые пластыри или
гликопиррония бромид
Кататония……………………… Электрошоковая терапия
Психические нарушения…………………. Вальпроат натрия для лечения
биполярных аффективных расстройств, селективные ингибиторы
обратного захвата серотонина при депрессиях
Диарея……………………………….. Диета, лоперамид
72.
Видыдислипидемий.
73.
Определение.Дислипопротеидемии – нарушение
нормального соотношения ЛП, ХС, ТГ. М.б.
первичные (наследственные) и вторичные
(в ответ на факторы окружающей среды/
основное заболевание).
74.
75.
76.
77.
Если ХМ есть, то – I или V.Если ↑ ЛППП, то – III.
α – ЛП - ЛПВП (антиатерогенное действие)
β - ЛП – ЛПНП (самые атерогенные)
пре-β - ЛП – ЛПОНП
ЛППП – ЛП промежут. плотности (образуются при
нарушении превращения ЛПОНП)