











 law
lawSimilar presentations:



")

")

")


Изменения в подходах к работе с документацией на АФС по новым правилам регистрации ЕАЭС
1.
Изменения в подходах к работе сдокументацией на АФС по новым
правилам регистрации ЕАЭС
ОРЗ
Август 2021
2.
2Содержание
Цель презентации
Нормативная база/использованные источники
Где мы сейчас
Сравнение старого и нового подходов к работе с док-ми на АФС
Выводы
Правовые проблемы вне нашего влияния
Опасности игнорирования новых правил
3
4
5
6
9
17
18
3.
Цель презентацииЧТО
Обзор текущих регуляторных требований к работе с документацией на
АФС в сравнении с предыдущими правилами
ЗАЧЕМ
Установить для всех участвующих подразделений общее видение
текущих регуляторных правил и требований законодательства
относительно работы с АФС в части спецификаций, аналитических
методик и управления изменениями в них.
Для того, чтобы совместными усилиями привести существующие на
предприятии процессы и процедуры к состоянию, в котором они будут
адекватны текущим регуляторным требованиям
3
4.
Нормативная база / Использованные источникиПравила регистрации и экспертизы лекарственных средств для
медицинского применения УТВЕРЖДЕНЫ Решением Совета
Евразийской экономической комиссии от 3 ноября 2016 г. № 78
Приложение №10 к Правилам «Процедура работы с мастер-файлом
на активную фармацевтическую субстанцию»
Guideline on Active Substance Master File Procedure, EMEA/CVMP/134/02
Rev 4. European Medicines Agency.
4
5.
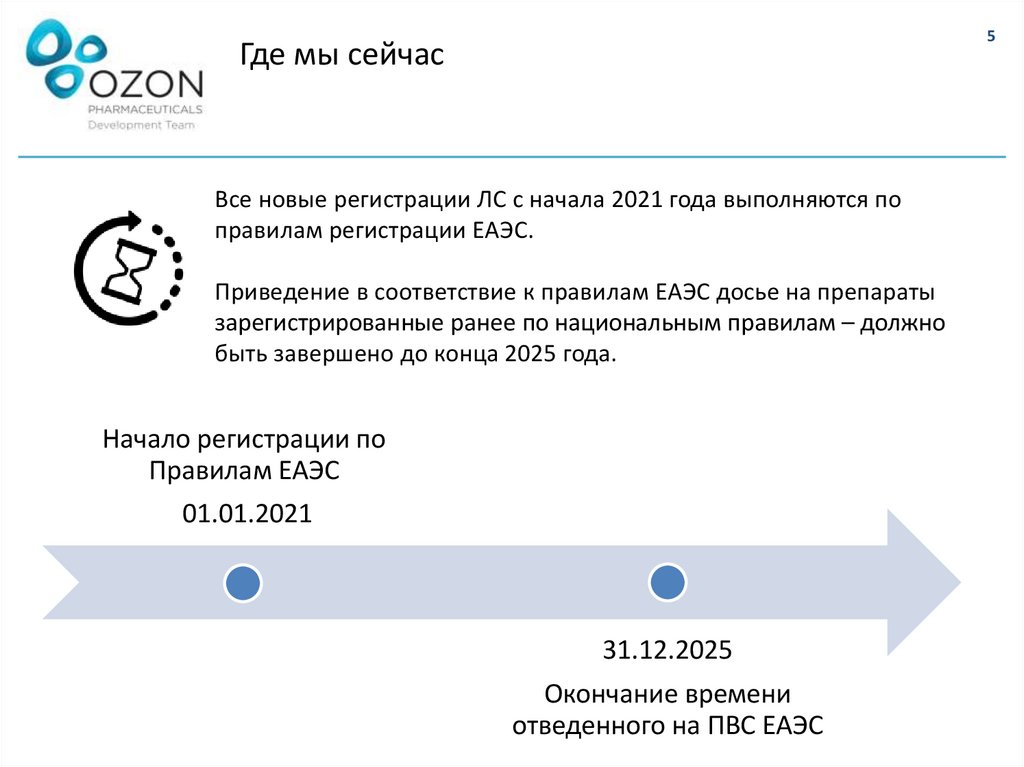
5Где мы сейчас
Все новые регистрации ЛС с начала 2021 года выполняются по
правилам регистрации ЕАЭС.
