

; 47,ХХ,18+ или 47,ХY,18+")
; 47,ХХ,21+ или 47,ХY,21+")

")
; 47, ХХХ")
; 46,XX(5p-) или 46,XY(5p-)")




























 medicine
medicineSimilar presentations:










Наследственные заболевания человека
1.
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯЧЕЛОВЕКА
Смирнова С.Н.
СМИРНОВА С.Н.
2.
3. Цитогенетический метод исследования
Половой хроматин(тельце Барра)
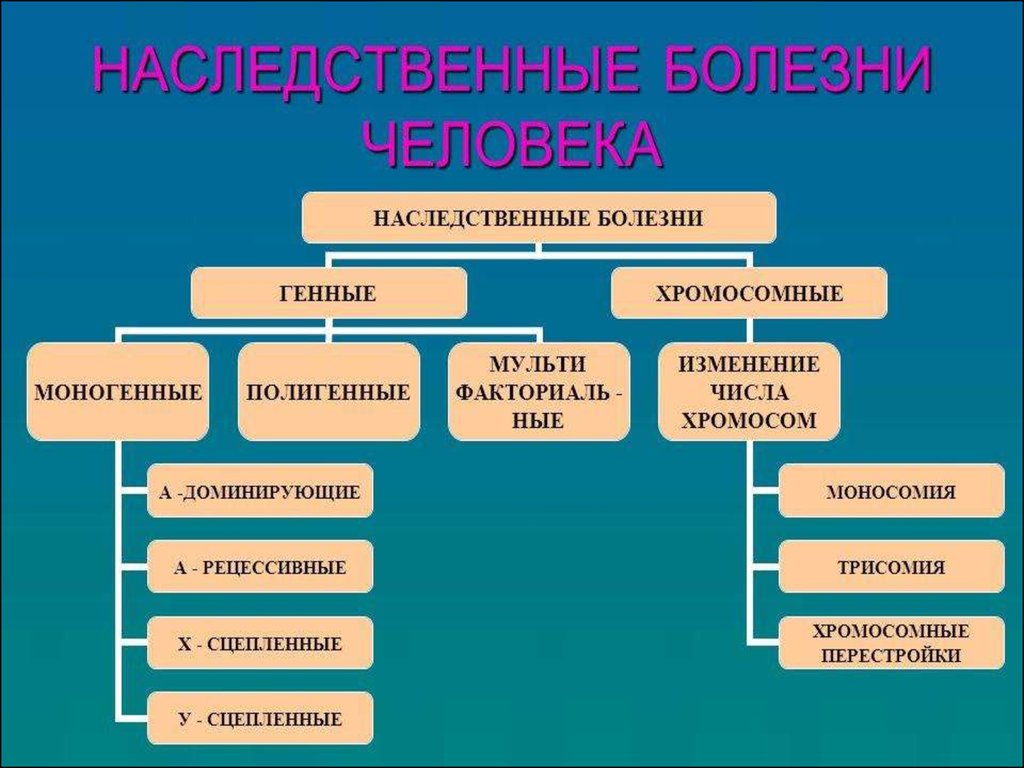
4. Хромосомные болезни - заболевания, в основе развития которых лежат нарушения числа или структуры хромосом.
Болезни, обусловленные изменением числа аутосомСиндром Патау (синдром трисомии 13); 47,ХХ,13+ или 47,ХY,13+
-Симптомы:
- микроцефалия,
расщелины губы и неба,
- аплазия костей носа,
- множественные грубые пороки
развития внутренних органов
5. Синдром Эдвардса (синдром трисомии 18); 47,ХХ,18+ или 47,ХY,18+
Синдром Эдвардса (синдром трисомии 18);Симптомы:
- аномалии мозгового и лицевого
черепа,
- недоразвитие нижней челюсти,
- короткая грудина,
- ушные раковины деформированы и
низко расположены
- множественные грубые пороки
развития внутренних органов
47,ХХ,18+ или 47,ХY,18+
6. Синдром Дауна (синдром трисомии 21); 47,ХХ,21+ или 47,ХY,21+
Цитогенетические варианты:1. Трисомный – 90-93%.
2. Транслокационный – 3-4%. 46, 14t(21/14).
3. Мозаичный – 3-4%. 47,21+/46; 46, 13t(21/13)/46.
