


































")

")











 medicine
medicineSimilar presentations:
")
")



")




Гемобластозы. Классификация по FAB
1. АО «Медицинский Университет Астана» Кафедра внутренних болезней интернатуры Гемобластозы
Астана 20182.
ОпределениеГемобластозы (лат. haemoblastosis; др.-греч. αἷμα «кровь» + βλαστός росток,
зародыш + -osis) — опухолевые (неопластические) заболевания кроветворной и
лимфатической ткани. Они делятся на две большие группы: лейкозы (системные
опухолевые заболевания кроветворной ткани) и лимфомы (регионарные
опухолевые заболевания кроветворной или лимфатической ткани).
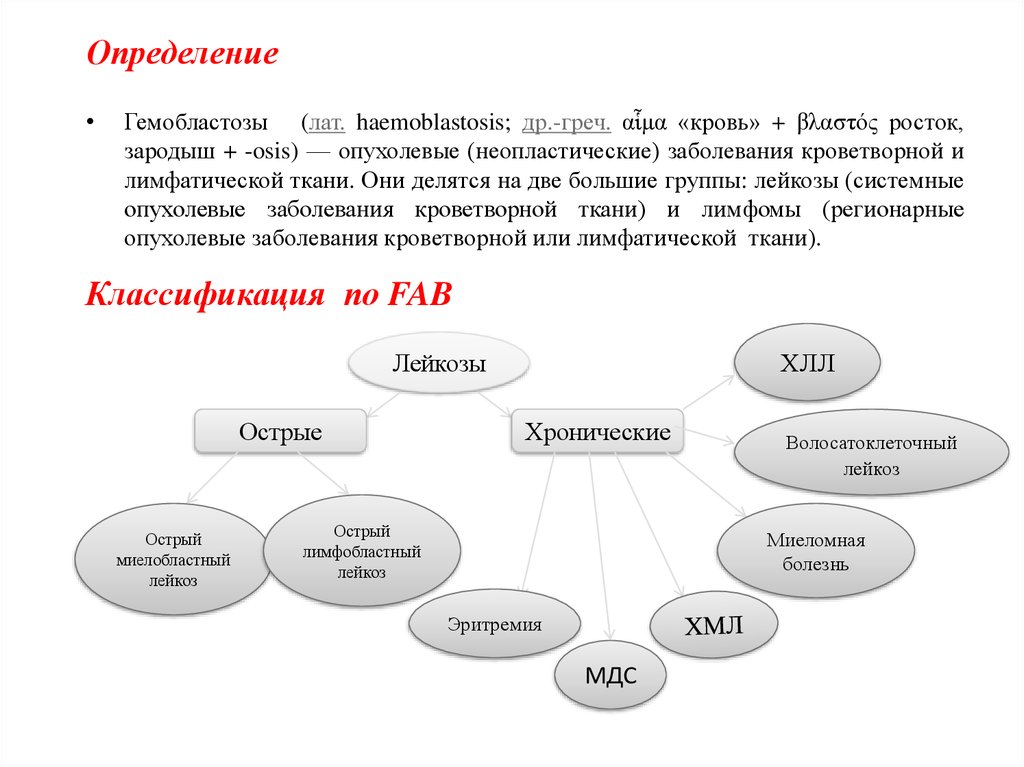
Классификация по FAB
Лейкозы
Острые
Острый
миелобластный
лейкоз
ХЛЛ
Хронические
Острый
лимфобластный
лейкоз
Волосатоклеточный
лейкоз
Миеломная
болезнь
Эритремия
МДС
3.
Острый лимфобластный лейкозОпределение
Острые лимфобластные лейкозы представляют собой гетерогенную группу
опухолевых заболеваний системы крови, характеризующуюся неконтролируемой
пролиферацией в костном мозге, периферической крови и других органах незрелых
лимфоидных клеток.
Эпидемиология
Скорригированная по возрасту заболеваемость ОЛЛ составляет 1,6 на 100 000
населения в год. ОЛЛ является наиболее частым видом острого лейкоза у детей,
составляя 70-80%. С возрастом его частота снижается. В возрасте старше 45 лет
диагностируется около 23% случаев ОЛЛ.
Стандартное возрастное распределение частоты заболеваний гемобластозами у детей.
1 - ОЛЛ; 2 - ОНЛЛ; 3 - ХМЛ; 4 - ЛГ; 5 – НХЛ.
4. Этиология
Ионизирующая радиация
Химические вещества
Вирусная инфекция
Генетические и наследственные факторы, хромосомные аномалии (трисомия
21, анемия Фанкони, синдром Клайнфельтера (47XXY), наследственная
нейтропения, наследственная телеангиэктазия и т.п. )
5. Патогенез
Бензол, пестициды,цитостатики и др
химические вещества
Ионизирующая
радиация
онковирусы
Наследственные факторы,
хромосомные аномалии
протоонкогены
онкогены
Соматическая мутация клеток-предшественниц
кроветворной или лимфоидной ткани
Нарушение пролиферации клеток с сохранением дифференцировки
(доброкачественный опухолевый рост)
Вторичные мутации, связанные с
нестабильностью клеточного генома
Нарушение
апоптоза
Нарушение
системы
иммунитета
Нарушение пролиферации и дифференциации (злокачественный
опухолевый рост)
Пролиферация в
костном мозге,
накопление массы
опухолевых клеток
Лейкемическая инфильтрация органов
и тканей, лимфоидных органов
Вытеснение нормального
кроветворения
Аутоиммунные
осложнения
Инфекционновоспалительные
осложнения
Клинико-гематологические проявления лейкоза
А.Н.Окороков. Руководство. Диагностика болезней внутренних органов. Том 4. Москва. Медицинская литература 2007 г.
6. Классификация острого лимфобластного лейкоза по ИФТ
В-клеточный ОЛЛТ-клеточный ОЛЛ
В - І (pro В)
T - І (pro T)
В – II (Common B)
T – II (pre T)
B – III (pre-В)
T– III (contrical T)
B – IV (mature B)
T – IV (mature T)
FAB-классификация
ОЛЛ разделяется на три класса:
L1 - с малыми размерами бластов;
L2 – с крупным размерами бластов;
L3 – с бластными клетками типа клеток при лимфоме Беркитта.
7. Клиническая картина
I. Гиперпластический синдром :Увеличение лимфатических узлов (периферических и
нередко в средостении со сдавленней органов, в нем
расположенных — пищевода, верхней полой вены с
развитием синдрома верхней полой вены и др);
Увеличение печени и селезенки;
Большая частота оссалгии (они обусловлены эрозиями
костей или вовлечением в лейкозный инфильтративный
процесс надкостницы);
Достаточно частое вовлечение в патологический процесс
центральной нервной системы (нейролейкемия наблюдается
у 25-30% больных);
II. Интоксикационный синдром:
Слабость
Потливость
Головная боль
Головокружение
Отсутствие аппетита
Снижение массы тела
Атрофия мускулатуры
8.
III. Иммунодефицитный синдром:Меньшая частота язвенно-некротических поражений кожи
и слизистых оболочек, реже встречаются язвеннонекротический стоматит, гиперплазия десен и миндалин;
поражение кожи чаще ассоциируется с пре-В-клеточным
вариантом острого лимфолейкоза;
Наличие у 80% больных гипертермического синдрома, не
связанного
с
присоединением
инфекционновоспалительного процесса (лихорадка обусловлена
влиянием
эндогенного
пирогена,
продуцируемого
лимфобластами);
IV. Анемический синдром:
Утомляемость
Снижение работоспособности
Шум в ушах
Сердцебиение
Бледность кожных покровов
V. Геморрагический синдром:
Наличие геморрагического синдрома (у 50% больных), но
выраженность геморрагических явлений значительно
меньшая, чем при других вариантах острого лейкоза
(например, при остром промиелоцитарном лейкозе);
9. Диагностика
ГемограммаОбщее количество лейкоцитов ниже 5 *109/л
(у 30 % больных)
Общее количество лейкоцитов превышает 50100 *109/л (у 50 % больных)
При
значительном
лейкоцитозе
в
периферической
крови
одновременно
выявляются в большом количестве и
лимфобласты, при нормо- и алейкемических
вариантах
бласты
практически
не
определяются. У больных с Т-клеточным
вариантом острого лимфобластного лейкоза
количество лейкоцитов обычно значительно
увеличено по сравнению с В-клеточными
вариантами. Часто отмечается нейтропения.
Анемия и тромбоцитопения характерны для
всех больных, у 40-50% больных анемия
тяжелой степени (уровень гемоглобина
падает до величин ниже 50 г/л); количество
тромбоцитов обычно снижается до уровня 50
х109/л и ниже.
Пример:
Эритроциты – 2,4*10^12/л
Гемоглобин – 60 г/л
Цветовой показатель – 0,75
Ретикулоциты – 0,1%
MCV – 84
МСН – 30
МСНС – 40 г/л
Тромбоциты – 72 х109/л
Лейкоциты - 87 *109/л
Бластные клетки - 65%
Эозинофилы – 0 %
Базофилы – 0
Нейтрофилы – 10%
Лимфоциты – 20%
Моноциты – 5%
СОЭ –25 мм/ч
Заключение: бластемия, гиперлейкоцитоз,
нормохромная анемия тяжелой степени,
тромбоцитопения, ускорение СОЭ
10. Миелограмма
В костном мозге при остром лейкозе
выраженная инфильтрация бластными
клетками более 20%
Гипреплазия костного мозга
Редуцирование нормального гемопоэза:
эритро- и тромбоцитопоэзом.
Лейкемический провал
11.