Приведение в соответствие к правилам ЕАЭС досье на препараты
зарегистрированные ранее по национальным правилам – должно
быть завершено до конца 2025 года.
Начало регистрации по
Правилам ЕАЭС
01.01.2021
31.12.2025
Окончание времени
отведенного на ПВС ЕАЭС
6.
Сравнение подходов к работе с документами наАФС
Основное изменение в подходе к работе с АФС в новых правилах регистрации,
это возложение полной ответственности за обладание всей актуальной
информацией о производстве субстанции, ее спецификации и аналитических
методиках – на держателя РУ.
Обладание держателем РУ этой информацией, и поддержание ее в актуальном
состоянии – рассматривается как необходимое условие обеспечения
держателем РУ качества готового лекарственного средства, производимого с
использованием этой субстанции.
6
7.
Сравнение подходов к работе с документами наАФС
ЧТО
БЫЛО
7
СТАЛО
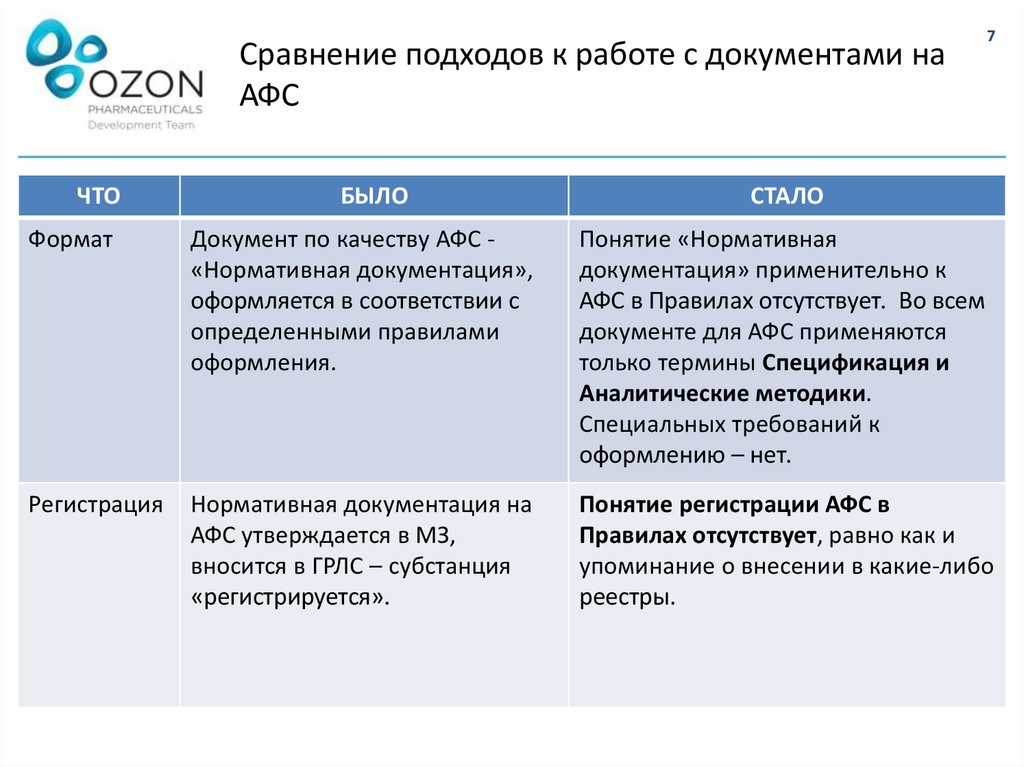
Формат
Документ по качеству АФС «Нормативная документация»,
оформляется в соответствии с
определенными правилами
оформления.
