













































 medicine
medicine biology
biologySimilar presentations:










Генетические основы болезней. Наследственные ферментопатии
1.
Лекция № 1Генетические основы болезней
Наследственные ферментопатии
ПМФИ кафедра патологии
к.фармац.наук Саджая Л.А.
2.
План лекции1.
Молекулярные основы наследственности: структура ДНК,
репликация, структура хромосом, транскрипция.
2.
Характеристика генома человека.
3.
Наследственные патологии и их классификация.
4.
Наследственные ферментопатии
3.
Молекулярные основынаследственности: структура ДНК,
репликация, структура хромосом,
транскрипция.
4.
Дезоксирибонуклеиновая кислота - ДНКДезоксирибонуклеиновая кислота является местом хранения генетической информации
организмов. Можно сказать, что это «самая главная молекула».
Роль ДНК стала понятна после того, как Дж. Уотсон и Ф. Крик в 1953 г. предложили модель ее
структуры и характер репликации.
Согласно этой модели, молекула ДНК состоит из двух полинуклеотидных цепей, спирально
закрученных одна относительно другой (Watson J., Crick F., 1953). Открытие «двойной
спирали» было одним из самых волнующих событий в истории биологии. Только через 5 лет
были получены первые экспериментальные подтверждения модели в работах М.
Мезельсона и Ф. Сталя
Дж. Уотсон и Ф. Крик
5.
Строение ДНКВ составе нуклеотидов ДНК
встречаются 4 типа основных
азотистых оснований:
А – аденин;
Т – тимин;
Г – гуанин;
Ц – цитозин.
Углевод
нуклеотида
ДНК
дезоксирибоза (С5Н10О4).
Две полинуклеотидные цепочки
объединяются в единую молекулу
ДНК при помощи водородных
связей
между
азотистыми
основаниями нуклеотидов разных
цепей
–
6.
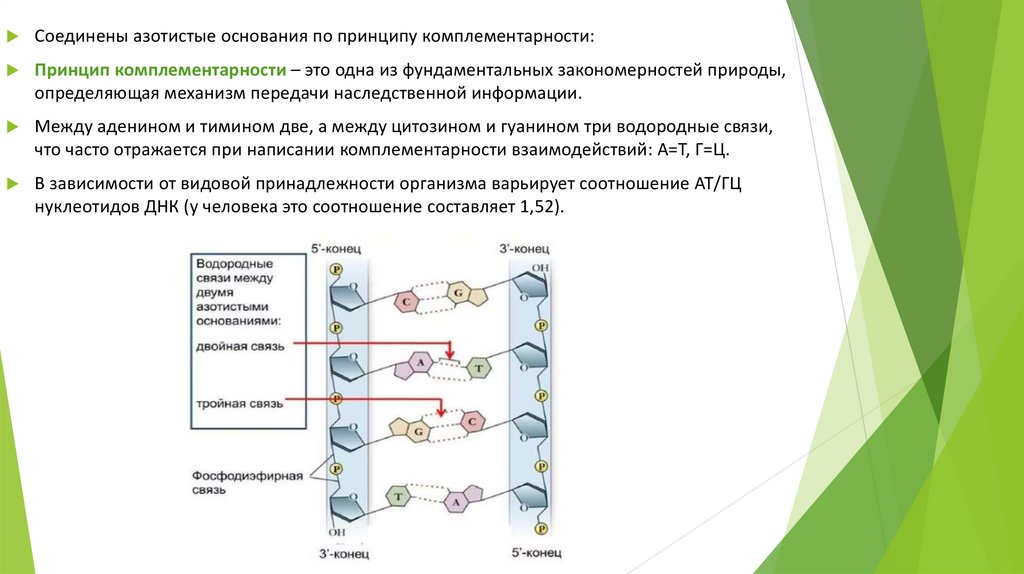
Соединены азотистые основания по принципу комплементарности:Принцип комплементарности – это одна из фундаментальных закономерностей природы,
определяющая механизм передачи наследственной информации.
Между аденином и тимином две, а между цитозином и гуанином три водородные связи,
что часто отражается при написании комплементарности взаимодействий: А=Т, Г=Ц.
В зависимости от видовой принадлежности организма варьирует соотношение АТ/ГЦ
нуклеотидов ДНК (у человека это соотношение составляет 1,52).
7.
Рибонуклеиновая кислота - РНКРибонуклеиновая кислота имеет множество разновидностей, но все ее молекулы
построены по общим структурным принципам.
Они состоят из одной полинуклеотидной цепочки, значительно более короткой, чем
цепочка ДНК.
В нуклеотидах РНК имеются 4 типа азотистых оснований: А, Г, Ц, У(урацил).
РНК чаще, чем ДНК, содержит нетипичные нуклеотиды, которые обычно
модифицируют ее функции. Углевод РНК – рибоза (С5Н10О5).
8.
Основные виды РНК в клеткеИнформационная (матричная) РНК – и-РНК (м-РНК).
Содержит от нескольких сотен до десятков тысяч
нуклеотидов. Молекула и-РНК представляет собой
незамкнутую цепочку. Она переносит информацию о
структуре белка с ДНК на рибосомы – место
непосредственного синтеза полипептидной цепочки.
Рибосомальная РНК – р-РНК. Входит в состав рибосом.
Помимо
структурной
функции,
принимает
непосредственное участие в синтезе полипептидной
цепочки. Составляет 85 % всей РНК клетки.
Транспортная РНК – т-РНК. Переносит аминокислоты к
месту синтеза белков на рибосомы. Каждая молекула тРНК содержит немногим более 80 нуклеотидов.
Гетерогенная ядерная РНК – гя-РНК. Является
предшественником и-РНК у эукариот и превращается в иРНК. Обычно гя-РНК значительно длиннее и-РНК.
Малая ядерная РНК – мя-РНК. Принимает участие в
процессе преобразования гя-РНК.
РНК-праймер – крошечная РНК (обычно 10 нуклеотидов),
участвующая в процессе репликации ДНК.
9.