Цитохимическое исследованиеДля
лимфобластов
характерны
положительная реакция на гликоген (он
расположен в цитоплазме в виде глыбок)
и
отрицательные
реакции
на
миелопероксидазу и липиды.
Цитогенетическое исследование
Лабораторный анализ, при котором
образец крови или костного мозга
исследуют под микроскопом с целью
обнаружить определенные изменения в
хромосомах лимфоцитов.
12. Иммунологическое фенотипирование
Этот метод позволяет уверенно различать В- и Т-лимфобласты и выделять
фенотипы В- и Т-лимфобластов.
Иммунологические маркеры
B-клеточные
ОЛЛ
HLA-DR
TdT
CD 10
CD 19
CD 20
CD 22
Cyt Ig
Ранний В
+
+
-
+
-
-
-
Общий
+
+
+
+
+/-
-
-
Пре-В
+
+
+
+
+/-
+
-
В
+
-
+/-
+
+
-
+
Т-ОЛЛ
HLA-DR
TdT
CD 10
CD 1a
CD 2
S CD 3
CD 7
Ранний Т
+/-
+
+/-
-
-
-/Cyt +
+
Промежуточный
-
+
+/-
+
+
+/Cyt -
+
Зрелый
-
+/-
-
-
+
+/Cyt -
+
13. Группы риска
Группа стандартного риска:1.
Исходное число лейкоцитов ниже 30х10^9/л для ОЛЛ из В-предшественников
2.
Исходное число лейкоцитов ниже 100х10^9/л для ОЛЛ из Т-предшественников
3.
Иммунофенотипический вариант : общий и пре-В ОЛЛ, тимический и зрелый Т-ОЛЛ
4.
ЛДГ ниже 2-х норм
5.
Отсутствие транслокации t(4;11)
6.
Число бластных клеток в костном мозге на 8 день лечения (после предфазы ГКС)
равно или менее 25%
7.
Ремиссия на 36 день лечения (на момент завершения первой фазы индукции)
Группа высокого риска:
1.
Исходное число лейкоцитов выше 30х10^9/л для ОЛЛ из В-предшественников
2.
Исходное число лейкоцитов выше 100х10^9/л для ОЛЛ из Т-предшественников
3.
Иммунофенотипический вариант : ранний пре-В ОЛЛ, ранний Т-ОЛЛ
4.
ЛДГ выше 2-х норм
5.
Наличие транслокации t(4;11)
6.
Число бластных клеток в костном мозге на 8 день лечения (после предфазы ГКС)
равно или более 25%
7.
Отсутствие ремиссия на 36 день лечения (на момент завершения первой фазы
индукции)
14. Лечение
Протокол ALL-2013 KzПредфаза:
• 1-6 дни - Дексаметазон – 10 мг/м 2 в/в, Циклофосфамид 300 мг/м 2 в/в (3 ч)
• 6 день -Ритуксимаб 375 мг/м 2 в/в
Индукция I фаза
• 7-16 дни - Дексаметазон – 10 мг/м 2 в/в
• 7 день - Даунорубицин 60 мг/м 2 R(24 часа ИЛИ 2 часа), Винкристин 2 мг в/в
• 14 день - Винкристин 2 мг в/в
• 21 день - Пункция костного мозга
Индукция II фаза
• 22 день - Циклофосфамид 1000 мг/м 2 в/в (1 ч)
• 22 день - Ритуксимаб 375 мг/м 2 в/в.
• 24-27, 31-34 дни - Цитарабин 75 мг/м 2 в/в 1 раз в день (1 ч)
• 22-49 дни - Меркаптопурин 50 мг/м 2 внутрь ежедневно
• 36 день - Пункция костного мозга (+МРБ)
Консолидация I
• 50 день - Метотрексат 1 г/м 2 (в течение 24 часов) + лейковорин по схеме
• 50 день - Ритуксимаб 375 мг/м 2 в/в
• 52 день - L-аспарагиназа 2 500 Ед/м 2 в/в
• 52 день - ИЛИ ПЭГ-L-аспарагиназа 1000 ЕД/м 2 в/в
• 55, 58, 61, 64, 67 дни - L-аспарагиназа 5 000 Ед/м 2 в/в (2 ч) или в/м
• 67-87 - Меркаптопурин 50 мг/м 2 внутрь ежедневно
15.
Консолидация II: Реиндукция I88 день - Ритуксимаб 375 мг/м 2 в/в.
88-91, 98-101, 108-111 дни - Дексаметазон 10 мг/м2
88-90 дни - Циклофосфамид 300 мг/м2 в/в (3 ч)
91, 98 дни - Даунорубицин 45 мг/м2 в/в (15 мин) ИЛИ Доксорубицин 30 мг/м2 в/в (15 мин), Винкристин 2 мг
в/в болюсно
92, 95, 98, 101, 104, 107,110 дни - L-аспарагиназа 5 000 Ед/м 2 в/в (2 ч) или в/м
92 день - ИЛИ ПЭГ-L-аспарагиназа 1000 ЕД/м 2 в/в
112-126 дни - Меркаптопурин 50 мг/м 2 внутрь
112, 119 дни - Метотрексат 20 мг/м 2 в/м или в/в
126 день - Пункция костного мозга (+МРБ)
Консолидация III
127 день - Ритуксимаб 375 мг/м 2 в/в.
127 день - Цитарабин 2 г/м 2 в течение 1,5 часов х 2 раза в день
128, 131, 134, 137, 140, 143, 146 дни - L-аспарагиназа 5 000 Ед/м 2 в/в (2 ч) или в/м
128 день - ИЛИ ПЭГ-L-аспарагиназа 1000 ЕД/м 2 в/в
127-130 дни - Дексаметазон 10 мг/м 2
Консолидация IV: Реиндукция II
149 день - Ритуксимаб 375 мг/м 2 в/в.
149-163 дни - Меркаптопурин 50 мг/м 2 внутрь
149 день - Циклофосфамид 1000 мг/м 2 в/в (1 ч)
151-154, 158-161 дни - Цитарабин 75 мг/м 2 в/в 1 раз в день (1 ч)
150, 153, 156,159, 162 дни - L-аспарагиназа 5 000 Ед/м 2 в/в (2 ч) или в/м
150 день - ИЛИ ПЭГ-L-аспарагиназа 1000 ЕД/м 2 в/в
Консолидация V
164 день - Ритуксимаб 375 мг/м 2 в/в.
164 день - Метотрексат 1 г/м 2 (в течение 24 часов) + лейковорин по схеме
165, 168, 171, 174 дни - L-аспарагиназа 5 000 Ед/м 2 в/в (2 ч) или в/м
165 день - ИЛИ - Пегаспаргаза1000 ЕД/м 2 в/в
164-180 дни - Меркаптопурин 50 мг/м 2 внутрь ежедневно
179 дни - Пункция костного мозга (+МРБ)
16.
Поддерживающая терапия со 180 дня (в течение двух лет)• Ритуксимаб 375 мг/м 2 в/в при CD20+ В-ОЛЛ.
• Альтернативой L-аспарагиназе - ПЭГаспаргаза1000 ЕД/м 2 в/в. меркаптопурин 60 мг/м 2
внутрь ежедневно с коррекцией дозы в зависимости от уровня лейкоцитов и тромбоцитов.
• Метотрексат 20 мг/м 2 в/м или в/в 1 раз в неделю.
• Люмбальная пункция (метотрексат 15 мг, цитарабин 30 мг, дексаметазон 4 мг), пункция
костного мозга 1 раз в 3 месяца, всего 8 пункций.
• Профилактические люмбальные пункции (inth. метотрексат 15 мг, Цитарабин 30 мг,
дексаметазон 4 мг) - 0/1, 7, 14, 21, 28, 35/36, 80, 126 дни и далее 1 раз в 3 месяца во время
поддерживающей терапии (всего 8+8=16 пункций).
Трансплантация костного мозга
• Трансплантация стволовых гемопоэтических клеток от родственного донора показана всем
больным в первой полной ремиссии острого лимфобластного лейкоза после завершения им
программы индукции/консолидации, у кого есть HLA-идентичные сиблинги, за исключением
группы стандартного риска (уровень доказательности В) .
• Пациентам, у которых не удается получить полную ремиссию ни на одном этапе лечения,
ТКМ при наличии донора может обсуждаться в качестве терапии «спасения».
• Трансплантация костного мозга проводится согласно показаниям, указанным в протоколе
лечения пациента. Если проведение трансплантации не предусмотрено протоколом, пациент
не является кандидатом для ТКМ.
17. Сопроводительная терапия
Профилактика инфекционных осложнений ( 6 раз в день полоскание полости рта
дезинфицирующими растворами, дезинфекция кожи, уход за зубами)
При наличии очага инфекции и повышения температуры тела >38,0°С :
Антибактериальные средства:
амикацин, порошок для инъекций, 500 мг/2 мл или порошок для приготовления раствора для инъекций,
0,5 г;
амоксициллин/клавулановая кислота, таблетка, покрытая пленочной оболочкой, 1000мг, порошок для
приготовления раствора для внутривенного и внутримышечного введения 1000 мг+500 мг;
ванкомицин, порошок/лиофилизат для приготовления раствора для инфузий 1000 мг;
гентамицин, раствор для инъекций 80мг/2мл 2мл;
имипинем, циластатин порошок для приготовления раствора для инфузий, 500 мг/500 мг;
колистиметат натрия*, лиофилизат для приготовления раствора для инфузий 1 млн ЕД/флакон;
метронидазол таблетка, 250 мг, раствор для инфузий 0,5% 100мл, гель стоматологический 20г;
левофлоксацин, раствор для инфузий 500 мг/100 мл, таблетка 500 мг;
линезолид, раствор для инфузий 2 мг/мл;
меропенем, лиофилизат/порошок для приготовления раствора для инъекций 1,0 г;
цефепим, порошок для приготовления раствора для инъекций 500 мг, 1000 мг;
ципрофлоксацин, раствор для инфузий 200 мг/100 мл, 100 мл, таблетка 500 мг;
18.