Вероятность рождения больного
ребенка в зависимости от возраста
матери:
20-24 года – 1:1562
24-30 лет – 1:1000
35-39 лет – 1:214
>45 лет – 1:19
Частота рождения детей с синдромом
Дауна: 1 на 800-1000.
Американский актер Крис Бурк
7. Болезни, обусловленные изменением числа половых хромосом
Синдром Шерешевского-Тернера (моносомия поХ-хромосоме); 45, Х0
• Клинические признаки: низкий
рост, первичная аменорея,
бесплодие, стертые вторичные
половые признаки, крыловидные
кожные складки на шее,
врожденные пороки сердца,
гипоплазия ногтей, снижение
остроты зрения и слуха,
поперечная ладонная складка,
незначительное снижение
умственного развития.
• Популяционная частота – 2 : 10
000
8. Синдром Клайнфельтера (синдром полисомии Х)
Цитогенетические варианты:- дисомия – 47, XXY
- трисомия – 48, XXXY
- тетрасомия – 49, XXXXY
Симптомы:
- Высокий рост,
- Евнухоидное телосложение,
- Гинекомастия,
- Слабый рост волос на лице и теле,
- Оволосение лобка по женскому типу.
9. Синдром трипло-Х (трисомия Х); 47, ХХХ
- 2 тельца Барра в буккальном соскобе- Нормальное физическое, психическое и
половое развитие
- Высокий риск спонтанных абортов и
хромосомных нарушений у потомства
Полисомия Х: умственная отсталость,
судороги, недоразвитие гениталий,
пороки развития
Синдром дисомии по Y; 47, XYY
В большинстве случаев клинически не проявляется.
10. Синдром Лежёна (синдром «кошачьего крика»); 46,XX(5p-) или 46,XY(5p-)
Болезни, обусловленные изменением структуры хромосомСиндром Лежёна (синдром «кошачьего крика»); 46,XX(5p-) или
46,XY(5p-)
-Симптомы:
- Своеобразный крик при рождении,
- Лунообразное лицо,
- Эпикант,
- Уплощенная спинка носа,
- Короткая шея,
- Низко расположенные уши,
- Высокое небо,
- Гипотония мышц,
- Умственная отсталость.
11. Генные болезни – группа заболеваний, обусловленных мутациями на генном уровне
ГенБелок
Признак
Мутация гена
Нарушение
синтеза
фермента
Ферментопатия
12.
13. Синдром Марфана
Корней Иванович ЧуковскийНикколо Паганини
Ганс Христиан Андерсен
Аутосомно-доминантное
заболевание из группы
наследственных патологий
соединительной ткани
Арахнодактилия
Джованни Казелли, Смерть Арахны
14. Нарушение аминокислотного обмена
Фенилкетонурия – аутосомно-рецессивное заболеваниеМутация в гене
в длинном плече
12-й хромосомы
Дефицит
фенилаланингидроксилазы
Накопление фенилаланина и его производных
в жидкостях и тканях
Частота встречаемости
Ирландия
1:4500
Россия
1:8-10000
Италия
1:12000
Швейцария
1:16000
Афроамериканцы
1:50000
Финлядния
1:100000
Диагностика: скрининг-тест в роддоме
Лечение: диетотерапия
При ранней диагностике возможна полная
реабилитация больного и его полноценная
адаптация к социальной жизни
15. Нарушение углеводного обмена
Галактоземия – непереносимость лактозыМиссенс-мутации в гене
в коротком плече 9-й
хромосомы
Дефицит галактозо-1фосфат-уридилтрансферазы
Накопление галактозы
и ее метаболитов
Частота встречаемости
в России – 1:16242
Поражение ЦНС, печени,
хрусталика
Диагностика: скрининг-тест
Лечение: пожизненная диета
(исключение из рациона молочных
продуктов)
16. Нарушение углеводного обмена
Фруктоземия – непереносимость фруктозыМутация гена
в длинном плече 9-й
хромосомы
Дефицит фруктозо-1фосфатальдолазы
Накопление фруктозо-1фосфата в печени,
кишечнике и почках
Частота встречаемости
1 случай на 100-130 тысяч
Лечение: пожизненная диета.
Исключение из рациона продуктов,
содержащих фруктозу:
- фруктов и продуктов,
изготавливаемых из них;
- меда и изделий из него;
- всех продуктов, содержащих
тростниковый и свекловичный сахар
17. Нарушение минерального обмена
Нарушение липидного обмена – амавротическая идиотия (синдром ТеяСакса), сфинголипидоз (синдром Нимана-Пика), глюкоцереброзидоз(болезнь Гоше).
