




































 physics
physics chemistry
chemistrySimilar presentations:



")



 строения и реакционной способности органических соединений")


Радиоспектроскопические методы исследования, часть 2
1. Радиоспектроскопические методы исследования
2.
ЯМР-спектр.Как он выглядит и как его читать
Химсдвиг
Интегральная интенсивность
Мультиплетность сигнала. Треугольники Паскаля
и биномиальное распределение
Константа спин-спинового взаимодействия КССВ. Как
их считать?
Шкала ЯМР и миллионные доли
Выбор эталонного сигнала
Спиновые системы разного типа. Спектры первого
порядка
2
3.
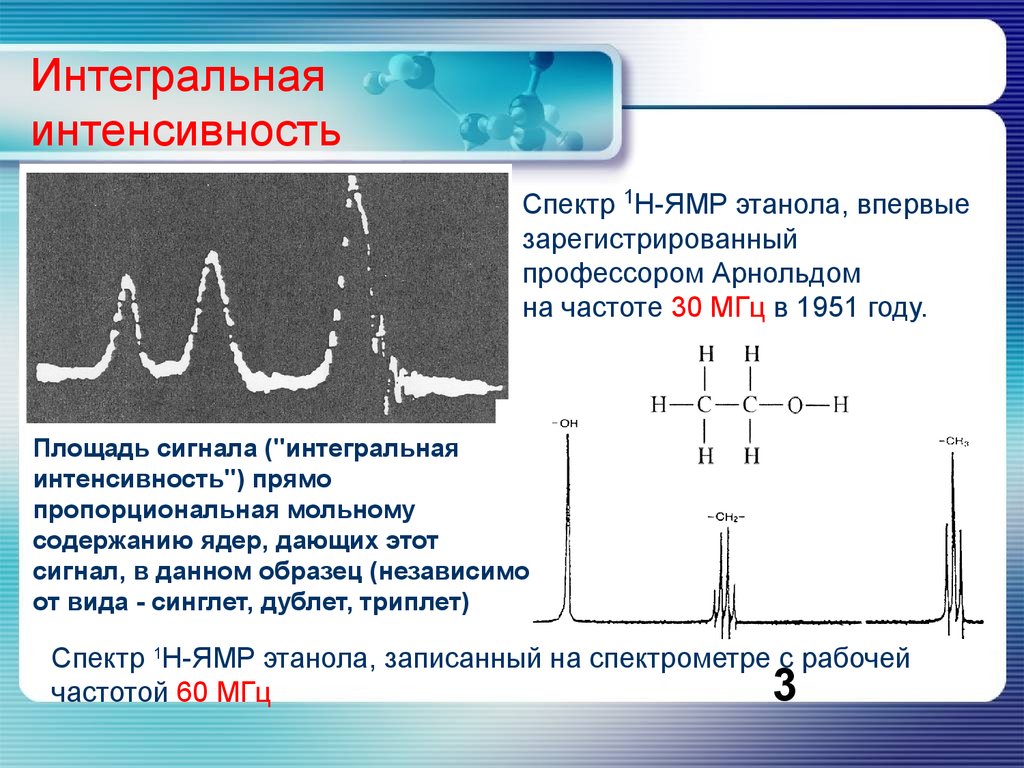
Интегральнаяинтенсивность
Спектр 1Н-ЯМР этанола, впервые
зарегистрированный
профессором Арнольдом
на частоте 30 МГц в 1951 году.
Площадь сигнала ("интегральная
интенсивность") прямо
пропорциональная мольному
содержанию ядер, дающих этот
сигнал, в данном образец (независимо
от вида - синглет, дублет, триплет)
Спектр 1Н-ЯМР этанола, записанный на спектрометре с рабочей
частотой 60 МГц
3
4.
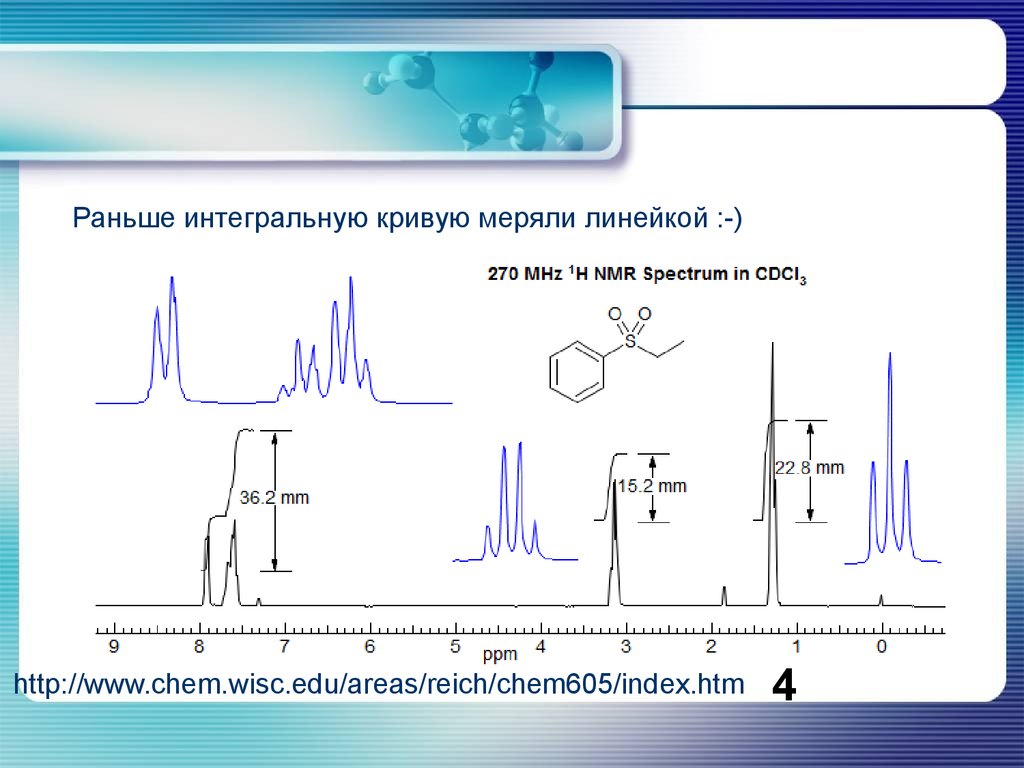
Раньше интегральную кривую меряли линейкой :-)http://www.chem.wisc.edu/areas/reich/chem605/index.htm
4
5.
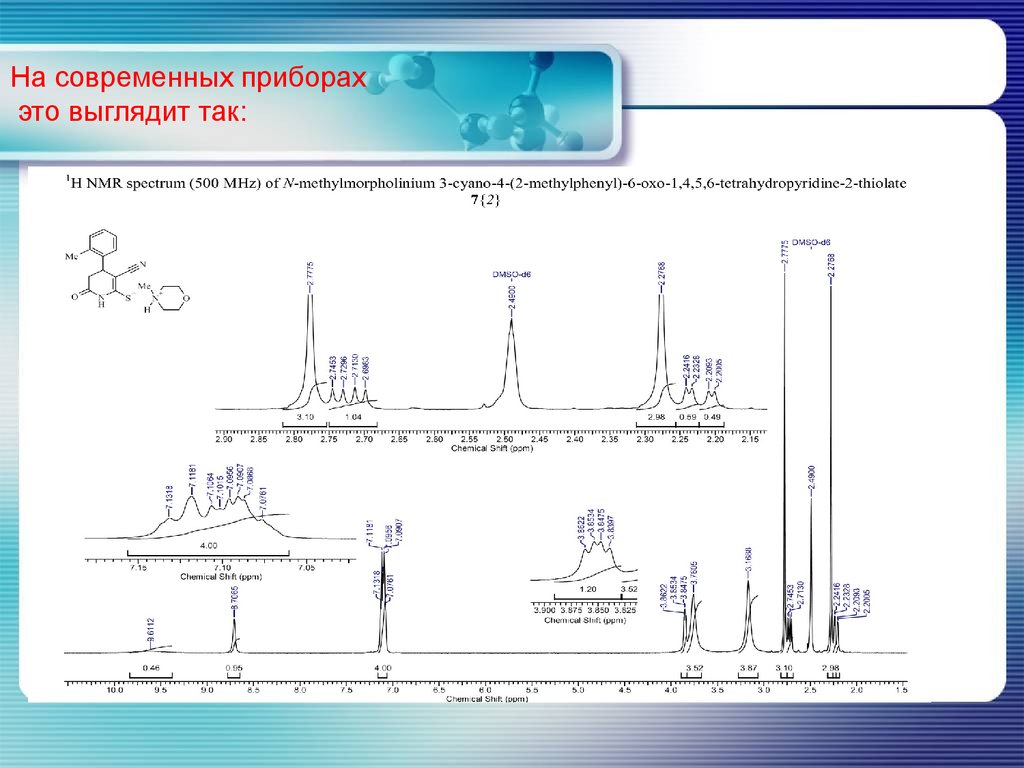
На современных приборахэто выглядит так:
5
6.