ОСТРЫЙ МИЕЛОБЛАСТНЫЙ ЛЕЙКОЗОпределение
Острый миелобластный лейкоз представляет собой опухоль, исходящую из
клетки-предшественницы миелопоэза и состоящую преимущественно из
родоначальных клеток гранулоцитарного ряда - миелобластов.
Эпидемиология
Встречается в любом возрасте, но наиболее часто у подростков (пик в 15-20
лет).
FAB (French-American-British)-классификация
• М0 — с недифференцированными бластными клетками
• М1 — ОМЛ без признаков созревания бластов
• М2 — ОМЛ с признаками созревания бластов
• М3 — острый промиелоцитарный лейкоз
• М4 — острый миеломоноцитарный лейкоз
• М5— острый моноцитарный (монобластный) лейкоз
• М6 — острый эритролейкоз
• М7 — острый мегакариобластный лейкоз
19.
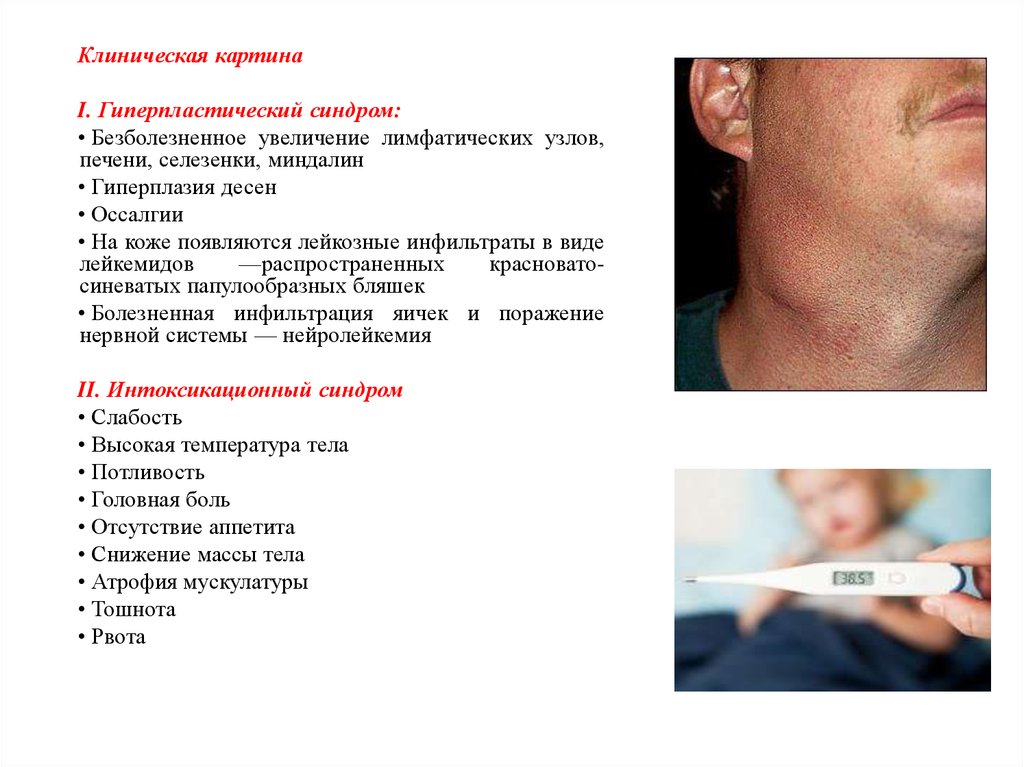
Клиническая картинаI. Гиперпластический синдром:
• Безболезненное увеличение лимфатических узлов,
печени, селезенки, миндалин
• Гиперплазия десен
• Оссалгии
• На коже появляются лейкозные инфильтраты в виде
лейкемидов
—распространенных
красноватосиневатых папулообразных бляшек
• Болезненная инфильтрация яичек и поражение
нервной системы — нейролейкемия
II. Интоксикационный синдром
• Слабость
• Высокая температура тела
• Потливость
• Головная боль
• Отсутствие аппетита
• Снижение массы тела
• Атрофия мускулатуры
• Тошнота
• Рвота
20.
III.Иммунодефицитный синдром:•Развитие различных инфекционно-воспалителых процессов
•Развитие септического состояния
•Неопластическая лихорадка:
-температура тела ежедневно выше, чем 38.7 °С;
- длительность лихорадки более 2 недель;
- отсутствуют клинические проявления инфекции;
-отсутствуют аллергические механизмы лихорадки;
-отсутствует положительная терапевтическая реакция на эмпирическую антибактериальную
терапию;
- посевы крови и мочи на бактерии, грибковую флору, вирусную инфекцию отрицательные;
- быстро исчезает после приема напроксена и других нестероидных противовоспалительных
средств;
-программное лечение острого лейкоза цитостатическими средствами вызывает стойкую
нормализацию температуры тела.
21.
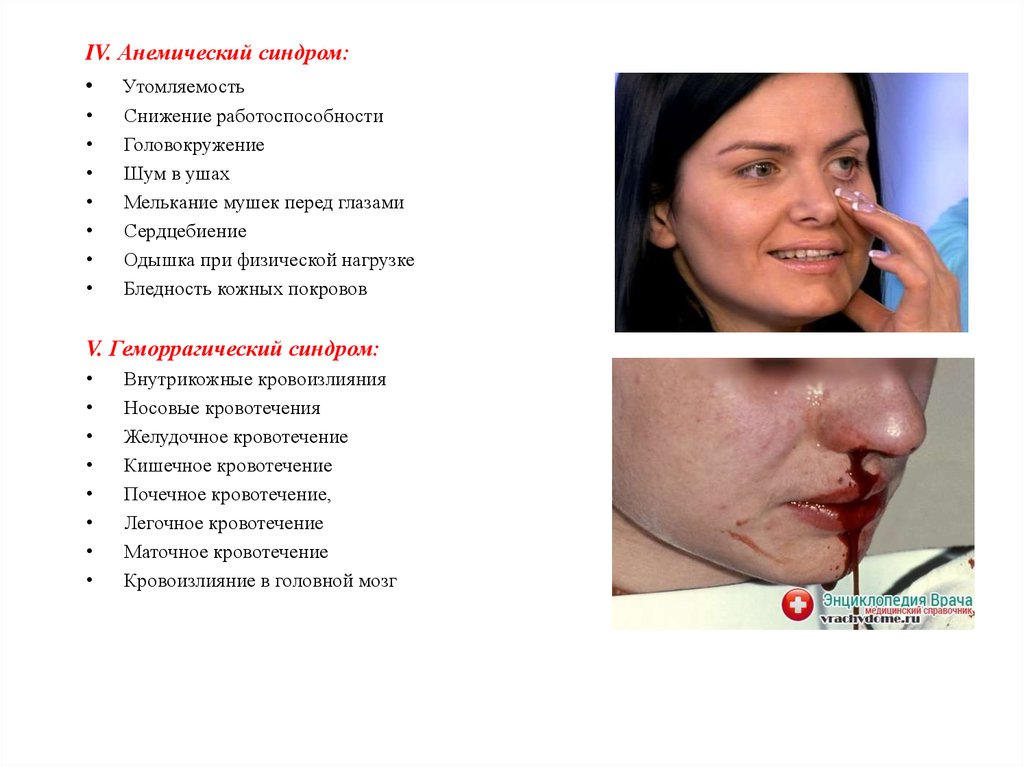
IV. Анемический синдром:• Утомляемость
Снижение работоспособности
Головокружение
Шум в ушах
Мелькание мушек перед глазами
Сердцебиение
Одышка при физической нагрузке
Бледность кожных покровов
V. Геморрагический синдром:
Внутрикожные кровоизлияния
Носовые кровотечения
Желудочное кровотечение
Кишечное кровотечение
Почечное кровотечение,
Легочное кровотечение
Маточное кровотечение
Кровоизлияние в головной мозг
22. Гемограмма:
Анемия (нормохромная, нормоцитарная,
редко макроцитарная)
Ретикулоцитопения
Тромбоцитопения
Изменение общего количества
лейкоцитов(лейкемия,сублейкемия,алейке
мия )
Бластемия
Феномен ≪провала≫ (≪лейкемического
зияния≫)
Исчезновение эозинофилов и базофилов;
Увеличение СОЭ.
Пример:
Нв-66г/л (ж 120-140 г/л; м 130-160 г/л )
Эритроциты-2,2*10¹²/л (ж 3,7-4,7x1012/л ;
м 4-5,1x1012/л )
Цветовой показатель-0,9 (0,85-1,15)
Ретикулоциты-1,5% (0,2-1,2% )
MCV-86 фл (80-100фл)
MCH-27 пг ( 26-34пг)
MCHC-30 % (31-37%)
Лейкоциты-54,5*10 9/л (4-9x109 /л)
Бластные клетки -69%,
Базофилы-0% (0-1%)
Эозинофилы-0% (0-5%)
Нейтрофилы-0,12*109/л
Лимфоциты-20%
Моноциты-1%
Тромбоциты-28,0*109/л (180-400x109/л)
СОЭ-67 мм/час ( ж 2-15 мм/ч; м 1-10 мм/ч )
Заключение: Бластемия, гиперлейкоцитоз,
нейтропения, нормохромная анемия, тяжелой
степени тяжести, тромбоцитопения.
Увеличение СОЭ.
