


































 medicine
medicineSimilar presentations:










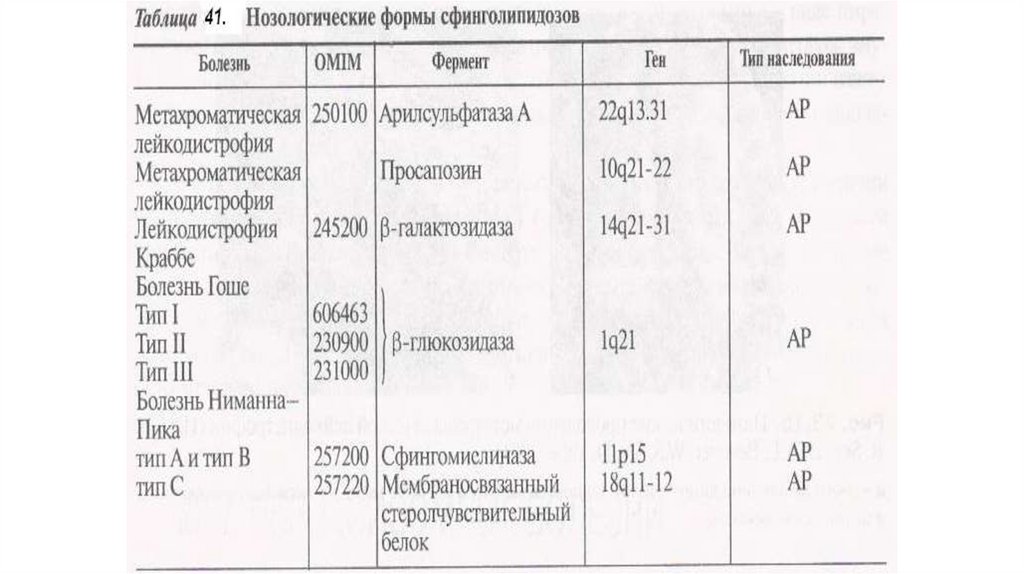
Наследственные болезни накопления (сфинголипидозы)
1. Лекция 12
.НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ(СФИНГОЛИПИДОЗЫ). ГЕМОГЛОБИНОПАТИИ.
НАСЛЕДСТВЕННЫЕ ГЕМОЛИТИЧЕСКИЕ АНЕМИИ,
2.
План лекции1. Лизосомные болезни ( сфиноголипидозы)
2. Метахромати́ческая лейкодистрофи́я
3. Болезнь Тея-Сакса
4. Болезнь Гоше
5. Наследственные гемолитические анемии, гемоглобинопатии,
6. Болезни нарушения свертываемости крови
7. Митохондриальные болезни
3.
ЛИЗОСОМНЫЕ БОЛЕЗНИ. СФИНГОЛИПИДОЗЫСфинголипидозы - врожденные нарушения метаболизма
липидов, главным образом сфинголипидов, входящих в состав
клеточных мембран головного мозга и других органов.
Нарушения обусловлены отсутствием лизосомных ферментов,
катализирующих процессы распада сфинголипидов.
В клиническом плане болезни накопления сфинголипидов
характеризуются
прогрессирующими
умственными
и
двигательными расстройствами вследствие изменений головного
мозга, поражениями костей, паренхиматозных органов (печень,
селезенка, почки), кожи и сетчатки глаз.
4.
Сфинголипидозы (СЛ) — группа наследственных заболеваний,которая обусловлена снижением активности ферментов,
обеспечивающих деградацию сфингомиелинов — галактозидов и
цереброзидов. Сфингомиелины — сложные липиды, основным
структурным компонентом которых является церамид.
5.
6.
Метахромати́ческаялейкодистрофи́я(англ.