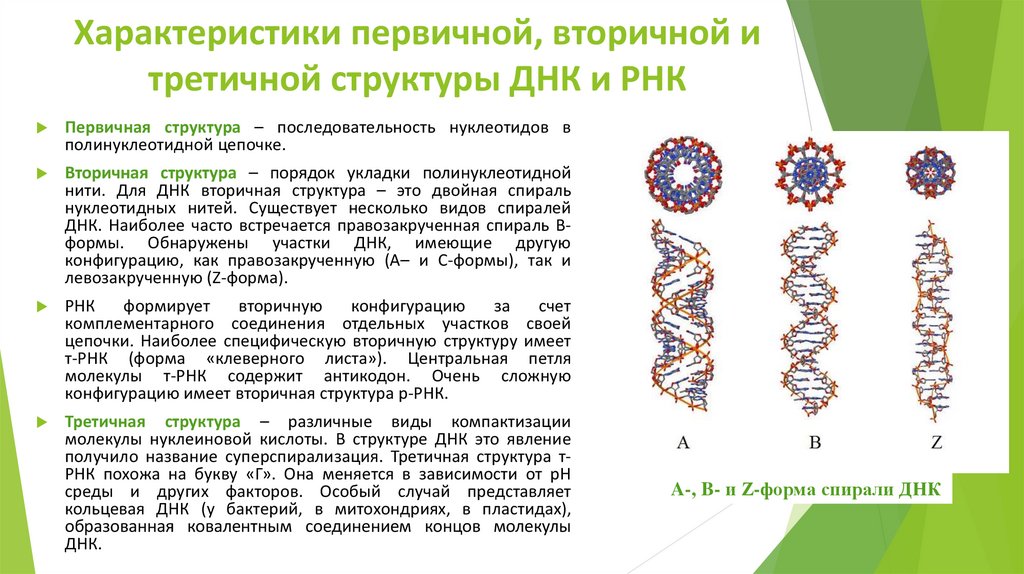
Характеристики первичной, вторичной итретичной структуры ДНК и РНК
Первичная структура – последовательность нуклеотидов в
полинуклеотидной цепочке.
Вторичная структура – порядок укладки полинуклеотидной
нити. Для ДНК вторичная структура – это двойная спираль
нуклеотидных нитей. Существует несколько видов спиралей
ДНК. Наиболее часто встречается правозакрученная спираль Вформы. Обнаружены участки ДНК, имеющие другую
конфигурацию, как правозакрученную (А– и С-формы), так и
левозакрученную (Z-форма).
РНК
формирует
вторичную
конфигурацию
за
счет
комплементарного соединения отдельных участков своей
цепочки. Наиболее специфическую вторичную структуру имеет
т-РНК (форма «клеверного листа»). Центральная петля
молекулы т-РНК содержит антикодон. Очень сложную
конфигурацию имеет вторичная структура р-РНК.
Третичная структура – различные виды компактизации
молекулы нуклеиновой кислоты. В структуре ДНК это явление
получило название суперспирализация. Третичная структура тРНК похожа на букву «Г». Она меняется в зависимости от рН
среды и других факторов. Особый случай представляет
кольцевая ДНК (у бактерий, в митохондриях, в пластидах),
образованная ковалентным соединением концов молекулы
ДНК.
A-, B- и Z-форма спирали ДНК
10.
Репликация ДНКРепликацией называется процесс удвоения молекул ДНК.
3 этапа репликации:
1.
Двойная спираль ДНК (под действием ферментов ДНК-геликазы, ДНК-топоизомеразы и др.) раскручивается,
водородные связи разрываются и цепи расходятся. В результате образуется структура, названная репликативной
вилкой.
2.
Происходит матричный синтез. К образовавшимся свободным связям присоединяются по принципу
комплементарности (А-Т, Г-Ц) свободные нуклеотиды. У каждой дочерней молекулы ДНК одна нить происходит
от материнской молекулы, а другая является вновь синтезированной – полуконсервативная модель репликации.
Поскольку синтез возможен только в направлении 5’-3', на одной нити идет быстрый синтез, а на другой –
медленный, короткими фрагментами (1000–2000 нуклеотидов) - фрагментами Оказаки. Свободный 3'-конец,
необходимый для начала синтеза фрагмента Оказаки, обеспечивает РНК-праймер, синтезируемый при помощи
особой РНК-полимеразы – праймазы. После выполнения своей функции РНК-праймер удаляется, а ДНК-лигаза
соединяет фрагменты Оказаки и восстанавливает первичную структуру ДНК.
3.
Закручивание спирали и восстановление вторичной структуры ДНК при помощи ДНК-гиразы. Слаженная работа
ферментов позволяет осуществлять репликацию с огромной скоростью: у прокариот – около 3000 п. н. (пар
нуклеотидов) в секунду, у эукариот – 100–300 п. н. в секунду. Две новые молекулы ДНК представляют собой
точные копии исходной молекулы.
11.
Репликация ДНК12.
Структура хромосомВ настоящее время принята нуклеосомная модель организации
хроматина эукариот (Kornberg R., 1974; Olins А., Olins D., 1974).
Согласно этой модели, белки-гистоны (они практически одинаковы у
всех эукариот) формируют особые глобулы из 8 молекул в каждой
глобуле (по две молекулы гистонов Н2а, Н2б, Н3, Н4). Нить ДНК делает
по два витка вокруг каждой глобулы.
Структура, состоящая из гистонового октамера, обвитого участком ДНК
(размером 140–160 п. н.), называется нуклеосомой. Такая укладка ДНК
сокращает ее длину в 7 раз. Нуклеосомная модель получила название
«бусинки на нитке».
Положительно заряженные гистоны и отрицательно заряженная ДНК
образуют относительно прочный ДНК-гистоновый комплекс. Участок
ДНК между нуклеосомами содержит гистон Н1. Он играет важную роль в
спирализации нуклеосомной нити и образовании второго уровня
организации хромосом – винтообразной структуры соленоида.
Степень компактизации хроматина различается в разных участках
хромосом и зависит от периода клеточного цикла. Важную роль в этом
процессе играют разнообразные негистоновые белки. Благодаря
процессу компактизации, гигантские молекулы ДНК упакованы в клетке
в небольшом объеме. Например, ДНК хромосом человека общей
длиной около 1,8 м упакована в ядре диаметром менее 1 микрометра.
13.
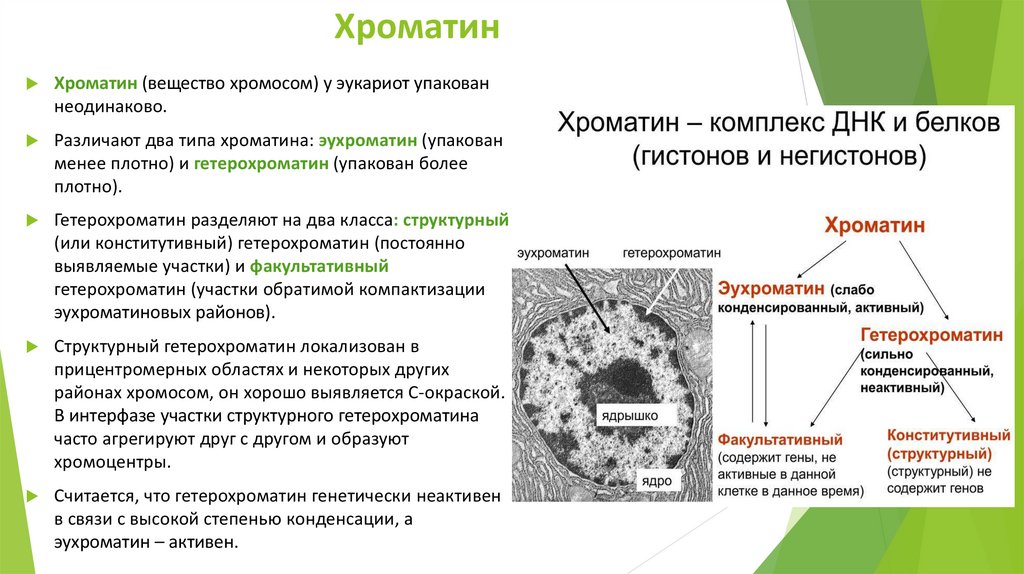
ХроматинХроматин (вещество хромосом) у эукариот упакован
неодинаково.