Мутация гена ATP7B,
расположенного в
13-й хромосоме
Нарушение минерального обмена
Болезнь Вильсона-Коновалова
Дефицит транспортирующего медь АТФазного
протеина Р-типа
Накопление Cu в клетках
печени, ЦНС, почках,
радужке
Кольцо Кайзера-Флейшера
18.
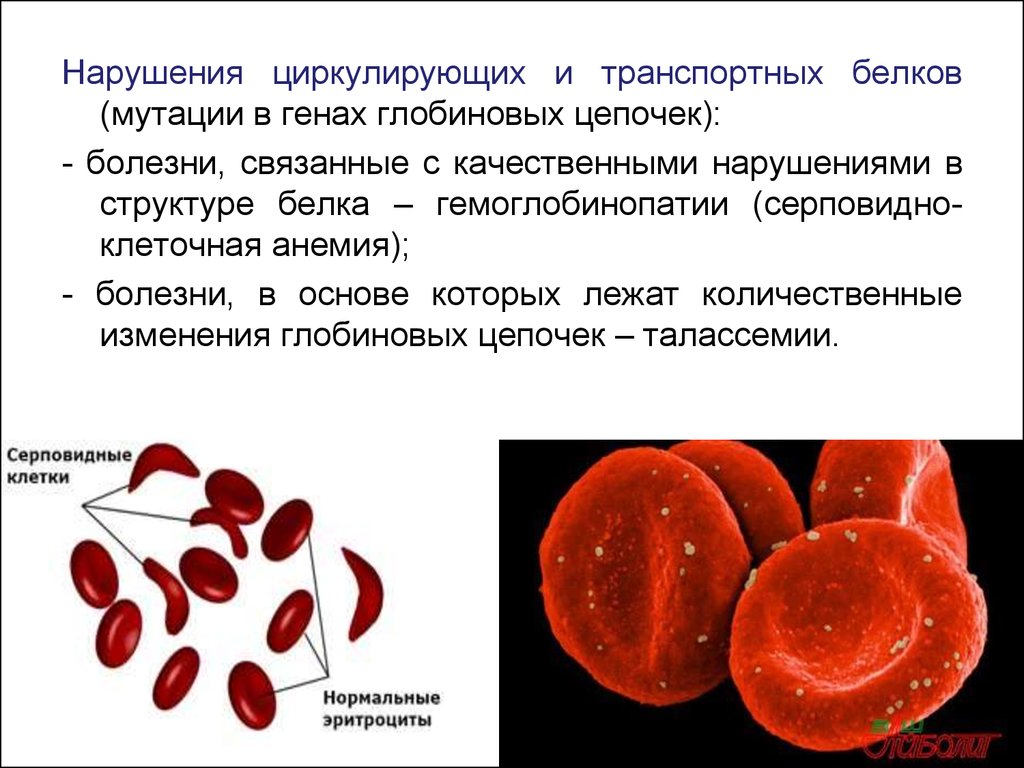
Нарушения циркулирующих и транспортных белков(мутации в генах глобиновых цепочек):
- болезни, связанные с качественными нарушениями в
структуре белка – гемоглобинопатии (серповидноклеточная анемия);
- болезни, в основе которых лежат количественные
изменения глобиновых цепочек – талассемии.
19.
Анемия серповидно-клеточная – это гемоглобинопатия изразряда качественных. Этот вид анемии считается
региональным, и распространен более всего среди жителей
Африканского континента, в странах с тропическим
климатом, второй по распространенности данной анемии
считается регион Ближнего и Среднего Востока. И затем,
более редко, серповидно-клеточную анемию можно
встретить в странах Средиземноморья и Южной Америки.
20.