Metachromatic
leukodystrophy (MLD), также сульфати́дный липидо́з) —
редкое наследственное заболевание из группы лизосомных болезней
накопления с аутосомно-рецессивным механизмом наследования
нарушения обмена веществ [ Le, Tao.,2012]. Данная нозологическая
единица из разряда лейкодистрофий (патология роста и/или
развития
миелиновой
оболочки,
покрывающей
большинство нервных волокон центральной и периферической
нервной системы) относится к сфинголипидозам. Лейкодистрофии это группа заболеваний, характеризующихся прогрессирующим
поражением белого вещества головного мозга и глии (в слове
«лейкодистрофия» корень «лейко» происходит от латинского слова
leucos - белый). Первое описание МЛД было сделано Альцгеймером
в 1919 году. Тип наследования заболевания аутосомно-рецессивный.
7.
Метахроматическая лейкодистрофия встречается с частотой от 1 на 40 000[Т. Р. Харрисон.,1996] до 1 на 160 000 в зависимости от региона
Метахроматическая
лейкодистрофия
развивается
в
результате дефицита лизосомного фермента арилсульфатазы А
(АРСА, англ. ARSA) [Poeppel P, Habetha M, Marcão A, et al., (March
2005)] или цереброзидсульфатазы. На фоне развёртывания клинической
симптоматики уровень активности фермента в лейкоцитах существенно
снижается и составляет менее 10 % от нормальных величин. При
возникновении блока этих обменных превращений происходит
накопление субстратов в лизосомах, приводящее к нарушению функции
нейронов. Наряду с классическим генетическим вариантом МЛД описано
несколько случаев со снижением активности арилсульфатазы А
вследствие мутации в гене просапозина, локализованном на хромосоме 10
и содержащем 15 экзонов. Показано, что продуктом этого гена являются
четыре активаторных белка: сапозин А, В, С, D - один из которых, а
именно сапозин В, регулирует активность арилсульфатазы А.
8.
Клинические проявления МЛД полиморфны. Выделяют несколько вариантов,отличающихся возрастом начала, характером клинических симптомов и
тяжестью течения. Первый вариант встречается довольно редко и называется
врожденным; клиническая картина разворачивается в возрастном диапазоне
от рождения до 4-месячного возраста. Характерные симптомы: мышечная
гипотония, снижение сухожильных рефлексов, эпизоды апноэ, цианоз и
тонико-клонические судороги. Смерть больных наступает спустя несколько
месяцев или даже дней от момента появления первых симптомов. Второй
вариант - наиболее распространенный, его называют ювенильным. Симптомы
при этой форме заболевания становятся заметными в возрасте от 6 месяцев
до 2 лет, после периода нормального психомоторного развития. У ребенка
возникает мышечная гипотония; потеря ранее приобретенных двигательных
навыков; прогрессирующая умственная отсталость; атрофия дисков
зрительных нервов; корковая слепота, дизартрия, переходящая в афазию;
атаксия; бульбарный или псевдобульбарный паралич и судороги.
9.
В терминальных стадиях заболевания мышечная гипотония сменяетсямышечным гипертонусом с появлением спастического тетрапареза. Смерть
больных наступает спустя 2—3 года от начала заболевания. Взрослая форма
начинается на втором-третьем десятилетии жизни. Клинические проявления
заболевания характеризуются наличием выраженных психических нарушений
в виде психозов, шизофреноподобных симптомов в виде бреда, слуховых
галлюцинаций и деперсонализации. По мере прогрессирования заболевания к
этим симптомам присоединяется очаговая неврологическая симптоматика в
виде пирамидных и экстрапирамидных симптомов, атрофии дисков
зрительных нервов, энуреза и энкопреза. Часто такие больные оказываются
пациентами психиатрических клиник. Смерть больных наступает через пять
— десять лет от манифестации заболевания. Лечение не разработано — не
существует лекарства от метахроматической лейкодистрофии.
10.