23.
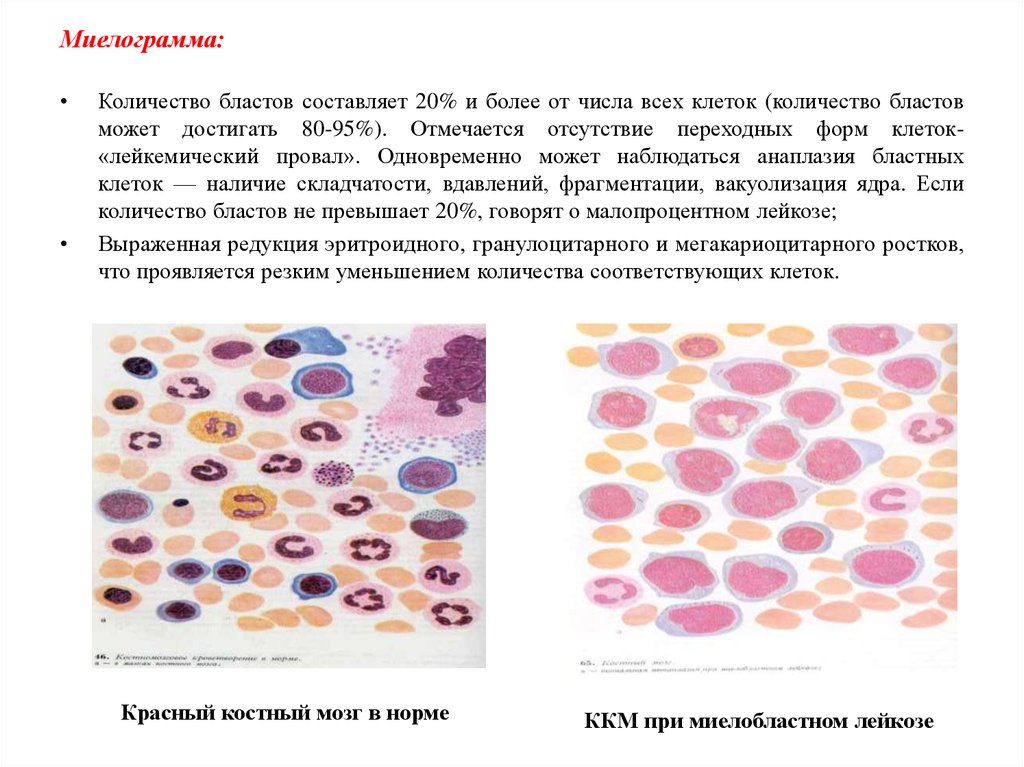
Миелограмма:Количество бластов составляет 20% и более от числа всех клеток (количество бластов
может достигать 80-95%). Отмечается отсутствие переходных форм клеток«лейкемический провал». Одновременно может наблюдаться анаплазия бластных
клеток — наличие складчатости, вдавлений, фрагментации, вакуолизация ядра. Если
количество бластов не превышает 20%, говорят о малопроцентном лейкозе;
Выраженная редукция эритроидного, гранулоцитарного и мегакариоцитарного ростков,
что проявляется резким уменьшением количества соответствующих клеток.
Красный костный мозг в норме
ККМ при миелобластном лейкозе
24.
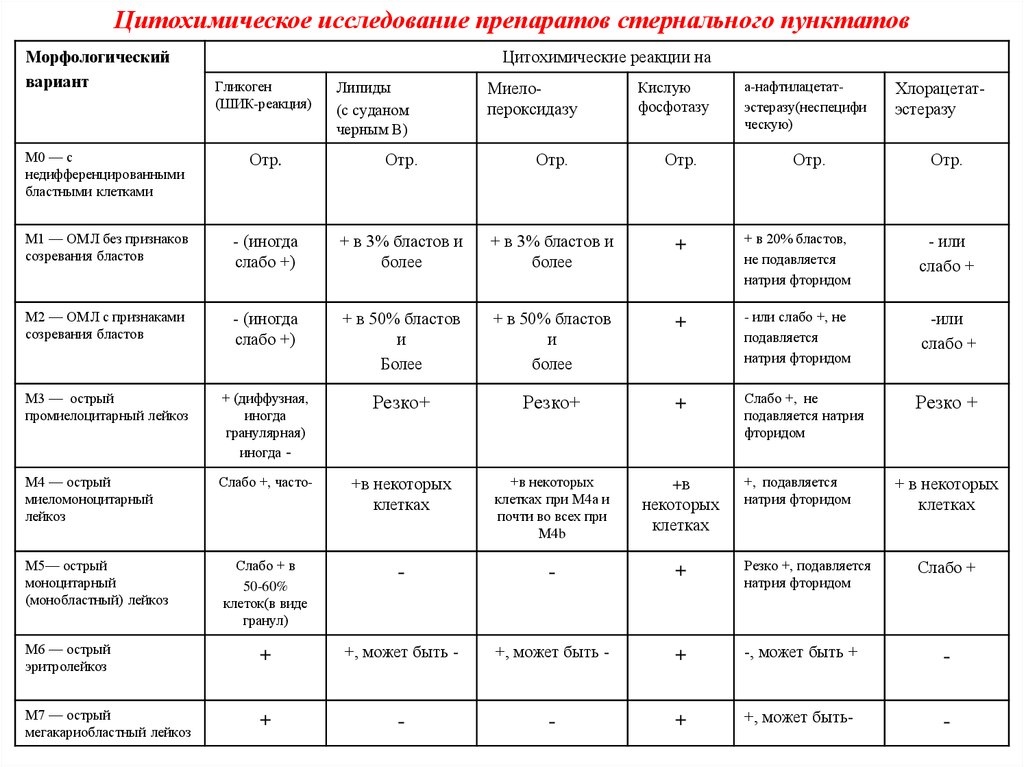
Цитохимическое исследование препаратов стернального пунктатовМорфологический
вариант
Цитохимические реакции на
Гликоген
(ШИК-реакция)
Липиды
(с суданом
черным В)
Миелопероксидазу
Кислую
фосфотазу
а-нафтилацетатэстеразу(неспецифи
ческую)
Хлорацетатэстеразу
М0 — с
недифференцированными
бластными клетками
Отр.
Отр.
Отр.
Отр.
М1 — ОМЛ без признаков
созревания бластов
- (иногда
слабо +)
+ в 3% бластов и
более
+ в 3% бластов и
более
+
+ в 20% бластов,
не подавляется
натрия фторидом
- или
слабо +
М2 — ОМЛ с признаками
созревания бластов
- (иногда
слабо +)
+ в 50% бластов
и
Более
+ в 50% бластов
и
более
+
- или слабо +, не
подавляется
натрия фторидом
-или
слабо +
М3 — острый
промиелоцитарный лейкоз
+ (диффузная,
иногда
гранулярная)
иногда -
Резко+
Резко+
+
Слабо +, не
подавляется натрия
фторидом
М4 — острый
миеломоноцитарный
лейкоз
Слабо +, часто-
+в некоторых
клетках
+в некоторых
клетках при М4а и
почти во всех при
М4b
+в
некоторых
клетках
Слабо + в
50-60%
клеток(в виде
гранул)
-
-
+
Резко +, подавляется
натрия фторидом
М6 — острый
эритролейкоз
+
+, может быть -
+, может быть -
+
-, может быть +
-
М7 — острый
мегакариобластный лейкоз
+
-
-
+
+, может быть-
-
М5— острый
моноцитарный
(монобластный) лейкоз
Отр.
+, подавляется
натрия фторидом
Отр.
Резко +
+ в некоторых
клетках
Слабо +
25. Иммунофенотипирование по FAB
Морфологические типы острого лейкозаХарактерные иммунологические
фенотипы
Острый недифференцируемый лейкоз M0
MPO+, HLA-DR+, CD13+, CD33+, CD34+, CD117+,
CD7-/+, TdT-/+
Острый миелобластный лейкоз без созревание
бластов Μ1
MPO+, HLA-DR+, CD13+, CD33+, CD34+
(слабее, чем при М0), CD117+,CD7-/+, TdT-/+,
CD15-/+
Острый миелобластный лейкоз с созреванием
бластов Μ2
MPO+, HLA-DR+, CD13+, CD33+, CD117+, CD34+, TdT-/+,
CD15+, CD65+/-, CD11b+/-
Острый промиелоцитарный лейкоз (Μ3)
MPO+, HLA-DR-, CD13+, CD33+, CD117-/+, CD15+,
CD2-/+
Острый миеломоноцитарный лейкоз (Μ4)
MPO+, HLA-DR+, CD13+, CD33+, CD117-/+, CD15+,
CD14-/+, СD34-/+, CD38+, CD4-/+, CD11b+/, CD64+
Острый монобластный лейкоз( М5)
MPO+, HLA-DR+, CD13+, CD33+, CD117-/+, CD15+,
CD14-/+, CD36+, CD11b+/-, CD11c, CD4-/+
Острый эритролейкоз (М6)
MPO+, HLA-DR+, CD13+, CD33+, CD117-/+, СD34-/+,
CD38+, CD71+, CD235+ (гликофорин А)
Острый мегакариобластный лейкоз (М7)
MPO+, HLA-DR+, CD13+, CD33+, CD117-/+, СD34-/+,
CD38+, CD61+, HLA-DR+/-, CD41+, CD42b+
Клинический протокол диагностики и лечения ОМЛ у взрослых. Протокол №6 от 9 июля 2015 года
26. Прогностические факторы при остром миелоидном лейкозе
Факторы прогнозаБлагоприятный прогноз
Неблагоприятный прогноз
< 45 лет
De novo (впервые
возникшая)
< 25 000 в мм3
--------
< 2 лет, > 60 лет
Предшествовавшая развитию
лейкемии миелодисплазия
> 100 000 в мм3
----------
Быстрый
Замедленный
+
+
+
-
+
М3,М4
М5,М6,М7
CD14 (-), CD 13 (-)
CD14 (+),CD13(+),CD34 (+)
-
+
-
+
• лимфоидные
CD2, CD19
Бифенотипические (а2 лимфоидных
маркеров)
• цитогенетические
t(15,17)
t(8,21)
ιην(16)
del(16q)
-7, del (7q)
-5, del (5q)
Патология в области 11q23,
Аномалии 3q21 и 3q26
Клинические
• возраст
• особенности развития
лейкемии
• лейкоцитоз
• поражение центральной
нервной системы
• темп снижения цитоза
Морфологические
• палочки Ауэра в властных
клетках
• зозинофилы
• мегалобластоподобные
клетки
• FAB-тип
Поверхностные маркеры
• миелоидные
•. HLA-DR
• TdT
27.