Важное отличиеспектров 1Н от 13С:
помимо прочего, в 13С
спектрах интегральная
интенсивность сигнала
не оценивается
6
7.
Немного о погрешности интегрирования.The integration of NMR spectra can be carried out with high accuracy, but this is
only possible if a number of sources of error are properly handled. On a modern
spectrometer accuracy of ±5% can be achieved easily if relaxation issues are
handled properly. To get errors of <1% a number of factors have to be considered
and optimized.
1. Signal to Noise. The spectrum must have adequate signal to noise to support
the level of accuracy required for the experiment.
2. Saturation Effects. NMR spectroscopy has a feature unique among
spectroscopic methods, that relaxation processes are relatively slow (on the order of
seconds or tenths of seconds), compared to milli, micro, and pico seconds for IR
and UV. In other words, once the spectrometer has perturbed the equilibrium
population of nuclei by scanning over the resonance frequency or pulsing the nuclei,
it takes from 0.1 to 100s of seconds (typically several seconds) for them to return to
their original populations (T1 the spin-lattice relaxation time). If power settings are
too high (for CW spectra) or pulse angle and repetition rates too high (for FT
spectra) then spectra can become saturated, and integrations less accurate,
because the relaxation rates of various protons in the sample are different.
Saturation effects are particularly severe for small molecules in mobile solvents,
because these typically have the longest T1 relaxation times.
7
8.
It is important to recognize that integration errors caused by saturation effects willdepend on the relative relaxation rates of various protons in a molecule. Errors will be
larger when different kinds of protons are being compared (such as aromatic CH to a
methyl group), than when the protons are similar or identical in type.
3. Line Shape Considerations. NMR signals in an ideally tuned instrument are
Lorenzian in shape, so the intensity extends for some distance on both sides of the
center of the peak. Integrations must be carried out over a sufficiently wide frequency
range to capture enough of the peak for the desired level of accuracy. Thus, if the
peak width at half height is 1 Hz, then an integration of ±2.3 Hz from the center of the
peak is required to capture 90% of the area, ±5.5 Hz for 95%, ±11 Hz for 98% and
±18Hz for >99% of the area. This means that peaks that are closely spaced cannot
be accurately integrated by the usual method, but may require line-shape simulations
with a program like NUTS or WINDNMR to accurately measure relative peak areas.
4. Digital Resolution. A peak must be defined by an adequate number of points if
an accurate integration is to be obtained. The errors introduced are surprisingly
small, and reach 1% if a line with a width at half height of 1 Hz is sampled every 0.5
Hz.
8
9.
5. Isotopic Satellites. All C-H signals have 13C satellites located ±JC-H/2 from the center ofthe peak (JC-H is typically 115-135 Hz, although numbers over 250 Hz are known) Together
these satellites make up 1.1% of the area of the central peak (0.55% each). They must be
accounted for if integration at the >99% level of accuracy is desired. Larger errors are
introduced if the satellites from a nearby very intense peak fall under the signal being
integrated. The simplest method to correct this problem is by 13C decoupling, which
compresses the satellites into the central peak. A number of other elements have significant
fractions of spin ½ nuclei at natural abundance, and these will also create satellites large
enough to interfere with integrations. Most notable are 117/119Sn, 29Si, 77Se, 125Te, 199Hg.
6. Spinning Sidebands. These can appear at ± the spinning speed in Hz in spectra run on
poorly tuned spectrometers and/or with samples in low-quality tubes. They draw intensity
from the central peak.
7. Baseline Slant and Curvature. Under some conditions spectra can show significant
distortions of the baseline, which can interfere with obtaining high-quality integrations.
8. Decoupling. When decoupling is being used, as is routinely done for 13C NMR spectra
and occasionally for 1H NMR spectra, peak intensities are distorted by Nuclear Overhauser
Effects. Integrations of such spectra will not give accurate ratios of peak areas.
9
10.
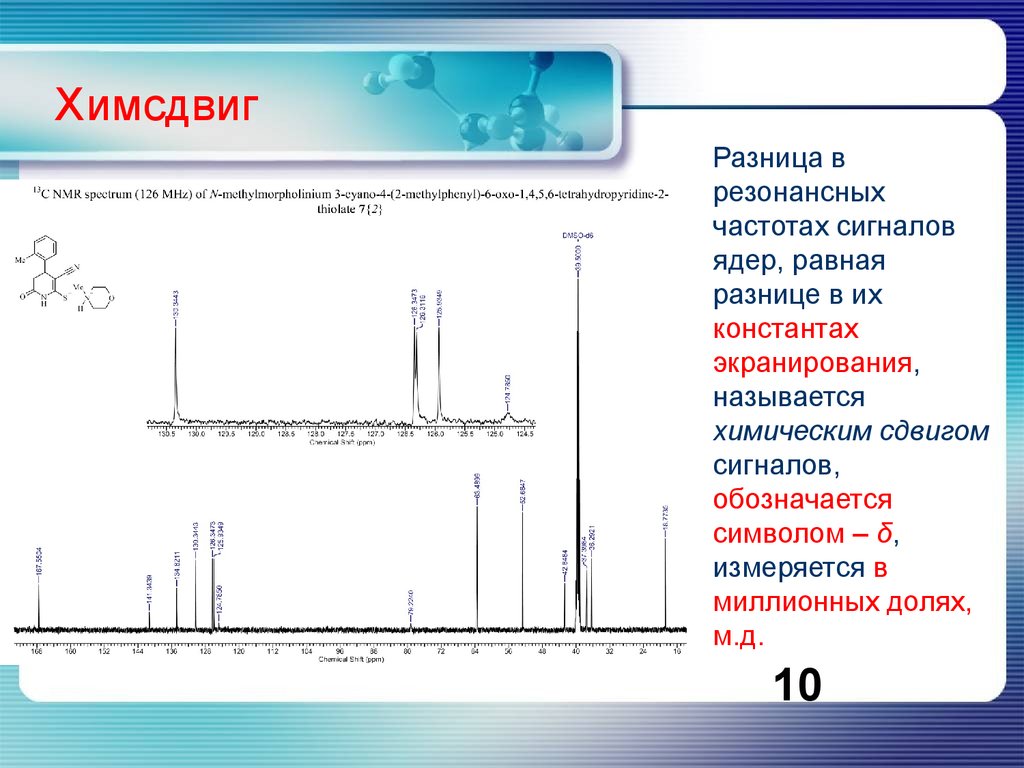
ХимсдвигРазница в
резонансных
частотах сигналов
ядер, равная
разнице в их
константах
экранирования,
называется
химическим сдвигом
сигналов,
обозначается
символом – δ,
измеряется в
миллионных долях,
м.д.
10
11.
Откуда берутся химсдвиги? Почему сигналы разных ядер"разъезжаются" по спектру?