Болезнь Тея-Сакса (Tay-Sachs disease; MIM 272800 ) наследственное нейрометаболическое, связанное с накоплениемв головном мозге ганглиозида GM2. Болезнь Тея-Сакса является
аутосомно-рецессивным, прогрессирующим нейродегенеративным заболеванием, которое, в классической инфантильной
форме, как правило, приводит к смертельному исходу в возрасте
2 или 3-х лет. Бета-гексозаминидаза ( EC 3.2.1.52 .) существует в
двух формах - A и B. Гексозаминидаза А (hexosaminidase A;
HEXA) - лизосомный фермент, катализирующий катаболизм
GM2 ганглиозида. При отсутствии данного фермента в
лизосомах клеток накапливается субстрат реакции - GM2
ганглиозид, главным образом в центральной нервной системе.
11.
При патолого-анатомическом исследовании обнаруживаютсяувеличение мозга, явления гидроцефалии, диффузная атрофия
отдельных областей больших полушарий, а также истончение
зрительных нервов. Лечение симптоматическое.
12.
Болезнь Гоше (Gaucher disease; MIM 230800 ) описана Gaucher в1882 году. Представляет собой форму внутриклеточных
липоидозов, характеризующуюся накоплением цереброзидов в
клетках
ретикулоэндотелиальной
системы.
Патологоанатомически
наиболее
тяжелые
изменения
наблюдаются в селезенке, печени, а также в лимфатических
узлах, костном мозге, легких, почках. Ретикуло-эндотелиальная
система (РЭС) — устаревший термин для обозначения
тканевых макрофагов (например: микроглия, клетки Купфера в
печени, альвеолярные макрофаги, клетки Лангерганса).
Клинически различают три формы болезни Гоше:
1) Тип I ( MIM 230800 ) - взрослый тип
2) Тип II ( MIM 230900 ) - инфантильный тип
3) Тип III ( MIM 231000 ) - ювенильный тип.
13.
Болезнь Гоше типа I вызвана гомозиготной или сложнойгетерозиготной мутацией в гене, кодирующем кислую бетаглюкозидазу (GBA, 606463 ). Ген глюкоцереброзидазы (бетаглюкозидазы - GBA) локализуется на 1-ой хромосоме человека в
позиции q21 (Cormand B.,1997). Gene: [01q21/GCGD]
glucocerebrosidase (D-glucosyl-N-acylsphingosine glucohydrolase).
14.
Одна из главных причин инвалидизации при 1 и 3 типе болезниГоше —- поражение костной ткани. Нарушение нормальных
физиологических процессов происходит из-за накопления
липидов в остеокластах и замещении инфильтратами клеток
Гоше нормальных элементов костного мозга. Несмотря на
увеличение печени и её дисфункцию, случаи тяжелой печеночной
недостаточности встречаются редко.
15.
Наследственныегемолитические
анемии,
гемоглобинопатии,
гемофилии
Анемия Фанкони
Анемия Фанкони наследственная апластическая, конституциональная
инфантильная панмиелопатия, врожденная панмиелопатия Фанкони, (MIM
227650 ; MIM 227660 ) впервые описана в 1937 году Fankoni и позднее
названа детским семейным апластическим миелозом. Анемия Фанкони болезнь с аутосомно-рецессивным типом наследования, сопровождающаяся
множественными хромосомными аномалиями. Клинически проявляется
прогрессирующей панцитопенией, пороками развития , патологией скелета (
отсутствие лучевой кости , недоразвитие большого пальца кисти ),
косоглазием , микроцефалией , низкорослостью и гипогонадизмом (
Chaganti R.S.K.,1991 ; Glanz A.,1982 ). А также предрасположенностью к
злокачественным новообразованиям , ломкостью хромосом и повышенной
чувствительностью к химическим мутагенам, в том числе к диэпоксибутану
( бутандиендиэпоксиду ) и циклофосфамиду. Часто развиваются ОМЛ и рак
16.
К настоящему времени выявлено восемь комплементационных групп прианемии Фанкони: FA1, FA-B, FA-C, FA-D, FA-G, FA-E, FA-H [Joenje
H.,1997 ; Joenje H.,2000].
У четырех комплементационных групп анемии Фанкони (FA-A, FA-C, FAG, FA-E) клонирована кДНК. Идентифицированы мутации в генах FA-A,
FA-C и FA-G в соответствующих комплементационных группах анемии
Фанкони.