Различают два типа хроматина: эухроматин (упакован
менее плотно) и гетерохроматин (упакован более
плотно).
Гетерохроматин разделяют на два класса: структурный
(или конститутивный) гетерохроматин (постоянно
выявляемые участки) и факультативный
гетерохроматин (участки обратимой компактизации
эухроматиновых районов).
Структурный гетерохроматин локализован в
прицентромерных областях и некоторых других
районах хромосом, он хорошо выявляется С-окраской.
В интерфазе участки структурного гетерохроматина
часто агрегируют друг с другом и образуют
хромоцентры.
Считается, что гетерохроматин генетически неактивен
в связи с высокой степенью конденсации, а
эухроматин – активен.
14.
Типы хромосомХромосомы типа «ламповых щеток» - растянутый и раскрученный вариант
обычных хромосом ооцитов во время длительного мейоза. Длина таких
хромосом в 30 раз превышает их длину в обычном состоянии. Хромосомы типа
«ламповых щеток» получили свое название из-за наличия петель - участков
хромосомной нити, которые выступают из более компактного материала и
являются местом активной транскрипции. В конце мейоза хромосомы типа
«ламповых щеток» возвращаются к обычному состоянию.
Политенные хромосомы образуются в некоторых клетках в результате
максимальной деспирализации и многократной репликации без последующего
расхождения хромосом. Это явление называется эндомитозом. Перед
эндомитозом гомологичные хромосомы соединяются попарно – конъюгируют.
Такая конъюгация не характерна для других соматических клеток.
Все политенные хромосомы кариотипа объединяются центромерами в общий
хромоцентр. Поскольку политенные хромосомы содержат более 1000 нитей,
они в 1000 раз толще обычных хромосом и у них хорошо видны участки более
плотной спирализации – диски. В геноме дрозофилы выявлено около 5000
дисков – все они пронумерованы и формируют цитологические карты
хромосом.
Каждый диск представляет собой самостоятельную функциональную единицу,
содержащую от одного до нескольких генов. Во время экспрессии активные
диски «вздуваются» и образуют пуфы, которые появляются и исчезают в
определенной последовательности, в зависимости от активности генов на
разной стадии онтогенеза.
Хромомеры – это участки временно конденсированной неактивной ДНК.
Хромомеры могут деконденсироваться и переходить в активное состояние,
формируя петли, на которых происходит синтез РНК.
Хромосомы типа
«ламповых щеток»
Политенные хромосомы
15.
Мутации геновГеномные мутации: увеличение полного набора хромосом –
полиплоидии, или изменения количества хромосом одной
пары – анеуплоидии.
У человека описано два вида полиплоидий – триплоидии и
тетраплоидии – трех- и четырехкратное увеличение числа
гаплоидного набора. Подобные аномалии встречаются только
у спонтанных абортусов или мертворожденных.
Хромосомные мутации могут быть числовыми (анеуплоидии)
или структурными, то есть затрагивать число хромосом или их
структуру.
Наиболее частыми числовыми аномалиями являются
моносомии – отсутствие одной из гомологичных хромосом и
трисомии – существование добавочной третьей копии одной
из гомологичных хромосом, причем эта добавочная
хромосома может быть как материнского, так и отцовского
происхождения. Трисомии найдены не для всех хромосом, и
наиболее частыми из них являются синдромы Дауна, Эдвардса
и Патау – трисомии по 21, 18 и 13 хромосомам соответственно.
Иногда количество добавочных хромосом может быть еще
больше, эти аномалии называются полисомиями. Моносомии
и полисомии описаны, главным образом, для половых
хромосом. Другие геномные мутации несовместимы с жизнью
и приводят к ранней эмбриональной гибели.
16.
Структурные мутации затрагивают не целые хромосомы, а их фрагменты, ичаще они возникают в области гетерохроматина.
К структурным перестройкам относятся делеции (отсутствие), инсерции
(вставки), дупликации (удвоения) , инверсии (переворот на 1800 фрагмента
одной из хромосом), а также транслокации – перенос фрагмента одной
хромосомы на другую.
Среди структурных мутаций очень важно различать сбалансированные и
несбалансированные хромосомные перестройки.
При сбалансированных перестройках не происходит утраты генетического
материала, и носители подобных мутаций, как правило, клинически здоровы.
Однако в их потомстве велика вероятность рождения ребенка с
несбалансированной хромосомной перестройкой, а значит, с хромосомной
болезнью.
Типичным примером сбалансированных хромосомных перестроек являются
реципрокные и робертсоновские транслокации. В первом случае взаимный
обмен между участками двух хромосом происходит настолько точно, что весь
генетический материал сохраняется, однако меняется расположение генов в
хромосомах. В робертсоновских транслокациях участвуют только такие
хромосомы, у которых центромеры локализованы на концах, так называемые
акроцентрики, и именно в этих местах происходит слияние двух хромосом. В
результате этого слияния в кариотипе носителя робертсоновской транслокации
присутствует не 46, а 45 хромосом. Несмотря на это они здоровы. Однако у
носителей как реципрокных, так и робертсоновских транслокаций повышена
частота бесплодия, выкидышей и мертворождений, а также повышена
вероятность рождения детей с хромосомными болезнями
17.
Генные (точковые) мутации - небольшие структурные перестройки,затрагивающие от одного до несколько нуклеотидов или даже целых
экзонов, такие как делеции, инсерции, дупликации и инверсии.
Последствия таких внутригенных перестроек зависят от протяженности
нарушения, но еще в большей степени от его кратности величине
кодона, то есть трем нуклеотидам.
В случае делеции, некратной 3 нуклеотидам, происходит сдвиг рамки
считывания, при котором очень велика вероятность случайного
формирования стоп-кодона, следствием чего будет преждевременное
прекращение трансляции, образование укороченного белка и
дальнейшая его деградация.
Так, например, основным типом нарушений при мышечной дистрофии
Дюшенна/Беккера являются протяженные внутригенные делеции,
захватывающие один или несколько соседних экзонов. Подобные
перестройки встречаются у 65-70% больных. Однако при тяжелой форме
миодистрофии Дюшенна делеции, как правило, приводят к сдвигу рамки
считывания, и у больных продукт соответствующего гена (белок
дистрофин) полностью отсутствует. При гораздо более мягкой форме
миодистрофии Беккера делеции чаще всего оказываются кратны трем
нуклеотидам, и потому сдвига рамки считывания не происходит. В
результате дистрофин у больных синтезируется, хотя его структура,
конечно, нарушена.