Истинное значение напряженности поля в точке, где находится ядро, не равно
напряженности поля магнита В0. Атомные ядра окружены плотной электронной шубой.
Под действием магнитного поля электроны начинают совершать циркуляции (курс
физики в средней школе), которые приводят к возникновению вторичных,
индуцированных внешним магнитным полем В0, магнитных полей. Возникает
экранирование ядра. В результате эффективное поле в точке расположения ядра
оказывается отличным от приложенного поля В0. Его компонента по оси Z может
увеличивать или уменьшать В0. Электронные плотности на атомах одного типа
(например, на протонах), занимающих разные структурные положения в молекуле,
различаются. Вследствие этого эффективные поля Вэфф у разных атомов также будут
различны.
11
12.
Откуда берутся химсдвиги? Почему сигналы разных ядер"разъезжаются" по спектру?
σ – «константа экранирования» данного
ядра. Ее можно вычислить.
ρ(r) – функция распределения электронной
плотности, а r –расстояние от ядра. Величина σ
может быть и положительной, и отрицательной.
Чем больше электронная плотность на нем, тем
больше по модулю σ.
Для протонов σ положительна. Распределение электронной плотности около протонов в
органических молекулах близко к сферически симметричному, а поэтому создаваемое
электронными циркуляциями вторичное поле направлено против поля В0. Электронная
плотность на атомах водорода в молекуле этанола наибольшая на протонах CH3 группы,
для них константа экранирования σ самая большая, и поэтому сигнал протонов
метильной группы находится в спектре этанола в самом сильном поле. А сигнал OH
протона, электронная плотность на котором сильно понижена, поскольку она смещена к
электроотрицательному кислороду, расположен в самом слабом поле.
12
13.
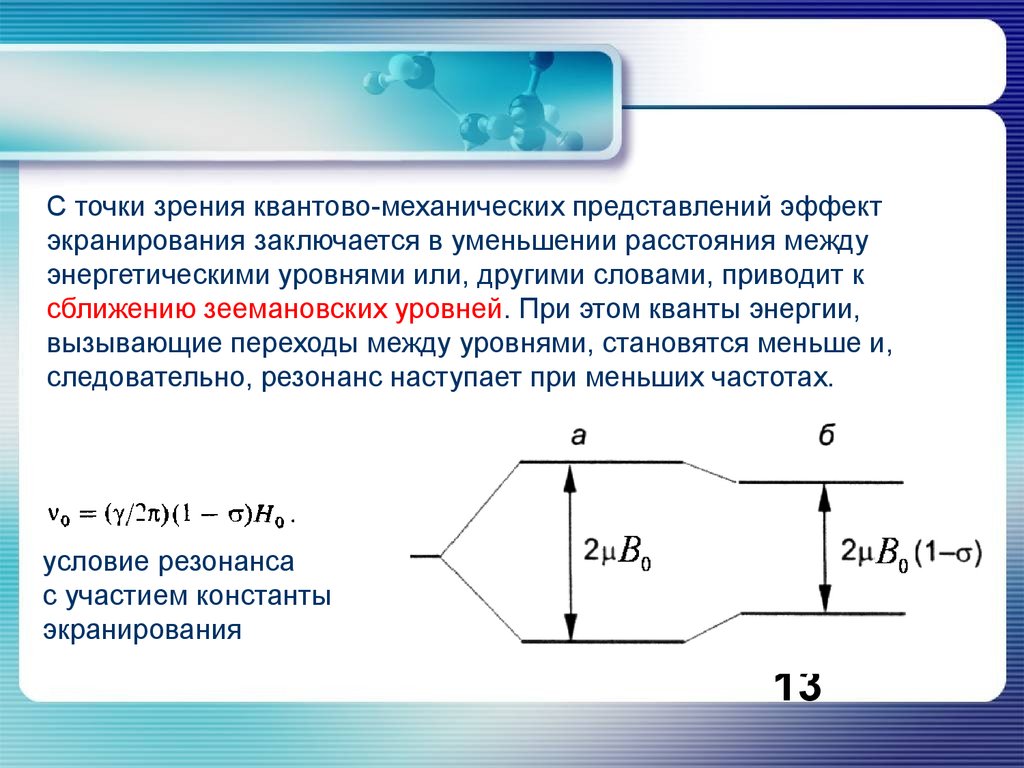
С точки зрения квантово-механических представлений эффектэкранирования заключается в уменьшении расстояния между
энергетическими уровнями или, другими словами, приводит к
сближению зеемановских уровней. При этом кванты энергии,
вызывающие переходы между уровнями, становятся меньше и,
следовательно, резонанс наступает при меньших частотах.
условие резонанса
с участием константы
экранирования
13
14.
Резюмируем: различие резонансных частот ядер Н, занимающихструктурно неэквивалентные положения в молекуле, имеет чисто
химическую природу. Это явление получило название «химического
сдвига». Величины σ очень чувствительны даже к очень малым
изменениям в структуре молекул. Они чутко реагируют на любые
факторы, оказывающие влияние на распределение электронной
плотности в молекуле.
Спектр ЯМР каждого соединения может содержать столько
разных сигналов, сколько в нем типов неэквивалентных
магнитных ядер.
Если зафиксировать частоту электромагнитного
поля V0 и плавно повышать напряженность
постоянного магнитного поля (развертка по полю),
то условия резонанса наступят раньше (т. е. при
более слабом поле) для протонов Me групп
диметилового эфира (ДМЭ), затем – Me3N
(ТМА) и, наконец -тетраметилсилана (ТМС)
14
15.
Шкала ЯМР и миллионные доли. Зачем миллионныедоли, если можно указывать Герцы или Теслы?
спектр ЯМР представляет
собой график зависимости в
координатах «интенсивность
поглощения – частота
радиочастотного поля»
Спектр фенилэтилового спирта PhCH2CH2OH (фрагмент), рег. в поле
9.4 Тесла. Шкала в дана Герцах. Различие в 6 знаке. Неудобно.
Аналогично будет, если градуировать шкалу в Теслах.
15
16.
Однако в этом случае возникнутсложности, связанные с тем, что в
разных спектрах сигналы не будут
совпадать, т.к. рабочая частота
прибора может быть разной, и за 0
принимаются разные точки. Нужен
стандарт и единая шкала.
16
17.
В спектроскопии ЯМР 1H и 13C в качестве эталонного соединенияиспользуется тетраметилсилан (Me4Si или ТМС). Удобство использования
ТМС состоит в том, что
• все двенадцать протонов в нем эквиваленты,
• показывают в спектре единственный узкий сигнал в самой
высокопольной части спектра относительно основного большинства
сигналов протонов органических соединений.
• Кроме того, ТМС химически инертен, магнитно изотропен, имеет низкую
температуру кипения (26.50С), добавляется в образец в малых
количествах (2-3 капли) и легко удаляется из образца после записи
спектра.
17
18.
Использование ТМС в качестве внутреннего эталона не всегда возможно. Егонельзя использовать в водных растворах, поскольку ТМС совершенно не
растворим в воде. В этом случае вместо него чаще всего используют хорошо
растворимую в воде натриевую соль 3-(триметилсилил)-3,3,2,2тетрадейтеропропионовой кислоты
(СН3)3SiCD2CD2COONa. Химический сдвиг мощного сигнала протонов группы
(СН3)3Si в этом соединении в δ-шкале точно совпадает с химическим сдвигом
ТМС (0 м.д.), а сигналы метиленовых групп отсутствуют в спектре.
Иногда в качестве эталонного можно использовать один из сигналов
растворителя. Хотя для измерения спектров обычно используют
дейтерированные растворители очень высокого качества, степень
дейтерирования в них редко превышает 99,9%. В связи с этим в спектре 1НЯМР все же присутствуют сигналы «остаточных протонов» растворителя,
которые можно использовать для эталонирования.
18
19.
Химические сдвиги «остаточных протонов» в спектрах 1Н-ЯМР наиболеечасто используемых дейтерированных растворителей.