Лечение ОМЛ28.
Индукция7+3
Цитарабин
100мг/м2 2 раза в день каждые 12
часов в/в 30-минутная инфузия
или 100-200мг/м2 непрерывная
круглосуточная инфузия в 1-7
дни курса
Даунорубицин
45 или 60мг/м2 в/в инфузия в
течение 10 минут на 50мл физ.
Раствора в 1-3дни курса, через 2
часа после введения цитарабина.
Двойная индукция
Цитарабин
Даунорубицин
Малые дозы цитозара
Цитарабин
Два курса 7+3, проводимых по
принципу двойной
индукции(второй курс
начинается на 22 или 29день от
начала первого, то есть, на 14
или21 день перерыва)
100мг/м2 в день в виде
постоянной инфузии, 1-7 день
45 или 60мг/м2 в/в инфузия в
течение 10 минут на 50 мл физ.
10мг/м2 2 раза в сутки п/к в
Раствора 1-3 дни курса , через 2
течение 14-28 дней
часа после введения цитарабина.
29.
Консолидация7+3Mito
Цитарабин
100мг/м2 2 раза в день каждые 12
часов в/в 30-минутная инфузия в
1-7 дни курса
Митоксантрон
10мг/м2 в/в инфузия в течение 30
минут на 50мл физ. Раствора в13 день курса, через 2 часа после
введения циторабина
5+2
НА
Цитарабин
100мг/м2 2 раза в день каждые 12
часов п/к или в/в 1-5 дни курса
Даунорубицин
45 или 60мг/м2 в/в инфузия в
течение 10 минут на 50мл физ.
Раствора в 1-2дни курса
Цитарабин
2000-3000мг/м2 2 раза в день
каждые 12 часов 3-х часовая в/в
инфузия в 1,3,5 дни курса
30.
Терапевтическая тактика в ходе проведения индукционного лечения.Выбор курса индукции ремиссии для всех пациентов определяется согласно возрасту:
• 18-45 лет;
• 46-55 лет;
• >55 лет.
Всем пациентам первой возрастной группы (18-45 лет) проводится курс индукции ремиссии по схеме:
7+3 (DNR 60 мг/м2). Если после первой индукции на 14 или 21 день констатирована ремиссия, то
пациент переводиться на этап консолидации-I. Если ремиссии нет, то пациент ведется по программе
«двойной индукции».
Пациентам второй возрастной группы (46-55лет) рекомендовано проводить 2 курса по схеме: 7+3 (DNR
45 мг/м2). Если после первой индукции на 14 или 21 день констатирована ремиссия, то пациент
переводиться на этап консолидации-I. Если ремиссии нет, то пациент также ведется по программе
«двойной индукции».
Пациентам >55 лет рекомендуется проведение 2 курсов индукции ремиссии малыми дозами Цитарабин
(AraC) – в течение 14-28 дней. Исключение: пациентам в возрасте 55-60 лет – при удовлетворительном
соматическом статусе и отсутствиитяжелой сопутствующей патологии возможно проведение курса
ПХТ по схеме: 7+3 (DNR 45 мг/м2).
Всем пациентам до начала ПХТ необходимо проведение иммунофенотипирования, стандартного
цитогенетического исследования костного мозга и молекулярно-цитогенетического исследования
методом FISH.
Расчет доз цитостатических препаратов - цитозин-арабинозида Цитарабин, даунорубицина проводится в соответствии с площадью поверхности тела больного. Дозы цитостатических препаратов
пересчитываются после каждого курса индукции и консолидации, так как многие больные худеют во
время лечения. Снижение доз препаратов во время курса недопустимо ни в одном случае, за
исключением ниже оговоренных ситуаций.
31. Хронический лимфолейкоз
ОпределениеКлональное лимфопролиферативное неопластическое заболевание, характеризующееся
пролиферацией и увеличением в периферической крови количества зрелых лимфоцитов
на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов,
селезенки и других органов.
Эпидемиология
Заболевание регистрируется с частотой 2,7: 100 000 населения.
Мужчины болеют в 1,5—2 раза чаще, чем женщины. В основном болезнь людей
пожилого возраста, средний возраст заболевших составляет 65—69 лет. Более 70%
заболевают в возрасте старше 60 лет, менее 10% — до 40 лет.
Этиология
Этиология на данный момент неизвестна;
В настоящее время не ассоциируется с воздействием ионизирующей радиации,
лекарств, химических веществ;
Наиболее очевидна роль вирусной инфекции (ретровирусов, Эпштейн-Барра);
Генетические аномалий (у 50% больных аномалии в области 12, 13, 14-й хромосом);
Частота развития ХЛЛ в семье, где есть больные с ХЛЛ в 30 раз выше.
32. Классификация
Стадии ХЛЛ по Rai (1975)Стадии ХЛЛ по Binet (1981)
Только лимфоцитоз в крови более 15,0•109/л,
в костном мозге – более 40%; прогноз –
Стадия 0
Стадия А
хороший,
продолжительность
жизни
соответствует популяционной
Лимфоцитоз сочетается с увеличением
лимфоузлов;
Стадия I
Стадия В
прогноз
–
промежуточный,
медиана
выживаемости – 9 лет
Лимфоцитоз + спленомегалия и (или)
увеличение печени, независимо от размеров
Стадия II лимфоузлов;
_
прогноз
–
промежуточный,
медиана
выживаемости – 6 лет
Лимфоцитоз и снижение уровня гемоглобина
менее 110 г/л, независимо от увеличения
Стадия III лимфоузлов, селезенки, печени; прогноз – Стадия С
плохой, медиана выживаемости – менее
3 лет
Лимфоцитоз плюс тромбоцитопения ниже
100•109/л, независимо от анемии и размеров
Стадия IV
лимфоузлов, селезенки и печени; прогноз –
плохой, медиана выживаемости – 1,5 года
_
Содержание
гемоглобина
более
100 г/л, тромбоцитов более 100•109/л,
увеличение лимфатических узлов в 1–
2 областях; медиана выживаемости –
как в популяции.
Содержание
гемоглобинов
и тромбоцитов выше тех же
показателей,
но
лимфоузлы
увеличены в 3 и более областях;
медиана выживаемости – 7 лет.
_
Содержание
гемоглобина
менее
100 г/л, тромбоцитов – менее
100•109/л при любом количестве зон
с увеличенными узлами и независимо
от увеличения селезенки и печени;
медиана выживаемости – 2 года.
_
33. Клиническая картина
I. Гиперпластический, или лимфопролиферативный (связанный с ростомопухоли):
Увеличение периферических и центральных лимфоузлов(эластично – тестоватая
консистенция, безболезненны, не спаяны с кожей и между собой, не изъязвляются и не
нагнаиваются);
Отеки шеи, лица, рук — появляются при сдавлении увеличенными внутригрудными
лимфоузлами верхней полой вены (сосуд, приносящий кровь к сердцу от верхней
половины тела);
Боль и тяжесть в левой верхней части живота (спленомегалия);
Параллельное увеличение печени и селезенки(→ анемия и тромбоцитопения);
Инфильтрация всех отделов и органов ЖКТ(язвы, кровотечение, диспепсия,
мальабсорбция);
Инфильтрация легких(пневмонии, ДН по рестриктивному типу, очень частые
воспалительные процессы, фибринозный или экссудативный плеврит);
Инфильтрация всех отделов нервной системы(менингит, менингоэнцефалит, паралич
ЧМН);
Инфильтрация проводящей системы сердца(миокардиодистрофия, различные нарушения
сердечного ритма и АВ проводимости);
Полиорганная недостаточность.
II. Иммунодефицитный (синдром инфекционных осложнений): Присоединение любых
инфекций (бактерия, вирус) связано с недостаточным образованием нормальных
лейкоцитов обеспечивающих защиту от микроорганизмов. На фоне изменение иммунной
системы организма развитие аутоиммунной гемолитической анемии, тромбоцитопении.
34.
III.Интоксикационный синдром (отравление организма продуктами распадаопухоли):
Выраженная общая слабость;
Утомляемость;
Снижение массы тела;
Потливость;
Повышение температуры тела.
IV.
Анемический синдром:
Слабость, снижение работоспособности;
Головокружение;
Обморочные состояния;
Шум в ушах, мелькание «мушек» перед глазами;
Одышка и сердцебиение при незначительной физической нагрузке;
Колющие боли в грудной клетке.
V. Геморрагический синдром (наличие кровоизлияний и кровотечений):
При хроническом лимфолейкозе обычно слабо выражен.
Возможны: подкожные и подслизистые (например, в полости рта) кровоизлияния;
Десневые, носовые, маточные и другие кровотечения.