Понятие «Нормативная
документация» применительно к
АФС в Правилах отсутствует. Во всем
документе для АФС применяются
только термины Спецификация и
Аналитические методики.
Специальных требований к
оформлению – нет.
Регистрация
Нормативная документация на
АФС утверждается в МЗ,
вносится в ГРЛС – субстанция
«регистрируется».
Понятие регистрации АФС в
Правилах отсутствует, равно как и
упоминание о внесении в какие-либо
реестры.
8.
Сравнение подходов к работе с документами наАФС
8
ЧТО
БЫЛО
СТАЛО
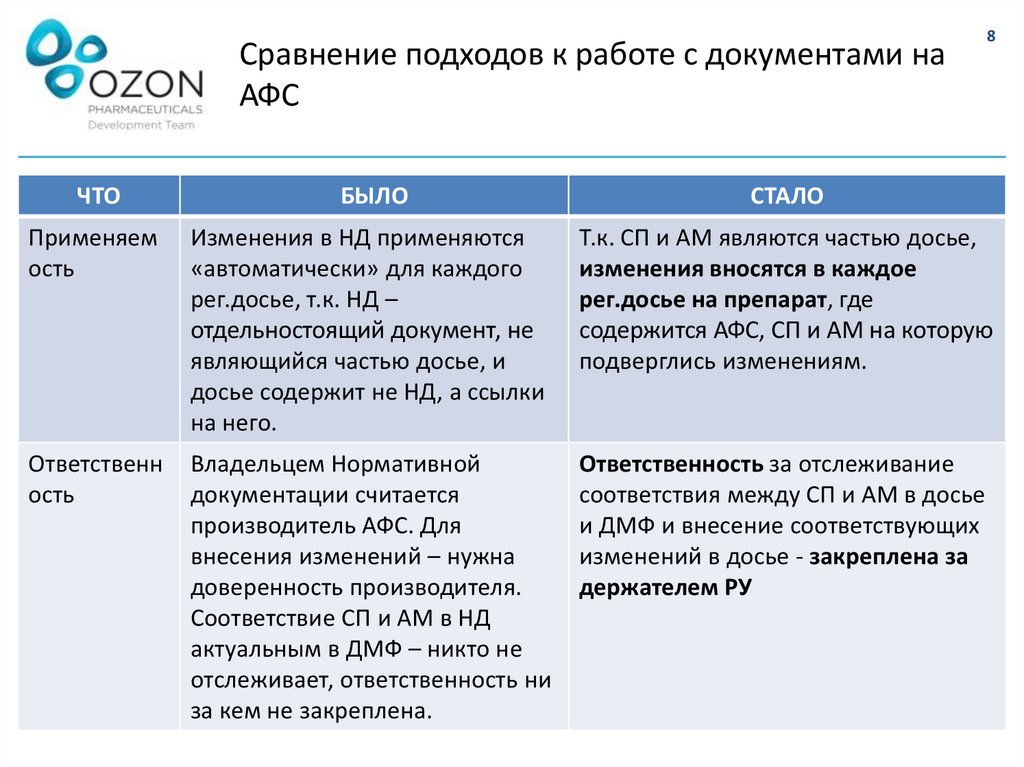
Применяем
ость
Изменения в НД применяются
«автоматически» для каждого
рег.досье, т.к. НД –
отдельностоящий документ, не
являющийся частью досье, и
досье содержит не НД, а ссылки
на него.
Т.к. СП и АМ являются частью досье,
изменения вносятся в каждое
рег.досье на препарат, где
содержится АФС, СП и АМ на которую
подверглись изменениям.
Ответственн
ость
Владельцем Нормативной
документации считается
производитель АФС. Для
внесения изменений – нужна
доверенность производителя.
Соответствие СП и АМ в НД
актуальным в ДМФ – никто не
отслеживает, ответственность ни
за кем не закреплена.