19
20.
Me-O-MeЗа единицу химического сдвига принимается одна миллионная доля
напряженности поля или резонансной частоты (м.д.), поэтому в уравнение
вводится фактор 106. В зарубежной литературе этому сокращению
соответствует ppm (parts per million).
20
21.
2122.
Теперь самое сложное и вкусное.• Почему некоторые сигналы
расщепляются?
• Треугольники Паскаля и
биномиальное распределение
• Константа спин-спинового
взаимодействия КССВ
22
23.
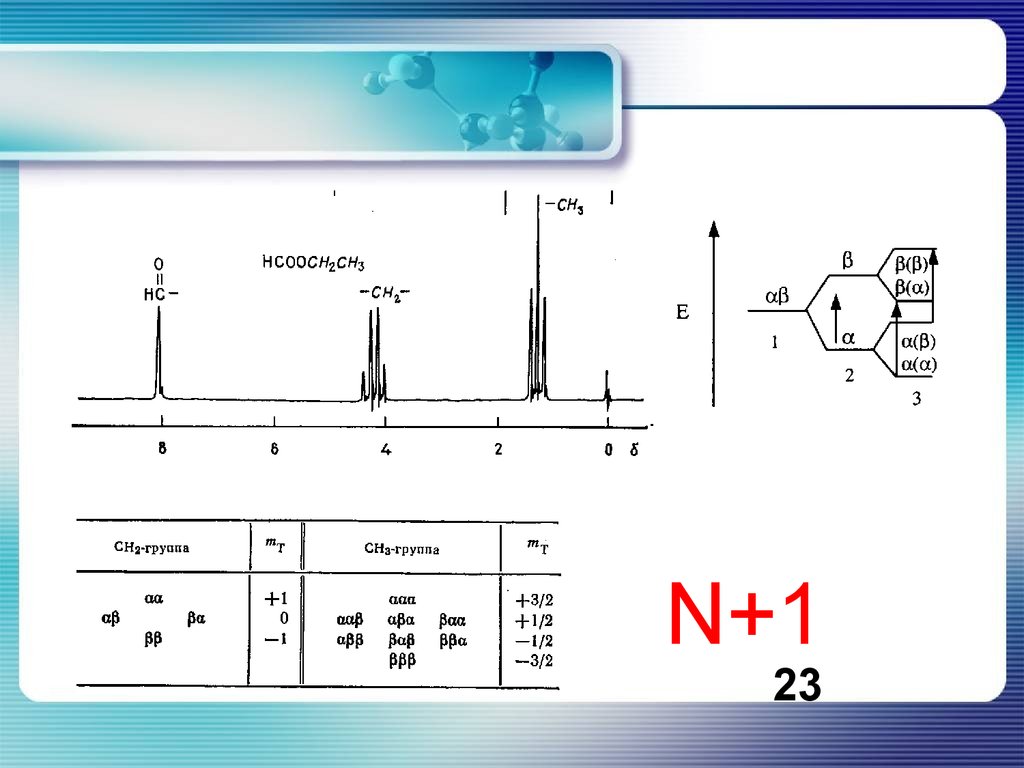
N+123
24.
В молекулах, где спины обоих протонов метиленовой группы направлены пополю (αα) или против него (ββ), протоны метильной группы будут испытывать
влияние дополнительного поля и их резонанс будет смещен в более сильное
или более слабое поле по сравнению с предыдущим случаем. Сигнал
протонов метильных групп всех молекул в образце будет состоять из трех
линий, причем средняя, отвечающая резонансу той части молекул, где
реализуется ситуация αβ или βα для метиленовых протонов, будет иметь
вдвое большую интенсивность, чем крайние. Отметим, что наличие трех
протонов в самой резонирующей группе никак не отражается на виде ее
сигнала.
24
25.
Метильная группа. Для каждой конкретной молекулы возможна одна из восьмиситуаций в отношении спинов метильной группы (табл. 5.4). При ориентации
спинов всех протонов по полю (ααα) суммарная проекция спинов на
направление Но составляет 3/2, при противоположной ориентации (βββ) -3/2. В
остальных случаях проекция составляет 1/2 или -1/2; поскольку эта ситуация
реализуется тремя различными способами, вероятность встретить молекулу с
таким суммарным спином для протонов метильной группы втрое больше, чем
со спином 3/2 или -3/2. В результате сигнал метиленовой группы будет
выглядеть как квартет с соотношением интенсивностей линий 1:3:3:1.
25
26.
2627.
2728.
1. Nuclei must be chemical shift nonequivalent to show obvious coupling to eachother. Thus the protons of CH2Cl2, Si(CH3)4, Cl-CH2-CH2-Cl, H2C=CH2 and benzene
are all singlets. Equivalent protons are still coupled to each other, but the spectra
do not show it. There are important exceptions to this rule: the coupling between
shift equivalent but magnetically inequivalent nuclei can have profound effects on
NMR spectra 2. J coupling is mutual, i.e. JAB = JBA always. Thus there is never just
one nucleus which shows J splitting - there must be two, and they must have the
same splitting constant J. However, both nuclei need not be protons - fluorine (19F)
and phosphorus (31P) are two other common nuclei that have spin ½ and 100%
abundance, so they will couple to all nearby protons (the other 100% spin 1/2 nuclei
are 89Y,103Rh and 169Tm). If these nuclei are present in a molecule, there are likely to
be splittings which are present in only one proton multiplet (i.e. not shared by two
multiplets).
28
29.
И еще немногоо химсдвигах и КССВ
J-прямая константа через одну связь, 2J-геминальная константа
через две связи, 3J - вицинальная константа через три связи, 4J
и т. д.-дальние константы.
1
Прямая константа 1J. О прямой протон-протонной КССВ можно
говорить лишь в случае молекулы водорода. Эту константу нельзя
наблюдать экспериментально из-за совпадения химических
сдвигов, двух протонов в молекуле, но ее можно вычислить,
определив КССВ в молекуле HD. Константы взаимддействия
пропорциональны гиромагнитным отношениям ядер:
29
30.
Прямые КССВ 1JC13-H являются причиной расщепления сигналов ЯМР 13Сметиновых, метиленовых и метильных групп в дублеты, триплеты и
квартеты. Константы 1JC13-H в наибольшей степени зависят от
гибридизации атома углерода (возрастают с увеличением s-характера
связи), а также от числа и электроотрицательности заместителей при
атоме углерода (возрастают с ростом электроотрицательности
заместителей и их числа)
30
31.
Protons two (2J, geminal) or three bonds (3J, vicinal) apart are usuallycoupled to each other, more remote protons (4J, 5J) may be if geometry is
right, or if π-systems (multiple bonds) intervene. Long range couplings
(4J or greater) are usually small, typically <0.5 Hz, but up to 3 Hz in some
cases where there are intervening bonds.
31
32.