Клетки
больных
анемией
Фанкони
проявляют
хромосомную
нестабильность ( German J.,1987 ) и гиперчувствительны к ДНКсшивающим агентам
17.
Недостаточность глюкозо-6-фосфат-дегидрогеназыГемолитическая несфероцитарная анемия ( MIM 305900 ) обусловлена
дефицитом глюкозо-6-фосфат дегидрогеназы (G6PD) и нестойкостью
глютатиона эритроцитов ( Beutler E.,1996 ; Luzzatto L.,1995 ; Luzzatto
L.,1985 ; Mason P.J.,1996 ; Vulliamy T.J.,1992 ). Фермент G6PD
представляет собой олигомер (или димер или тетрамер в зависимости
от условий), состоящий из субъединиц с молекулярным весом 56000
D ( Persico M.G.,1986 ; Boyer S.H.,1962 ;Cohen P.,1969 ).
Г-6-ФД - первый фермент пентозофосфатного гликолиза . Основная
функция фермента заключается в восстановлении НАДФ до НАДФН ,
необходимого для перехода окисленного глутатиона (GSSG ) в
восстановленную форму.
18.
Эритроцитоз, элиптоцитоз, сфероцитоз, овалоцитоз - группанаследственных заболеваний ( MIM 182860 ; MIM 182870 ; MIM 182900 ;
MIM 130500 ; MIM 166900 ; MIM 130500 ), характеризующихся
измененной овальной формой эритроцитов, обусловленной дефектами ряда
генов - гена альфа спектрина (SPTAI), гена бета спектрина (SPTB), гена
анкирина (ANKI), белка band 4.1 (EPBA41). У людей, имеющих в крови до
80-90% эритроцитов овальной формы, какой-либо специфической
патологии не наблюдается, в связи с чем элиптоцитоз считается безвредной
для организма аномалией.
19.
20.
Гемоглобинопатия — наследственное или врождённое изменениеили нарушение структуры белка гемоглобина, обычно приводящее
к клинически или лабораторно наблюдаемым изменениям в его
кислород-транспортирующей функции либо в строении и
функции эритроцитов.
К наиболее часто встречающимся и известным гемоглобинопатиям
относятся
серповидно-клеточная
анемия,
бетаталассемия, персистенция фетального гемоглобина.
21.
Гемоглобинопатии - группа наследственных заболеваний,этиологическим фактором которых являются мутации в
генах, кодирующих полипептидные цепи глобиновых
белков. При различных вариантах гемоглобинопатии
гемоглобин либо приобретает неправильную форму, либо в
его составе меняется соотношение глобиновых цепей. В
норме у взрослого человека 95%—98% всего гемоглобина
составляет НbА, в состав которого входях две α и две β
цепи. Таким образом, его формула записывается как НbА
(α2; β2). От 2% до 2,5% приходится на долю НЬ А2,
имеющего формулу (α2; δ2) и лишь 0,1-2% гемоглобинов у
взрослого человека составляет фетальный HbF(α2; γ2).
22.
В период эмбриогенеза соотношение гемоглобинов бываетсовершенно другим и, кроме того, присутствуют такие его формы,
которые никогда не встречаются в постнатальном периоде.
Например, у плода до 18-недельного возраста имеется Hb Gower 2
(α2; ε2), в состав которого входит отсутствующая у взрослых εполипептидная цепь. С 20-ой недели внутриутробной жизни плода
происходит постепенная замена этого типа гемоглобина на HbF
(α2; γ2), который доминирует у плода и новорожденных детей.
Замена фетального гемоглобина на гемоглобин А происходит в
течение первого года жизни ребенка. Существует множество
генетических механизмов возникновения заболеваний этой
группы, обусловленных особенностями расположения генов
глобиновых белков и сложным контролем их транскрипции.
23.