Ответственность за отслеживание
соответствия между СП и АМ в досье
и ДМФ и внесение соответствующих
изменений в досье - закреплена за
держателем РУ
9.
9Выводы
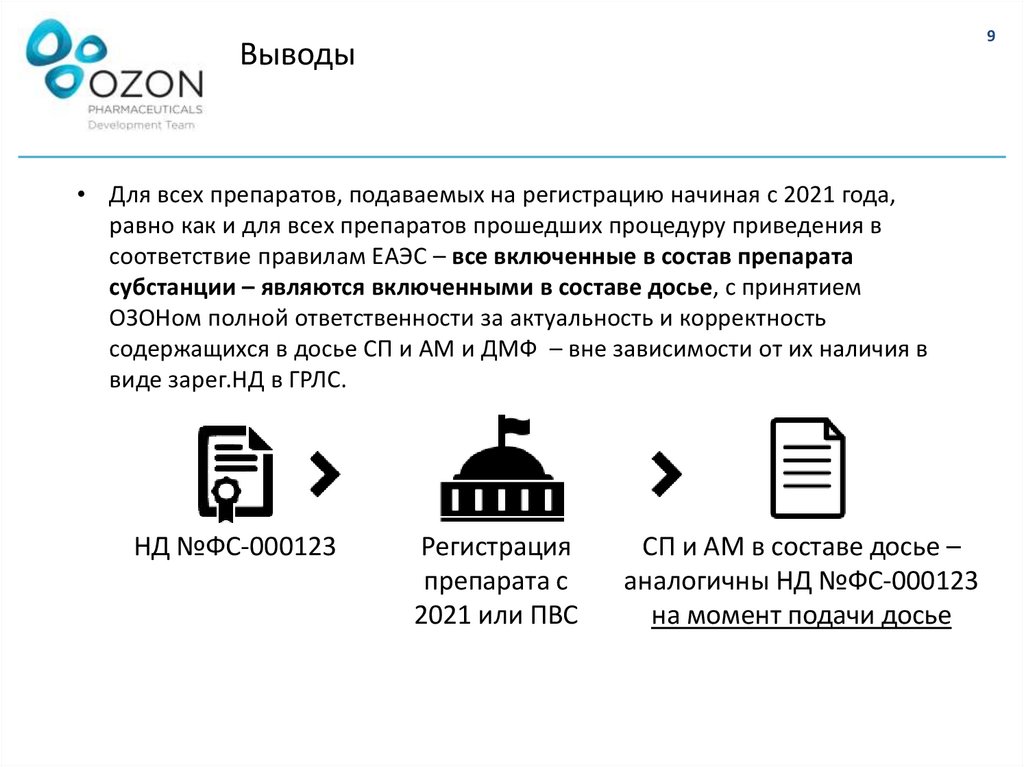
• Для всех препаратов, подаваемых на регистрацию начиная с 2021 года,
равно как и для всех препаратов прошедших процедуру приведения в
соответствие правилам ЕАЭС – все включенные в состав препарата
субстанции – являются включенными в составе досье, с принятием
ОЗОНом полной ответственности за актуальность и корректность
содержащихся в досье СП и АМ и ДМФ – вне зависимости от их наличия в
виде зарег.НД в ГРЛС.
НД №ФС-000123
Регистрация
препарата с
2021 или ПВС
СП и АМ в составе досье –
аналогичны НД №ФС-000123
на момент подачи досье
10.