Симптомы талассемииКлинические признаки разнообразны, зависят от формы талассемии. Чаще
проявляются в детстве. Для таких детей характерны следующие симптомы:
своеобразный “ башенный” череп (практически квадратной формы);
•сильно сплющенная переносица (седловидная);
•монголоидный разрез глаз (сужение глазной щели);
•увеличенная верхняя челюсть;
•увеличение печени и селезенки является ранним признаком заболевания. Они
увеличиваются из-за внемозгового кроветворения и гемосидероза (избыточное
отложение в тканях гемосидерина – пигмента, который образуется в результате
распада гемоглобина);
•желтушность и бледность кожных покровов;
•язвы в области голеней;
•образование билирубиновых камней в желчных путях;
•водянка плода, как правило, несовместима с жизнью (избыточное скопление
цереброспинальной жидкости в желудочковой системе головного мозга);
•отставание в физическом и половом развитии;
•слабость;
•повышенная утомляемость;
•снижение сопротивляемости организма к инфекционным заболеваниям
(вирусам гриппа, герпеса).
21. Мультифакториальные болезни
Характер их наследования не может быть объясненменделевскими законами.
Для мультифакториальных заболеваний характерно
наследование предрасположенности, зависящей от
значительного числа генов с суммарным эффектом и от
факторов внешней среды.
Частота широко распространенных мультифакториальных
заболеваний: гипертоническая болезнь (10-20%), ИБС (510%), алкоголизм (1,4-10%), язвенная болезнь желудка и
12-перстной кишки (2-5%), сахарный диабет и
шизофрения (1-2%).
22. Болезни с нетрадиционным типом наследования
1. Болезни импритинга.2. Митохондриальные.
3. Пероксисомные.
4. Болезни экспансии тринуклеотидных
повторов.
23.
ИМПРИНТИНГ И БОЛЕЗНИ ИМПРИНТИНГАХарактеристика геномной памяти
Молекулярно-генетический процесс, в ходе которого
модифицируются (маркируются) аллели родительских
генов в локусах хромосом отцовского и материнского
происхождения и обеспечивается моноаллельный характер
их экспрессии, называется импринтом или эпигеномным
процессом. Этот процесс также называют импринтингом
или геномной памятью, а сами маркированные гены
называются импринтированными генами.
В настоящее время в геноме человека предполагается
наличие от 100 до 200 генов, связанных с геномной
памятью
(уже
выделено
около
70
генов).
Импринтированные гены локализованы в локусах
большинства аутосомных хромосом (хромосомы 1-3, 5-7,
11, 13-15, 18, 19, 20 и 22) и локусах Х-хромосомы.
24.
ОБЩАЯ ХАРАКТЕРИСТИКА БОЛЕЗНЕЙ ИМПРИНТИНГАС нарушениями процессов геномной памяти связаны
болезни импринтинга или болезни геномной памяти (БГП),
объединяющие 24 нозологии, которые в зависимости от
этиологической причины делятся на три класса:
• болезни генного импринтинга - 14 нозологий (58,3% всех
БГП);
• болезни хромосомного импринтинга - 10 нозологий (31,7%);
болезни
ошибок
импринтинга,
обусловленные
микроделециями
в
регуляторных
областях
импринтированных генов или центрах импринтинга (ЦИ) две нозологии (входят в предыдущие два класса).
25.
Болезни генного импринтингаЭтиология
При болезнях генного импринтинга наблюдается моноаллельная
экспрессия в локусах хромосом одного из родителей. Причина - точковые
мутации в генах, дифференцированно экспрессирующихся в зависимости
от материнского и отцовского происхождения и приводящих к
специфическому метилированию цитозиновых оснований в молекуле ДНК.
Эти мутации обусловливают развитие заболеваний, для которых большое
значение имеют характер наследования и происхождение хромосом. К
таким заболеваниям относятся:
• болезнь Гиршпрунга, обусловленная мутацией в гене RET (10q11.2);
чаще всего наследуется по материнской линии;
• нейрофиброматоз Реклингаузена (тип 2) - мутация в гене SCH (22q12);
наследуется по материнской линии;
• псориаз - проявляется тяжелее, если наследуется по отцовской линии;
семейная гипертрофическая кардиомиопатия - наследуется по
материнской линии;
26.
Болезнь Гиршпрунга - врождённый аганглиоз толстой кишки (отсутствиесобственно нервных клеток в мышечном сплетении Ауэрбаха и
подслизистом сплетении Майсснера. с отсутствием сокращения в
пораженной зоне кишки, застоем каловых масс в вышележащих
отделах, в результате чего возникают значительное расширение и
удлинение кишки.
Частота встречаемости болезни Гиршпрунга: 1:5 000 новорождённых.
Преобладающий пол - мужской (4-5:1).
27.
Нейрофиброматоз РеклингхаузенаНейрофиброматоз Реклингхаузена относится к группе факоматозов.