Для α-цепи имеется два дуплицированных гена, расположенныхна хромосоме 16p13.3-pter. Гены глобиновой цепи образуют на
хромосоме 11p15.4-15.5 кластер. Последовательность генов
соответствует этапам их экспрессии от периода эмбриогенеза до
взрослого возраста -первым располагается ген ε-цепи, затем два
гена γ-цепей (γ-G и γ-А). Продукты этих генов вместе с αглобиновой цепью формируют различные типы гемоглобинов.
Очередность такой экспрессии, по-видимому, регулируется
генами
контролирующих
регионов,
расположенных
в
промоторной области на некотором расстоянии от 5'-конца этого
кластера.
24.
Заболевания, при которых количество α-или β-глобиновуменьшено или они полностью отсутствуют и заменены в
молекуле
гемоглобина
другими
цепями,
называют
средиземноморскими лихорадками, или талассемиями (от
греческого слова «таласса», которым в древности называли
Средиземное море). В зависимости от нарушения синтеза α- или
β-глобиновых цепей выделяют α- или β-талассемии. Эти
заболевания составляют значительную часть в структуре
гемоглобинопатии и представлены множеством аллельных
вариантов, возникающих в результате точковых мутаций в генах
глобиновых цепей. К настоящему времени описано более 300
таких мутаций, большинство из них очень редки.
25.
Наиболее выраженные клинические проявления обусловленымутациями, затрагивающими функционально значимые участки
белковой молекулы (т.е. теми мутациями, которые нарушают
формирование вторичной и третичной структуры глобина или
аминокислотную последовательность в местах прикрепления гема
или контактах цепей друг с другом. К наиболее распространенным
аллельным вариантам гемоглобинопатии, помимо талассемий
можно
отнести
серповидноклеточную
анемию,
метгемоглобинемию и эритроцитоз.
26.
Альфа-талассемия: общие сведенияАльфа талассемия ( MIM 141800 ) может проявляться в гомозиготной и
гетерозиготной формах. Гетерозиготная форма характеризуется
гипохромной анемией, микроцитозом с наличием в пуповинной крови
гемоглобина Барта. Гомозиготная форма ведет к развитию водянки плода
и к его гибели до рождения. Альфа-талассемия - гемолитическая анемия,
вызванная дефицитом синтеза альфа-глобина в результате потери или
повреждения одного или нескольких альфа-глобиновых генов. Снижение
синтеза альфа-глобиновых цепей приводит к накоплению свободных
гамма-глобиновых цепей и бета-глобиновых цепей и формированию из
них тетрамеров-гамма4 ( Hb Bart's ) и нестабильного бета4 ( Hb H ) с
последующим ускорением разрушения эритроцитов. Обладая очень
высоким сродством к кислороду, эти тетрамеры не могут выполнять
функцию переноса кислорода.
27.
Клиническая картина тяжелой альфа-талассемии характеризуетсякомбинацией гипохромной анемии, гемолиза и дефектного
транспорта
кислорода
вследствие
различных
количеств
физиологически неэффективного Hb в эритроцитах. В результате
степень тканевой гипоксии значительно превышает ожидаемую при
соответствующей степени анемии.
Выделяют 4 группы клинических синдромов альфа-талассемии:
- немое носительство;
- альфа-талассемию с минимальными изменениями;
- гемоглобинопатию Н ;
- альфа-талассемическую водянку плода.
Тяжесть фенотипического проявления альфа-талассемии прямо
пропорциональна снижению альфа-глобинового синтеза.
28.
Бета-талассемия (OMIM: 141900). Группа заболеваний, обусловленныхмутациями в генах глобиновых цепей. В зависимости от типа мутации и
наличия или отсутствия β-глобиновых цепей выделяют несколько
клинико-генетических вариантов β-талассемий. Первый вариант
обозначается как β0-талассемия. Он возникает вследствие нонсенсмутаций и сплайсинговых мутаций, а также мутаций, приводящих к
сдвигу рамки считывания. Второй вариант -β′ талассемия. Его причиной
служат мутации в промоторной области гена, в 5'- и 3'-нетранслируемых
регионах, TATA-, СААТ- и САССС-боксах. В результате таких мутаций
происходит уменьшение или прекращение синтеза глобиновых цепей и
замена их в молекуле гемоглобина. Один из редких вариантов бетаталассемии связан с персистированием фетального гемоглобина, при
котором не происходит его замена на гемоглобин взрослого.