35. Миелограмма
Выраженная лимфоидная инфильтрация. Лимфоциты составляют более 30% (иногда
50-60% и даже больше) от общего количества миелокариоцитов.
В III стадии по Rai на фоне лимфоидной инфильтрации угнетение эритроидных
ростков.
IV стадии – угнетение мегакариоцитарного ростка.
Значительное уменьшение количества клеток гранулоцитарного ряда.
В норме
При ХЛЛ
36. Гемограмма:
Пример:
50х109/л
Количество лейкоцитов от
до 100
9
9
х10 /л и даже 200х10 /л;
Резкое увеличение количества лимфоцитов
от 10 до 100 х 109/л или до 80-90% в
лейкоцитарной формуле;
Клетки Боткина-Гумпрехта—
полуразрушенные ядра лимфоцитов;
Нормохромная нормоцитарная анемия;
Ретикулоциты нормальные или повышенные;
Тромбоцитопения;
Увеличение СОЭ.
В норме
При ХЛЛ
•Эритроциты – 3,6х1012/л
•Hb -105 г/л
•ЦВ- 0,875
•Гематокрит – 0,42
•MCV – 85фм
•MCH – 28пг
•MCHC – 32%
•Ретикулоциты – 1,1
•Лейкоциты – 88,2х109/л
•Палочкоядерные – 1%
•Сегментоялерные – 27%
•Эозинофилы –1%
•Базофилы – 0%
•Моноциты – 1%
•Лимфоциты – 70%
•Клетки Боткина-Гумпрехта
•Тромбоциты – 155х109/л
•СОЭ – 27 мм/ч.
Заключение: гиперлейкоцитоз с абсолютным
лимфоцитозом, нормохромная, нормоцитарная
анемия, тромбоцитопения, повышенное СОЭ.
37.
ИммунофенотипированиеВ 95% случаев ХЛЛ имеет В-клеточный иммунологический фенотип с экспрессией
поверхностных В-клеточных антигенов CD 19, CD20, CD24, CD79a и активационных
антигенов CD5, CD23, CD43;
Экспрессия CD5 считается обязательной для иммунологического подтверждения ВХЛЛ;
Однако описаны редкие наблюдения ХЛЛ (7%) с отсутствием CD5 на В-лимфоцитах;
CD23 используют для дифференциальной диагностики В-ХЛЛ и лейкемизации
лимфомы из клеток зоны мантии лимфатического узла, имеющей аналогичный В-ХЛЛ
иммунофенотип, но без экспрессии CD23;
Для ХЛЛ в отличие от нормальных В-лимфоцитов и лимфосарком характерна слабая
экспрессия поверхностных иммуноглобулинов (чаще slgM, реже IgM +IgD с
одинаковыми легкими цепями).
Цитогенетическое исследование
del13q14 выявляется в ~55 % случаев, делеция может быть моно- и биаллельной,
заболевание, как правило, диагностируется на ранней стадии и развивается медленно,
прогноз благоприятный;
трисомия по хромосоме 12 выявляется в ~15 % случаев, прогноз обычный;
del11q выявляется в ~15 % случаев, болезнь диагностируют на более поздних стадиях,
выше вероятность проявления конституциональных симптомов, болезнь быстро
прогрессирует, прогноз неблагоприятный, данная мутация может ассоциироваться с
резистентностью к алкилирующим химиопрепаратам;
del6q21 характеризуется неблагоприятным прогнозом.
38. Лечение по протоколу
ЛЕЧЕНИЕ РАННИХ СТАДИЙ ХЛЛ БЕЗ ПРИЗНАКОВ ПРОГРЕССИРОВАНИЯ (СТАДИИ A И В ПО BINET, СТАДИИ 0-IIПО RAI)
Терапия ранних стадий ХЛЛ не увеличивает выживаемость. Стандартная тактика при ранних стадиях – стратегия
«watchandwait» (англ. наблюдать и ждать). Контрольное клинико-лабораторное обследование с обязательным
исследованием развернутого ОАК должно проводиться каждые 3-6-12 месяцев.
ЛЕЧЕНИЕ ПРОДВИНУТЫХ СТАДИЙ ХЛЛ СТАДИИ A И B ПО BINET С ПРИЗНАКАМИ АКТИВНОСТИ, СТАДИЯ С ПО
BINET; СТАДИИ 0–II ПО RAI С СИМПТОМАМИ, СТАДИИ III–IV ПО RAI
В данной группе у пациентов имеются показания к проведению химиотерапии. У пациентов моложе 70 лет без
сопутствующих заболеваний терапией первой линии являются FCR (Флударабин + Циклофосфамид +
Ритуксимаб), BR (Бендамусти+Ритуксимаб). Пентостатин и кладрибин могут использоваться в качестве терапии
первой линии ХЛЛ, но комбинация FCR является более предпочтительной. Использование Бендамустина в составе
первой линии терапии является менее токсичным вариантом лечения в сравнении с FCR, более эффективным, чем
Хлорамбуцил и может быть рекомендовано при наличии противопоказаний к Флударабину.
У пациентов старше 70 лет и/или с тяжелыми сопутствующими заболеваниями стандартной терапией первой
линии является Хлорамбуцил. Наиболее частыми альтернативами могут быть Бендамустин, монотерапия
Ритуксимабом.
ЛЕЧЕНИЕ РЕЦИДИВОВ И РЕФРАКТЕРНЫХ ВАРИАНТОВ ХЛЛ
При рецидиве ХЛЛ, если он происходит через 12-24 месяца после монотерапии или 24-36 месяцев после
хемоиммунотерапии, может быть повторена терапия первой линии, на которой был достигнут ответ. В случае
рефрактерности к терапии первой линии или рецидиве ранее указанных сроков проводят лечение по одному из
Salvage-режимов (анг. Спасение). Из Salvage-режимов в зависимости от клинической ситуации рекомендуются
следующие:
Алемтузумаб с последующей аллогенной трансплантацией костного мозга у физически сохранных пациентов
молодого возраста, имеющих потенциального донора;
Терапия по схеме FCR у пациентов, получивших в первой линии лечение, базирующееся на алкилирующем агенте;
Бендамустин-содержащий курс для пациентов, нечувствительных к FCR или физически несостоятельных без
del(17p).
Аллогенная трансплантация костного мозга является основным методом лечения при рефрактерности и/или
вариантах с del(17p) и мутациями р53. Аутологичная трансплантация не улучшает результаты в сравнении с
хемоиммунотерапией. Эффективность консолидации или поддерживающей терапии при ХЛЛ недоказана.
39.
Группа пациентовПервая линия терапии
Терапия при рецидиве/ рефрактерности
Пациенты моложе 70 лет и без тяжелых
сопутствующих заболеваний
Хемоиммунотерапия;
Флударабин + Циклофосфамид +
Ритуксимаб (FCR);
Флударабин + Ритуксимаб (FR)
Пентостатин + Циклофосфамид +
Ритуксимаб (PCR);
Бендамустин + Ритуксимаб (BR);
Обинутузумаб + Хлорамбуцил.
Ибрутиниб;
Иделалисиб + Ритуксимаб;
FCR;
PCR;
Бендамустин ± Ритуксимаб;
Флударабин ± Алемтузумаб;
R-CHOP (Циклофосфамид, Доксорубицин,
Винкистрин, Преднизолон);
OFAR (Оксалиплатин,
Флударабин,Цитарабин, Ритуксимаб);
Офатумумаб;
Леналидомид ± ритуксимаб; Алемтузумаб±
ритуксимаб;
Высокие дозы
метилпреднизолона±Ритуксимаб.
Пациенты старше 70 лет, или с
тяжелыми сопутствующими
заболеваниями
Обинутузумаб+ Хлорамбуцил;
Ритуксимаб + Хлорамбуцил;
Бендамустин (70 мг/м2 в 1 цикле с
повышением до 90 мг/м2) +Ритуксимаб
(BR); Циклофосфамид+Преднизолон±
Ритуксимаб;
Флударабин±Ритуксимаб; Кладрибин;
Хлорамбуцил.
Ибрутиниб;
Иделалисиб + ритуксимаб;
FCR с редукцией дозы;
PCR с редукцией дозы;
Бендамустин ± ритуксимаб;
Высокие дозы
метилпреднизолона±Ритуксимаб;
Ритуксимаб + Хлорамбуцил ;
Офатумумаб;
Леналидомид ± ритуксимаб;
Алемтузумаб± ритуксимаб;
40. Основные схемы химиотерапии хронического лимфолейкоза
ПрепаратыРежим введения
Флударабин+Циклофосфамид+Ритуксимаб
(FCR)
1 раз в 4 недели Х 6 курсов
Флударабин
25 мг/м2 в/в 1-3 дни
Циклофосфамид
250 мг/м2 в/в 1-3 дни
Ритуксимаб
375 мг/м2 в/в в 1 день 1го курса, 500 мг/м2
в/в в 1 день 2-6 курсов
Циклофосфамид + Винкристин +
Преднизолон (СVP)
1 раз в 3 недели до 18 месяцев
Циклофосфамид
300 мг/м2 внутрь 1-5 дни
Винкристин
1,4 мг/м2 (max 2 мг) в/в 1 день
Преднизолон
100 мг/м2 внутрь 1-5 дни
Бендамустин+Ритуксимаб (BR)
1 раз в 4 недели Х 6 курсов
Бендамустин
90 мг/м2 в/в в течение 30 мин 1-2 дни 1 раз в
месяц Х 6 курсов
Ритуксимаб
375 мг/м2 в/в в 1 день 1го курса, 500 мг/м2
в/в в 1 день 2-6 курсов.