10Выводы
Регистрация по
нац.правилам
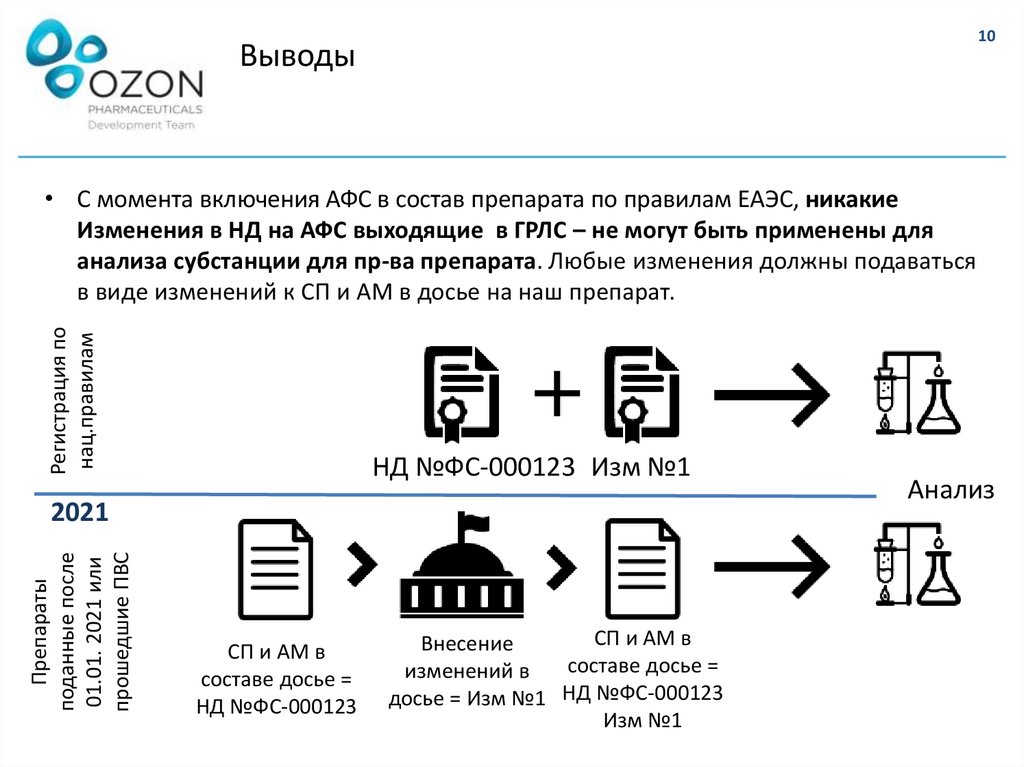
• С момента включения АФС в состав препарата по правилам ЕАЭС, никакие
Изменения в НД на АФС выходящие в ГРЛС – не могут быть применены для
анализа субстанции для пр-ва препарата. Любые изменения должны подаваться
в виде изменений к СП и АМ в досье на наш препарат.
НД №ФС-000123 Изм №1
Препараты
поданные после
01.01. 2021 или
прошедшие ПВС
2021
СП и АМ в
составе досье =
НД №ФС-000123
СП и АМ в
Внесение
составе досье =
изменений в
досье = Изм №1 НД №ФС-000123
Изм №1
Анализ
11.
11Выводы
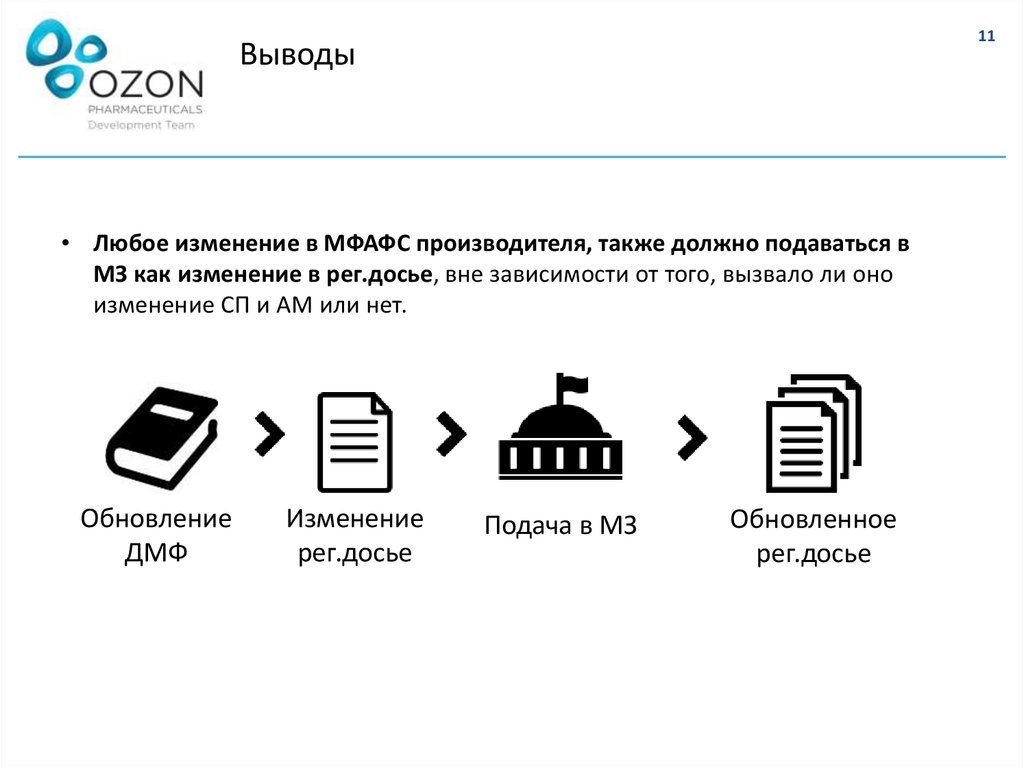
• Любое изменение в МФАФС производителя, также должно подаваться в
МЗ как изменение в рег.досье, вне зависимости от того, вызвало ли оно
изменение СП и АМ или нет.
Обновление
ДМФ
Изменение
рег.досье
Подача в МЗ
Обновленное
рег.досье
12.
Выводы12
Очевидно, что для выполнения требований регулятора, в компании
потребуется, как минимум, организация процедур, систем и процессов,
обеспечивающих следующее:
Хранение спецификаций и методик анализа АФС в разрезе препаратов,
с контролем соответствия их актуальным МФАФС
Контроль актуальности МФАФС, хранение МФАФС, регистрация
изменений и контроль версионности МФАФС, подачу изменений
МФАФС в рег.досье на препараты.
Оценку вносимых в МФАФС изменений в отношении необходимости
внесения изменений в спецификацию и методики анализа на
предприятии
Разработку, апробацию и внесение изменений в спецификации и