29.
Известно несколько типов персистирующего фетального гемоглобина: 1)гемоглобин представлен двумя γ-цепями (G и А), при этом δ- и β-цепи
отсутствуют; 2) гемоглобин в основном представлен γ-G- и δ-цепями, при
этом ген β-цепи функционально активен; 3) гемоглобин представлен γG- и
γА-цепями, а бета-цепи отсутствуют. Показано, также, что возникновение
симптомов талассемии (так называемых γ-δ-, β-талассемий) может быть
обусловлено делецией в регуляторном элементе глобиновых генов.
Предполагается, что этот регион, обозначаемый аббревиатурой LCR,
находится на расстоянии 100 т.п.н. от кластера глобиновых генов и
контролирует его экспрессию в ходе развития.
Клинические симптомы бета-талассемий разнообразны и зависят от типа
мутаций в гене β-глобиновых цепей. Большая талассемия (анемия Кули), как
правило, проявляется в возрасте 2-6 лет.
30.
Убольных
отмечается
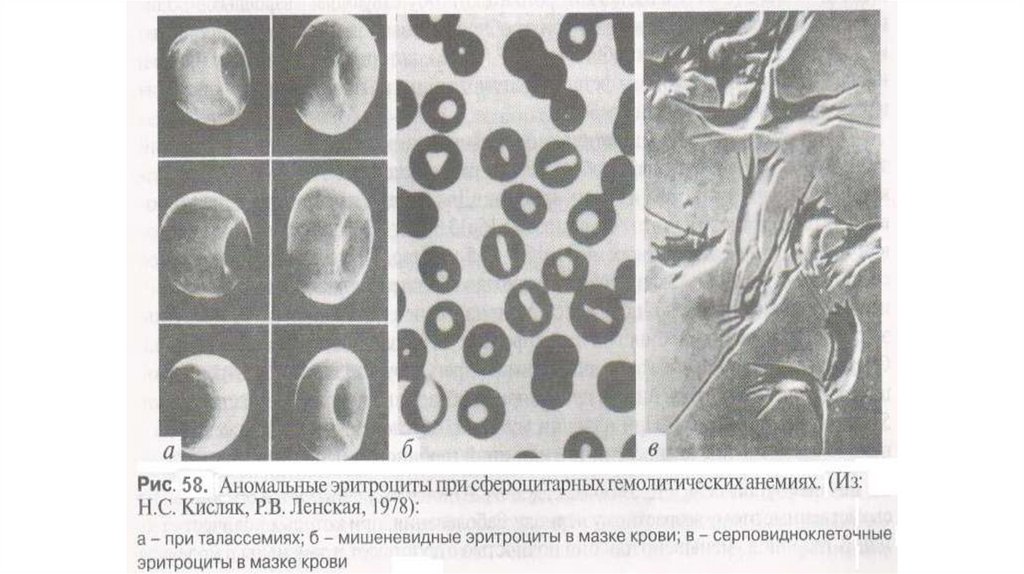
задержка
роста,
гепатоспленомегалия,
дизморфические черты строения черепа и лица – результат остеопороза во
внутриутробном периоде. Гемолиз проявляется желтушной окраской кожи и
слизистых оболочек, приступами лихорадки. Анализ крови демонстрирует
тяжелую гипохромную анемию, снижение содержания гемоглобина,
уменьшение количества эритроцитов и изменение их морфологии
(мишеневидные эритроциты, пойкилоцитоз, анизоцитоз, шизоцитоз,
фрагментация и нормобластоз. Часто при большой талассемии возникают
осложнения в виде трофических язв, цирроза печени, мочекислого диатеза и
гемосидероза. Для определения генетического варианта α- и β-талассемий и
выявления
гетерозиготного
носительства
проводят
молекулярногенетический анализ мутаций в гене α- и β-глобиновых цепей. Возможна
дородовая диагностика талассемии на раннем сроке беременности на
основании анализа ДНК, выделенной из клеток плода.