41. Миеломная болезнь (множественная миелома, генерализованная плазмоцитома, болезнь Рустицкого-Каллера)
ОпределениеПарапротеинемический гемобластоз, характеризующийся злокачественной
опухолевой пролиферацией плазматических клеток одного клона с
гиперпродукцией моноклонального иммуноглобулина или свободных
моноклональных легких цепей иммуноглобулинов.
Эпидемиология
Заболеваемость миеломной болезнью во всем мире примерно одинакова и
составляет 4:100000 в год; с возрастом она растет (после 25 лет - 30:100000).
Негры болеют примерно вдвое чаще белых; мужчины - несколько чаще женщин.
Этиология
Специфические этиологические факторы миеломной болезни неизвестны,
но установлены факторы: ионизирующей радиации в развитии заболевания,
генетическая предрасположенность, цитогенетические нарушения
(гиперэкспрессия с-тус и H-ras онкогенов). Описана мутация супрессорных генов
р53 и Rb-1, хроническая антигенная стимуляции В-лимфоцитов и их
трансформации в плазматические клетки с последующей продукцией парапротеинов,
длительные контакты с нефтепродуктами, бензолом, асбестом.
А.Н.Окороков: Руководство диагностики болезней внутренних
органов. Том 4. Москва. Медицинская литература, 2006 г.
42. Система стадирования множественной миеломы по Durie–SalmonPLUS.
Система стадирования множественной миеломы по Durie–SalmonPLUS.
Стадия
Кальций,
ммоль/л
Гемоглобин, г/л
Уровень М
компонента
Количество очагов деструкции по
данным ЯМРТ/ПЭТ
IА
<2,6
>100
IgG<50 г/л
IgA<30г/л
Белок BJ в
моче<4г/сут
Может быть солитарная
плазмацитома или одиночный
очаг деструкции
IВ
<2,6
>100
0–4
IIA или В
2,6 – 3,0
85 – 100
5 – 20
IIIA или В
≥3,0
<85
IgG>70 г/л
IgA>50г/л
Белок BJ в
моче>12г/су
Более 20 или тяжелое диффузное
поражение костей
Субклассификация: А – сохранная функция почек (уровень креатинина ≤177 мкмоль/л)
В – нарушение функции почек (уровень креатинина ≥177 мкмоль/л)
43. Международная система стадирования множественной миеломы (ISS)
СтадияАльбумин в сыворотке крови, г/л
β2-микроглобулин сыворотки крови
мг/л
I
≥35
<3
II
≥35
3,5-5,5
<35
<3
-
≥5,5
III
«КЛИНИЧЕСКИЙ ПРОТОКОЛ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
МНОЖЕСТВЕННАЯ МИЕЛОМА» Рекомендовано Экспертным советом
РГП на ПХВ «Республиканский центр развития здравоохранения»
Министерства здравоохранения и социального развития Республики
Казахстан от «9» июля 2015 года Протокол № 6
44. Формы множественной миеломы в зависимости от активности заболевания
Формы множественноймиеломы
Критерии
«Тлеющая» (англ.
Smoldering) Синоним:
асимптомная
М-градиент в сыворотке более 30 г/л
И/ИЛИ Клональных плазматических
клеток в костном мозге ≥10% Нет
органной дисфункции, остеолитических
очагов или симптомов
Активная (англ. Active)
Синоним: симптомная
CRAB (см. ниже п.12) или другие
признаки активности заболевания: ALамилоидоз, рецидивирующие инфекции,
синдром повышенной вязкости крови с
клиническими проявлениями
45.
Выделяют несколько вариантов миеломной болезни в зависимости от характера
распространения миеломных инфильтратов в костном мозге, от характера миеломных
клеток и от типа синтезируемого парапротеина.
По характеру распространённости опухолевого инфильтрата в костном
мозге выделяют:
– диффузную,
– диффузно-очаговую,
– очаговую формы миеломы;
По клеточному составу:
– плазмоцитарную,
– плазмобластную,
– полиморфно-клеточную,
– мелкоклеточную миелому (по Струкову А. И.)
В зависимости от способности секретировать различные типы парапротеинов
различают несколько вариантов миеломной болезни:
– несекретирующие,
– диклоновые миеломы,
– миелому Бенс-Джонса,
– G-, A-, D-, M-, E-миеломы.
46.
Клиническая картинаI.Синдром костной патологии:
• Боли
• Опухоли
• Переломы
• Рентгенограмма пораженных костей:
Очаги деструкции (очаговые,
диффузные - от нескольких
миллиметров до 2-5 см и больше).
На рентгенограммах черепа «дырявый череп», «симптом
пробойника».
Очаги деструкции и остеолизиса
выявляются и в других плоских
костях — лопатках, ребрах, тазовых
костях. («пчелиные соты»)
Деструктивно-остеопоротический
процесс в позвонках приводит к их
уплощению, изменению формы
(клиновидная, чечевицеподобная,
«рыбьи» позвонки), компрессионным
переломам.
47.
II.Синдром белковой патологии:Гиперпротеинемия (90-180 г/л);
Снижение содержания в крови нормального γ-глобулина;
Электофорез белков сыворотки крови и мочи: обнаружение моноклонального
иммуноглобулина в крови и белка Бенс-Джонса в моче. Иммунофиксационный
метод позволяет обнаружить наличие белка Бенс-Джонса и расчитать его
количество (в г/л). Белки разделяются с помощью электрофореза, затем
проводится иммунофиксация в присутствии антител к тяжелым и легким цепям
иммуноглобулинов. При связывании белков с антителами образуются иммунные
комплексы, которые можно увидеть после окрашивания. Иммунные комплексы,
содержащие нормальные иммуноглобулины, откладываются в виде широкой
полосы, моноклональные (белок Бенс-Джонса) — в виде более узкой и четко
очерченной. (http://www.analizmarket.ru/tests/id/10040/)
Наличие М-компонента (градиента)
Электрофорез позволяет выявить М-градиент
(полосу моноклонового белка в зоне миграции
глобулинов и снижение фракции вне этой зоны)
Стойкая протеинурия (белок Бенс-Джонса)
Развитие амилоидоза.
48.
III. Синдром поражения почек:В I стадии (доклинической) изменений
клубочков и почечного интерстиция еще
нет, но имеются явления белковой
дистрофии эпителия почечных канальцев.
Во II стадии имеются выраженная
белковая дистрофия и умеренная атрофия
части канальцев; изменения клубочков
минимальные; в моче определяются
белок,
лейкоциты,
эритроциты,
цилиндры.
В III стадии большинство почечных
канальцев
блокировано
белковыми
цилиндрами,
эпителий
канальцев
атрофирован,
имеется
выраженный
склероз интерстиция почек, значительно
уменьшено
количество
клубочков,
отмечается
отложение
кристаллов
кальция в интерстициальной ткани.
Клиническая картина в этой стадии
характеризуется
симптоматикой
прогрессирующей
почечной
недостаточности.
49.
IV. Синдром вторичного иммунодефицита:При миеломной болезни развивается вторичное иммунодеффицитное
состояние вследствие резкого уменьшения продукции нормальных
иммуноглобулинов, чему способствует высокая активность
трансформирующего ростового фактора β. Синдром недостаточности антител
проявляется частыми инфекционными осложнениями со стороны легких и
бронхов, мочевыводящих путей.
V. Синдром висцеральной патологии:
Гепатомегалия
Спленомегалия
Плевриты
Поражение ЖКТ.
VI. Синдром повышенной вязкости крови:
Неврологические симптомы
Нарушения зрения
Нарушение периферического кровотока в руках и ногах с трофическими изменениями кожи вплоть до
акрогангрены в наиболее тяжелых случаях
Геморрагический синдром.
VII. Неврологический синдром:
Периферическая нейропатия
Расстройства по типу корешковому типу
Параплегия
Симптоматика поражения ЧМН
VIII.Гиперкальциемический синдром:
Повышение уровня сывороточного кальция более 2,8 ммоль/л (11,5 мг/дл)
50.
Гемограмма:Пример:
Эитроциты 2,1 х 1012/л
Гемоглобин 70 г/л
ЦП 0,8
MCV 85 фл
MCH 30 пг
MCHC 35 %
Ht 40%
Лейкоциты 2,5 х 109/л
Сегменто-ядерные нейтрофилы 61%
Палочко-ядерные нейтрофилы 6%
Эозинафилы 1%
Моноциты 2%
Базофилы 1%
Лимфоциты 19%
Плазмациты 10%
Тромбоциты 120 х 109/л
СОЭ 73 мм/час
Заключение: нормохромная анемия, средней
степени, лейкопения, плазмациты,
тромбоцитопения, увеличение СОЭ.
Нормоцитарная-нормохромная
анемия
Панцитопения
Плазматические клетки
Стойкое повышение СОЭ
Плазматичсекие
клетки
51.