Факоматозы - это наследственные заболевания, которые относятся к
эктодермомезодермальным дисплазиям с поражением кожи, глаз,
нервной системы и внутренних органов. Обязательным симптомом этой
группы заболеваний является наличие пигментных пятен на коже или
сетчатке глаза.
Нейрофиброматоз
Реклингхаузена наследуется по аутосомнодоминантному типу, однако 50% случаев возникают вследствие новой
мутации. Распространенность заболевания оценивается как 1:3000. Оно
поражает в равной степени и мужчин, и женщин.
Патогенез
нейрофиброматоза
связан
с
дефектом
гена
нейрофибромина.
28.
Болезни генного импринтингаЭтиология
• синдром Ангельмана (СА) - делеция критического района, находящегося
в материнской хромосоме 15 (15q11.2-q13);
синдром Вильямса - проявляется более выраженной задержкой
физического и умственного развития и микроцефалией, если делеция
затрагивает материнскую хромосому 7 (7q11.23);
• синдром «крика кошки» - проявляется более выраженно, если делеция
захватывает отцовскую хромосому 5 (5р15.3);
• синдром Корнелии де Ланге (3q26) - проявляется более выраженно, если
наследуется по материнской линии;
синдром Прадера-Вилли (СПВ) - делеция критического района,
находящегося в отцовской хромосоме 15 (15q11.2-q13);
• синдром Турета и поликистоз почек - проявляются раньше и тяжелее,
если наследуются по материнской линии;
тяжелая (злокачественная) шизофрения - проявляется более
выраженно, если наследуется по отцовской линии;
• spina bifida - наследуется по материнской линии (в 2 раза чаще, чем по
отцовской линии);
• эпилепсия - проявляется тяжелее, если наследуется по материнской
линии.
29.
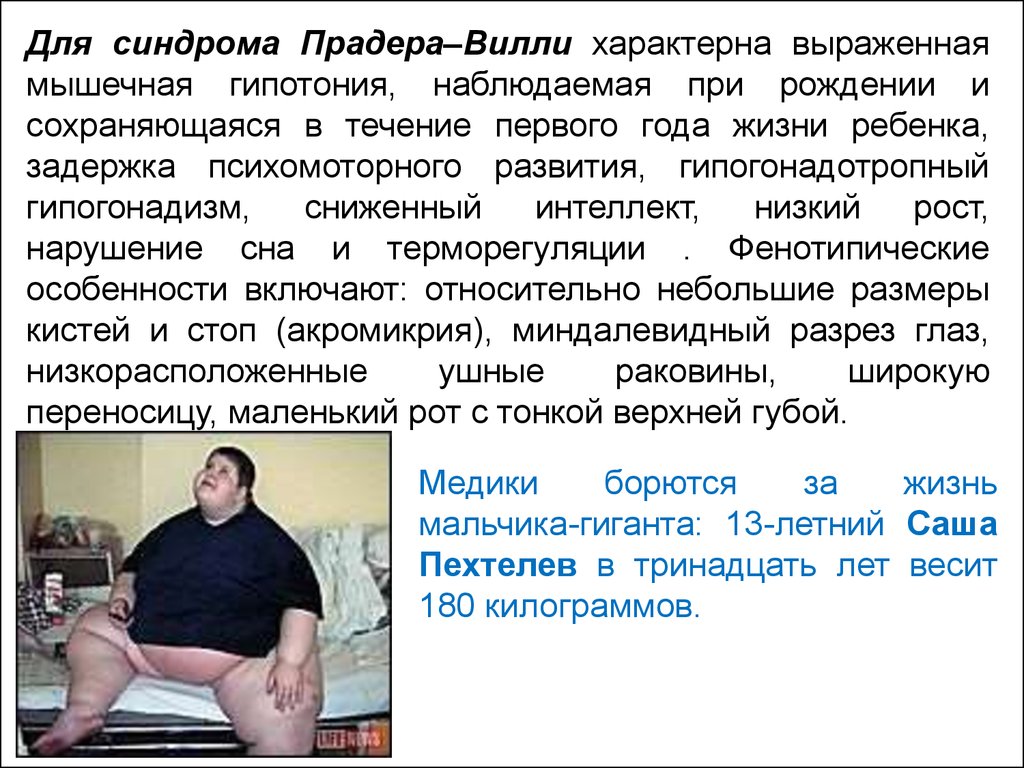
Для синдрома Прадера–Вилли характерна выраженнаямышечная гипотония, наблюдаемая при рождении и
сохраняющаяся в течение первого года жизни ребенка,
задержка психомоторного развития, гипогонадотропный
гипогонадизм,
сниженный
интеллект,
низкий
рост,
нарушение сна и терморегуляции . Фенотипические
особенности включают: относительно небольшие размеры
кистей и стоп (акромикрия), миндалевидный разрез глаз,
низкорасположенные
ушные
раковины,
широкую
переносицу, маленький рот с тонкой верхней губой.