методики анализа на АФС на предприятии
13.
13Выводы
При изменении системы работы, возможно стоит рассмотреть следующее:
o Уйти от ранее принятого формата Нормативного документа на АФС и
термина как такового, и перейти к работе с раздельными документами –
Спецификация и Аналитические методики. (Возможно, так будет удобнее
ими управлять)
НД
СП
АМ
o Перейти к именованию производителей АФС в документации на
английском языке. (Требования переводить/транскрибировать названия
производителей нигде нет. Записи из реестра ЛС ЕАЭС косвенно
свидетельствуют о допустимости такого подхода. Потенциально поможет
избежать путаницы в наименованиях одного и того же производителя типа
ЧжЕцзян – ЧжЭцзян, которые переходят по наследству от кривых записей в
ГРЛС). Цзянсу Энхуа Фармасьютикал Ко., Лтд.
Jiangsu Nhwa Pharmaceutical Co., Ltd.
14.
14Выводы
На данный момент в портфеле предприятия:
РУ на разных жизненных этапах - более
430
Записей на АФС в РУ: более 1200
Из них уникальных связок АФС-Производитель – более 700
И эти количества растут с каждым годом.
8-9
РУ ежемесячно должны приводиться в соответствие, чтобы успеть
завершить ПВС до конца 2025 года.
Очевидно, что поддержание силами предприятия в актуальном
состоянии такого количества спецификаций и наборов аналитических
методик - в существующей системе работы невозможно.
Действующая структура предприятия в принципе не предусматривает
проведение такой работы на регулярной основе.
15.