31.
Серповидноклеточная Анемия. Серповидноклеточная анемия (СА) группа генетически гетерогенных заболеваний, возникающих у гомо- игетерозигот по мутации в гене β-глобина, детерминирующей серповидную
форму эритроцитов. Это заболевание было впервые описано в 1910 году.
Наибольшее количество гетерозиготных носителей аномального
гемоглобина (HbS) обнаружено в эндемичных по малярии регионах
планеты — в Центральной Африке, Средиземноморье, Индии, странах
Ближнего и Среднего Востока, что связано с устойчивостью обладателей
такого генотипа к малярии. Около 8% афроамериканцев гетерозиготны по
HbS. Причиной образования гемоглобина S является точковая мутация, в
результате которой в 6-ом положении полипептидной цепи β-глобина
аминокислота глутамин заменяется на валин. Эта замена приводит к
пониженной
растворимости
гемоглобина
и
повышению
его
полимеризации, что в свою очередь обусловливает изменение формы
эритроцитов на серповидную.
32.
Такие эритроциты становятся ригидными, теряют пластичность,закупоривают мелкие сосуды и гемолизируются. Закупорка сосудов
часто приводит к образованию очагов ишемии, а при длительном
течении заболевания — инфарктов во внутренних органах, костном
мозге и головном мозге. Заболевание часто имеет приступообразное
течение. Появление ишемических кризов часто провоцируется тканевой
гипоксией, снижением рН крови, повышением температуры тела и
замедлением кровотока. Обычно выделяют несколько фаз криза:
ишемическую, длительностью до шести часов; инфарктную,
длительностью до 3 суток; микроэмболическую, длительность которой
варьирует у различных больных от недели до месяца. Типичными
симптомами ишемической фазы являются сильные боли в позвоночнике,
крупных суставах, костях. Если в начале этой фазы начать интенсивное
лечение, то возникновение других фаз заболевания можно
предотвратить.
33.
При отсутствии лечения или недостаточной терапевтическойэффективности используемых препаратов возникает инфарктная фаза, во
время которой боли усиливаются. Появляются лихорадка и симптомы
вегето-сосудистой дистонии. Для микроэмболической фазы характерны
симптомы закупорки кровеносных сосудов, клинические проявления
варьируют в зависимости от локализации участка эмболии. В
межприступный период у больных регистрируется умеренная
нормохромная анемия. К наиболее грозным осложнениям заболевания
можно отнести инсульты мозга, остеомиелит, переломы конечностей,
калькулезный холецистит. Диагностика заболевания основывается на
типичных клинических симптомах, выявлении серповидных эритроцитов
в крови, а также обнаружении мутаций в гене β-цепи гемоглобина при
проведении молекулярно-генетического анализа.
34.
Анемия Минковского-ШофараОдна
из
наиболее
распространенных
мембранопатий
микросфероцитарная анемия Минковского-Шоффара (ОМIМ :182900). Ее
частота составляет 2:10000 новорожденных. Это генетически
гетерогенная группа гемолитических сфероцитарных анемий, две из
которых встречаются чаще других: первая обусловлена мутациями в гене
белка спектрина, вторая связана с мутациями в гене белка анкирина. Оба
эти белка обеспечивают целостность мембраны эритроцитов и ее
устойчивость к деформации. Спектрин представляет собой тетрамер из
2137 аминокислот, состоящий из двух неидентичных цепей - α и β.
Анкирин, состоящий из 1880 аминокислот, обеспечивает связь с
липидными слоями мембраны и пространственную ориентацию
спектрина и его взаимодействие с цитоплазматическими белками. Ген αцепи спектрина локализован на хромосоме 1q21, β-цепи - на хромосоме
14q22-q23, а ген анкирина -в области хромосомы 8р 11.2,
35.
Гемофилия A: ГенетикаГен коагуляционного фактора VIII (F VIII) локализуется на
дистальном конце длинного плеча X хромосомы в позиции Xq28
[Patterson M.N.,1987 ; Patterson M.N.,1989 ; Mandel J.L.,1989].