18.
К точковым мутациям относятся замены нуклеотидов,которые могут приводить к различным нарушениям белков, а
могут и не иметь таких последствий.
Например,
замена
нуклеотида,
которая
в
силу
вырожденности генетического кода (то есть способности
кодировать одну и ту же аминокислоту различными
вариантами триплетов) не привела к изменению
аминокислоты в соответствующем белке. Это значит, что и на
уровне фенотипа никаких изменений не произошло.
Такие нейтральные замены относятся к классу нормальных
аллелей или полиморфизмов, и они могут с высокими
частотами встречаться в разных популяциях.
Практически в каждом гене можно найти полиморфные
аллели.
Поэтому после идентификации у больного замены какого-то
нуклеотида в гене, ответственном за определенное
наследственное заболевание, на следующем этапе
необходимо
исследовать,
насколько
эта
замена
функциональна, встречается ли и как часто у здоровых членов
семьи и в общей популяции.
Однонуклеотидный полиморфизм
19.
Характеристика генома человека20.
ДНК-уровеньОбщее количество ДНК в соматической клетке составляет
6,2х109 пар оснований, следовательно, гаплоидный набор
состоит из 3,1х109 пар нуклеотидов. Основное количество
ДНК локализовано в хромосомах (99,5%). Внехромосомная
часть генома человека - это ДНК митохондрий (0,5%).
В ядерной или хромосомной ДНК только 25-35% составляют
гены и их регуляторные участки (это уникальные
последовательности). Лишь 10% относящейся к генам ДНК
является кодирующей.
Следовательно, 2,5-3,5% всей ядерной ДНК имеют
отношение к синтезу белков. Что делает остальная часть
генома, пока неизвестно. Однако вряд ли она не имеет
функций.
21.
Повторяющиеся последовательности ДНКВ составе геномной ДНК выделяют
повторяющихся последовательностей.
несколько
классов
Участки ДНК различаются по длине каждого повтора и числу
повторяющихся единиц (их называют тандемными). Различают
умеренно повторяющиеся последовательности (до 1000 повторов в
одном локусе) и высокоповторяющиеся (свыше 1000 повторов). Они
могут быть локализованы в одном локусе или во многих локусах
одной или разных хромосом.
Одна и та же последовательность может повторяться в разных
локусах
разное
число
раз.
Такие
повторы
называют
гипервариабельными тандемными повторами. Если повтор состоит
из 2-6 пар нуклеотидов, то такие повторы называют
микросателлитами. Число повторяющихся копий микросателлитов
варьирует от 5 до 50, а суммарная протяженность может достигать
несколько сотен нуклеотидов.
Другая группа повторов - мини-сателлиты, представлена
повторяющимися элементами размером от 10 до 100 пар
нуклеотидов. Этот умеренно повторяющийся класс повторов
формирует тракты протяженностью 102-105 нуклеотидов.
Значительная часть мини-сателлитов равномерно рассеяна по
геному. Некоторые гипервариабельные повторы этого класса
кластеризуются
в
субтеломерных
областях
хромосом.
Высокоповторяющиеся последовательности размером от 100 пар
нуклеотидов и более, формирующие тракты протяженностью до
103- 107 нуклеотидов, составляют фракцию сателлитной ДНК.
Данный класс повторов локализован преимущественно в областях
конститутивного гетерохроматина и в прицентромерных регионах
хромосом.
22.
Особый класс низкокопийных повторов составляют крупные блокирассеянных по геному дупликаций. Многие из них имеют достаточно
протяженные размеры (до 100 тыс. пар нуклеотидов) и обнаруживают
высокую
степень
идентичности
нуклеотидных
последовательностей(>95%).
Существует
две
категории
сегментных
дупликаций
внутрихромосомные и межхромосомные. Часто они кластеризуются в
прицентромерных и субтеломерных районах хромосом. В геноме
человека блоки сегментных дупликаций локализованы в хромосомах 7,
15, 17, 22, X.
Высокая степень гомологии нуклеотидных последовательностей в
пределах
сегментных
дупликаций
определяет
возможность
прохождения между ними неравного кроссинговера, что приводит к
возникновению микроделеций и микродупликаций в хромосомных
сегментах. Многие из этих нарушений проявляются хромосомными
или генными заболеваниями (в зависимости от размера
затрагиваемого участка).
Наконец, недавно в геноме человека было идентифицировано 255
областей с крупными, размером от 100 тыс. пар нуклеотидов до 2,2
млн пар нуклеотидов, блоками повторов (CNV - Copy Number Variation).
В настоящее время ведутся исследования, направленные на
установление структуры и функции данного класса повторов. Получены
первые данные, указывающие на ассоциацию числа крупных блоков
повторов с системной красной волчанкой, псориазом, риском
инфицирования вирусом иммунодефицита человека (ВИЧ) I типа.
Сегментные дупликации в геноме человека
23.
Внехромосомные и кольцевые молекулыДНК
Обнаруживают в цитоплазме и ядре.
У человека они изучены еще недостаточно. В строгом смысле они
являются не составными элементами генома, а его продуктом.
Размер колеблется от 150 пар нуклеотидов до 20 тыс. пар
нуклеотидов. Являются ли эти молекулы продуктом фрагментации
хромосомной ДНК в клетке или они образуются в результате
других генетических процессов (гомологичной рекомбинации,
обратной транскрипции), пока не ясно.
Исследованные к настоящему времени у млекопитающих большие
кольцевые ДНК размером от 150 до 900 тыс. пар нуклеотидов,
локализованные только в ядрах, представляют собой
амплифицированные участки онкогенов или генов устойчивости к
ядам и антиметаболитам. С этими молекулами предположительно
связывают устойчивость клеток к лекарствам и способность клеток
к неограниченному росту. Их происхождение объясняют
делециями соответствующих областей хромосом.
24.
ПолиморфизмПолиморфизм - это такие варианты последовательностей ДНК, которые распространены в общей
популяции с частотой не менее 1%.
Эти изменения могут быть качественными, когда они обусловлены заменой или потерей нуклеотидов,
или количественными, когда в определенном локусе варьирует число нуклеотидных повторов
различной протяженности. И те, и другие варианты генетического полиморфизма встречаются как в
смысловых (внутриэкзонных), так и в несмысловых (внегенных или интронных) последовательностях
молекулы ДНК.
Существует несколько типов полиморфизма ДНК:
1)
полиморфизм по числу и распределению мобильных генетических элементов;
2)
полиморфизм по числу копий тандемных повторов ДНК (VNTR - variable number of tandem repeats);
3)
однонуклеотидные замены в последовательности ДНК (однонуклеотидные полиморфизмы - ОНП).
ОНП (однонуклеотидные полиморфизмы) - одна из наиболее частых форм генетического
полиморфизма. Под этим термином понимают варианты последовательностей ДНК у разных людей с
вовлечением одной пары оснований. ОНП - наиболее характерный источник вариаций между людьми.