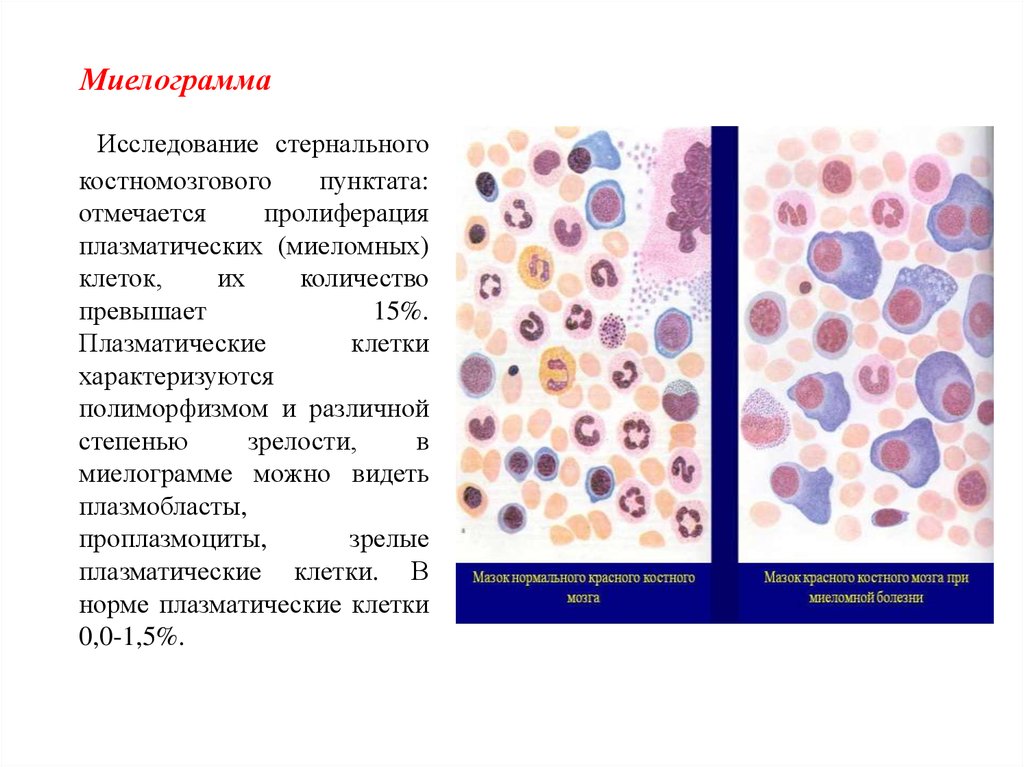
МиелограммаИсследование стернального
костномозгового
пунктата:
отмечается
пролиферация
плазматических (миеломных)
клеток,
их
количество
превышает
15%.
Плазматические
клетки
характеризуются
полиморфизмом и различной
степенью
зрелости,
в
миелограмме можно видеть
плазмобласты,
проплазмоциты,
зрелые
плазматические клетки. В
норме плазматические клетки
0,0-1,5%.
52.
Электрофореграмма белков сыворотки крови выявляет гомогенный пик (Мградиент) с электрофоретической подвижностью в зоне β- или γ-глобулинов.Иммуноэлектрофорез белков крови устанавливает IgM-природу макроглобулина — парапротеина с одним типом легких цепей к или λ, который
составляет 30-70% общего белка крови. Моноклональный IgM можно также
обнаружить с помощью иммунофлюоресцентного метода на поверхности
лимфоцитов.
53.
Цитогенетическое исследование• del 13, del 17p13, t(4;14), t(14;16), амплификация 1q21.
Иммунофенотипирование
• Миеломные клетки имеют следующий иммунофенотип:
CD38+++, CD44+, CD45-/+, CD54+, CD138+, CD56+, CD58+,
CD28-/+, clg, в то время как фенотип плазматических клеток в
норме и при М ГНГ представлен CD38+++, CD44+, CD45-/+,
CD54+, CD138+, CD56-, CD58-, CD28-, clg
«КЛИНИЧЕСКИЙ ПРОТОКОЛ ДИАГНОСТИКИ И ЛЕЧЕНИЯ МНОЖЕСТВЕННОЙ МИЕЛОМЫ»
Рекомендовано Экспертным советом РГП на ПХВ «Республиканский центр развития здравоохранения»
Министерства здравоохранения и социального развития Республики Казахстан от «9» июля 2015 года Протокол
№6
А.Н.Окороков: Руководство диагностики болезней внутренних органов. Том 4. Москва. Медицинская литература,
2006 г.
54.
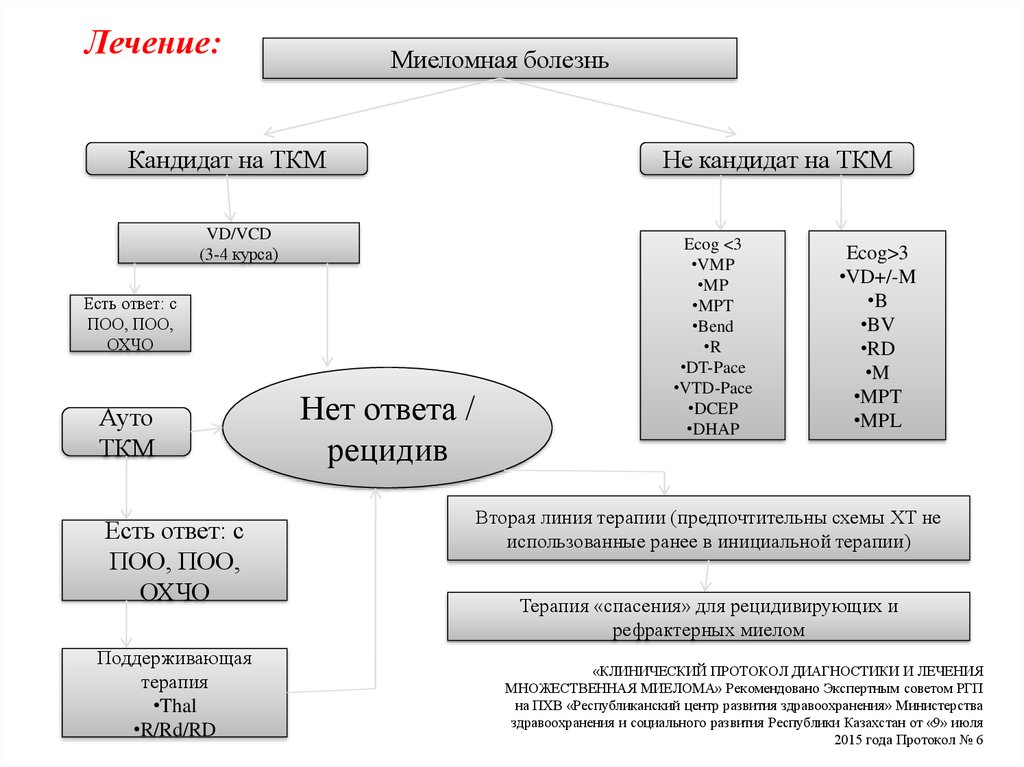
Лечение:Миеломная болезнь
Кандидат на ТКМ
VD/VCD
(3-4 курса)
Есть ответ: с
ПОО, ПОО,
ОХЧО
Ауто
ТКМ
Есть ответ: с
ПОО, ПОО,
ОХЧО
Поддерживающая
терапия
•Thal
•R/Rd/RD
Нет ответа /
рецидив
Не кандидат на ТКМ
Ecog <3
•VMP
•MP
•MPT
•Bend
•R
•DT-Pace
•VTD-Pace
•DCEP
•DHAP
Ecog>3
•VD+/-M
•B
•BV
•RD
•M
•MPT
•MPL
Вторая линия терапии (предпочтительны схемы ХТ не
использованные ранее в инициальной терапии)
Терапия «спасения» для рецидивирующих и
рефрактерных миелом
«КЛИНИЧЕСКИЙ ПРОТОКОЛ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
МНОЖЕСТВЕННАЯ МИЕЛОМА» Рекомендовано Экспертным советом РГП
на ПХВ «Республиканский центр развития здравоохранения» Министерства
здравоохранения и социального развития Республики Казахстан от «9» июля
2015 года Протокол № 6
55.
Первая линия терапии для пациентов являющихся кандидатами на ТКМ
(оценка ответа после 2 курсов ХТ): Бортезомиб/Дексаметазон (Vel/Dex или
VD), Бортезомиб/Циклофосфан/Дексаметазон (CyBorD или VCD),
Бортезомиб/Доксорубицин/Дексаметазо н (PAD)
Первая линия терапии для пациентов НЕ являющихся кандидатами на ТКМ
(оценка ответа после 2 курсов ХТ): Бортезомид/Дексаметазон (Vel/Dex или
VD), Мелфалан/преднизолон (МР), Мелфалан/Преднизолон/Бортезомиб
(МРВ/VMP)
Поддерживающая терапия: Бортезомиб (Bor), Леналидомид (R), Талидомид
(Thal)
Терапия рецидивирующей, рефрактерной миеломы (терапия «спасения»):
Бортезомиб , Карфилзомиб+дексаметазон+/- леналидомид (Car+d+\-R),
Бортезомиб/Талидомид/Дексаметазон (Thal+Dex+Vel) 1q,
Леналидомид/Дексаметазон (RD), Леналидомид/малые дозы дексаметазаона
(Rd), Мелфалан/Преднизолон/Леналидомид (МРL),
Мелфалан/Преднизолон/Талидомид (МРТ)
56.
Прогноз:Около 15% больных погибают в первые 3 мес после постановки диагноза; в
последующем летальность составляет 15% в год. После 2-5 лет хронического течения
развивается резистентная к терапии терминальная стадия, обычно с инфильтрацией
внутренних
органов
миеломными
клетками
и
панцитопенией
без
снижения
клеточности костного мозга. Продолжительность терминальной стадии не превышает 6
мес. Примерно 46% больных не доживают до этой стадии, погибая от роста опухоли
(16%), почечной недостаточности (10%),сепсиса (14%) или сочетания этих причин
(6%). В терминальной стадии погибают 26% больных; причиной смерти обычно
становится рост опухоли (13%) и сепсис (9%). Пять процентов больных умирают
от острого миелобластного лейкоза (или острого монобластного лейкоза); связь лейкоза
с
миеломной
болезнью
сомнительна,
скорее
он
обусловлен
длительным
лечением алкилирующими средствами. Остальные 23% больных погибают от
сопутствующих заболеваний (инфаркта миокарда, ХОЗЛ, сахарного диабета, инсульта),
которые связаны не столько с опухолью, сколько с возрастом.
http://humbio.ru/humbio/har/0038791d.htm