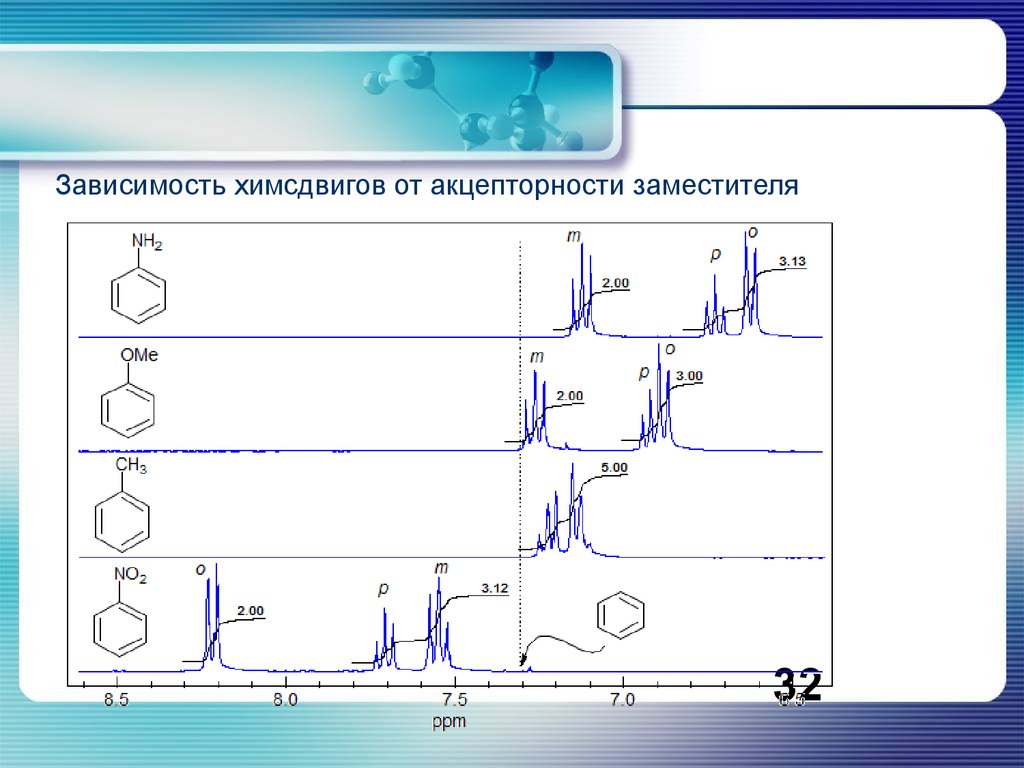
Зависимость химсдвигов от акцепторности заместителя32
33.
3334.
3435.
При низких температурах удаетсяразличить сигналы аксиальных и
экваториальных протонов, причем
сигнал аксиального протона
расположен на 0.5 м.д. правее
(более экранирован), чем сигнал
экваториального протона.
35
36.
3637.
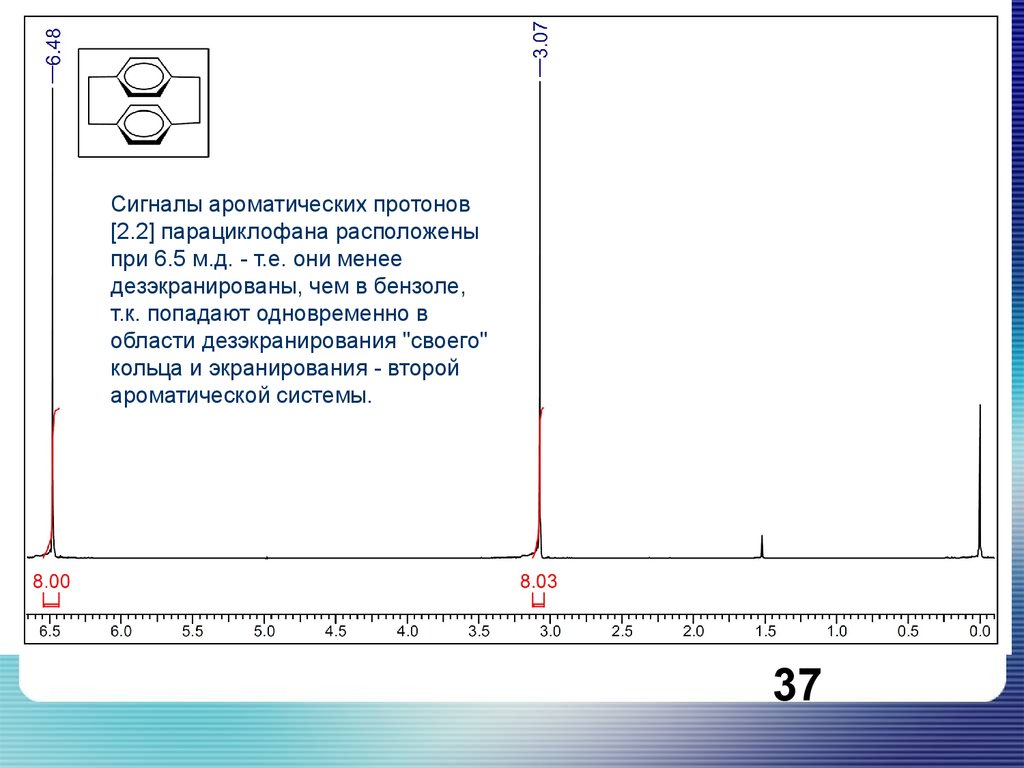
Сигналы ароматических протонов[2.2] парациклофана расположены
при 6.5 м.д. - т.е. они менее
дезэкранированы, чем в бензоле,
т.к. попадают одновременно в
области дезэкранирования "своего"
кольца и экранирования - второй
ароматической системы.
37
38.
3839.
В алкинах сигналы протонов C≡CH расположены при ~3 м.д. В то же время в алкенахсигналы винильных протонов -CH= расположены в области 5-6 м.д., т.е. протоны,
связанные с менее электроотрицательным атомом (sp2-гибридизованным), оказываются
более дезэкранированными. Для объяснения этого факта также привлекается понятие об
областях экранирования. В алкинах "в конусе" С≡С находится область экранирования:
в алкенах и карбонильных соединениях (и вообще, в случае sp2-гибридизованного атома
С) в плоскости sp2-гибридизованной системы расположена область
дезэкранирования:
39
40.
4041.
4142.
4243.
4344.
4445.
4546.
4647.
Sw ater
3.3319
NH
2.9779
3.2250
3.2136
3.1959
3.1845
3.6496
3.7410
3.0070
2.4900
DMSO-d6
3.7067
3.6839
CN O
3.0807
N
3.1098
NC
1.4560
1.3034
Me Me
w ater
3.7
3.5
3.0
7.25
7.20
Chemical Shif t (ppm)
7.15
7.10
2.98
13.5
13.0
12.5
12.0
11.5
11.0
3.1959
3.1845
3.1098
3.0070
3.7067
3.6839
7.2935
7.1627
7.1440
7.1440
2.00
1.04
14.0
3.1
3.7410
3.6496
7.30
3.2
1.00
7.3091
2.98
7.35
3.4
3.3
Chemical Shift (ppm)
7.2427
7.2551
7.1627
7.2748
7.2935
7.3091
13.8111
3.6
1.03
7.2748
3.8
1.95
3.3319
1.99
10.5
10.0
9.5
9.0
8.5
8.0
7.5
7.0
6.5
Chemical Shif t (ppm)
47
1.99
6.0
5.5
5.0
4.5
4.0
3.5
1.95
3.0
2.78
2.5
2.0
1.5
1.0
0.5
48.
DMSO-d6NC
39.5000
Me Me
S
NH
N
128.3775
128.1832
CN O
192
184
176
168
152
144
136
128
120
112
104
Chemical Shif t (ppm)
96
88
80
72
64
54.4332
58.2557
38.3184
39.0 38.5
Chemical Shift (ppm)
56
23.3138
20.2843
127.4734
115.9464
114.8718
160
52.0
38.3184
52.5
52.0064
53.5
53.0
Chemical Shif t (ppm)
51.9390
54.0
60.3771
54.5
51.9390
128.3775
128.1832
114
136.2882
116
54.4332
52.0064
200
118
164.5130
124
122
120
Chemical Shif t (ppm)
114.8718
115.9464
127.4734
126
200.5810
128
208
39.5000
DMSO-d6
48
48
40
32
24
16
49.