Секвенированием геномов или их частей у разных людей установлено, что однонуклеотидные различия
обнаруживаются в среднем с частотой 1 замена на 600-1200 нуклеотидов. Расчеты показывают, что 2
человека на 99,9% идентичны по нуклеотидным последовательностям, т.е. только 0,1% различий по
одному нуклеотиду создает огромные индивидуальные фенотипические вариации. Предполагают, что
различия по одному основанию между определенными отрезками геномов лежат в основе не только
генных болезней, но и чувствительности к возбудителям или защиты от них, в основе
приспособительных реакций и наследственной предрасположенности к многофакторным болезням.
В генах человека около 50% некодирующих ОНП, 25% синонимичных кодирующих (не изменяющих
аминокислоту в кодируемом белке) и 25% несинонимичных кодирующих ОНП.
С помощью карт ОНП выясняют вклад индивидуальных генов в болезни комплексной
(многофакторной) и полигенной природы. Сравнение частот определенных типов ОНП у пациентов и в
контрольных группах позволяет идентифицировать ОНП, с которыми ассоциируется заболевание.
25.
Митохондриальный геномМитохондрии содержат кольцевую двухцепочечную ДНК, которую иногда называют 25-й
хромосомой человека (мтДНК - митохондриальная ДНК). В каждой соматической клетке в среднем
содержится около 1000 митохондрий. Суммарно ДНК митохондрий составляет не более 0,5% общего
количества ДНК в организме. ДНК митохондрий реплицируется полуавтономно от ядерной ДНК.
Геном митохондрий человека содержит 16 569 пар оснований и кодирует 2 рибосомные РНК (рРНК)
[12S и 16S], 22 транспортные РНК (тРНК) и 13 полипептидов. Полипептиды являются субъединицами
ферментативных комплексов окислительного фосфорилирования. Другие 66 субъединиц
дыхательной цепи кодируются в ядре.
Митохондриальный геном как целое отличается от ядерного генома несколькими признаками:
1)
мтДНК наследуется по материнскому типу. В зиготе содержится от 1 до 4 отцовских митохондрий, а
материнских - 25 000. К тому же не исключается, что после оплодотворения репликация отцовских
митохондрий вообще блокируется.
2)
Комбинативная изменчивость мтДНК (мейоз) отсутствует. Нуклеотидная последовательность
меняется в поколениях только в результате мутаций.
3)
Митохондриальный геном непрерывен, т.е. не содержит интронов. В нем имеется всего лишь
несколько межгенных пар оснований (или их вообще нет). Известно только одно исключение - около
1000 пар нуклеотидов является интроном в области промоторов (Д-петля). В мтДНК нет защитных
гистонов и системы репарации ДНК. Такая организация определяет примерно в 10 раз большую
скорость мутирования по сравнению с ядерной ДНК.
4)
Большинство генов мтДНК чередуются с генами тРНК, которые служат разделяющими сигналами для
дальнейшего процессинга первичных транскриптов.
5)
Внутри одной клетки могут функционировать митохондрии с разными типами мтДНК. Это состояние
называют гетероплазмией. Присутствие в клетках митохондрий с одним типом мтДНК - гомоплазмия.
6)
В мтДНК транскрибируются или транслируются обе цепи. Код мтДНК лишь частично отличается от
универсального (UGA кодирует триптофан, AUA кодирует метионин, AGA и AGG являются стопкодонами).
Мутации генов мтДНК лежат в основе митохондриальных болезней, отличающихся от моногенных
болезней не только особенностями передачи из поколения в поколение по материнской линии, но и
своеобразными чертами клинической картины. Патологические мутации мтДНК открыты в каждом
типе митохондриальных генов.
26.
Генный уровеньГен - последовательность нуклеотидов в ДНК, кодирующих
определенную мРНК и соответствующий белок, либо РНК, несущие
структурные или регуляторные функции. Большинство генов являются
участками ДНК, которые несут информацию о последовательности
аминокислотных остатков в белке, однако некоторые гены кодируют
только РНК. Со всеми генами связаны регуляторные последовательности
ДНК, т.е. участки, к которым присоединяются белки, определяющие,
будет ли ген экспрессирован в данное время и в данном месте.
На основе данных по секвенированию определено, что в геноме
человека около 30 000 генов, а не 70 000-100 000, как считали ранее. По
уточненным данным Национального центра биотехнологической
информации США на март 2008 г. в геноме человека насчитывается 31
809 генов, включая псевдогены, гены, кодирующие микроРНК. В базу
OMIM (Gene Map) включены только гены, влияющие на различные
заболевания. На май 2009 г. зарегистрировано 10 752 таких генов. Сотни
генов, вероятно, получены человеком в результате горизонтальной
передачи, начиная от бактерий. Более 6500 генов человека (примерно
1/6 часть генома) охарактеризованы экспериментально (по функции
продукта, наличию мутаций, тканеспецифичности, размеру
транскрипта).
Динамика картирования генов человека
27.
Благодаря альтернативному сплайсингу, число синтезируемых белковыхпродуктов, очевидно, в 1,5-2 раза больше, чем число генов.
Из одного и того же первичного РНК-транскрипта в процессинге РНК в
разных тканях образуется не один, а несколько разных по длине мРНКтранскриптов. Соответственно синтезированные полипептиды также будут
различными.
Таким образом, одна и та же ДНК-последовательность может кодировать не
один, а несколько разных белковых продуктов. Предполагается, что 40-60%
генов человека подвергается альтернативному сплайсингу. Это
существенным образом увеличивает разнообразие кодируемых геном
продуктов.
Размер генов человека, число экзонов и интронов в них варьируют в
широких пределах.
Классификация генов человека по размеру
(примеры)
Большинство генов имеет размеры до 50 000 пар нуклеотидов. Средняя
длина участка хромосомы, приходящегося на ген, составляет 27 000 пар
нуклеотидов. Однако есть гены, размер которых в 100 раз меньше или в 100
раз больше этой средней величины.
Распределение генов человека по размеру
28.
Как известно из менделевской генетики, различные аллели могут проявляться вдоминантном, рецессивном и кодоминантном вариантах. В геноме человека это правило
в отдельных случаях нарушается.
В таблице приведены примеры доминантного или рецессивного проявления одних и тех
же фенотипов, обусловленных различными мутациями в одном и том же гене.
Эти данные необходимо принимать во внимание при медикогенетическом
консультировании, когда родословная может не укладываться в рамки привычных типов
наследования.
29.
Функции геновНакопленные сведения о генах человека позволяют выделить их группы по функциям
первичного продукта:
ферменты;
модуляторы белковой функции;
рецепторы; транскрипционные факторы;
белки внутриклеточного матрикса;
белки внеклеточного матрикса;
трансмембранные переносчики;
структуры ионных каналов;
молекулы клеточных сигналов; гормоны;
экстраклеточные переносчики;
иммуноглобулины.