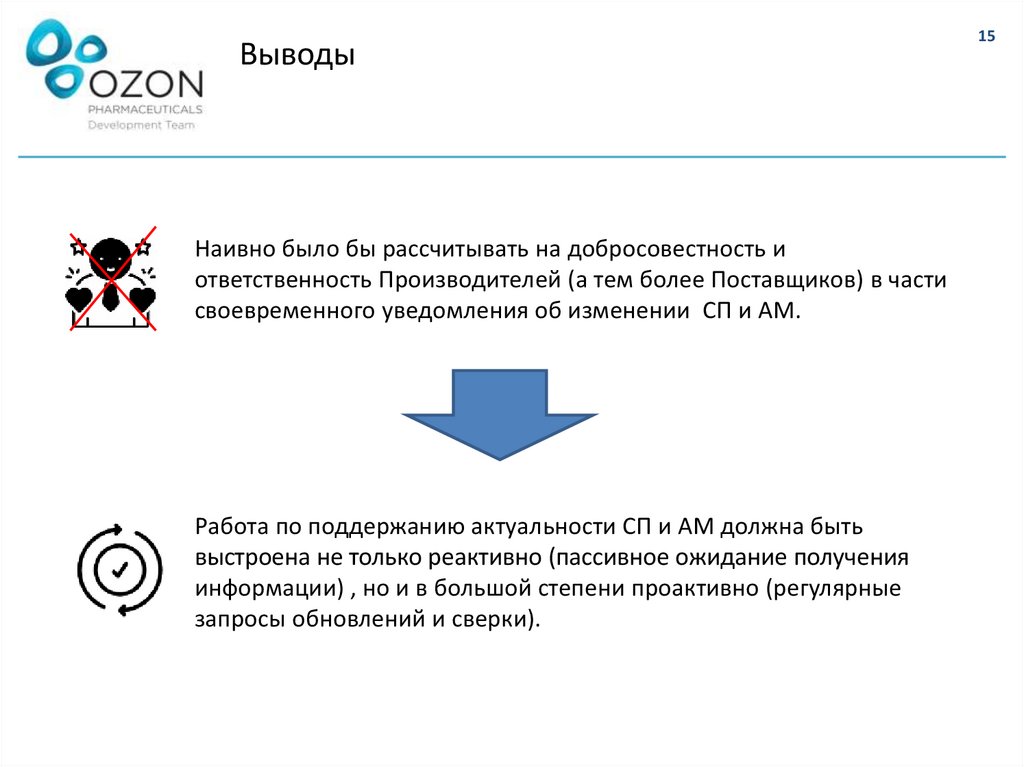
ВыводыНаивно было бы рассчитывать на добросовестность и
ответственность Производителей (а тем более Поставщиков) в части
своевременного уведомления об изменении СП и АМ.
Работа по поддержанию актуальности СП и АМ должна быть
выстроена не только реактивно (пассивное ожидание получения
информации) , но и в большой степени проактивно (регулярные
запросы обновлений и сверки).
15
16.