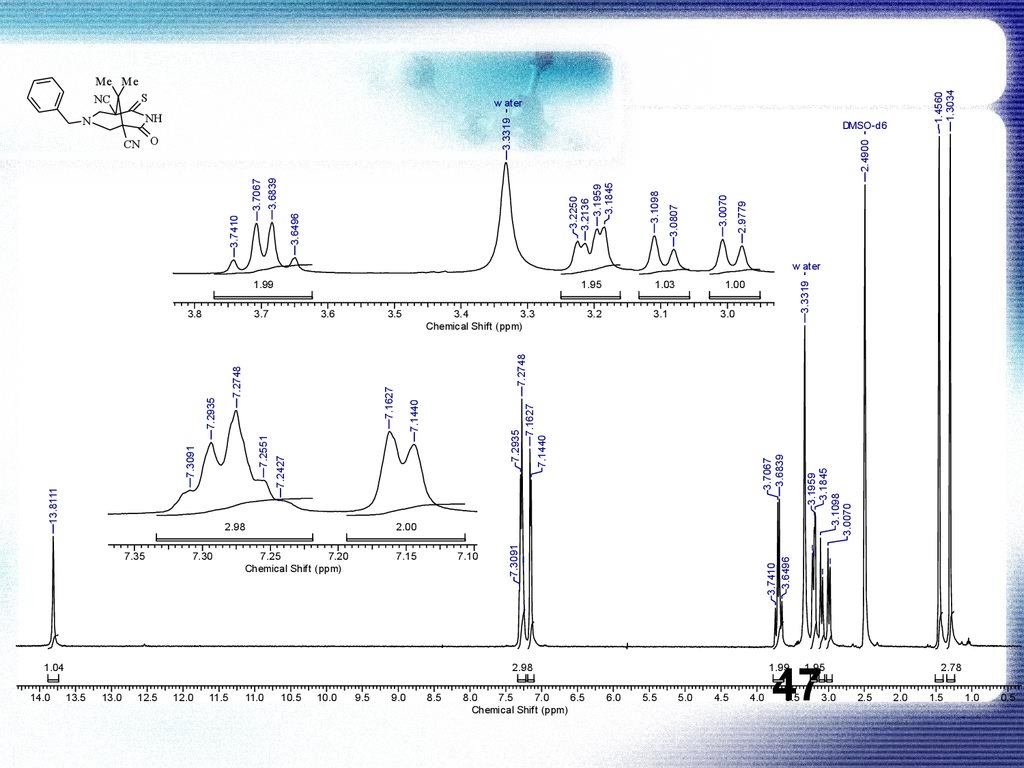
Спектр ЯМР 1Н (400 МГц, ДМСО-d6), , м. д. (J, Гц):1.30 (3Н, с, Me); 1.46 (3Н, с, Me); 3.00 (1H, д, 2J = 11.6,
СН2NCH2); 3.10 (1H, д, 2J = 11.6, СН2NCH2); 3.19-3.23
(2H, м, СН2NCH2); 3.70 (2Н, АВ-к, 2J = 13.7, NCH2Ph);
7.15 (2Н, д, 3J = 7.5, Ph); 7.24-7.31 (3H, м, Ph); 13.81
(1H, c, NH).
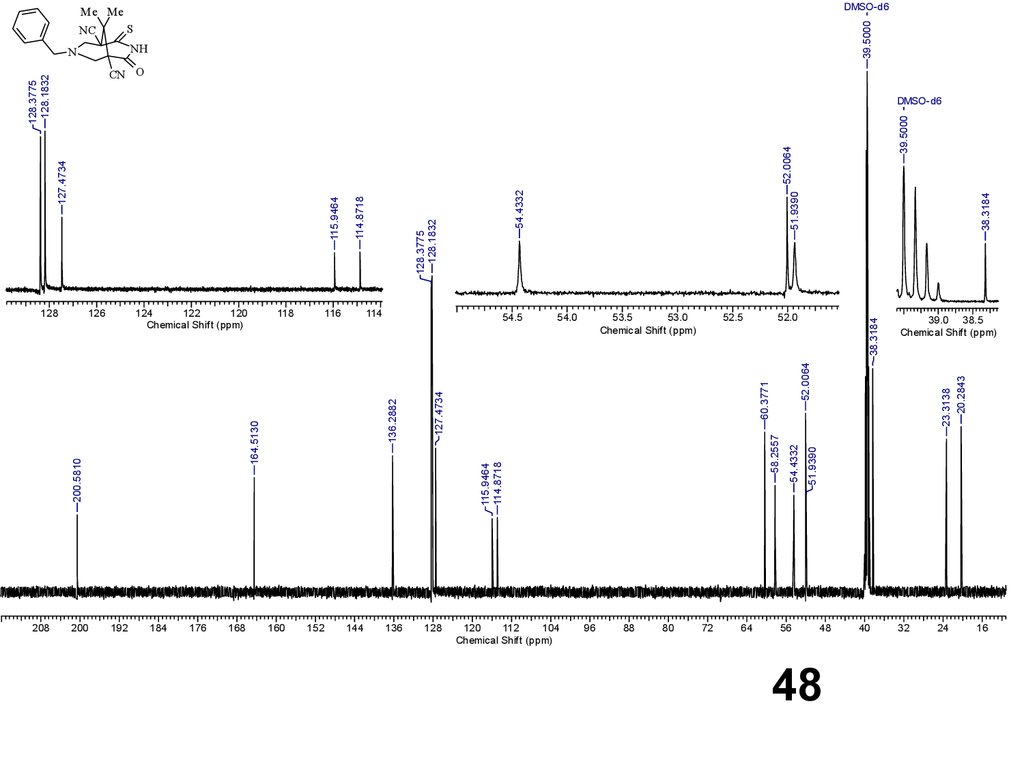
Спектр ЯМР 13C (126 МГц, ДМСО-d6), , м. д.: 20.3
(Me); 23.3 (Me); 38.3 (C-9); 51.9 (C-1 или С-5); 52.0 (С6 или С-8); 54.4 (С-5 или С-1); 58.3 (С-8 или С-6); 60.4
(NCH2Ph); 114.9 (C≡N); 115.9 (C≡N); 127.5 (C-Ar);
128.2 (C-Ar); 128.4 (C-Ar); 136.3 (C-Ar); 164.5 (C=O);
200.6 (C=S).
49
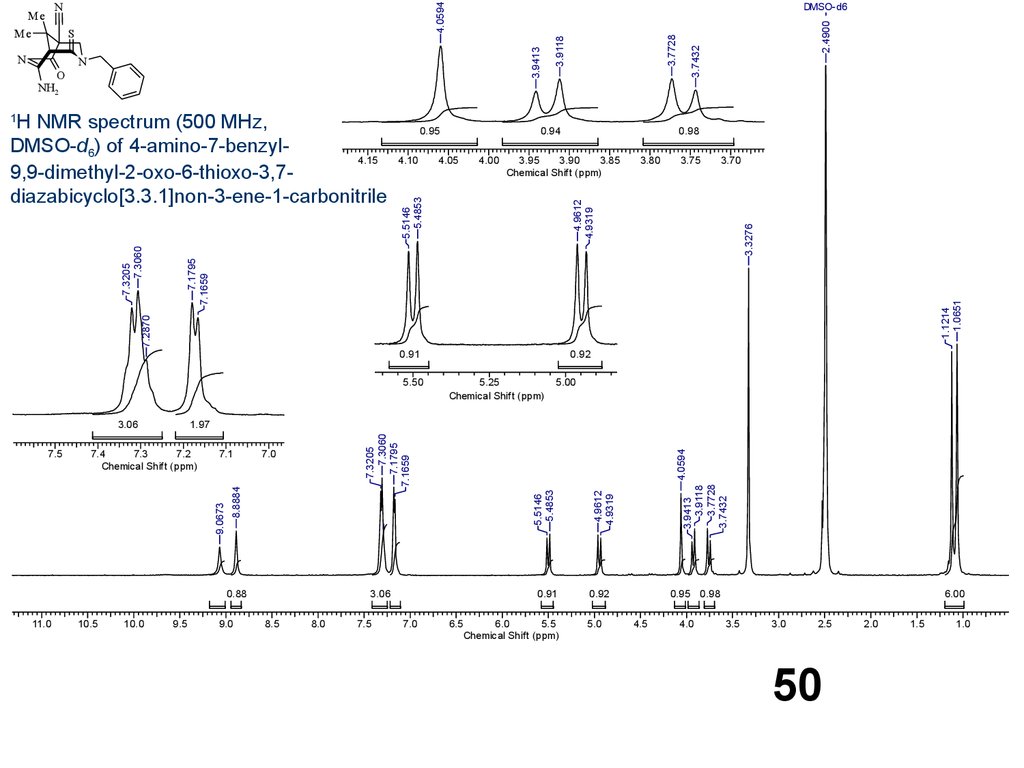
50.