Наибольшую функциональную категорию (31,2% общего числа идентифицированных
генов) составляют гены, кодирующие ферменты. В 2 раза меньше генов-модуляторов
белковой структуры и функции (13,6%). Они стабилизируют, свертывают полипептидные
цепи или влияют на функции белка. Каждая из остальных категорий генов составляет
менее 10% общего числа.
30.
Кроме того, необходимо отметить наличие генов, продуктом которых являются различные классы РНК(тРНК, рРНК, малые ядерные РНК).
В настоящее время идет активное изучение новых видов РНК, которые участвуют в синтезе белка,
посттранскрипционной регуляции, репликации ДНК. Например, микроРНК в эукариотических клетках и
малые интерферирующие РНК могут блокировать трансляцию мРНК или усиливать ее распад, что приводит
к подавлению экспрессии гена. Этот феномен назван РНК-интерференцией и по эффекту напоминает
эпигенетическую регуляцию экспрессии генов путем их метилирования. Малая ядерная РНК участвует в
сплайсинге путем удаления интронов из мРНК. Очевидно, что основная роль большинства видов РНК регуляция экспрессии генов и их продуктов. Полагают, что 30-50% регуляции транскрипционной активности
выполняется упомянутыми выше видами РНК.
Сроки развития наследственных болезней во многом зависят от функции вовлеченного в патологию гена.
Болезни, ассоциированные с генами, кодирующими белки во всех функциональных категориях, могут
проявляться в любом периоде жизни.
Особенно высока доля болезней с началом на 1-м году жизни, вызванных мутациями в генах, кодирующих
ферменты (47%). Развивающийся плод имеет доступ к материнской метаболической системе гомеостаза
через плаценту. Таким образом, дети с врожденными нарушениями, вызванными недостаточностью
ферментов, обычно нормальны при рождении, но симптомы нарушения гомеостаза развиваются после
рождения, когда у ребенка включается собственная дефектная система метаболизма.
Болезни, вызванные дефектами генов, кодирующих ферменты, наследуются по аутосомно-рецессивному типу,
а связанные с генами, кодирующими модуляторы белковой функции или рецепторы, - по аутосомнорецессивному или аутосомно-доминантному. Болезни, вызванные генами транскрипционных факторов,
обычно относятся к группе аутосомно-доминантных.
Таким образом, временные закономерности формирования наследственных болезней строго
соответствуют роли и месту первичных продуктов в онтогенезе. Болезни транскрипционных факторов
развиваются внутриутробно, патология ферментов - в течение 1-го года жизни, рецепторов - в возрасте от 1
года до пубертатного периода, модуляторов белковой функции - у взрослых до 50 лет.
31.
Клетка живет и работает благодаря строго скоординированным действиям генов.Количественное распределение функций генов, участвующих в основных процессах типичной
клетки человека, следующее:
неизвестная
функция
17%
синтез РНК и белков
22%
клеточные структуры
8%
клеточное деление
12%
обмен (метаболизм)
17%
клеточные сигналы
12%
защита клетки
12%
32.
Немаловажная роль в регуляции активности генов отводится эпигенетическиммеханизмам, которые обеспечивают наследуемые, но потенциально обратимые
изменения экспрессии генов, не связанные с нарушениями их нуклеотидной
последовательности.
Молекулярную основу эпигенетической регуляции составляют ковалентные
модификации ДНК (метилирование цитозина) и гистоновых белков (ацетилирование,
метилирование, фосфорилирование и ряд других) в составе хроматина,
обеспечивающие формирование уникальной для каждой клетки функциональной
организации ее генома, так называемого эпигенотипа.
Клетки организма, обладающие одинаковым генотипом, могут иметь бесконечное
множество эпигенотипов, при этом реализация генотипа в фенотип осуществляется
сквозь призму эпигенотипа.
Эпигенетические заболевания связаны с нарушениями эпигенотипа - стабильными и
наследуемыми изменениями генной экспрессии, приводящими к возникновению
заболевания при отсутствии структурных мутаций в кодирующих генах.
Примерами таких заболеваний являются хроматиновые болезни, болезни геномного
импринтинга.
Весомый вклад эпигенетических нарушений в развитие опухолевых процессов.
33.
Правило эквивалентности реципрокных скрещиваний - наследованиеравнозначной функции аллеля, полученного от отца или от матери.
Однако, было доказано несоблюдение этого правила. Функции генов
взаимосвязаны и могут изменяться вплоть до дифференциального
выключения одного из аллелей на протяжении всего онтогенеза. Случаи
наследования с выключением одного из аллелей (материнского или
отцовского) объясняют генетическим импринтингом.
Генетический
импринтинг
это
эпигенетический
процесс,
дифференциально помечающий локусы хромосом одного из родителей,
что приводит к выключению экспрессии генов, в них расположенных.
Следовательно, в участках генома, подверженных импринтингу,
обнаруживается моноаллельная (а не биаллельная) экспрессия генов, т.е.
если импринтирован материнский аллель, то экспрессируется только
отцовский, и наоборот.
Неэквивалентный вклад родителей в геном потомства обусловливает
отклонение от строгих менделевских законов, согласно которым вклад
каждого из родителей в наследственность потомков равнозначен.
Механизмом
импринтинга
в
большинстве
случаев
является
дифференциальное полоспецифическое метилирование цитозиновых
оснований ДНК, устанавливаемое при созревании половых клеток,
которое и выключает в конечном итоге транскрипцию гена у потомства.
Таким образом, фенотипические проявления конкретного гена могут
меняться из-за трех причин: не только из-за его делеции или мутации в
нем, но и за счет эпигенетического выключения экспрессии. Речь идет о
стойких функциональных различиях экспрессии гомологичных генов у
потомства.
Примеры генетического импринтинга
34.
Наследственные патологии и ихклассификация
35.
Генетическая классификациянаследственных болезней
В основу генетической классификации наследственных болезней
положен этиологический принцип, а именно тип мутаций, в том
числе эпимутации, тип клеток и характер взаимодействия со
средой.
Всю наследственную патологию можно разделить на 3 группы.
1. Болезни, обусловленные мутациями в половых клетках:
хромосомные (например, синдромы Дауна, Клайнфелтера,
«кошачьего крика»);
генные (например, гемофилия, ахондроплазия, фенилкетонурия);
многофакторные или болезни с наследственным
предрасположением (как правило, полигенные; например,
шизофрения, эссенциальная гипертензия, псориаз);
эпигенетические (например, синдромы Ретта, Коффина- Лоури,
Прадера-Вилли).
Пример наследования генной мутации при
фенилкетонурии
36.
2. Болезни, обусловленные мутациями в соматических клетках:хромосомные (например, мозаичные формы хромосомных
болезней, лейкозы);
генные (например, опухоли на фоне мутаций онкогенов);
многофакторные (например, вторичные
врожденные пороки развития, опухоли);
эпигенетические (например, опухоли пищевода, молочной
железы).