Доменная структура фактора VIII может быть представлена как A1a1-A2-B-a2-A3-C1-C2.
К настоящему времени удалось установить, что гемофилия A может
быть вызвана следующими поражениями гена фактора VIII:
дупликациями, большими делециями и инсерциями (более 100
нуклеотидов), малыми делециями и инсерциями (менее 100
нуклеотидов), заменами нуклеотидов (nonsense и missense
мутациями), сплайсинговыми мутациями. Описана два случая
дупликаций. Между экзонами 22 и 25 была выявлена дупликация из
22 килобаз IVS 22 [ Gitschier J.,1988]. Дупликация экзона 13
зарегистрирована [Murru S.,1990
36.
37.
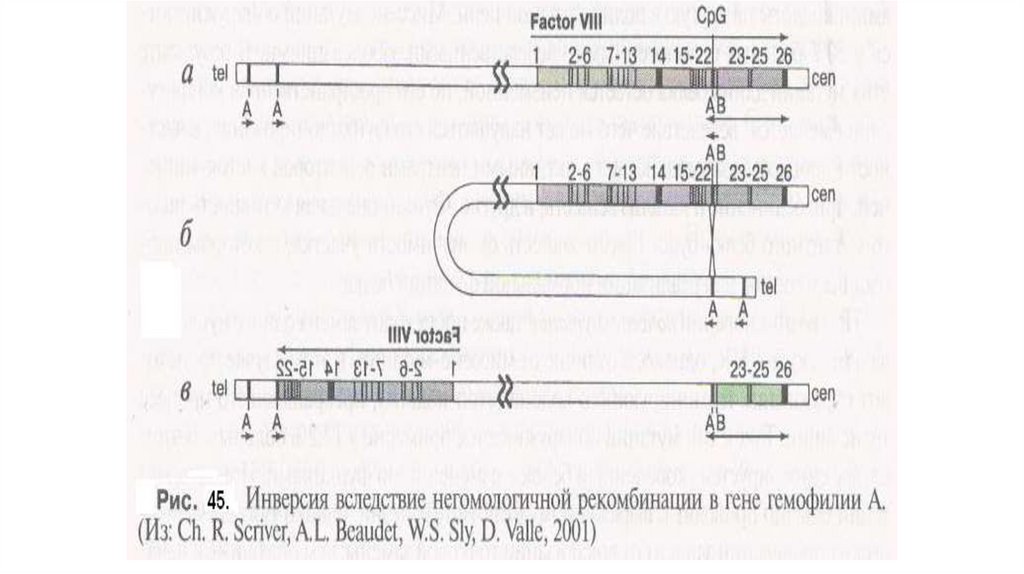
Около 50% тяжелой формы гемофилии A обусловлено большой инверсиейинтрона 22 гена фактора VIII. В интроне 22 локализована область int22h-1, а
дистальнее гена фактора VIII локализовано две копии этой
последовательности, обозначаемые int22h-2 и int22h-3. Внутрихромосомная
гомологичная рекомбинация между int22h-1 и экстрагенными копиями
int22h-2 и int22h-3 приводит к инверсиям. Впервые подобную инверсию у
больных гемофилией A описал Lakish D. в 1993. У 5% больных гемофилией
A отмечаются крупные делеции (более 100 нуклеотидов). К настоящему
времени зарегистрировано более 70 крупных делеций. Делеции могут
захватывать как ген целиком, так и отдельные его экзоны. Данный класс
мутаций приводит к тяжелой форме гемофилии A.
38.
Контрольные вопросы:1.Какие заболевания относятся к сфинголипидозам?
2.Охарактеризуйте метахромати́ческие лейкодистрофи́и.
3.Назовите наследственные болезни с внутриклеточными
липоидозами?
4..Какие заболевания относятся к сфероцитарным и
несфероцитарным анемиям?
5.Какие нарушения гемоглобина характерны для альфа и бетаталласемий