2.49003.7432
N
O
3.7728
N
3.9118
S
3.9413
Me
DMSO-d6
4.0594
N
Me
NH2
Н NMR spectrum (500 MHz,
DMSO-d6) of 4-amino-7-benzyl4.15
9,9-dimethyl-2-oxo-6-thioxo-3,7diazabicyclo[3.3.1]non-3-ene-1-carbonitrile
4.00
3.95
3.90
3.85
Chemical Shift (ppm)
0.91
0.92
3.80
3.75
3.70
1.1214
1.0651
7.1795
7.1659
4.9612
4.9319
4.05
0.98
5.5146
5.4853
4.10
0.94
3.3276
0.95
7.2870
7.3205
7.3060
1
5.50
11.0
10.5
10.0
9.5
9.0
4.0594
5.5146
5.4853
0.88
3.06
8.5
8.0
7.5
0.91
7.0
6.5
6.0
5.5
Chemical Shift (ppm)
3.9413
3.9118
3.7728
3.7432
7.0
4.9612
4.9319
7.1
7.3205
7.3060
7.1795
7.1659
7.4
7.3
7.2
Chemical Shift (ppm)
9.0673
7.5
5.00
1.97
8.8884
3.06
5.25
Chemical Shift (ppm)
0.92
5.0
0.95 0.98
4.5
4.0
6.00
3.5
3.0
2.5
50
2.0
1.5
1.0
51.
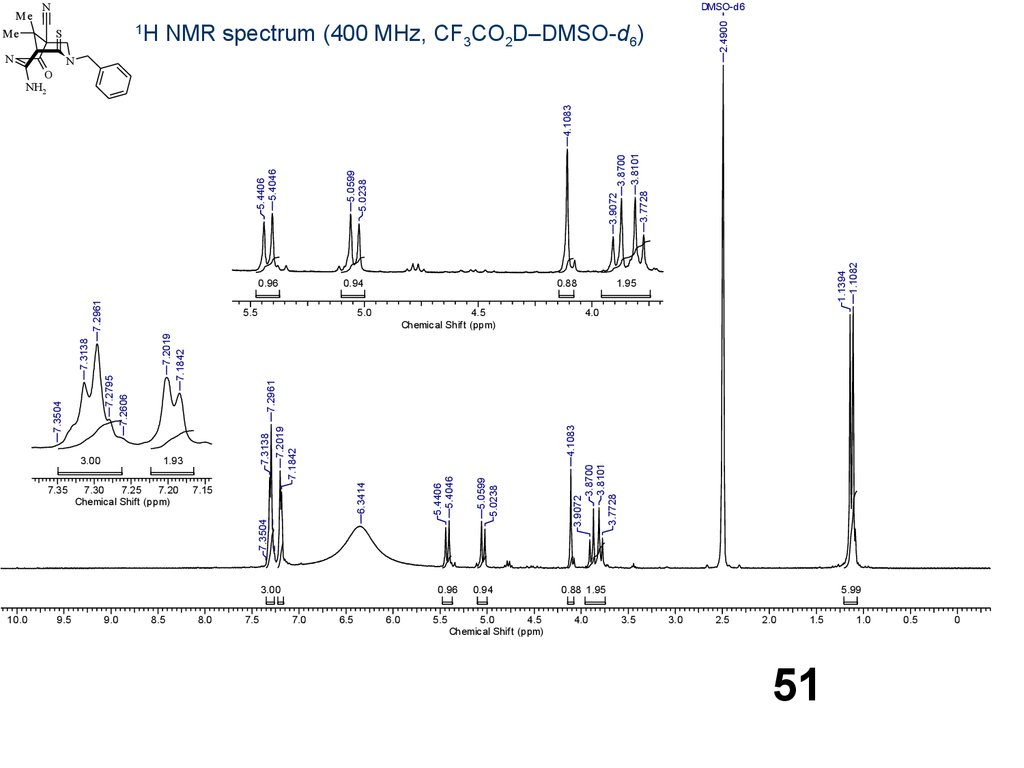
NDMSO-d6
Me
1
S
N
Н NMR spectrum (400 MHz, СF3CO2D–DMSO-d6)
2.4900
Me
N
O
9.5
9.0
8.5
3.7728
3.8700
3.8101
5.0599
5.0238
3.9072
5.4406
5.4046
4.0
8.0
7.5
4.1083
0.96
7.0
6.5
6.0
5.5
0.94
5.0
4.5
Chemical Shif t (ppm)
3.9072
3.8700
3.8101
3.7728
5.0599
5.0238
5.4406
5.4046
6.3414
7.3504
7.15
3.00
10.0
1.95
7.2961
4.5
Chemical Shift (ppm)
7.3138
7.2019
7.1842
1.93
7.30
7.25
7.20
Chemical Shift (ppm)
0.88
5.0
7.2019
7.1842
7.2795
7.2606
7.3138
7.3504
0.94
5.5
3.00
7.35
0.96
1.1394
1.1082
7.2961
4.1083
NH2
0.88 1.95
4.0
5.99
3.5
3.0
2.5
2.0
1.5
51
1.0
0.5
0
52.
3.958010.0
9.4478
0.93
0.95
9.5
w ater
3.3683
9.7970
2.4900
DMSO-d6
2.00
9.0
8.5
8.0
7.5
7.0
6.5
6.0
Chemical Shif t (ppm)
5.5
5.0
4.5
4.0
3.5
52
3.0
2.5
53.
NH2(E)
O
DMSO-d6
2.4900
Me
3.7337
CN
H
N
S
+
7.3226
7.3008
NH2
N
H
0.80
0.25 0.76
10.0
9.5
Chemical Shift (ppm)
7.3496
2.02
0.90 0.76 0.28 0.25
9.0
7.4
8.5
7.3
2.01
7.2
7.1
Chemical Shif t (ppm)
7.0
6.9
7.3226
7.3008
6.9634
6.9406
10.5
8.2279
8.3743
8.3410
8.6359
8.6047
9.0480
10.4371
10.4059
(Z)
8.5103
S
8.9587
O
Me
6.9634
6.9406
CN
13.5
0.80
13.0
12.5
12.0
11.5
11.0
10.5
0.76 0.90 0.28
10.0
9.5
9.0
3.3226
7.3496
9.0480
10.4371
10.4059
13.7176
13.6844
0.26
14.0
8.9587
8.6359
8.6047
8.5103
8.3743
8.3410
8.2279
w ater
2.02
8.5
8.0
7.5
Chemical Shift (ppm)
2.01
7.0
3.04
6.5
6.0
5.5
5.0
4.5
53
4.0
3.5
3.0
2.5
2.0
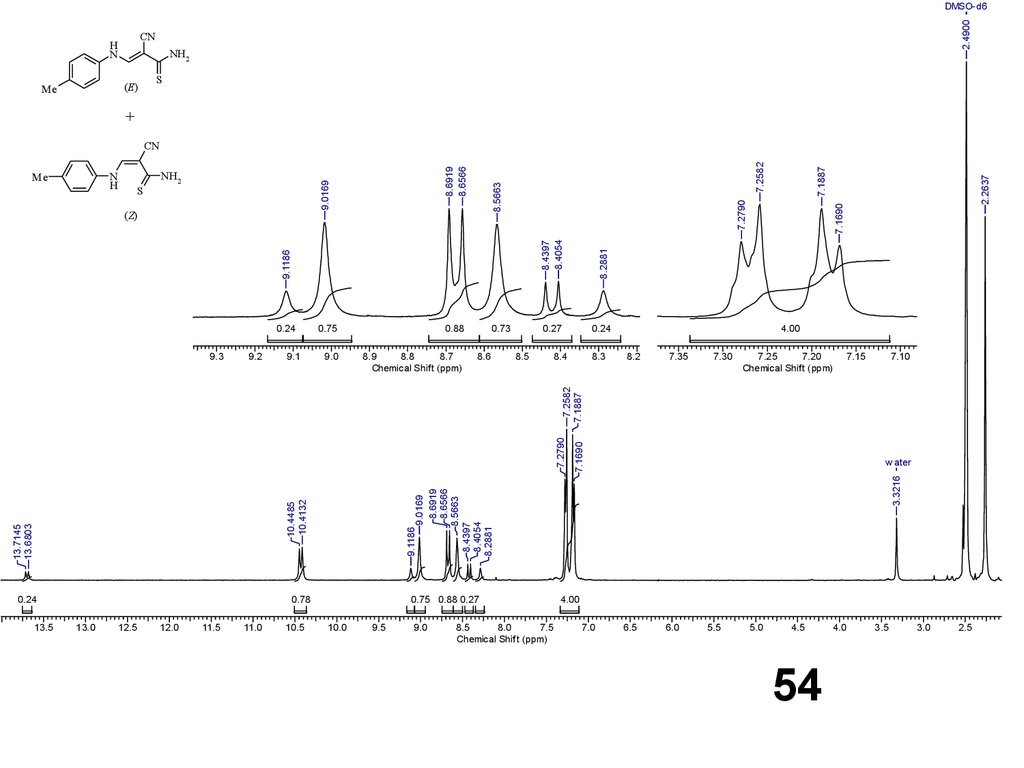
54.