иммунодефициты,
3. Болезни, обусловленные мутациями в половых и соматических
клетках:
хромосомные (например, лейкозы у больных с синдромом
Дауна);
генные (например, ретинобластома, опухоль Вильмса);
многофакторные (например, аутоиммунные заболевания);
эпигенетические
(например,
колоректальный рак).
семейный
Транслокация между 9 и 22 хромосомами, лежащая
в основе миелоидного лейкоза
неполипозный
Ретинобластома (вследствие генной мутации
инактивируется RB1 белок, в норме сдерживающий
деление клеток)
37.
Клиническая классификациянаследственных болезней
Клиническая классификация наследственных болезней по органному, системному
принципу или по типу обмена веществ очень условна.
Например,
больные
с
нейрофиброматозом
(аутосомно-доминантное
заболевание) встречается и в нейрохирургических клиниках (у больных
развиваются опухоли мозга), и в дерматологических клиниках, поскольку у этих
больных первоначально появляются обширные светло-коричневые пятна и
нейрофиброматозные узелки на коже, и в клиниках нервных болезней в связи с
глубокими нейрофибромами. Больные с хореей Гентингтона являются
пациентами и невропатолога, и психиатра, больные с гепатолентикулярной
дегенерацией - терапевта и невропатолога.
При очень немногих наследственных болезнях избирательно поражается одна
система. Даже моногенно детерминируемые болезни вследствие плейотропного
действия гена и вторичных патогенетических звеньев затрагивают разные органы
и системы. Большинство генных мутаций, а тем более хромосомные и геномные,
вызывают генерализованное повреждение какой-либо ткани (например, болезни
соединительной ткани) или захватывают несколько органов. В связи с этим
многие наследственные болезни проявляются в виде синдромов или комплекса
патологических признаков, на первый взгляд не связанных между собой.
«кофейные» пятна при
нейрофиброматозе
шваннома слухого нерва при
нейрофиброматозе
38.
Классификация по типу обмена веществБлагодаря данной классификации многие молекулярные болезни называются наследственные болезни
обмена (НБО). Среди них выделяют:
болезни аминокислот (алкаптонурия, альбинизм, гистидинемия, гомоцистинурия, лейциноз, ФКУ);
болезни биосинтеза кортикостероидов (АГС, гипоальдостеронизм);
болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз);
болезни липидов (ганглиозидозы, гиперлипидемия, гиперхолестеринемия, муколипидозы,
сфинголипидозы, цереброзидозы);
болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный периодический паралич);
болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглер-Найяра, Порфирии);
болезни пуринов и пиримидинов (оротовая ацидурия, подагра, синдром Леша-Найяна);
болезни транспорта систем почек (болезнь де Тони-Дебре- Фанкони, витамин D-резистентный рахит,
тубулопатии);
болезни углеводов (галактоземия, гликогенозы, мукополисахаридозы, непереносимость фруктозы);
болезни эритрона: анемия Фанкони, гемолитические анемии, недостаточность глюкозо-6фосфатдегидрогеназы.
Кроме того, в рамках НБО выделяют особую группу болезней накопления, тезаурисмозов. Эта группа
МБ объединяет НБО, связанные с дефицитом ферментов лизосом (наследуются с цитоплазмой по
материнской линии). Такие НБО проявляются прогрессирующим накоплением предшественников
метаболических реакций в клетках разных тканей, например, отложением макромолекул гликогена,
гликопротеинов, гликолипидов, мукополисахаридов, сфинголипидов, которые в условиях нормального
обмена веществ «самоперевариваются» лизосомальными ферментами, что необходимо при очищении
организма от продуктов жизнедеятельности и мертвых клеток.
муколипидоз
Болезнь Гоше
39.
Классификации молекулярных болезней наоснове мутационных спектров и оптимальных
алгоритмов ДНК-диагностики
Классификация базируется на двух принципах: это уникальность спектра мутационных изменений каждого
гена и оптимальные алгоритмы для их ДНКдиагностики. Авторами классификации предложено разделение
МБ по классам:
Первый класс- это болезни, вызванные немажорными мутациями, равномерно распределенными по всей
нуклеотидной последовательности гена. В их число включены замены, делеции, дупликации и инсерции
одного или нескольких нуклеотидов. Данный класс МБ представлен: аутосомно-доминантным
врожденным поликистозом почек, гемофилией А и В, множественными эндокринными неоплазиями (МЭН),
семейным раком грудной железы и раком яичников, семейным аденоматозно-полипозным раком толстого
кишечника, CRASH-синдромом (болезнь LI), синдромом Смита-Лемли-Опица, Х-сцепленной формой
синдрома Альпорта, Х-сцепленной гидроцефалией.
Для молекулярной диагностики этого класса МБ применяются методы ДНК-анализа для выявления
неизвестных мутаций, но чаще других используется комбинация методов SSCP (анализ однонитевого
конформационного полиморфизма) и методов анализа отдельных экзонов с последующим прямым
сиквенированием тех фрагментов ДНК, где подозревают наличие мутаций.
40.
Второй класс - это болезни, вызванные мажорными мутациями, которые принципиально важны для ДНКдиагностики. Среди таких патологий: ахондроплазия, болезни Вильсона-Коновалова, Леша-Найяна и Ретта,болезни накопления, МВ, рецессивные формы наследственной глухоты (несиндромальная несенсорная
тугоухость), ФКУ. Для идентификации каждого типа мажорной мутации предложен оптимальный метод из
общего арсенала методов ДНК-анализа, включающий мультиплексную ПЦР с анализом внутригенных
полиморфных сайтов рестрикции и др.
Третий класс - это болезни, вызванные протяженными дупликациями и делециями, возникшими в
результате неравного кроссинговера или неправильного спаривания гомологичных хромосом во время
мейоза, которое происходит по фланкирующим повторам участка ДНК, содержащего ген.
Такая мутация называется генной конверсией, и если в участок нуклеотидной последовательности ДНК
(область мутации) вовлечен, например, ген РМР22 (17р11.2), контролирующий синтез периферического
миелинового белка 22, то возникнет аутосомно-доминантная моторно-сенсорная нейропатия Шарко-МариТус.
Особенно много мутаций в результате генной конверсии возникает в псевдогенах. В этом случае происходит
перенос фрагмента одного аллеля в другой аллель гена или перенос фрагмента псевдогена в нормальный
ген., например, такая мутация происходит при переносе последовательности псевдогена в ген 21гидроксилазы (CYP21B). К данному классу мутаций принадлежит мутация, обусловливающая развитие
болезни Гоше - это самая частая аутосомнодоминантная болезнь накопления. В ее основе лежит мутация
гена GBA (1q21), гена глюкоцереброзидазы (лизосомной гидроксилазы), рядом с которым локализован
псевдоген psGBA.