Медики
борются
за
жизнь
мальчика-гиганта: 13-летний Саша
Пехтелев в тринадцать лет весит
180 килограммов.
30.
Болезни хромосомного импринтингаЭтиология
Для болезней хромосомного импринтинга характерна однородительская
дисомия (ОРД) или наличие двух копий хромосомы либо отцовского, либо
материнского происхождения. Термины: «однородительская дисомия» или
«изодисомия» впервые предложены Э. Энжелом в 1980 г. для обозначения
одного из типов анеуплоидии в гаметах млекопитающих. Эти термины
указывают на наличие у диплоидного потомства двух локусов, полученных
от одного и того же родителя, тогда как в норме наследуется только по
одному локусу от каждого родителя. Примерами таких заболеваний
служат:
ОРД по материнской хромосоме 2, сопровождающаяся
дизэмбриогенезом и задержкой развития;
ОРД по длинному плечу отцовской хромосомы 6 (6q23-q24),
сопровождающаяся неонатальным сахарным диабетом;
• ОРД по длинному плечу материнской хромосомы 7 при муковисцидозе;
• ОРД по короткому плечу материнской хромосомы 7 в области гена
GRB10 при синдроме Сильвера-Рассела;
частичная трисомия отцовской хромосомы 11 (11р15.5) или
сбалансированная транслокация с точками разрывов в материнской
хромосоме 11 при синдроме Беквитта-Видемана.
31.
Синдро́м Рассе́ла-Си́львера (карликовость РасселаСильвера)—
комплекс
наследственных
аномалий
(предполагается аутосомно-рецессивный тип наследования)
врождённый карликовый рост в результате нарушений
эмбрионального развития: малая масса тела при рождении,
низкий рост, задержка общего развития, треугольное лицо,
опущенные вниз уголки рта, укороченные и согнутые пальцы
рук, синдактилия
Эбби Кинг в возрасте 22-х месяцев
еще не доросла и до размеров
шестимесячного ребенка. Она
носит одежду для новорожденных,
но давно ходит и радует родных
своими успехами. Ее рост — 65
сантиметров,
а
вес
—
6
килограммов.
Она
прекрасно
сформированный
миниатюрный
человек — реальная Дюймовочка.
32.
Митохондриальные болезни (цитопатии) - гетерогенная группасистемных расстройств, обусловленных мутациями митохондриального
или ядерного генома, которые поражают преимущественно мышечную,
нервную и нервно-мышечную системы.
Наследование мутаций в митохондриальном геноме носит особый
характер. Если гены, заключенные в ядерной ДНК, дети получают поровну
от обоих родителей, то митохондриальные гены передаются потомкам
только от матери. Это связано с тем, что всю цитоплазму с
содержащимися в ней митохондриями потомки получают вместе с
яйцеклеткой, в то время как в сперматозоидах цитоплазма практически
отсутствует. По этой причине женщина с митохондриальным заболеванием
передаёт его всем своим детям, а больной мужчина - нет.
33.
На сегодня известно три синдрома, которые рассматривают какнаследственные
пероксисомные
болезни:
акаталаземия,
цереброгепаторенальный
синдром
Целлвегера
и
системная
недостаточность карнитина.
Акаталаземия - заболевание, в основе которого лежит резкое снижение
активности каталазы в печени и других органах вследствие ее
пониженной термостабильности. Основным клиническим синдромом
этого заболевания является гангренозная ротовая полость, покрыта
язвами.
34.