CNH
N
NH2
(E)
Me
2.4900
DMSO-d6
S
+
0.24
9.2
9.1
0.88
9.0
8.9
8.8
8.7
Chemical Shift (ppm)
8.6
0.27
8.5
7.1690
2.2637
7.1887
7.2582
0.73
0.24
8.4
8.3
4.00
8.2
7.35
7.30
7.25
7.20
Chemical Shif t (ppm)
7.15
7.10
13.5
12.5
12.0
11.5
11.0
10.5
3.3216
9.0169
8.6919
8.6566
8.5663
8.4397
8.4054
8.2881
0.78
13.0
w ater
9.1186
10.4485
10.4132
13.7145
13.6803
0.24
7.1690
7.2790
7.2582
7.1887
9.3
0.75
8.2881
9.1186
8.4397
8.4054
(Z)
7.2790
S
8.5663
NH2
9.0169
N
H
Me
8.6919
8.6566
CN
0.75 0.88 0.27
10.0
9.5
9.0
8.5
8.0
7.5
Chemical Shif t (ppm)
4.00
7.0
6.5
6.0
5.5
5.0
4.5
54
4.0
3.5
3.0
2.5
55.
5556.
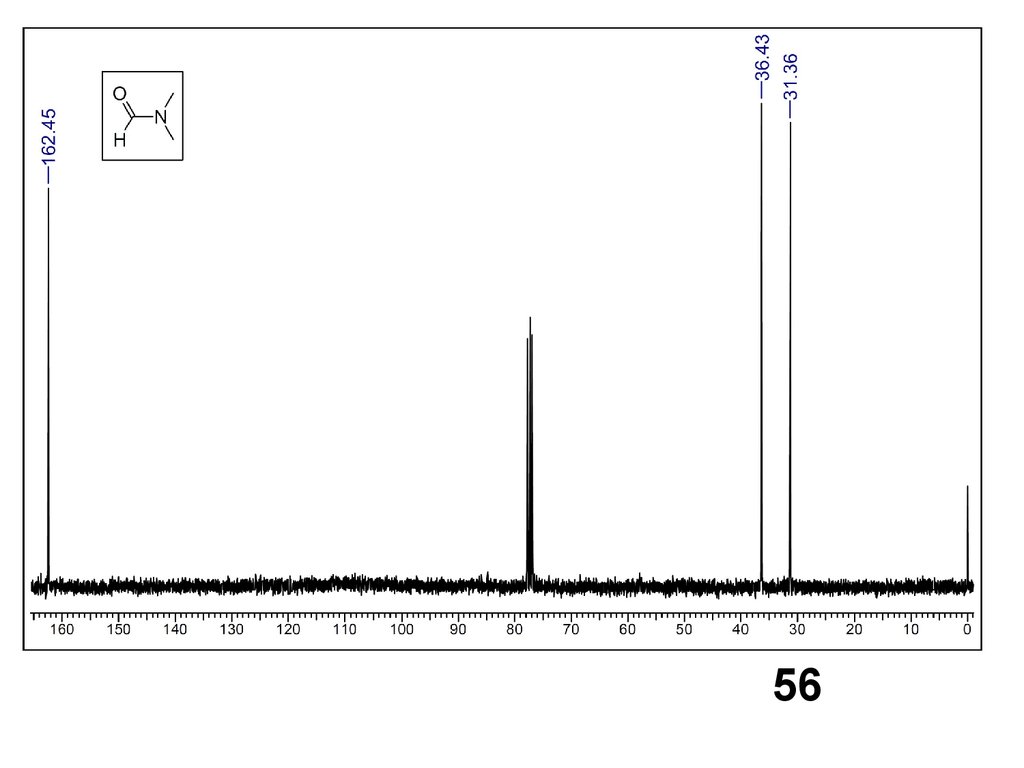
5657.
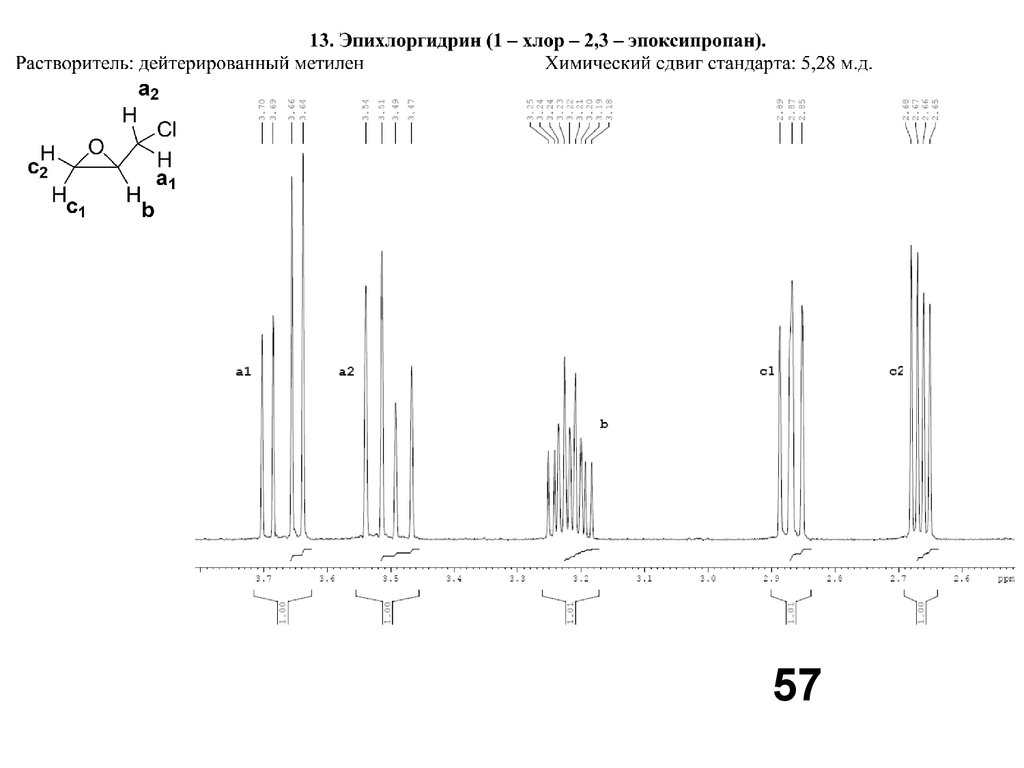
5758.
5859.
Thanks for yourpatience and attention
59