Также типичной патологией этого класса МБ считается сцепленная с полом миодистрофия ДюшеннаБеккера (Хр21), обусловленная мутациями (протяженными делециями и дупликациями, микроделециями и
нонсенс-мутациями) самого крупного гена человека, кодирующего белок - дистрофин, входящий в состав
сарколеммы мышечного волокна. Кроме того, сюда относятся семейная мышечная атрофия трех типов,
включающая болезнь Верднига-Гоффмана, болезнь Кугельбергера-Веландер и промежуточную форму. Все
типы мышечной атрофии представляют собой аллельные варианты мутаций одного дуплицированного гена
SMN1 (5q13). Копия этого гена - ген SMN2 - вариант псевдогена, располагается ближе к центромере и
отличается от гена SMN1 по 8 нуклеотидам, причем 5 из них находятся в интронах, а 3 - в экзонах (различия в
гене и псевдогене выявляются методом SSCP и методом ПДРФанализа).
41.
Четвертый класс- это болезни экспансии, в основе которых лежат динамические мутации.В последние годы была опубликована еще одна классификация, согласно которой все МБ
делят на три группы в соответствии с видом наследственной изменчивости:
болезни, обусловленные мутационной изменчивостью генома; в каталоге В. Мак-Кьюсика
(OMIM) перечислено 1100 мутаций идентифицированных, отчетливо проявляющихся в
фенотипах больных при 1,5 тыс.;
болезни, обусловленные вариационной (комбинативной) изменчивостью генома, в составе
которого выделены не менее 50% повторяющихся последовательностей ДНК, относящихся
к 5 классам: повторы, возникшие из транспозонов, неактивные перемещенные копии
клеточных генов, простые повторяющиеся последовательности, сегментарные дупликации,
блоки тандемноповторяющихся генов. Эти повторы ДНК перестраивают геном, вызывая
эктопические нарушения, приводящие к развитию болезней генома;
болезни, обусловленные эпигенетической (эпигеномной) изменчивостью, связанной не со
структурными нарушениями генома, а с изменениями регуляции генной активности,
которые, хотя и наследуются, но обратимы. К примерам эпигеномных нарушений
относятся: инактивация Х-хромосомы, эффект положения теломеры, сайлесинг гена,
тканевая специфичность и возрастзависимая модификация ДНК, парамутации,
трансфекции, импринтинг, цитодукция и гомологзависимые процессы;
болезни, обусловленные изменениями в геноме, в том числе гонадный мозаицизм,
мейотический драйв, прионизация, цитоплазматическая наследственность, экспансия
нуклеотидных повторов.
42.
Наследственные ферментопатии43.
Наследственные ферментопатииНаследственные
ферментопатии
(первичные
энзимопатии) - генетически детерминированные
нарушения обмена веществ вследствие ферментопатии
лежат в основе многих наследственных болезней.
При этом может полностью отсутствовать ген,
контролирующий синтез белковой молекулы фермента
(апофермента), либо апофермент синтезируется, но
активность фермента отсутствует или резко снижена.
В результате генных мутаций может изменяться
последовательность аминокислот в структуре активного
центра фермента или в регионе связывания
апофермента с коферментом (чаще всего витамином
или металлом).
Кроме того, могут синтезироваться нестабильные легко
распадающиеся молекулы ферментов. При этом дефект
синтеза фермента наследуется, в основном, по
аутосомно-рецессивному типу. Гетерозиготы, как
правило, не имеют фенотипических отклонений.
44.
Нарушение образования конечныхпродуктов
Недостаточное
количество
конечного
продукта
определенного метаболического пути (при отсутствии
других альтернативных путей синтеза) может приводить
к развитию клинических симптомов, характерных для
данного заболевания.
В качестве примера можно рассмотреть альбинизм.
При альбинизме в меланоцитах нарушен синтез
пигментов – меланинов. Меланин содержится в коже,
волосах, радужке, пигментном эпителии сетчатки глаза
и определяет их окраску.
Синтез меланина
При альбинизме наблюдается слабая пигментация
кожи, волос, красноватый цвет радужки глаза из-за
просвечивающихся
капилляров.
Возникновение
альбинизма связано с недостаточностью фермента
тирозиназы (тирозингидроксилазы) – одного из
ферментов,участвующих в синтезе меланинов.
Альбинизм
45.
Накопление субстратовпредшественниковПри недостаточности определенного фермента (Ех) будут накапливаться
метаболиты, во многих случаях также и предшествующие соединения,
которые в процессе метаболических превращений образуются до этапа
действия поврежденного энзима Ех.
Увеличение концентрации субстратов-предшественников для дефектного
фермента является ведущим звеном развития данных заболеваний.
В качестве примера можно привести алкаптонурию. При этом
заболевании нарушено окисление в тканях гомогентизиновой кислоты –
промежуточного метаболита катаболизма тирозина.
У таких пациентов наблюдается недостаточность фермента диоксигеназы
гомогентизиновой кислоты. В результате этого увеличивается
концентрация гомогентизиновой кислоты и её выведение с мочой.
В присутствии кислорода гомогентизиновая кислота превращается в
соединение черного цвета – алкаптон. Поэтому моча таких пациентов на
воздухе окрашивается в черный цвет. Алкаптон образуется также в
биологических жидкостях, накапливаясь в тканях, коже, сухожилиях,
суставах. При значительных отложениях алкаптона в суставах нарушается
их подвижность.
Алкаптонурия
Диоксигеназа гомогентизиновой кислоты
46.
Нарушение образования конечных продуктови накопление субстратов-предшественников
Имеются заболевания, когда одновременно недостаток
продукта и накопление исходного субстрата формируют
клиническую картину.
Примером является болезнь Гирке (гликогеноз I типа), при
которой развивается гипогликемия в перерывах между
приемами пищи. Это связано с нарушением распада
гликогена в печени и выхода из нее глюкозы вследствие
дефекта фермента глюкозо-6-фосфатазы.
Одновременно у таких пациентов увеличиваются размеры
печени
(гепатомегалия)
вследствие
накопления
в
нейгликогена.
Болезнь Гирке
47.
Общая характеристика моногенныхнаследственных болезней обмена
1. носят врожденный характер;
2. манифестируют в любом возрасте;
3. проявляются определенной, часто прогрессирующей клинической
симптоматикой;
4. сопровождаются грубыми нарушениями жизнедеятельности человека;
5. сопровождаются различной степенью умственной отсталости;
6. подлежат трудоемкому и дорогостоящему лечению;
7. имеют высокий закономерный риск передачи потомству;
8. в случаях точной диагностики и наличия подробной информации о типе
мутации и ее доклинических (биохимических) проявлениях подлежат
дородовой диагностике;
9. в случаях разработки эффективной терапии подлежат доклиническому
выявлению и лечению.
48.
Биохимическая классификация НБО1. болезни обмена аминокислот;
2. болезни углеводного обмена;
3. болезни обмена органических кислот;
4. болезни обмена жирных кислот;
5. болезни обмена пуринов и пиримидинов;
6. болезни обмена холестерина;
7. болезни обмена гема и порфиринов;
8. болезни обмена металлов;
9. болезни обмена витаминов;
10. болезни клеточных органелл: лизосомные, пероксисомные, митохондриальные;
11. нарушения цикла мочевины.