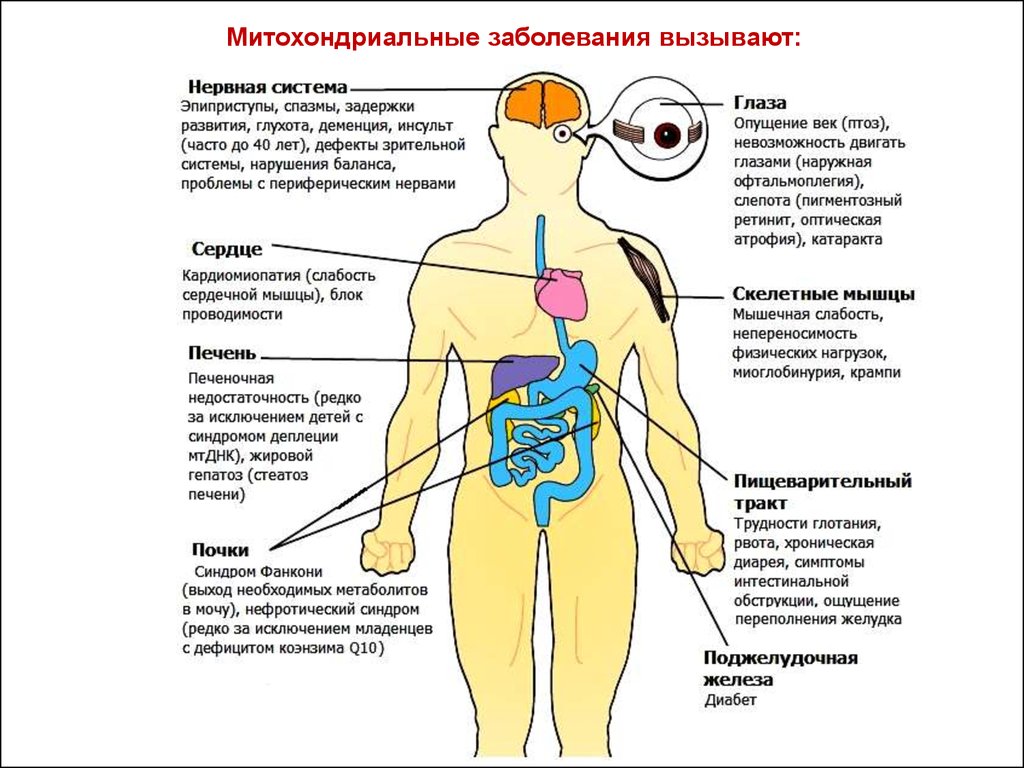
Митохондриальные заболевания вызывают:35.
Пероксисомные болезниСИНДРОМ ЦЕЛЛВЕГЕРА
Генетика: Мутации в генах 13 пероксинов могут приводить к наследственным
нарушениям биогенеза пероксисом.
Тип наследования: аутосомно-рецессивный.
Эпидемиология:Суммарная частота заболеваний 1:50 000 живых новорожденных.
Клинические проявления: В большинстве случаев обнаруживают изменения со
стороны органов зрения, такие как, помутнение роговицы, пятна "Брушфильда",
глаукому, катаракту, пигментную дегенерацию сетчатки, атрофия зрительного нерва.
Часто встречается крипторхизм, гипоспадия / эписпадия. Практически у всех
больных с СЦ наблюдается симптомокомплекс "вялого ребенка": выраженная
диффузная мышечная гипотония, сухожильная гипорефлексия/арефлексия, грубая
задержка психомоторного развития. Неблагоприятный исход на первом году жизни
от легочно-сердечной и печеночной недостаточности.
36.
Хорея Гентингтона - одно из самых тяжелых прогрессирующихнейродегенеративных наследственных заболеваний головного мозга . Хорея
(chorea; от греческого слова "choreia" - пляска) - форма гиперкинеза,
характеризуется непроизвольными, быстрыми, нерегулируемыми движениями,
возникающими в различных мышечных группах.
Его распространенность составляет около 10:100000. Отличительные признаки
- хорея и расстройства поведения . Заболевание может начинаться с любого из
этих симптомов или с обоих сразу. Болезнь Гентингтона может развиться в
любом возрасте - как в детстве, так и в 70 лет и старше, но чаще первые
симптомы появляются в 30-50 лет.
Хорея начинается исподволь. Первые признаки хореи Гентингтона проявляются
в возрасте 25-50 лет, реже в детском возрасте. Мужчины болеют чаще, чем
женщины; первыми симптомами могут быть неусидчивость , суетливость
движений , что не расценивается больным и его родственниками как
заболевание. Со временем, однако, двигательные нарушения нарастают и
могут привести к инвалидности. Характерны частые, внезапные, неритмичные
судорожные движения конечностей или туловища. Возможны спазмы лицевой
мускулатуры , всхлипывания ,нарушения артикуляции . Страдает координация
движений при ходьбе : походка становится "танцующей" (хореической) . Часто
наблюдаются депрессия , апатия , отчужденность , раздражительность ,период
ическая
расторможенность.