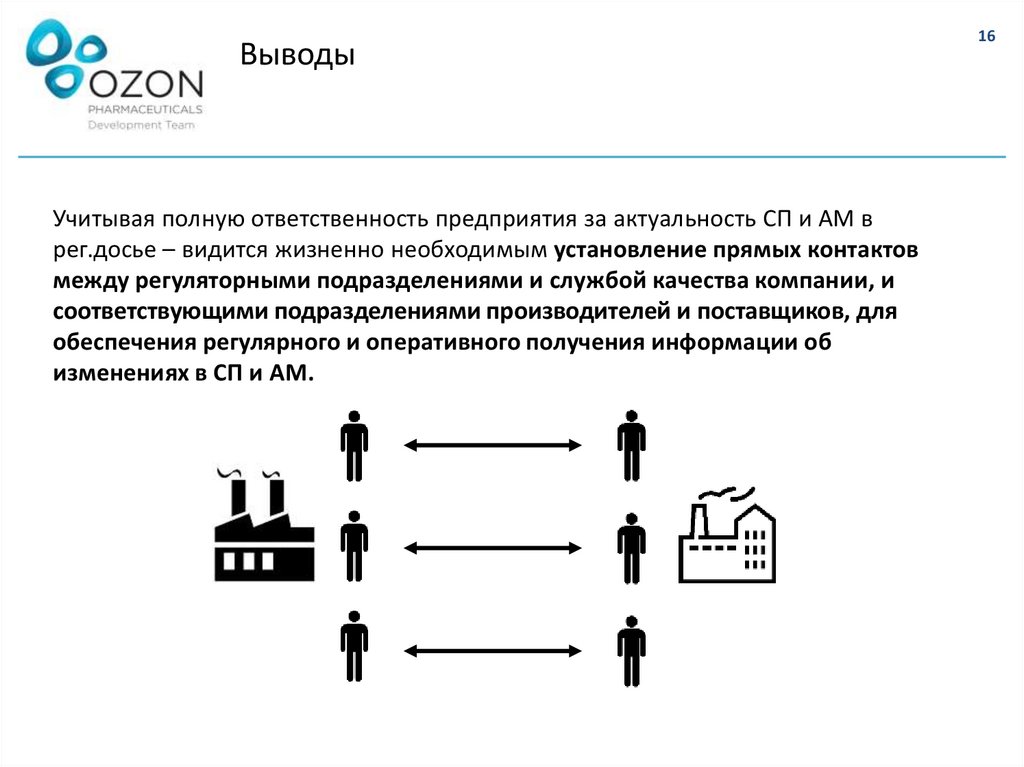
ВыводыУчитывая полную ответственность предприятия за актуальность СП и АМ в
рег.досье – видится жизненно необходимым установление прямых контактов
между регуляторными подразделениями и службой качества компании, и
соответствующими подразделениями производителей и поставщиков, для
обеспечения регулярного и оперативного получения информации об
изменениях в СП и АМ.
16
17.
Правовые проблемы вне нашего контроля• Отсутствие единого регулятора. В Европе есть ЕМА – единый европейский орган,
основная задача которого разработка и гармонизация единых регистрационных
процедур и подходов для всех стран-членов. В ЕАЭС, насколько мы понимаем,
такого органа нет, для ЕЭК это не основная деятельность, и регуляторы странчленов и заявители/держатели остались один на один с Правилами, без
разъяснений, поддержки и направления.
• Предполагаем, что в настоящее время не разработаны и не действуют
инструменты общей работы регуляторов стран-членов, предполагаемые
правилами. Например - нет общего реестра МФАФС, письмо на доступ к
которому должны выдавать заявителям держатели МФАФС, после
единовременной подачи МФАФС В МЗ, и получения референсного номера.
• Несогласованность действий ведомств, контролирующих фарм.деятельность в
РФ и правовые коллизии между правом РФ и правом ЕАЭС. Росздравнадзор по
прежнему требует утвержденные НД на АФС руководствуясь положениями 61ФЗ, которые на сегодня вступают в противоречие с действующим правилами
регистрации ЕАЭС.
17
18.
Опасности игнорирования новых правилОсновной опасностью нам видится то, что не контролируя, какие
СП и АМ включены в наше рег.досье, и выполняя анализ
субстанции, предназначенной для препарата зарегистрированного
по ЕАЭС или приведенного в соответствие по ЕАЭС, по НД из ГРЛС, а
не по СП и АМ в рег.досье – мы начинаем не соответствовать
собственному рег.досье. Возможно, ГП выпущенная по таким РУ,
может быть признана несоответствующей при проверках, т.к.
входной контроль АФС осуществлялся по документам не
соотв.досье.
Также, чем дольше мы откладываем переход на новые рельсы, тем
больше мы копим т.н. «технического долга», т.е. того, что нам
потом придется переделывать - во внутренней документации, в
досье, и проч.
18
19.
Опасности игнорирования новых правил19
20.
20Спасибо за
внимание