В
некоторых
случаях
развиваются бред и навязчивые состояния , в связи с чем сначала ошибочно
диагностируется шизофрения .
37.
38. Медико-генетическое консультирование
Советский генетик, невропатологС.Н. Давиденков организовал первые в
мире медико-генетические консультации
(Москва, 1920, Ленинград, 1934).
Показания для МГК:
- Рождение в семье ребенка с врожденными
уродствами и множественными пороками
развития
- Умственная отсталость у ребенка
- Повторные спонтанные аборты, выкидыши,
мертворождения у женщины
- Выявленная патология у ребенка при
проведении массовых скринирующих программ
- Близкородственные браки
- Сведения о неблагоприятном воздействии
мутагенов или тератогенов на ранних сроках
беременности
39.
ГЕНЕТИЧЕСКИЕ ОСНОВЫ ПРОФИЛАКТИКИНАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Общие положения
С профилактической точки зрения всю наследственную
патологию целесообразно подразделить на 3 категории:
• вновь возникающие мутации (в первую очередь это
анеуплоидии и тяжелые формы доминантных мутаций);
• унаследованные от предыдущих поколений (как генные, так
и хромосомные);
• болезни с наследственной предрасположенностью.
Различают 3 вида профилактики наследственной патологии.
40.
Первичная профилактикаПод первичной профилактикой понимают действия, которые должны
предупредить
зачатие
больного
ребенка;
это
планирование
деторождения и улучшение среды обитания человека.
Планирование деторождения включает 3 основные позиции:
• оптимальный репродуктивный возраст, который для женщин составляет
21-35 лет (более ранние или поздние беременности увеличивают
вероятность рождения ребенка с врожденной патологией и
хромосомными болезнями);
• отказ от деторождения в случаях высокого риска наследственной и
врожденной патологии (при отсутствии надежных методов дородовой
диагностики, лечения, адаптации и реабилитации больных);
• отказ от деторождения в браках с кровными родственниками и между
двумя гетерозиготными носителями патологического гена.
Улучшение среды обитания человека должно быть направлено
главным образом на предупреждение вновь возникающих мутаций путем
жесткого контроля содержания мутагенов и тератогенов в окружающей
среде. Это особенно важно для профилактики всей группы соматических
генетических болезней (врожденные пороки развития, злокачественные
новообразования, иммунодефицитные состояния и т.п.).
41.
Вторичная профилактикаВторичная
профилактика
предполагает
прерывание
беременности при высокой вероятности заболевания плода
или пренатально-диагностированной болезни. Прервать
беременность можно только в установленные сроки и с
согласия женщины. Основанием для элиминации эмбриона
или плода является наследственная болезнь.
Прерывание беременности - не самое лучшее решение, но
пока это единственный метод для вторичной профилактики
большинства тяжелых и смертельных генетических дефектов.
42.
Третичная профилактикаПод третичной профилактикой наследственной патологии понимают коррекцию
проявления
патологических
генотипов.
Это
можно
назвать
и нормокопированием, поскольку при патологическом генотипе стремятся
получить нормальный фенотип.
Третичная профилактика проводится как при наследственных болезнях, так и
(особенно часто) при болезнях с наследственной предрасположенностью. С ее
помощью можно добиться полной нормализации функций или снижения
выраженности патологического процесса. Для некоторых форм наследственной
патологии она может совпадать с лечебными мероприятиями в
общемедицинском смысле.
Предотвратить развитие наследственного заболевания (нормокопирование)
можно внутриутробно или после рождения.
Для некоторых наследственных заболеваний возможно внутриутробное лечение
(например, при резус-несовместимости, некоторых ацидуриях, галактоземии).
Развитие заболевания в настоящее время можно предотвратить путем
коррекции (лечения) после рождения больного. Типичными примерами
болезней, для которых эффективна третичная профилактика, могут быть
галактоземия, фенилкетонурия, гипотиреоз (см. ниже) и др. Например, целиакия
проявляется с началом прикорма ребенка. В основе болезни лежит
непереносимость глютена. Исключение этого белка из пищи полностью
гарантирует избавление от тяжелейшей патологии ЖКТ.